


Abstract
Guanine-rich nucleic acid sequences can fold into four-stranded G-quadruplex (G4) structures. Despite growing evidence for their biological significance, considerable work still needs to be done to detail their cellular occurrence and functions. Herein, we describe an optimized core-extended naphthalene diimide (cex-NDI) to be exploited as a G4 light-up sensor. The sensing mechanism relies on the shift of the aggregate-monomer equilibrium towards the bright monomeric state upon G4 binding. In contrast with the majority of other ligands, this novel cex-NDI is able to discriminate among G4s with different topologies, with a remarkable fluorescent response for the parallel ones. We investigate this sensing by means of biophysical methods, comparing the lead compound to a non-selective analogue. We demonstrate that mitigating the affinity of the binding core for G4s results in an increased selectivity and sensitivity of the fluorescent response. This is achieved by replacing positively charged substituents with diethylene glycol (DEG) side chains. Remarkably, the limit of detection values obtained for parallel G4s are more than one order of magnitude lower than those of the parallel-selective ligand N-methyl mesoporphyrin IX (NMM). Interestingly, the classical fluorescent intercalator displacement (FID) assay failed to reveal binding of cex-NDI to G4 because of the presence a ternary complex (G4-TO-cex-NDI) revealed by electrospray-MS. Our study thus provides a rational basis to design or modify existent scaffolds to redirect the binding preference of G4 ligands.
INTRODUCTION
The last decades were characterized by extensive research on nucleic acids (NA) folded into G-quadruplex (G4) secondary structures (1). These are formed by guanine-rich DNA or RNA single strands, which can assemble into quartets via Hoogsteen hydrogen bonds (2). Such quartets then stack on top of each other constituting the core of the G4 structure. This is further stabilized by monovalent cations sitting in the inner channel (3,4). Despite this common backbone, G4s are highly polymorphic as for molecularity (mono, bi or tetramolecular), topology (parallel, anti-parallel or hybrid), glycosidic bond angle (syn or anti), sugar conformation (ring puckering), loops and grooves size (5,6). Reasons for this widespread interest in G4s reside in mounting evidence of their regulatory roles in key biological processes, such as telomere maintenance, replication, transcription and translation (7–10). Interestingly, many putative G4 sequences (PQS) are located in close proximity to genes involved in a wide range of pathologies. These include cancer (11–13), neurodegenerative diseases (14,15), viral (16,17) and parasitic (18,19) infections. Therefore, G4s hold great potential as novel therapeutic and theranostic targets (20,21).
In order to validate them as such, however, their formation and functions must be proven in cells. In recent years, monoclonal antibodies were engineered to signal G4 formation in cellular models (22–24). However, imaging biases arising from cell fixation and chaperone activity of such proteins cannot be excluded. Among the investigation tools, molecular probes undergoing a fluorescence light-up provide a complementary method to address the issue (25,26). A number of fluorescent small molecules were already reported to specifically emit upon G4 binding (27–30). Structures of such sensors usually mimic the scaffolds of powerful G4 ligands, to ensure high affinity (31–34). Unfortunately, most ligands are unable to discriminate among different topological classes of G4s, not to mention recognize a specific sequence. Specific recognition is nonetheless essential to thoroughly understand G4-related processes. Identifying probes able to recognize specific structural motifs is thus of primary importance and relevant efforts have already been made in this direction (29,35–38).
Herein, we exploit a fluorescence light-up mechanism based on core-extended naphthalene diimides (cex-NDIs). cex-NDIs have high G4 affinity and form non-emitting aggregates under physiological conditions (39,40). Their fluorescence can be restored upon monomerization induced by interaction with the G4. Despite proving the sensing mechanism both in vitro and in fixed cells, the prototype cex-NDIs displayed sub-optimal properties: (i) the aggregation of those compounds is only partial at μM concentration, leading to non-negligible background fluorescence and thus modest sensitivity; (ii) the compounds excitation and emission bands are both placed at the lower edge of Red-NIR (near infrared) spectroscopic window; (iii) finally, little or no selectivity for a peculiar G4 topology was observed.
Herein, we present novel cex-NDI probes designed to overcome these issues. Concerning selectivity among different G4s, we propose a new strategy, counterintuitively based on the reduction of the overall ligand affinity for G4s. In fact, the most potent ligands produce a high and possibly saturated binding response with all structures, regardless of their identity and conformation. We thus propose to remove the less specific interactions resulting from the electrostatic attraction between the phosphate backbone and the protonated amine groups (41). In this way, we would indeed lose some of the affinity, but at the same time we would let the core selectivity emerge. We demonstrate this hypothesis by testing two cex-NDIs with different substituents (Scheme 1), one displaying positively charged dimethyl amine groups (1) and the other neutral DEG pendals (2).
Scheme 1.

4-Substituted cex-NDIs (1 and 2) investigated in the present study and unsubstituted cex-NDI (3) or 11-substituted cex-NDIs (4 and 5), previously investigated as G4 sensors (39,40).
The newly employed functionalization pattern is also aimed at improving the sensing mechanism. In particular, we introduce an additional hydrophilic and electron-donating side chain at position 4 in contrast with previously investigated cex-NDIs (3–5; Scheme 1). On the one hand, this change should produce a significant red-shift in both absorption and emission, as previously observed for NDIs (42). On the other hand, the positioning of the hydrophilic substituents away from the lipophilic core-extension should enhance the aggregation propensity of the core. Moreover, DEG functionalization should likely favour the aggregation. It would in fact maintain charge neutrality at physiological pH, while still ensuring sufficient water solubility.
MATERIALS AND METHODS
General materials and methods
Reagents, solvents and chemicals were purchased from Alfa Aesar or Sigma-Aldrich and were used as supplied without further purification. For in vitro studies 5 × 10−3 M stock solutions of cex-NDI 1 and 2 were prepared in MilliQ water and DMSO respectively and were stored at −20°C. The DNA and RNA oligonucleotides (Supplementary Tables S1–S3) were purchased from Sigma Aldrich or Eurogentec and dissolved in MilliQ water to prepare 5 × 10−4 M stock solutions. The exact concentrations were then determined by absorption measurements at 260 nm. The solutions were stored at −20°C. The NAs were annealed by incubation at 95°C for 5 min, in the presence of the relevant amounts of salt (see specific protocols for details) and of 1 × 10−2 M buffer (pH 7.2, either lithium cacodylate or Tris-HCl). Solutions were then let to equilibrate for at least 2 hours at room temperature. For the fluorophore-labelled oligonucleotides, the heating time was reduced to 2 min and the equilibration was performed in ice for 30 min. The folded samples were then stored at 0°C.
TLC analysis was carried out on silica gel (Merck 60F-254) with visualization at 254 and 366 nm. Reverse phase HPLC analysis was performed using an Agilent system SERIES 1260. The column was XSelectHSS C18 (2.5 Mm) (50 × 4.6 mm) (Waters). Flow was 1.4 ml/min. The following method (Method 1) was used: Aqueous solvent: 0.1% trifluoroacetic acid in water; Organic solvent: Acetonitrile; Gradient: 95% aqueous, gradually to 40% aqueous over 8 min, then isocratic flow for 4 min. Cex-NDI 2 was purified by reverse phase HPLC, specifically an Agilent Technologies 1260 Infinity preparative HPLC provided with a diode array UV–vis detector. The preparative column was XSelect CSH Prep Phenyl-Hexyl 5 μm (150 × 30 mm) (Waters). The flow was 30 ml/min. Purifications were performed through three different methods, (Method 2) Aqueous solvent: 0.1% trifluoroacetic acid in water; Organic solvent: Acetonitrile; Gradient: Isocratic flow over 2 min at 80% of aqueous solvent, gradually to 60% aqueous over 14 min, then isocratic flow for 2 min (λ detection: 254, 360 and 520 nm); (Method 3) Aqueous solvent: 0.1% trifluoroacetic acid in water; Organic solvent: Acetonitrile; Gradient: Isocratic flow over 2 min at 80% of aqueous solvent, gradually to 60% aqueous over 8 min, then isocratic flow for 4 min (λ detection: 590, 254 and 520 nm); (Method 4) Aqueous solvent: 0.1% trifluoroacetic acid in water; Organic solvent: Acetonitrile; Gradient: Isocratic flow over 2 min at 95% of aqueous solvent, gradually to 60% aqueous over 13 min, then isocratic flow for 1 min (λ detection: 630, 254 and 310 nm). 1H-, 13C-NMR spectra were recorded on a Bruker ADVANCE 300 MHz. HRMS were recorded on Agilent G6550A ESI Q-TOF instrument coupled with an UPLC Agilent 1290 Infinity II. UV/Vis spectra were run on an Agilent Cary 60 UV-Vis or a SAFAS spectrophotometer. Emission experiments were run on Cary Eclipse Fluorescence Spectrophotomer and on a JobinYvon Fluorolog-3 (HORIBA) fluorimeter. All spectrometers were equipped with Peltier temperature controllers. Quartz cuvettes of 0.4–10 cm path length were used.
Synthesis of compound 1
Compound 1 was synthesized, as reported in Scheme 2, according to a published procedure (43). Analytical HPLC (method 1) and comparison of NMR data with those available in the literature confirmed the identity and purity of the compound.
Scheme 2.

General synthetic scheme of 4-substituted cex-NDIs: (a) amine, acetic acid as solvent, 120°C, 30 min; (b) 1,2-phenylendiamine, acetonitrile, MW, closed vessel, 110°C, 20 min, 250 psi; (c) amine, N,N-dimethylacetamide, MW, closed vessel, 150°C, 10 min, 250 psi.
Synthesis of compound 2 (Scheme 2)
Compound 6 was synthesized according to a published procedure (44). Compound 8 was obtained heating compound 6 (0.25 mmol) and 2-(2-aminoethoxy)ethanol (1 mmol) in 3 mL of acetic acid at the microwave in a closed vessel (12 min, 110°C, 250 psi). The desired compound 8 was obtained in a mixture with compounds 9 and 10, produced by partial or total dehalogenation of the starting material and product 8. The mixture was used as such for the following steps, verifying its composition by TLC (CHCl3 95: MeOH 5). Acetic acid was stripped away and the residues were removed by filtration on silica (pure chloroform, then 2% MeOH in chloroform). 100 mg of the mixture were then suspended in 3 mL of acetonitrile and 1,2-phenylendiamine (1.25 mmol) was added. The suspension was heated at 110°C for 20 min (MW, closed vessel, 250 psi). The reaction progress was checked at the analytical HPLC (Method 1). The desired product 12 was obtained together with by-product 13, which in turn results from the reaction of the previous by-product 8 with 1,2-phenylendiamine. Compounds 12 and 13 exclusively precipitated from the reaction mixture (purple solid). The products remaining in fine suspension were recovered by filtration. Again, compound 12 was not purified, since by-product 13 did not affect the following step. Finally, the mixture was submitted to nucleophilic aromatic substitution. It was dissolved in 3 mL of 1:1 solution of DMA and 2-(2-aminoethoxy)ethanol and heated at the MW in a closed vessel (10 min, 150°C, 250 psi). The reaction progress was evaluated by analytical HPLC (Method 1). The mixture was diluted in acidic water (0.1% TFA) and purified at the preparative HPLC (Method 4). Possible residues of compound 13 were removed by subsequent column chromatography purification (pure CHCl3, then gradually to 5% MeOH), reaching an overall yield of 7%. All the intermediates (8, 9, 10, 12 and 13) were purified by preparative HPLC to perform a full characterization. The reported yields correspond to these purification steps. 8: purified by preparative HPLC (Method 2). Yellow solid. Yield = 42%. 1H NMR (300 MHz, DMSO-d6), δ = 8.69 (s, 1H); 8.64 (s, 1H); 4.26 (m, 4H); 3.71-3.65 (m, 12 H). 13C NMR (75 MHz, DMSO-d6), δ = 170.2; 162.5; 160.7; 160.5; 137.1; 130.5; 127.5; 126.5; 126.2; 126.1; 126.0; 125.6, 124.0; 72.1; 68.0; 66.7; 66.7; 66.5; 63.0; 60.2. 9: purified by preparative HPLC (Method 2). Yellow solid. Yield = 23%. 1H NMR (300 MHz, DMSO-d6), δ = 8.63 (s, 4H); 4.24 (m, 4H); 3.74–3.62 (m, 12 H). 13C NMR (75 MHz, DMSO-d6), δ = 160.8; 160.7; 131.1; 130.5; 126.5; 125.6, 120.0; 70.0; 65.3; 61.3. 10: purified by preparative HPLC (Method 2). Yellow solid. Yield = 4%. Identity of the compound was verified by NMR analysis. Data were in good agreement with those available in the literature (45). 12: purified by preparative HPLC (Method 3). Blue solid. Yield = 24%. 1H NMR (300 MHz, DMSO-d6), δ = 12.77 (s, 1H); 12.38 (s, 1H); 7.94 (s, 1H); 7.15 (m, 4H); 4.67 (s, 2H); 4.18 (m, 4H); 3.69–3.57 (m, 12 H). The product exhibited too low solubility for 13C NMR acquisition. HRMS m/z calcd. for C28H25BrN4O8 [M+1]+ 625.09, 627.09 Found: 625.0885, 627.0897. 13: purified by preparative HPLC (Method 3). Blue solid. Yield = 48%.1H NMR (300 MHz, DMSO-d6), δ = 12.2 (s, 2H); 7.75 (s, 2H); 7.06 (m, 2H); 6.99 (m, 2H); 4.59 (s, 2H); 4.05 (m, 4H); 3.58 (t, 4H); 3.47 (m, 8H). 13C NMR (75 MHz, DMSO-d6), δ = 164.5; 161.9; 141.7; 127.2; 126.1; 125.2; 124.3; 121.2; 117.0; 95.1; 75.3; 66.9; 60.3. 2: purified by preparative HPLC (Method 4). Green solid. Yield: 82%. 1H NMR (300 MHz, DMSO-d6), δ = 11.8 (s, 1H); 11.0 (s, 1H); 8.83 (s, 1H); 6.71 (m, 2H); 6.66 (s, 1H); 6.43 (m, 2H); 4.62 (s, 3H); 3.86 (m, 4H); 3.68–3.46 (m, 18H). The product exhibited too low solubility for 13C NMR acquisition. HRMS m/z calcd. for C32H35N5O10 [M+1]+ 650.24, Found: 650.2419.
Aggregation studies
Solvent dependence
Compounds solutions were prepared diluting the appropriate amount of the stock solutions in the various solvents. Temperature dependent measurements were performed by gradually heating the solution and recording the absorption spectrum after each step. Concentration dependent measurements were performed adding aliquots of 5 × 10−4 or 5 × 10−3 M stock solutions to the tested ones. When recording spectra in the micelles, solution at three cex-NDI concentrations (5, 10, 20 × 10−6 M) were prepared containing 2 × 10−2 M SDS. When recording the spectra at variable organic solvent – water compositions, solutions in the pure solvents already at the final concentrations were mixed in the appropriate percentages.
Aggregation constant calculation
Compound 1 was dissolved in buffered water (Tris–HCl, pH 7.2) at a concentration of 5 × 10−8 M and its absorption spectrum was measured using a 10 cm path length cuvette. The concentration was then gradually increased, recording the spectrum after each addition of aliquots of a 1 × 10−3 M solution of the same compound. The same was done for compound 2 in buffered water and then in 1:1 mixture of water and methanol. Molar absorptivity data in the maxima were plotted as a function of the concentration and fitted according to a isodesmic model with GraphPad 7.0 (Prism). The data are presented as an average of three independent replicates.
Ionic strength dependence
1 × 10−5 M solutions of compounds 1 and 2 were prepared in water (1.0 × 10−2 M lithium cacodylate buffer, pH 7.2) and aliquots of 1 M MCl (M = Li, K, Na) were added, varying the salt concentration from 0 to 2.0 × 10−1 M. Absorption spectra were recorded after each addition (T = 25°C). Spectra were corrected for the dilution.
Temperature dependence
2 × 10−5 M solutions of compounds 1 and 2 were prepared in water (1.0 × 10−2 M lithium cacodylate buffer, pH 7.2, 0.1 M KCl) and their absorption spectra were recorded raising the temperature from 25 to 95°C, with 5°C steps (1 min equilibration time).
pH dependence
1 × 10−5 M solutions of compound 2 were prepared in buffered water (1.0 × 10−2 M buffer, either phosphoric acid-sodium dihydrogenphosphate, or sodium dihydrogenphosphate-sodium monohydrogenphosphate, or sodium monohydrogenphosphate-sodium phosphate) and their absorption was subsequently recorded (T = 25°C). Compound 1 was instead dissolved in phosphoric acid-sodium dihydrogenphosphate buffered solution (pH 3.5, 1 × 10−3 M buffer, 1 × 10−2 M NaCl) and the pH was then gradually increased up to 12 upon addition of 0.1 M NaOH. The absorption spectrum was recorded after each addition of base. Normalized absorption data in the two maxima (617 and 670 nm) were plotted as a function of pH. The presented results are the average of three replicates.
FRET melting
FRET melting experiments were run on a Stratagene Mx3005P real-time PCR equipment in 96 wells plates, on the DNA sequences reported in Supplementary Table S2. Experiments were performed in 1 × 10−2 M lithium cacodylate buffer (pH 7.2) and either 1 × 10−2 M KCl and 9 × 10−2 M LiCl (F21T, F32KRAST, F21CTAT, FBom17T, FTBAT, FdxT) or 1 × 10−3 M KCl and 9.9 × 10−2 M LiCl (FcmycT, F25CebT) concentrations, depending on the Tm of the G4s alone. The DNA concentration was 2 × 10−7 M. The stabilisation (ΔTm) induced by compounds 1 and 2 was calculated as the difference between the mid-transition temperature of the nucleic acid (NA) alone and measured with the relevant ligand concentration (2.5 × 10−7 M for compound 1, 5 × 10−6 M for compound 2). Data are presented as an average of three independent measurements, each conducted in duplicate conditions (λexc = 492 nm, λem = 516 nm, T interval = 25–95°C, ramp: 25°C for 5 min, then 1°C/min, measurements every 1°C, 8x magnification of the fluorescence signal). The G4 selectivity of the ligands with respect to duplex and single stranded DNA was evaluated adding to the mixture increasing amounts of the competitors (Supplementary Table S3) and measuring the new ΔTm.
Thiazole orange displacement assay (46)
Fluorescence displacement assay was run on a TECAN plate reader, Infinite M1000 PRO, in 384 wells plates. Thiazole orange (TO) displacement was performed adding increasing amounts of ligands 1 or 2 to the pre-folded NA (2.5 × 10−7 M) – TO mixture (5 × 10−7 M). Ligands concentration was incremented from 0 to 2.5 × 10−6 M, corresponding to 10 molar equivalents with respect to the NA. Experiments were performed in 1 × 10−2 M lithium cacodylate buffer (pH 7.2) and 0.1 M KCl. The fluorescence spectra were measured for each ligand addition between 500 and 800 nm on 4 × 10−5 L of solution (λexc = 485 nm, bandwidth = 5 nm, z position and gain were adapted for each specific NA-TO complex, T = 25°C). The % of displacement was calculated as 100 × (1 – F/F0), where F is the TO fluorescence in the maximum (535 nm) and F0 is the initial fluorescence. Data are presented as an average of three independent replicates, all conducted in duplicate conditions.
Mass spectrometry experiments
Electrospray ionization mass spectrometry (ESI-MS) experiments were performed on a Thermo- Exactive Orbitrap mass spectrometer in the negative ion mode, equipped with a standard ESI source. Samples were injected at 3.5 μl/min by a syringe pump. The full scan mass range was [200–4000]. The Exactive was tuned to soft conditions using the bimolecular quadruplex d[G4T4G4]2 in 0.1 M ammonium acetate (47). G4 solutions were prepared in 0.1 M trimethyl ammonium acetate (TMAA), pH 7.0, and 1 × 10−3 M KCl. The nucleic acids were analysed at a concentration of 5 × 10−6 M, adding the relevant amounts of compounds 1 or 2 and/or thiazole orange. The mixture was supplemented with 10% of methanol just before measurement. Data were analysed using Xcalibur 2.2.0 software (Thermo Scientific).
In vitro sensing studies
1 × 10−3 L of 4 × 10−6 M solutions of compounds 1 and 2 were titrated with 1 × 10−4 M solutions of the relevant nucleic acid, after annealing (0.1 M KCl, 0.01 M Tris–HCl buffer, pH 7.2). All measurements were performed at 25°C. Absorption or emission (λexc = 650 nm) spectra were recorded after each NA aliquot addition (NA concentration: 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, 2.0, 2.4, 2.8, 3.2, 3.6, 4.0, 4.8, 5.6, 6.4, 7.2, 8.0 × 10−6 M). They were then corrected for the dilution. Data are presented as an average of three independent replicates.
Fitting
Fitting was performed according to the following equation (48), using GraphPad 7.0:
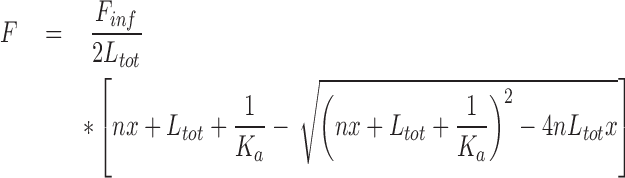 |
where F is the normalized fluorescence response, Finf is the fluorescence at infinite G4 concentration, Ltot is the analytical ligand concentration, n is the number of binding sites and Ka is the affinity constant per site (Table 1). The equation thus optimizes both the binding stoichiometry and the binding constant based on the supplied dataset. Such fitting relies on the assumption, usually met with G4s, that all binding sites display similar affinity and thus they are involved as first binding sites according to a statistical distribution.
Table 1.
Affinity constant values (Ka /M−1) obtained from the fitting of the data presented in Supplementary Figure S16. n is the number of binding sites and Ka is the affinity constant per site.
| cex-NDI 2 | cex-NDI 1 | |||
|---|---|---|---|---|
| NA | Ka (M−1) | n | K a (M−1) | n |
| 25Ceb | 5.8 ± 0.5 × 106 | 1 | a | a |
| c-myc | 4.1 ± 0.5 × 106 | 1 | a | a |
| 32KRAS | 1.0 ± 0.1 × 105 | 1 | a | a |
| 22AG | 2.20 ± 0.50 × 104 | 1 | 2.5 ± 0.3 × 106 | 2 |
| ds26 | 2.84 ± 0.06 × 103 | 1 | 3.2 ± 0.8 × 106 | 2 |
a) Data not yielding a reliable fitting with our method.
Fluorescence quantum yield measurements
Fluorescence quantum yield values for the compounds alone or upon G4 complexation were obtained using rhodamine 800 as primary reference [φ = 0.25 ± 0.03 in ethanol (49)]. Emission was measured exciting the compounds at three different excitation wavelengths (600, 605 and 610 nm) and integrating the resulting spectrum between 630 and 900 nm. Φ values were calculated applying the following formula:
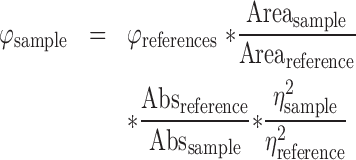 |
where Area is the area subtended by the curve, Abs is the absorption value at the excitation wavelength and η is the refractive index of the solution. All data are presented as a mean of the values obtained at the three wavelengths and are the average of three replicates. Residual φ values in water were measured at a 3 × 10−6 M concentration of cex-NDIs. cex-NDI-G4 complex φ was measured after the addition of three molar equivalents of the relevant G4 (25Ceb, c-myc or 22AG).
LOD measurements
Limit of detection (LOD) / sensitivity assay was run on a TECAN plate reader, Infinite M1000 PRO, in 384 wells plates. Solutions containing equimolar amounts of the relevant G4 (25Ceb or c-myc) and compound (2 or NMM porphyrin) at decreasing concentration were obtained by subsequent dilution of a 4 × 10−6 M one. Blank was measured on a 4 × 10−6 M solution of either compound 2 or NMM alone. All solutions contained 1 × 10−2 M lithium cacodylate buffer (pH 7.2) and 0.1 M KCl. Experiments on 2 were carried out exciting at 650 nm for 2 and at 400 nm for NMM and collecting the emission respectively in the 670–900 nm and 500–800 nm intervals. Sensitivity was assessed by plotting the fluorescence in the maxima (695 and 600 nm respectively) as a function of the concentration and calculating the LOD according to the following formula (50):
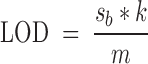 |
where sb is the standard deviation calculated out of 20 independent measurements on blank solutions; k is 3, according to IUPAC recommendations; m is the curve slope, obtained from data linear fitting. Data are presented as an average of three independent replicates, each conducted in duplicate conditions.
Circular dichroism studies
CD experiments were run on a Jasco J-1500 spectropolarimeter, equipped with a Peltier temperature controller. Titrations were performed on 3 × 10−6 M solutions of the relevant NAs (0.1 M KCl, 0.01 M lithium cacodylate buffer, pH 7.2) upon addition of aliquots of a 5 × 10−4 M solution of compound 1 or compound 2. Parameters: scan speed 50 nm/min, three acquisitions, bandwidth 2 nm, integration time 1 s, T = 25°C.
Native gel electrophoresis
Twelve percent polyacrylamide gels were prepared diluting 40% acrylamide-bisacrylamide (19:1) with water and adding the relevant amounts of KCl 100×, TBE 5× (Tris-Borate, EDTA buffer), APS (ammonium persulfate) solution and TEMED (tetramethylenediamine). The solution was poured between the glass slides and let polymerize (0.4 cm thickness, 20 wells comb). Samples were prepared diluting the appropriate amount of 5 × 10−5 M pre-folded NA solutions and compounds stock solutions in buffer (0.01 M lithium cacodylate, pH 7.2, and 0.1 M KCl). 20 μl of each sample were supplemented with 6 μl of sucrose solution (50% m/v) and loaded. A ladder was prepared containing coloured markers (0.1% xylene cyanol and 0.1% bromphenol blue) and 5 × 10−6 M single stranded NAs (dT9, dT15, dT21, dT30, dT57). The gels were run at 4 W (110 V, 37 mA) for 2 h or until the front marker dye had reached the lower end of the gel. The gels were then extracted and imaged with a Typhoon Trio variable-mode imager (GE Healthcare) (λexc = 633 nm, emission: 670 nm filter, PMT = 600, 200 μm resolution, + 3 mm focal plane). Subsequently they were exposed to SYBR Gold staining solution (1:10 000 stock solution dilution in water, containing 1 × 10−3 M lithium cacodylate buffer, pH 7.2, 40 min exposure), washed twice and reimaged (λexc = 473 nm, emission: 530 nm filter, PMT = 600 V, 200 microns resolution, + 3 mm focal plane).
RESULTS AND DISCUSSION
Synthesis and characterization of the aggregation process
Compound 1 was synthesized as already reported (43), via imidation of 6 with N,N-dimethylpropandiamine, yielding 7, subsequent nucleophilic aromatic substitution (SNAr) with 1,2-phenylendiamine, ring closure and final SNAr with N,N-dimethylpropanediamine at position 4 (Scheme 2). The same synthetic scheme was applied for compound 2, replacing N,N-dimethylpropanediamine with 2-(2-aminoethoxy)ethanol. The conditions, mostly for purification, were however adapted to the increased hydrophobicity of compound 2 and intermediates. In fact, compound 8 could not simply be recovered, unlike 7, by acetic acid neutralization and extraction, since this latter step was inefficient. We thus decided not to isolate compound 8, using the crude for the following step. Similarly, the poor solubility of intermediate 12 in both water and organic solvents hampered its purification. This was instead achieved in good yields for 11 by reverse phase HPLC. Nevertheless, such low solubility made possible the co-precipitation of a 50:50/12:13 mixture, leaving the by-products in the acetonitrile solution. Finally, the last SNAr step, proved to be quantitative for both compounds 11 and 12. However, compound 2 had to be purified by HPLC to eliminate the aforementioned by-product 13.
To assess self-aggregation, we next evaluated the spectroscopic characteristics of 1 and 2 as a function of solvent, concentration and temperature. Both cex-NDIs displayed an intense charge transfer (CT) band with maxima at 595 and 647 nm in methanol (Figure 1A and B), consistently red-shifted in comparison to the spectra of compounds 3–5 (Figure 1C) (39,40). We assigned the CT band observed in methanol to the monomer of cex-NDI 1, as its shape was not affected by compound concentration and temperature (Supplementary Figure S1A and B). In contrast, compound 2 displayed evidence of aggregation in methanol as its absorptivity decreased and the band broadened upon concentration increase or cooling (Supplementary Figure S1C and D). We thus ran the same experiments in DMSO, a highly disaggregating solvent. Effectively, cex-NDI 2 was present mostly in the monomeric form under these conditions (Supplementary Figure S2).
Figure 1.

Absorption and emission (λexc = 620 nm in A and B, 550 nm in C) of A) compound 1, (B) compound 2 and (C) compound 3 in water (pH 7.2, 0.01 M lithium cacodylate buffer) and methanol. Absorption spectra were recorded at 2 × 10-5 M, while emission spectra at 5 × 10-6 M concentration.
Moreover, the absorption bands for both 1 and 2 have a significantly different shape and lower molar absorptivity in buffered aqueous solvent than in organic media. Such bands were assigned to the H-aggregates. In addition, the absorption behaviour reflected in high fluorescence in organic solvent and negligible emission in water (Figure 1A and B, dashed line). More specifically, in aqueous solvent, emission of compound 1 was low but still detectable, whereas that of compound 2 was almost completely quenched. On the contrary, emission was restored in methanol, resulting in 0.25 ± 0.01 and 0.33 ± 0.01 fluorescent quantum yields (φ) for cex-NDIs 1 and 2, respectively. The lower value obtained for compound 1 might be due to partial quenching via back-electron transfer to the amine moieties.
The residual φ in buffered water (pH 7.2) were 0.017 ± 0.002 and 0.0051 ± 0.0006 for cex-NDIs 1 and 2, respectively. This, together with the broader band shape observed in absorption for cex-NDI 2 likely indicates tighter aggregation. In order to quantify this difference, we measured aggregation constants for the two compounds in buffered water (10−2 M Tris–HCl buffer, pH 7.2). For cex-NDI 1 we were able to measure an aggregation constant of 9.8(6) × 105 M−1 (number in parenthesis indicates uncertainty in the measurement; Supplementary Figure S3A and B). However, this was not possible for cex-NDI 2, as the aggregate was the only species observed even at the lower detection limit (Supplementary Figure S3C). We thus measured this latter aggregation constant in a 1:1 mixture of water and methanol. The measured constant [7.1(5) × 105 M−1] was comparable to that of cex-NDI 1 in neat water (Supplementary Figure S3D and E).
Finally, we ran experiments detailing the aggregation process dependence on the various environmental conditions, including solvent composition, ionic strength, temperature and pH. Concerning the solvent composition, the disaggregation process was observed in both absorption and emission when increasing the organic solvent content in the mixture with water (Supplementary Figure S4). In fact, the spectrum of the monomer reappeared for both compounds upon addition of MeOH to the mixture and so did the emission band. Interestingly, a much higher amount of organic solvent was required to obtain effective monomerization of compound 2 with respect to 1, confirming its tighter aggregation. Finally, the monomer spectrum could be observed in water for both compounds in the presence of SDS micelles (Supplementary Figure S5).
When testing compound 1, the environmental conditions significantly affected the equilibrium position in water. In particular, the aggregate was favoured upon increase the ionic strength and pH (Figures S6 and S7), while the monomer emerged upon heating in neat water (Supplementary Figure S8). Among these, pH proved to be the most influential factor, as aggregation was massively triggered by amine deprotonation. Plotting the normalized molar absorptivity coefficient data as a function of pH (Supplementary Figure S7B) revealed a mid-transition pH of 8.3. Smaller changes were observed for compound 2, when varying the ionic strength or the temperature (Figures S9 and S10), and pH variation did not prove meaningful (Supplementary Figure S11). Once again, this is due to the higher aggregation propensity of 2, reducing the effect of external factors.
G4 affinity
We first evaluated cex-NDIs 1 and 2 affinities for G4s by FRET melting assays on a small panel of fluorescently labelled NAs (Supplementary Table S2). This high-throughput test quantifies the stabilization imparted by the ligand to the NA secondary structure, by comparing its melting alone and upon complexation (51).
When testing compound 1, we observed an overall high stabilization for G4s, even at compound concentration as low as 2.5 × 10−7 M (Figure 2A). In more detail, telomeric F21T was the most stabilized G4, with a ΔTm of 19.4°C. Lower ΔTm values (∼10°C) were observed with FmycT, F25CebT, F21CTAT and F32KRAST. Finally, FTBAT and F17BomT displayed the smallest ΔTm values, between 4 and 5°C. Hairpin duplex DNA FdxT did not show any increase in stabilization at this compound concentration, although measurable stabilizations were found with higher ligand quantities (data not shown). In order to obtain comparable ΔTm values with compound 2 we had to increase its concentration to 5 × 10−6 M, corresponding to 20-fold the amount of cex-NDI 1 (Figure 2A). This confirms our preliminary hypothesis that the replacement of protonable amine groups by uncharged DEG moieties reduces the overall ligand affinity for the G4. Besides, the radar plot for cex-NDI 2 gives interesting information on its selectivity for G4s over duplex DNA. In fact, no stabilization was observed for FdxT even at such a high ligand concentration. To get a clearer picture on G4/ds selectivity, we performed FRET competition experiments in the presence of the duplex competitor ds26 (0, 3 and 10 μM). Interestingly, whereas compound 1 underwent a dramatic drop in stabilization for all analysed G4s already at low competitor concentration, compound 2 did not (Figure 2B and C). This held true also at higher competitor concentration (5 × 10−5 M) and with different duplex and single strand competitors (Supplementary Table S3, Supplementary Figure S12). Only when confronted with single stranded DNA, compound 1 did not show any decrease in stabilization activity (Supplementary Figure S12). These results are particularly important, because they confirm that positive charges on compound 1 enhance the affinity for NAs, but in a highly unspecific manner.
Figure 2.

(A) Results of FRET melting assays on a panel of labelled NAs (2 × 10−7 M, Supplementary Table S2), in the presence of 1 (2.5 × 10−7 M) or 2 (5 × 10−6 M); lithium cacodylate buffer 1 × 10−2 M, pH 7.2, KCl 1 × 10−2 M and LiCl 9 × 10−2 M (FTBAT, FBom17T, F21T, F21CTAT, F32KRAST) or KCl 1 × 10−3 M and LiCl 9.9 × 10−2 M (F25CebT, FmycT); (B, C) results of FRET melting assays carried out in the same conditions but adding increasing amounts of ds26 competitor (0, 3, 10 μM) with ligands 1 (B) or 2 (C).
An additional assay addressing ligands affinity for G4s is fluorescent intercalator displacement assay (G4-FID), in which the ligand complexation drives the displacement of thiazole orange (TO), a fluorescent probe (46). Affinity of the competing ligand can be assessed by monitoring the changes in TO emission. When tested by G4-FID assay, compound 1 proved capable of displacing TO from all NAs, including ds26, although to different extents (Figure 3A). In fact, more than 95% of displacement was obtained only with parallel G4s 25Ceb, c-myc and 32KRAS and with the hybrid 22AG. The displacement was complete upon addition of only two molar equivalents (eq.) of 1 for the parallel G4s, whereas around five eq. were required for 22AG, suggesting a lower affinity. Concerning anti-parallel G4s (21CTA and Bom17) and ds26, displacement could not be higher than 80%. The curve shapes suggest a less efficient competition with TO for their binding sites, particularly when considering 21CTA. The ability to displace TO from ds26 highlights once again the lack of selectivity for G4s over duplex DNA, as already observed in FRET melting assays.
Figure 3.

FID assays of (A) cex-NDI 1 and (B) cex-NDI 2 on a panel of G4s (c-myc, 25Ceb, 32KRAS, 22AG, Bom 17, 21CTA) and on control ds26 (pH 7.2, 1 × 10−2 M lithium cacodylate buffer, 1 × 10−1 M KCl, T = 25°C). Dotted lines indicate 50% displacement. Curves in A are only a guide for the eye. (C) Native MS analysis of a Pu24, TO, cex-NDI 2 mixture (concentrations 5, 10, 50 μM respectively, 0.1 M TMAA, pH 7, 1 mM KCl). All charge states shown are 5-. The annotated peaks correspond to complexes with 2 specific K+ ions bound. Asterisks indicate extra non-specific K+ adducts.
Surprisingly, compound 2 was completely unable to displace TO even at high concentrations (Figure 3B), although high ΔTm values were found in FRET-melting experiments. We reasoned that the absence of displacement might be due to the simultaneous binding of TO and cex-NDI. To verify this hypothesis, we analysed mixtures of Pu24 G4, TO and 2 by native mass spectrometry, at fixed stoichiometric ratios resembling FID conditions (Supplementary Figure S13 for FID). When analysing 5 μM Pu24 in the presence of 2 eq. of TO (10 μM) and increasing concentrations of 2 (0, 10, 25 and 50 μM; Supplementary Figure S14), we detected G4 complexes with the cex-NDI or TO individually, and also relatively abundant ternary complexes: [Pu24·2·TO]5− and [Pu24·(2)2·TO]5− (highlighted in green in Figure 3C), with the latter becoming more abundant at higher concentrations of 2. These complexes were observed at charge states 5- (Figure 3C), 4- and 6- (Supplementary Figure S14). The native mass spectrometry experiment illustrates that negative FID results do not always mean that a compound does not interact with the nucleic acid target: lack of TO displacement by compound 2 may simply result from binding to two different sites on the G-quadruplex. In this case, FID gave a false negative result regarding the binding capabilities of 2.
G4 fluorescent sensing
After evaluation of G4 affinity and fluorescent light up upon monomerization for both 1 and 2, we investigated their G4 sensing activity. We thus tested both cex-NDIs on a panel of NA sequences in fluorescence titration experiments. This NA list includes three parallel G4s (25Ceb, c-myc and 32KRAS), three anti-parallel ones (Bom17, TBA 15 and hRAS1), a hybrid G4 (22AG in K+), plus a single strand (ss SCR) and a duplex (ds26) as controls. Both compounds had their fluorescence restored upon interaction with the G4 NAs, such as 25Ceb (Figure 4A and B). We confirmed by spectrophotometric titrations with the same G4 that this was due to monomerization induced by G4 binding (Figure 4C and D). Such process induced changes in the absorption spectra, producing monomer-like bands. These were significantly redshifted with respect to those of the monomer alone (675 and 617 nm, compared to 661 and 607 nm in SDS). However, remarkable differences were observed for the two compounds, when analysing the whole NA panel. In fact, cex-NDI 1 displayed a moderate emission light-up (up to 14-fold) at 687 nm after a modest initial quenching, upon gradual NA addition (Figure 5A). Moreover, a robust turn-on effect was also detected in the presence of duplex and single-strand controls and no real selectivity was observed for any G4 topology. In contrast, cex-NDI 2, displayed much higher fluorescence light-up (up to 250-fold). This was selectively observed for parallel G4s, compared to the moderate responses to other G4s (hybrid and anti-parallel) or the negligible ones to the controls (Figure 5B). Fluorescent quantum yields values (φ) were calculated for the complexes of the two cex-NDIs with G4s. In particular, in the presence of three eq. of 25Ceb and c-myc, 2 displayed φ = 0.090 ± 0.005 and 0.084 ± 0.005, respectively, whereas only 0.032 ± 0.004 with 22AG. The latter confirms the incomplete complexation by the telomeric G4 even when present in large excess. 1 exhibited comparable φ regardless to the G4 topology [0.095 ± 0.002 with 25Ceb, 0.086 ± 0.003 with c-myc and 0.099 ± 0.005 in the presence of 22AG].
Figure 4.

Fluorometric (A and B) and spectrophotometric (C and D) titrations of 4 × 10−6 M solutions of (A, C) cex-NDI 1 and (B, D) cex-NDI 2 (1 × 10−2 M Tris–HCl buffer, pH 7.2, 1 × 10−1 M KCl) with 25Ceb G4 (from 0 M in black to 8 × 10−6 M in red, start and end curves of the titrations are highlighted in bold, see in vitro sensing studies subsection for 25Ceb concentration details).
Figure 5.

Normalized fluorescence in the maximum (685-95 nm), calculated from fluorescence titrations data of 4 × 10−6 M water solutions of (A) cex-NDI 1, B) cex-NDI 2 (pH 7.2, 1 × 10−2 M Tris–HCl, 1 × 10−1 M KCl, T = 25°C) with a panel of NAs, c = 0–8 × 10−6 M (c-myc, 25Ceb, 32KRAS, 22AG, hRAS1, Bom17, TBA 15, ss SCR, ds26).
In order to probe the selectivity of 2 for parallel G4s, we decided to expand the NA panel, adding five parallel DNA G4s (vav1, VEGF, c-kit2, c-kit1, bcl2) and two parallel RNA G4s (TERRA and NRAS). Compound 2 displayed a remarkable light-up with all of these sequences, although to variable levels (from 85-fold with c-kit1 to 260-fold with 25Ceb, Supplementary Figure S15). More specifically, 25Ceb, c-myc and bcl2 proved to be the highest responding G4s. ckit-2, VEGF, 32KRAS, NRAS, TERRA and Vav1 gave slightly lower light-up factors and ckit-1 was the least responding one, although still higher than any other non-parallel G4s. Similar effects were found with RNA and DNA parallel G4s, suggesting that the parallel topology itself is the key for this selective fluorescent turn on. Statistical analysis of the enhancement factors in the presence of 2 eq. of NAs revealed that the average response recorded for parallel G4s is significantly different from those observed with non-parallel G4s and controls (P < 0.001, Supplementary Table S4). This preference was also highlighted by apparent binding constants (Supplementary Figure S16, Table 1). In fact, Ka values for the most responsive G4s c-myc [4.1(5) × 106 M−1] and 25Ceb [5.8(5) × 106 M−1] were two and three orders of magnitude higher than those for 22AG and ds26, respectively [2.20(1) × 104 M−1 and 2.38(6) × 103 M−1]. Interestingly, they were also more than one order of magnitude higher than the Ka calculated for 32KRAS [1.0(1) × 105 M−1]. Moreover, comparison of the fits for the two cex-NDIs revealed comparable affinity of cex-NDI 1 for two binding sites (1 G4: 2 ligands), in contrast with cex-NDI 2 (1 G4: 1 ligand). Finally, cex-NDI 1 bound both 22AG and ds26 with Ka values [2.5(3) × 106 M−1 and 3.2(5) × 106 M−1, respectively] considerably higher than those of cex-NDI 2. Such values were comparable in magnitude to those calculated for cex-NDI 2 with the most responsive G4s 25Ceb and c-myc.
Finally, we addressed the sensitivity of compound 2 in parallel G4s signalling by limit of detection (LOD) measurement. We measured the fluorescence of solutions containing equimolar amounts of ligand 2 and parallel G4s 25Ceb and c-myc over a range of concentrations. We did the same with N-methyl mesoporphyrin IX (NMM), which is a renowned selective ligand for parallel G4s (52,53). Interpolation of the resulting curves revealed that cex-NDI 2 has LOD values more than one order of magnitude lower than NMM (Table 2, Supplementary Figure S17). LOD values in the low nanomolar range were also reported for green-emitting triaryl-imidazole based ligands, selective for parallel G4s (54). Because of this high sensitivity, together with the considerably more red-shifted excitation and emission wavelengths necessary for the cex-NDI, our new ligand provides a significant improvement of the available parallel G4 probes.
Table 2.
LOD values for 25Ceb and c-myc sensing by 2 and NMM, measured by fluorescence emission analysis with a plate reader
| Probe | 25Ceb | c-myc |
|---|---|---|
| 2 | 10 nM | 8 nM |
| NMM | 120 nM | 104 nM |
Binding selectivity or sensing selectivity?
Next, we tried to prove whether this selectivity of compound 2 observed for parallel G4s really concerned the binding event. Alternatively, it could arise from the formation of non-fluorescent complexes with non-parallel quadruplexes and thus be limited to the sensing. We started by qualitatively assessing the complexation by an emission-independent method. We thus monitored the CD signature of some selected G4s upon gradual addition of the two ligands. Even in this case, the two compounds displayed quite different behaviours. In fact, compound 1 strongly affected the signatures of all the tested NAs, while compound 2 affected only those of parallel G4s. For example, both cex-NDIs induced a small decrease in the absolute intensities of both the negative and positive maxima for c-myc, respectively at 240 nm and 265 nm (Figure 6A and B), maintaining however the overall shape of the signal. On the other side, only cex-NDI 1 induced significant changes on 22AG signature (Figure 6C and D). In fact, the 264 and 295 nm positive maxima intensities increased considerably upon complexation. Contextually, the whole band was slightly blue-shifted and the positive 250 nm maximum disappeared. Similar considerations can be made about the other NAs (Supplementary Figure S18), highlighting once again cex-NDI 2 selectivity for parallel G4s.
Figure 6.

Circular dichroism spectra of 3 × 10−6 M solutions (pH 7.2, 0.01 M lithium cacodylate buffer, 0.1 M KCl, T = 25°C) of (A, B) c-myc; (C, D) 22AG in the presence of 0–9 × 10−6 M cex-NDI 1 (A and C) or cex-NDI 2 (B and D), corresponding to 0–3 molar equivalents. Red: 0 eq; green: 0.5 eq; blue: 1 eq; yellow: 1.5 eq; light blue: 2 eq; purple: 3 eq.
Additional proofs of such binding preference were obtained by native gel electrophoresis. Complexes formed in solutions containing equimolar amounts of selected NAs and ligand 1 or 2 were let to migrate in non-denaturing conditions, so as to assess their stabilities. We tested a wide panel of NAs containing G4s of different topologies (parallel 25Ceb and c-myc, anti-parallel TBA 15 and hybrid 22AG), an RNA parallel G4 (TERRA), single- and double-stranded controls (DNA: ss37, ds26, RNA: SL2, mir122). Interestingly, only complexes of 2 with parallel G4s, either DNA or RNA, could effectively be observed (Figure 7A, see Supplementary Figure S19A for SYBR Gold staining). Instead, none could be visualized with G4s having other topologies, although some complex was shown to be formed at least with 22AG by fluorescence titrations. The same outcome was observed with control NAs. This suggested that only the complexes with the parallel G4s are stable enough to survive during migration in a native gel. On the other hand, compound 1 was found to stain all NAs, except TBA (Figure 7B; see Supplementary Figure S19B for SYBR Gold staining), providing a positive control for the selectivity of the DEGylated analogue 2.
Figure 7.

Native gel electrophoretic analysis of solutions containing either the NA alone or the NA with 1 equivalent of cex-NDI 2 (A) or 1 (B) (NA: 30 pmol, compound: 0–30 pmol, 12% polyacrylamide, 18°C, 2 h, 150 V). (A) cex-NDI 2 fluorescence (λexc = 633 nm; λem = 670 nm); (B) cex-NDI 1 fluorescence (λexc = 633 nm; λem = 670 nm). Green: parallel G4s; these are the only fluorescent bands revealed with 2 on the left gel. blue: non-parallel G4s, red: controls. L = ladder and fluorescent markers.
The modest solubility of ligand 2 and to lower extent of ligand 1, at concentration much lower than mM, prevented us from performing 1D-NMR titrations in the presence of parallel G4s. Therefore, in order to provide an additional and conclusive proof of 2 binding selectivity to parallel G4s over other topologies, we monitored competition experiments by mass spectrometry. We chose Pu24, a parallel G4 resembling c-myc, and 26TTA, a hybrid one related to 22AG, which are well characterized with respect to their behaviour in native MS conditions (55). Moreover, they display sufficient differences in mass to clearly visualize and identify all free and complexed species. Fluorescence (Supplementary Figure S20) and CD titrations (Supplementary Figure S21) confirmed that they behave as expected in the usual conditions, as well as in trimethylammonium acetate (TMAA) buffer + 1 mM KCl, used for MS experiments. A 1:1 mixture of Pu24 and 26TTA (5 μM each) was analysed in the presence of increasing amounts of compound 2 (0, 1, 25, 50 μM; Figure 8). When zooming on the 5- peaks, we detect the selective formation of the 1:1 and 1:2 complexes of NDI 2 with the parallel Pu24 G4, while the 1:1 complexes with hybrid 26TTA appeared only at high cex-NDI concentration. Concurrently, the signal of free Pu24 considerably decreases compared to that of free 26TTA, as the complexes with Pu24 increase. This is in line with the different extent of involvement in the binding. As a control, we ran the same experiment on compound 1 (Supplementary Figure S22). In that case, complexes with both G4s were observed from the very first ligand aliquot addition, suggesting a comparable affinity for both G4s and confirming the reliability of the methodology. We were thus able to confirm that cex-NDI 2 selectivity extends beyond G4 sensing, being an actual binding preference.
Figure 8.

Mass spectra of a 1:1 mixture of Pu24 and 26TTA G4s (5 × 10−6 M each, 1 × 10−1 M TMAA, pH 7, 1 × 10−3 M KCl) in the presence of increasing amounts of cex-NDI 2 (0–5 × 10−5 M, corresponding to 0, 2, 5 and 10 molar equivalents).
CONCLUSION
In this work, we presented two core-extended naphthalene diimides with a novel functionalization pattern, which we exploited as G4 fluorescent sensors. Such rational design improved the sensing effectiveness by affecting the ligand binding selectivity. Sensing was greatly enhanced through the increased self-aggregation and the shift of the compounds emission in the red-NIR. The most remarkable result is the relevant selectivity for parallel G4s versus other conformations obtained for compound 2, achieved by DEG functionalization. Polyethylene glycol (PEG) preferential interaction with parallel G4 structures was previously reported for the telomeric sequence in K+ rich buffer, for which a structural shift from hybrid to parallel topology was observed (56). Initially presented as a result of molecular crowding or dehydration (56,57), the effect was later associated to specific CH-π and lone pair-π interactions of the PEG chains with the G-quartets and loops (58,59). The observed binding preference could rely on similar effects.
Several complementary techniques were used to obtain a consistent view on the different selectivity of compounds 1 and 2. All methods converged to the same conclusion, with the notable exception of FID, for which no competition could be found for 2. This false negative result was explained by native mass spectrometry experiments, which unambiguously demonstrate that the same quadruplex may simultaneously accommodate TO and one or two 2. Finally, compound 2 affinity and turn-on properties allow it to reveal the presence of parallel G-quadruplexes with limits of detection in the low (8-10) nM range.
Achieving this level of G4 selectivity is still rare among G4 sensors and ligands. Our study provides a rational basis to design or modify existent scaffolds to redirect the binding preference. We demonstrated that reducing the unspecific electrostatic interactions was key to let the core (and possibly DEG-substituents) selectivity emerge. Interestingly, the compound NMM, exhibiting a clear preference for parallel structures, does not bear any positive charges, and is actually negatively charged at neutral pH. Our results thus argue against the use of cationic chains in order to increase affinity towards parallel G4 structures. We anticipate that the outlined strategy could be more widely applied to the design of novel G4 ligands and optimization of existent ones, beyond naphthalene diimides. We envisage this would provide remarkable benefits to the G4 field. Differentiated and selective targeting by small fluorescent probes would certainly improve the understanding of G4s biological roles.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council [ERC Consolidator grant 615879 to M.F.]; Italian Association for Cancer Research [14708 to M.F.]; European Regional Development Fund [SYMBIT project reg. no. CZ.02.1.01/0.0/0.0/15_003/0000477 to J.L.M.]. Funding for open access charge: ERC Consolidator [615879].
Conflict of interest statement. None declared
REFERENCES
- 1. Hänsel-Hertsch R., Di Antonio M., Balasubramanian S.. DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat. Rev. Mol. Cell. Biol. 2017; 18:279–284. [DOI] [PubMed] [Google Scholar]
- 2. Huppert J.L. Four-stranded nucleic acids: structure, function and targeting of G-quadruplexes. Chem. Soc. Rev. 2008; 37:1375–1384. [DOI] [PubMed] [Google Scholar]
- 3. Largy E., Mergny J.-L., Gabelica V.. Sigel A, Sigel H, Sigel RKO. The Alkali Metal Ions: Their Role for Life. 2016; Cham: Springer International Publishing; 203–258. [Google Scholar]
- 4. Bhattacharyya D., Mirihana Arachchilage G., Basu S.. Metal cations in G-Quadruplex folding and stability. Front. Chem. 2016; 4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Phan A.T., Luu K.N., Patel D.J.. Different loop arrangements of intramolecular human telomeric (3+1) G-quadruplexes in K+ solution. Nucleic Acids Res. 2006; 34:5715–5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S.. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006; 34:5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rhodes D., Lipps H.J.. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015; 43:8627–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu Y. Chemistry in human telomere biology: structure, function and targeting of telomere DNA/RNA. Chem. Soc. Rev. 2011; 40:2719–2740. [DOI] [PubMed] [Google Scholar]
- 9. Bugaut A., Balasubramanian S.. 5′-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res. 2012; 40:4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dexheimer T.S., Sun D., Hurley L.H.. Deconvoluting the structural and drug-recognition complexity of the G-quadruplex-forming region upstream of the bcl-2 P1 promoter. J. Am. Chem. Soc. 2006; 128:5404–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brooks T.A., Hurley L.H.. Targeting MYC expression through G-quadruplexes. Genes Cancer. 2010; 1:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moye A.L., Porter K.C., Cohen S.B., Phan T., Zyner K.G., Sasaki N., Lovrecz G.O., Beck J.L., Bryan T.M.. Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun. 2015; 6:7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Balasubramanian S., Hurley L.H., Neidle S.. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy. Nat. Rev. Drug Discov. 2011; 10:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dai J., Liu Z.-Q., Wang X.-Q., Lin J., Yao P.-F., Huang S.-L., Ou T.-M., Tan J.-H., Li D., Gu L.-Q. et al. Discovery of small molecules for Up-Regulating the translation of antiamyloidogenic secretase, a disintegrin and metalloproteinase 10 (ADAM10), by binding to the G-Quadruplex-Forming sequence in the 5′ untranslated region (UTR) of its mRNA. J. Med. Chem. 2015; 58:3875–3891. [DOI] [PubMed] [Google Scholar]
- 15. Zhou B., Liu C., Geng Y., Zhu G.. Topology of a G-quadruplex DNA formed by C9orf72 hexanucleotide repeats associated with ALS and FTD. Sci. Rep. 2015; 5:16673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amrane S., Kerkour A., Bedrat A., Vialet B., Andreola M.-L., Mergny J.-L.. Topology of a DNA G-Quadruplex structure formed in the HIV-1 Promoter: a potential target for anti-HIV drug development. J. Am. Chem. Soc. 2014; 136:5249–5252. [DOI] [PubMed] [Google Scholar]
- 17. Artusi S., Nadai M., Perrone R., Biasolo M.A., Palu G., Flamand L., Calistri A., Richter S.N.. The herpes simplex Virus-1 genome contains multiple clusters of repeated G-quadruplex: Implications for the antiviral activity of a G-quadruplex ligand. Antiviral Res. 2015; 118:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Belmonte-Reche E., Martínez-García M., Guédin A., Zuffo M., Arévalo-Ruiz M., Doria F., Campos-Salinas J., Maynadier M., López-Rubio J.J., Freccero M. et al. G-Quadruplex identification in the genome of protozoan parasites points to naphthalene diimide ligands as new antiparasitic agents. J. Med. Chem. 2018; 61:1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smargiasso N., Gabelica V., Damblon C., Rosu F., De Pauw E., Teulade-Fichou M.-P., Rowe J.A., Claessens A.. Putative DNA G-quadruplex formation within the promoters of Plasmodium falciparum var genes. BMC Genomics. 2009; 10:362–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Balasubramanian S., Hurley L.H., Neidle S.. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy. Nat. Rev. Drug Discov. 2011; 10:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Collie G.W., Parkinson G.N.. The application of DNA and RNA G-quadruplexes to therapeutic medicines. Chem. Soc. Rev. 2011; 40:5867–5892. [DOI] [PubMed] [Google Scholar]
- 22. Biffi G., Antonio, M. Tannahill, D., Balasubramanian S.. Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat. Chem. 2014; 6:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Biffi G., Tannahill D., McCafferty J., Balasubramanian S.. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013; 5:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu H.-Y., Zhao Q., Zhang T.-P., Wu Y., Xiong Y.-X., Wang S.-K., Ge Y.-L., He J.-H., Lv P., Ou T.-M. et al. Conformation selective antibody enables genome profiling and leads to discovery of parallel G-Quadruplex in human telomeres. Cell Chem. Biol. 2016; 23:1261–1270. [DOI] [PubMed] [Google Scholar]
- 25. Largy E., Granzhan A., Hamon F., Verga D., Teulade-Fichou M.-P.. Chaires JB, Graves D. Quadruplex Nucleic Acids. 2013; Berlin, Heidelberg: Springer; 111–177. [Google Scholar]
- 26. Vummidi B.R., Alzeer J., Luedtke N.W.. Fluorescent probes for G-quadruplex structures. ChemBioChem. 2013; 14:540–558. [DOI] [PubMed] [Google Scholar]
- 27. Bhasikuttan A.C., Mohanty J.. Targeting G-quadruplex structures with extrinsic fluorogenic dyes: promising fluorescence sensors. Chem. Commun. 2015; 51:7581–7597. [DOI] [PubMed] [Google Scholar]
- 28. Faverie A., Guédin A., Bedrat A., Yatsunyk L.A., Mergny J.-L.. Thioflavin T as a fluorescence light-up probe for G4 formation. Nucleic Acids Res. 2014; 42:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grande V., Doria F., Freccero M., Würthner F.. An aggregating amphiphilic squaraine: a light-up probe that discriminates parallel G-Quadruplexes. Angew. Chem. Int. Ed. Engl. 2017; 56:7520–7524. [DOI] [PubMed] [Google Scholar]
- 30. Laguerre A., Wong J.M., Monchaud D.. Direct visualization of both DNA and RNA quadruplexes in human cells via an uncommon spectroscopic method. Sci. Rep. 2016; 6:32141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doria F., Oppi A., Manoli F., Botti S., Kandoth N., Grande V., Manet I., Freccero M.. A naphthalene diimide dyad for fluorescence switch-on detection of G-quadruplexes. Chem. Commun. 2015; 51:9105–9108. [DOI] [PubMed] [Google Scholar]
- 32. Zuffo M., Doria F., Spalluto V., Ladame S., Freccero M.. Red/NIR G-quadruplex sensing, harvesting blue light by a coumarin–naphthalene diimide dyad. Chem.– Eur. J. 2015; 21:17596–17600. [DOI] [PubMed] [Google Scholar]
- 33. Arthanari H., Basu S., Kawano T.L., Bolton P.H.. Fluorescent dyes specific for quadruplex DNA. Nucleic Acids Res. 1998; 26:3724–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang P., De Cian A., Teulade-Fichou M.-P., Mergny J.-L., Monchaud D.. Engineering bisquinolinium/thiazole orange conjugates for fluorescent sensing of G-Quadruplex DNA. Angew. Chem. Int. Ed. Engl. 2009; 48:2188–2191. [DOI] [PubMed] [Google Scholar]
- 35. Zhang L., Er J.C., Li X., Heng J.J., Samanta A., Chang Y.-T., Lee C.-L.K.. Development of fluorescent probes specific for parallel-stranded G-quadruplexes by a library approach. Chem. Commun. 2015; 51:7386–7389. [DOI] [PubMed] [Google Scholar]
- 36. Hu M.-H., Chen S.-B., Wang Y.-Q., Zeng Y.-M., Ou T.-M., Li D., Gu L.-Q., Huang Z.-S., Tan J.-H.. Accurate high-throughput identification of parallel G-quadruplex topology by a new tetraaryl-substituted imidazole. Biosens. Bioelectron. 2016; 83:77–84. [DOI] [PubMed] [Google Scholar]
- 37. Jin B., Zhang X., Zheng W., Liu X., Zhou J., Zhang N., Wang F., Shangguan D.. Dicyanomethylene-functionalized squaraine as a highly selective probe for parallel G-quadruplexes. Anal. Chem. 2014; 86:7063–7070. [DOI] [PubMed] [Google Scholar]
- 38. Zhang L., Er J.C., Ghosh K.K., Chung W.J., Yoo J., Xu W., Zhao W., Phan A.T., Chang Y.-T.. Discovery of a structural-element specific G-quadruplex “light-up” probe. Sci. Rep. 2014; 4:3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zuffo M., Doria F., Botti S., Bergamaschi G., Freccero M.. G-quadruplex fluorescence sensing by core-extended naphthalene diimides. Biochim. Biophys. Acta (BBA) - Gen. Sub. 2017; 1861:1303–1311. [DOI] [PubMed] [Google Scholar]
- 40. Doria F., Nadai M., Zuffo M., Perrone R., Freccero M., Richter S.N.. A red-NIR fluorescent dye detecting nuclear DNA G-quadruplexes: in vitro analysis and cell imaging. Chem. Commun. 2017; 53:2268–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cuenca F., Greciano O., Gunaratnam M., Haider S., Munnur D., Nanjunda R., Wilson W.D., Neidle S.. Tri- and tetra-substituted naphthalene diimides as potent G-quadruplex ligands. Bioorg. Med. Chem. Lett. 2008; 18:1668–1673. [DOI] [PubMed] [Google Scholar]
- 42. Sakai N., Mareda J., Vauthey E., Matile S.. Core-substituted naphthalenediimides. Chem. Commun. 2010; 46:4225–4237. [DOI] [PubMed] [Google Scholar]
- 43. Perrone R., Doria F., Butovskaya E., Frasson I., Botti S., Scalabrin M., Lago S., Grande V., Nadai M., Freccero M. et al. Synthesis, binding and antiviral properties of potent Core-extended naphthalene diimides targeting the HIV-1 long terminal repeat promoter G-Quadruplexes. J. Med. Chem. 2015; 58:9638–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doria F., Di Antonio, M., Benotti M., Verga D., Freccero M. Substituted heterocyclic naphthalene diimides with unexpected acidity. Synthesis, properties, and reactivity. J. Org. Chem. 2009; 74:8616–8625. [DOI] [PubMed] [Google Scholar]
- 45. Bevers S., O’Dea P.T., McLaughlin L.W.. Perylene- and naphthalene-Based linkers for duplex and triplex stabilization. J. Am. Chem. Soc. 1998; 120:11004–11005. [Google Scholar]
- 46. Monchaud D., Allain C., Bertrand H., Smargiasso N., Rosu F., Gabelica V., De Cian A., Mergny J.L., Teulade-Fichou M.P.. Ligands playing musical chairs with G-quadruplex DNA: a rapid and simple displacement assay for identifying selective G-quadruplex binders. Biochimie. 2008; 90:1207–1223. [DOI] [PubMed] [Google Scholar]
- 47. Marchand A., Gabelica V.. Native electrospray mass spectrometry of DNA G-quadruplexes in potassium solution. J. Am. Soc. Mass Spectrom. 2014; 25:1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stootman F.H., Fisher D.M., Rodger A., Aldrich-Wright J.R.. Improved curve fitting procedures to determine equilibrium binding constants. Analyst. 2006; 131:1145–1151. [DOI] [PubMed] [Google Scholar]
- 49. Alessi A., Salvalaggio M., Ruzzon G.. Rhodamine 800 as reference substance for fluorescence quantum yield measurements in deep red emission range. J. Lumin. 2013; 134:385–389. [Google Scholar]
- 50. Hu M.-H., Chen S.-B., Guo R.-J., Ou T.-M., Huang Z.-S., Tan J.-H.. Development of a highly sensitive fluorescent light-up probe for G-quadruplexes. Analyst. 2015; 140:4616–4625. [DOI] [PubMed] [Google Scholar]
- 51. De Rache A., Mergny J.-L.. Assessment of selectivity of G-quadruplex ligands via an optimised FRET melting assay. Biochimie. 2015; 115:194–202. [DOI] [PubMed] [Google Scholar]
- 52. Nicoludis J.M., Barrett S.P., Mergny J.-L., Yatsunyk L.A.. Interaction of human telomeric DNA with N-methyl mesoporphyrin IX. Nucleic Acids Res. 2012; 40:5432–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sabharwal N.C., Savikhin V., Turek‐Herman J.R., Nicoludis J.M., Szalai V.A., Yatsunyk L.A.. N‐methylmesoporphyrin IX fluorescence as a reporter of strand orientation in guanine quadruplexes. FEBS J. 2014; 281:1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hu M.H., Chen X., Chen S.B., Ou T.M., Yao M., Gu L.Q., Huang Z.S., Tan J.H.. A new application of click chemistry in situ: development of fluorescent probe for specific G-quadruplex topology. Sci. Rep. 2015; 5:17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marchand A., Gabelica V.. Folding and misfolding pathways of G-quadruplex DNA. Nucleic Acids Res. 2016; 44:10999–11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xue Y., Kan Z.-Y., Wang Q., Yao Y., Liu J., Hao Y.-H., Tan Z.. Human telomeric DNA forms parallel-stranded intramolecular G-quadruplex in K+ solution under molecular crowding condition. J. Am. Chem. Soc. 2007; 129:11185–11191. [DOI] [PubMed] [Google Scholar]
- 57. Miyoshi D., Sugimoto N.. Molecular crowding effects on structure and stability of DNA. Biochimie. 2008; 90:1040–1051. [DOI] [PubMed] [Google Scholar]
- 58. Buscaglia R., Miller M.C., Dean W.L., Gray R.D., Lane A.N., Trent J.O., Chaires J.B.. Polyethylene glycol binding alters human telomere G-quadruplex structure by conformational selection. Nucleic Acids Res. 2013; 41:7934–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tateishi-Karimata H., Ohyama T., Muraoka T., Podbevsek P., Wawro A.M., Tanaka S., Nakano S.I., Kinbara K., Plavec J., Sugimoto N.. Newly characterized interaction stabilizes DNA structure: oligoethylene glycols stabilize G-quadruplexes CH-pi interactions. Nucleic Acids Res. 2017; 45:7021–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.