

Abstract
Sigma receptors are a fascinating receptor protein class whose ligands are actually under clinical evaluation for the modulation of opioid analgesia and their use as positron emission tomography radiotracers. In particular, peculiar biological and therapeutic functions are associated with the sigma-2 (σ2) receptor. The σ2 receptor ligands determine tumor cell death through apoptotic and non-apoptotic pathways, and the overexpression of σ2 receptors in several tumor cell lines has been well documented, with significantly higher levels in proliferating tumor cells compared to quiescent ones. This acknowledged feature has found practical application in the development of cancer cell tracers and for ligand-targeting therapy. In this context, the development of new ligands that target the σ2 receptors is beneficial for those diseases in which this protein is involved. In this paper, we conducted a search of new potential σ2 receptor ligands among a database of 1517 “small” marine natural products constructed by the union of the Seaweed Metabolite and the Chemical Entities of Biological Interest (ChEBI) Databases. The structures were passed through two filters that were constituted by our developed two-dimensional (2D) and three-dimensional Quantitative Structure-Activity Relationship (3D-QSAR) statistical models, and successively docked upon a σ2 receptor homology model that we built according to the FASTA sequence of the σ2/TMEM97 (SGMR2_HUMAN) receptor.
Keywords: virtual screening, database marine products, sigma-2 receptor, sigma-2 receptor ligands
1. Introduction
Sigma (σ) receptors include a particular pharmacologically defined family of membrane-bound receptors that bind compounds belonging to a variety of structural classes. Discovered in 1976 and recognized in two distinct subtypes in the early 1990s, they represent a potential target for the diagnosis and therapy of cancer and central nervous system (CNS) diseases [1,2]. Indeed, both σ receptor subtypes are highly expressed in several tissues and distinguished from each other through their affinity for different ligands and biological profiles.
The sigma-1 (σ1) receptor is a 25.3-kDa chaperone protein that resides in the endoplasmic reticulum–mitochondrion interface as well as in nuclear and plasma membranes [3,4,5], firstly cloned from Guinea pig liver cells [6] (Uni-ProtID Q60492, Gene names SIGMAR1, CHEMBL4153). The human σ1 receptor was characterized by X-ray crystallography and the crystal structures in complex with two ligands have been reported (PDB ID 5HK1 and 5HK2) [7]. Several synthetic small molecules with different structures bind with high affinity and selectivity to the σ1 receptor. The σ1 receptor agonists have shown neuroprotective, antiamnestic, and antidepressant effects; in contrast, the σ1 receptor antagonists possess modulatory effects on opioid analgesia, as well as antiproliferative and antiangiogenic activities [8,9,10,11,12,13,14].
The sigma-2 (σ2) receptor is an enigmatic protein that has attracted significant attention due to its involvement in several diseases, such as cancer and neurological disorders. However, despite the increasingly apparent medical importance of the σ2 receptor, its biological role has been stymied since the gene that encodes the receptor has never been identified, and a crystal structure of the receptor has never been released. The σ2 receptor has been reported to have a molecular weight between 18–21.5 kDa. For a long time, it has been contemplated as part of the progesterone receptor membrane component one (PGRMC-1), which is a heme-binding protein that is involved in cell survival and apoptosis [15]. Recently, it has been purified, revealing its identity as the Transmembrane Protein 97 (TMEM97), which is an endoplasmic reticulum-resident transmembrane protein that regulates the sterol transporter Niemann-Pick disease protein (NPC-1), which is involved in regulating intracellular Ca2+ concentration [2,11,16,17].
The σ2/Tmem97 ligands can produce a transient rise in intracellular Ca2+ levels [17] or overcome Ca2+ influx in the presence of an inducer [18]. The σ2 receptor ligands determine tumor cell death through apoptotic and non-apoptotic pathways [2,16]. The apoptotic processes mediated by the σ2 receptor include cell cycle arrest in the G1 phase, which seems to be induced by an expression of cyclinD and CDK2 proteins, as well as by a reduction in intracellular ATP levels [19]. Meanwhile, non-apoptotic mechanisms may include DNA fragmentation, lysosomal leakage, and oxidative stress [20,21,22]. Overexpressed in several tumor cell lines, the σ2 receptor ligands are actually under clinical evaluation as positron emission tomography (PET) radiotracers and indicated for the ligand-targeting therapy and as fluorescence imaging agents [23,24,25,26,27,28]. In this context, the development of new ligands that target the σ2 receptor may be particularly beneficial; on the other hand, few selective ligands have been found for the σ2 receptor, and in some cases, their finding occurred through an accidental discovery [26].
The spreading of computer science, and in particular the possibility of consulting hundreds of chemical databases, gives the opportunity to find novel compounds that are able to bind a specific receptor, enabling a rational investigation. This task may be carried out by means of virtual screening, which helps the end user in filtering many compounds based on virtual model specifications. In absence of virtual methods that are able to evenly handle an unlimited amount of compounds, this task must be manually accomplished by medicinal chemists, which has obvious limitations [29]. On these grounds and motivated by our ongoing interest in developing new compounds that selectively target the σ2 receptor, we have recently developed a two-dimensional (2D) [30,31] and a three-dimensional Quantitative Structure–Activity Relationship (3D-QSAR) model [32], which was built using the whole set of selective σ2 receptor ligands as retrieved from the σ2 receptor selective ligand database (S2RSLDB) (http://www.researchdsf.unict.it/S2RSLDB) [33].
Since structure and ligand-based computer-aided drug design are nowadays effective and useful tools in rational drug design [34,35,36,37,38,39], we used them to allow the identification of new virtually potent and selective molecules that are able to interact with the σ2 receptor. Therefore, herein, we report an investigation of new potentially σ2/TMEM97 receptor ligands among a database of 1517 “small” marine natural products, here named the Blue DataBase (BDB, Table S1), which was composed by the merging of the Seaweed Metabolite (http://www.swmd.co.in/) [40], the Chemical Entities of Biological Interest (ChEBI, http://www.ebi.ac.uk/chebi/) [41] databases, and from the reference [42]. The chemical structures were passed through two filters constituted by our developed 2D and 3D-QSAR statistical models that showed high statistical quality and robust predictive potential capability, and successively docked upon the σ2/TMEM97 receptor homology model. To the best of our knowledge, this is the first report on the build of a 3D structure of the σ2/TMEM97, by mixing the classic homology modeling approach with the evolutionary coupling analysis. Moreover, the robustness of this model has been confirmed by docking on it 200 σ2-ligands that were randomly selected from the S2RSLDB. Finally, the results of the two filters and docking have been merged, ordering them by the mean of the obtained pKi (−log10Ki) to draw up a list of the 15 best candidates (mean of 2D, 3D-QSAR, and docking pKi results) as possible powerful σ2 receptor marine ligands. Four of them are already known in the literature for their antiproliferative and cytotoxic effects against A549 and HT29 cancer cell lines, which are two typical cancer cell lines characterized by σ2 receptor overexpression. In particular, compound 848 resembles progesterone, which in itself is a potent σ2 receptor ligand.
2. Results and Discussion
2.1. 2D Ligand-Based Filter
The BDB was filtered through an in-house hybrid σ2 receptor affinity filter [30], returning for each chemical entity a predicted σ2 receptor pKi. This 2D-QSAR model [30,31] has been built by using a Monte Carlo-based QSAR analysis employing the software CORAL (version 2016, http://www.insilico.eu/coral/index.html) [43,44]. CORAL allows for a hybrid representation of molecular structures that includes a simplified molecular input line entry system (SMILES) and a molecular graph. Hybrid representation using SMILES together with the molecular graph may give better models with higher statistical quality with respect to those models, and with a unique representation of the molecular structure [45,46]. The here-used 2D-QSAR model, being constructed with a large and structurally diverse set of 548 compounds, allows for a prediction of different populations of chemical compounds endpoints (σ2 receptor pKi).
The chemical structures of the 1517 “blue” compounds were transformed into SMILES and converted into freebase, while those compounds presenting a carboxylate were left as is. Eventually, the aromatic rings were converted into the kekulé forms. These conversions have been performed in order to generate SMILES with the same depiction as those used for building the hybrid σ2 receptor affinity filter, since this model works by chopping the SMILES or the molecular graphs into small fragments, and iteratively overmatching them with the SMILES and molecular graphs fragments that compose the training set [30,31].
Among 1517 compounds, 1313 have been defined by the model to be outliers, which means that their chemical structures were expressed as SMILES or a molecular graph that was not described by the model, entirely or for a significant part. Indeed, for these compounds, the type of SMILES or molecular graphs fragments generated do not overmatch, entirely or for a significant part, with the SMILES or molecular graphs fragments that compose the training set. For these molecules, the software still predicts an endpoint, but it highlights them for not falling in the field of applicability, and thus being outliers. The remaining 204 “blue” compounds were returned with a predicted endpoint, and were indicated as falling within the domain of applicability. From this subset, 42 compounds have been predicted to possess a σ2 receptor, Ki, which was higher or equal to 100 nM (pKi ≥ 7), and is empirically considered an appropriate value for processing a compound into more complex phenotypic assays. These compounds are reported in Table S2.
2.2. 3D Ligand-Based Filter
The same dataset of compounds was also evaluated using a second ligand-based filter. All of the 3D structures of the compounds were aligned to our previously published 3D-QSAR model for the σ2 receptor. The alignment of the molecules in the 3D-QSAR pharmacophore model was performed with Forge (v10.4.2, Cresset, New Cambridge House, Hertfordshire, UK) [47]. Once aligned, the compounds were scored assuming that if the fields (the local extrema of the electrostatic, van der Waals, and hydrophobic potentials of the molecule) of the newly evaluated molecules were very similar to those of the original compounds, the resulting compounds will have similar biological properties [47]. The 15 most potent compounds resulted from the 3D-ligand based filter are reported in Table 1, while the full set of compounds is present in Table S2. The selected compounds resulted in an excellent distance to the model (i.e., description by the model), which means that all or most of the features in the molecules were present in the training set of the 3D-QSAR model, and hence that the predicted activity is reliable. In the full set of evaluated compounds, there are also several compounds that are not well described by the QSAR equation; the external user should pay attention to the “3D Applicability” column in the supplementary material (Table S2). Values of “Excellent” or “OK” indicate the predictive reliability. Worse values (i.e., “Bad” or “Poor”) indicate that the molecule has field points in places that are not described by the equation, resulting in unreliable predicted activities.
Table 1.
Structure and predicted pKi values of the 15 most potent marine products resulted from the three-dimensional Quantitative Structure-Activity Relationship (3D-QSAR) filter. BDB: Blue DataBase.
| BDB ID | Structure | Predicted pKi |
|---|---|---|
| 621 |
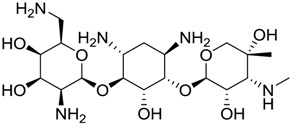
|
10.9 |
| 1612 |

|
9 |
| 848 |

|
8.6 |
| 670 |

|
8.3 |
| 1703 |
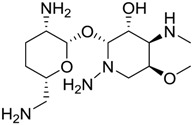
|
8.3 |
| 1144 |
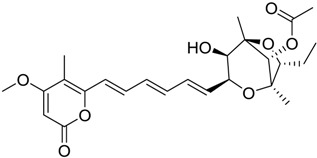
|
8.3 |
| 1342 |

|
8.3 |
| 55 |

|
8.2 |
| 972 |

|
8.2 |
| 933 |

|
8.1 |
| 1426 |

|
8.1 |
| 120 |

|
8.1 |
| 414 |

|
8.1 |
| 1453 |

|
8 |
| 84 |

|
8 |
2.3. Homology Model and Molecular Docking
To further reinforce the results obtained through the 2D and 3D filters, we decided to add a third filter based on molecular docking. Exploiting the recent identification of the σ2 receptor as the TMEM97, we built its 3D molecular structure by the homology modeling approach, starting from the Q5BJF2 (SGMR2_HUMAN) sequence deposited in the UniProt Knowledgebase (https://www.uniprot.org/uniprot/). To pursue this challenging task, due to the scarcity of crystallized structures possessing the sufficient sequence identity (>30%), we have chosen to employ an approach based on two strategies used in parallel. Indeed, this task has been developed by mixing the classical first three steps (i. finding the homologous template proteins of the known structure, ii. selecting the best template or set of templates, and iii. optimizing the multiple sequence alignment between the query and template protein sequences) [48] with the evolutionary coupling analysis [49].
Eventually, we performed the typical fourth step, which consisted of the build of the homology model for the query sequence that resembles the structures of the templates as closely as possible, accommodating for the deletions and insertions of query residues with respect to the template structures, in order to obtain a series of hybrid models that have been ordered by their overall z-score [50].
The obtained 3D model was docked with PB-28, which is a known σ2 receptor ligand (exp. Ki = 2.0 nM [51]); the best-obtained complex was then immersed in a simulated endoplasmic reticulum membrane, in physiological environment conditions, and subjected to a molecular dynamics (MD) simulation of 10 ns to accommodate the ligand, verified by the Root-mean-square deviation (RMSD) of the ligand. After the minimization of the frame with the best binding energy (belonging to the last 3 ns of MD simulation, where the ligand RMSD is constant), the re-docking of the ligand gave a calculated Ki of 1.5 nM, which was in excellent agreement with the experimental value. To additionally validate the 3D model, we docked 200 σ2 receptor ligands that were randomly selected from the S2RSLDB among those that possess a Ki in the range 0.01–1000 nM. The results (Table S3) are reported in Figure 1 as a plot of the experimental versus calculated Ki, and highlight a very good prediction power with an R2 of 0.91.
Figure 1.

Plot of experimental Ki vs. calculated ones for 200 σ2-ligands randomly chosen from the set of selective σ2 receptor ligands as retrieved from the σ2 receptor selective ligand database (S2RSLDB). In red, the straight line corresponding to the linear regression analysis.
On this model, we docked the best-predicted σ2 receptor ligands returned from the two 2D and 3D QSAR filters (524 compounds, being 13 in common). The 15 most potent compounds resulted from the docking analysis are reported in Table 2, while the full set of compounds is reported in Table S2.
Table 2.
Structure and calculated pKi values of the 15 most potent marine products resulted from docking.
| BDB ID | Structure | Calcd. pKi |
|---|---|---|
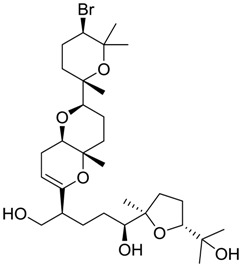
| 1169 |

|
10.26 |
| 1421 |
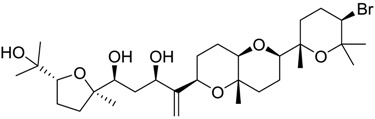
|
9.79 |
| 984 |

|
9.77 |
| 28 |
|
9.64 |
| 306 |
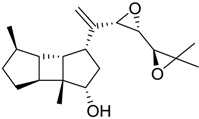
|
9.60 |
| 1333 |
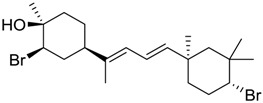
|
9.45 |
| 45 |

|
9.42 |
| 524 |
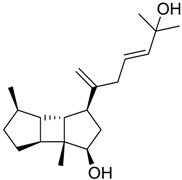
|
9.31 |
| 1172 |

|
9.31 |
| 1123 |
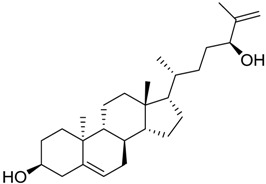
|
9.23 |
| 279 |

|
9.18 |
| 1581 |

|
9.14 |
| 798 |
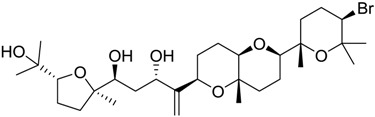
|
9.05 |
| 84 |

|
9.03 |
| 529 |
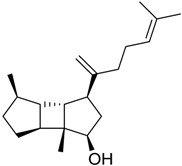
|
8.95 |
The 3D structures of the complex with the two best-docked molecules have been represented in Figure 2. Both molecules reside in the pocket constituted by the two ASP29 and ASP56 residues; in particular, compound 1169 shows two hydrogen bonds with ASP29.
Figure 2.

3D structures of the complex 1169/σ2 receptor (a) and 1421/σ2 receptor (b).
2.4. The Ultimate Filter
Finally, to account for all of the typology of obtained results, we sorted the BDB library according to the mean of the predicted pKi by 2D-QSAR, 3D-QSAR, and docking methodologies. The structures of the best 15 compounds have been reported in Table 3.
Table 3.
Structure, calculated mean pKi, and corresponding Ki (nM) values of the 15 most potent marine products resulted from the mean of the three combined filters.
| BDB ID | Structure | Mean pKi | Mean Ki |
|---|---|---|---|
| 1169 |
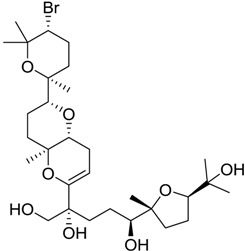
|
9.24 | 0.6 |
| 28 |
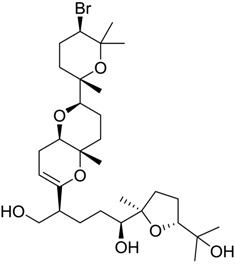
|
8.91 | 1.2 |
| 45 |
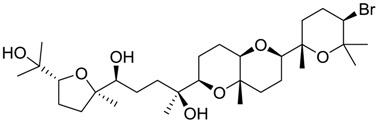
|
8.89 | 1.3 |
| 1172 |
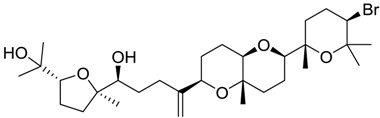
|
8.89 | 1.3 |
| 1421 |
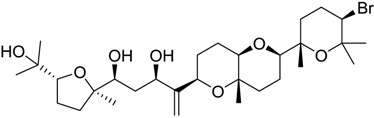
|
8.70 | 2.0 |
| 246 |

|
8.60 | 2.5 |
| 14 |

|
8.57 | 2.7 |
| 298 |

|
8.55 | 2.8 |
| 798 |

|
8.54 | 2.9 |
| 984 |

|
8.44 | 3.6 |
| 1179 |
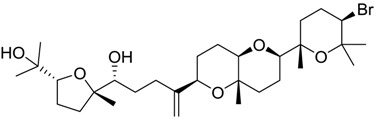
|
8.44 | 3.6 |
| 848 |

|
8.43 | 3.7 |
| 1333 |
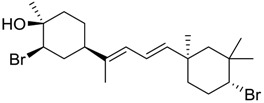
|
8.40 | 4.0 |
| 420 |
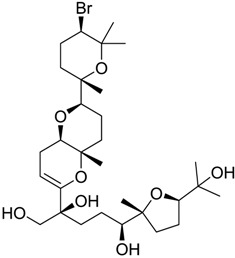
|
8.33 | 4.6 |
| 272 |
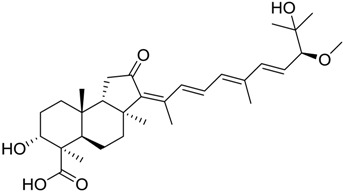
|
8.27 | 5.3 |
An accurate literature search for the 15 most promising hits highlighted that four of them (848, 984, 1169, and 1172) were reported to show clearly antiproliferative and cytotoxic effects against typical cancer cell lines such as A549 and HT29 (Table 4) [52,53,54,55,56,57,58]. These biological data were in agreement with specific σ2 receptors overexpression that was related to the same cell lines [59,60,61,62].
Table 4.
ED50 (μg/mL) of four BDB compounds on A549 and HT29 cell lines that are known to overexpress the σ2 receptor.
| BDB ID | A-549 | HT-29 |
|---|---|---|
| 848 | 1.00 | 0.63 |
| 984 | 2.5 | 2.5 |
| 1169 | 10 | 10 |
| 1172 | 2.5 | 2.5 |
In particular, compound 848 possess a steroidal structure resembling that of progesterone; this compound has been demonstrated to act as a potent σ2 receptor ligand with a Ki of 441 nM [63]. To further validate our model and the goodness of the predicted data, we docked progesterone, obtaining a calculated Ki of 749 nM that became 369 nM after allowing a best accommodation by 100 ps step-10 annealing and 100 ps steepest descent minimization, followed by a local re-docking, which is in good agreement with the experimental Ki.
3. Materials and Methods
3.1. Dataset of Marine Compounds
The chemical structures of the marine dataset were retrieved from the Seaweed Metabolite (http://www.swmd.co.in/), the Chemical Entities of Biological Interest (http://www.ebi.ac.uk/chebi/) databases, and from reference [42]. All of the molecules were manually checked, and the duplicates were removed to achieve a final number of 1517 compounds. The full list of molecules is available as SMILES for external users in the supplementary material (Table S1). In Figure 3, the workflow that was used has been schematized.
Figure 3.

Workflow of used filters.
3.2. Structures 2D to 3D Building and Minimization
The structures of the marine products were built using Marvin Sketch (v. 18.24, ChemAxon Ltd., Budapest, Hungary) [64]. The 2D structures were subjected to molecular mechanics energy minimization by Merck molecular force field (MMFF94) using the Marvin Sketch geometrical descriptors plugin [64]. The protonation states of the molecules were calculated assuming a pH of 7.0. Before the alignment for the 3D-QSAR filter, the geometry of the obtained 3D structures was further optimized at the semi-empirical level using the parameterized model number 3 (PM3) Hamiltonian as implemented in the MOPAC package (MOPAC2016 v. 18.151, Stewart Computational Chemistry, Colorado Springs, CO, USA) [65,66,67].
3.3. 2D-QSAR
The software CORAL (CORrelation And Logic, version 2016, Istituto di Ricerche Farmacologiche Mario Negri, Milano, Italy) was used for building the 2D-QSAR model using 548 σ2 receptor selective ligands over the σ1 receptor, as previously reported [33,34]. The unique SMILES composing the blue dataset were converted in order to have a SMILES depiction that was equal to that used for generating the model (vide supra). To each SMILES, a random endpoint value was associated in order for the software to compare this value with the predicted one. The following regression was used for predicting the endpoints:
where DCW is defined as the “descriptor of correlation weights”. The regression for the σ2 receptor pKi has been developed in a previously published 2D regression model [30].
3.4. Compound Alignment for the 3D-Ligand Based Filter
All of the optimized structures were imported into the software Forge (v10.4.2, Cresset, New Cambridge House, Hertfordshire, UK) for the evaluation of the dataset in the field-based 3D-QSAR model that was previously published [32]. All of the molecules were aligned with the training set of the 3D-QSAR model. The negative, positive, shape, and hydrophobic field points of each molecule were generated using the extended electron distribution (XED) force field in Forge. The molecules were then aligned with the training set of the 3D-QSAR model by a maximum common substructure algorithm using a customized set-up. The software’s parameters that were used for the conformation hunt and alignment are presented in the supplementary material (Figures S1 and S2). The maximum number of conformations that was generated for each molecule was set to 500, as suggested by the software. The root mean square deviation of atomic positions’ cutoff for duplicate conformers was set to 0.5 Å (the similarity threshold below which two conformers are assumed to be identical). The gradient cutoff for conformer minimization was set to 0.1 kcal/mol. The energy window was set to 2.5 kcal/mol. Conformers with a minimized energy outside the energy window were discarded.
3.5. Homology Model and Docking
All of the simulations and molecular modeling experiments have been conducted with YASARA software (v. 18.4.24, YASARA Biosciences GmbH, Vienna, Austria). The homology model was built starting from the Q5BJF2 (SGMR2_HUMAN) sequence deposited in the UniProt Knowledgebase (https://www.uniprot.org/uniprot/) and using the crystallographic structures corresponding to the following PDB IDs as templates: 4LGC, 1VT4, 4M58, 2PFF, 2MGY, AND 1T33. To these structures have been added the best two structures obtained by the evolutionary coupling analysis, which were executed with the EVfold web-server (http://evfold.org/evfold-web/newprediction.do), and the ensemble has been processed with the hm_build macro of YASARA. In the end, an optimized hybrid model was built through iteratively replacing bad regions in the top scoring model with the corresponding fragments from the other models.
This model was docked with the σ2 receptor ligand PB-28 (see below for details), and the best pose ligand/receptor complex structure was then immersed in a simulated endoplasmic reticulum membrane [68], in physiological environment conditions, and subjected to a molecular dynamics (MD) simulation of 10 ns to accommodate the ligand. The simulation was set up automatically by first scanning the protein for exposed transmembrane helices [i.e., helices longer than 16 residues, with more than seven hydrophobic residues and more than three exposed ones (accessible side-chain surface area >30% of maximum)]. The major axis vectors of these helices (i.e., the direction vectors of the least-squares lines through the Calpha atoms) were summed up to obtain the major axis of the protein, which was then oriented along the Y-axis, normally with respect to the plane of the membrane and the X–Z plane. The best shift of the membrane along this major axis was obtained by scanning the protein for the region with the largest number of exposed hydrophobic residues (see definition above) and a width of 28 Å (corresponding to the membrane core).
Having placed an equilibrated membrane structure (consisting of 55% of phosphatidylcholine, 30% of phosphatidylethanolamine, 10% of phosphatidylserine, and 5% of phosphatidylinositol molecules [68]) at this location named ‘MemCenterY’, the system was enclosed in a simulation cell of size [X*Y*Z] Å, the protein was temporarily scaled by 0.9 along the X–Z axes, and then, strongly clashing membrane lipids were deleted (lipids with an atom closer than 0.75 Å to a protein atom).
The temporary protein scaling, which was needed to avoid the deletion of too many lipids around the protein, was then slowly removed during a short simulation at 298 K in vacuum. The protein (with all of the atoms kept fixed) was scaled by 1.02 along the X–Z axes every 200 femtoseconds, while the membrane was allowed to move, but was restrained to ideal geometry (by pulling lipid residues with an atom further than 21.5 Å away from MemCenterY back into the membrane, and by pushing phosphorus atoms closer than 14 Å to MemCenterY back outwards). The force field was AMBER14, with Lipid17/GAFF2/AM1BCC parameters for non-standard residues. As soon as the protein had reached its original size again, the protein side-chain pKas were predicted, protonation states were assigned according to pH 7.4, and the simulation cell was filled with water, 0.9% NaCl, and counter ions (proteins 57, 678–683). The main simulation was then run with PME, and an 8.0 Å cutoff for non-bonded real space forces, a four fs time-step, constrained hydrogen atoms, and at constant pressure and temperature (NPT ensemble), as described in detail previously. During the initial 250 picoseconds, the membrane was restrained to avoid distortions while the simulation cell adapted to the pressure exerted by the membrane (see above, additionally water molecules that got closer than 14 Å to MemCenterY were pushed outside). The source code of this simulation protocol and visualizations of the individual steps can be found at www.yasara.org/membranemd.
Docking experiments were effectuated, employing the AutoDock (4.2.5.1, The Scripps Research Institute, San Diego, California Jupiter, Florida, US) software implemented in YASARA. The maps were generated by the program AutoGrid (4.2.5.1, The Scripps Research Institute, San Diego, California Jupiter, Florida, US) with a spacing of 0.375 Å, and dimensions that encompass all of the atoms extending 5 Å from the surface of the PM3 minimized structure of PB-28. All of the parameters were inserted at their default settings. In the docking tab, the macromolecule and ligand are selected, and Lamarckian Genetic Algorithm (LGA) parameters are set as ga_runs = 100, ga_pop_size = 150, ga_num_evals = 20,000,000, ga_num_generations = 27,000, ga_elitism = 1, ga_mutation_rate = 0.02, ga_crossover_rate = 0.8, ga_crossover_mode = two points, ga_cauchy_alpha = 0.0, ga_cauchy_beta = 1.0, number of generations for picking worst individual = 10.
4. Conclusions
In this study, we describe the screening of new potentially σ2/TMEM97 receptor ligands that were reported in a database of 1517 “small” marine natural products, and formulated by the union of the Seaweed Metabolite (http://www.swmd.co.in/) and the Chemical Entities of Biological Interest (ChEBI, http://www.ebi.ac.uk/chebi/). The structures were selected by our developed 2D and 3D-QSAR statistical models, and successively verified by a robust σ2/TMEM97 receptor homology model appropriately built by us. This work provided 15 best candidates as powerful σ2 receptor marine ligands; four of them are clearly reported in the literature as antiproliferative and cytotoxic compounds against typical cancer cell lines such as A549 and HT29, and are in agreement with the specific σ2 receptor overexpression that is related to the same cell lines. In particular, compound 848 resembles progesterone, which in itself is a potent σ2 receptor ligand. These findings will ensure prospectively advantageous applications to speed up the design and identification of new natural hit compounds as potent and selective σ2 receptor ligands. In vitro and biological screenings of the most promising compounds are in due course.
Acknowledgments
This work was supported by Research Funding from the University of Catania (FIR) 2014. Free academic licenses from Cresset and ChemAxon for their suites of programs are gratefully acknowledged.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/10/384/s1, Figure S1: Forge’s parameters used for the conformation hunt, Figure S2: Forge’s parameters used for the alignment, Figure S3: Homology model of the σ2-receptor immersed in the endoplasmic reticulum membrane, Table S1: Dataset of marine products, Table S2: Complete results of the three filters by color code, Table S3: Experimental and calculated Ki by docking.
Author Contributions
Conceptualization, G.F. and A.R.; Data curation, C.B., D.G. and R.T.; Formal analysis, C.B., D.G. and R.T.; Investigation, G.F., E.A. and A.R.; Methodology, G.F., E.A. and A.R.; Project administration, A.Z. and A.R.; Resources, G.F., D.G. and A.R.; Supervision, A.R.; Validation, G.F. and E.A.; Writing—original draft, G.F., E.A., C.B., A.M. and A.R.; Writing—review & editing, G.F., E.A., A.M. and A.R.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Martin W.R., Eades C.G., Thompson J.A., Huppler R.E., Gilbert P.E. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- 2.Hellewell S.B., Bruce A., Feinstein G., Orringer J., Williams W., Bowen W.D. Rat liver and kidney contain high densities of sigma 1 and sigma 2 receptors: Characterization by ligand binding and photoaffinity labeling. Eur. J. Pharmacol. 1994;268:9–18. doi: 10.1016/0922-4106(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 3.Su T.P., Hayashi T., Maurice T., Buch S., Ruoho A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 2010;31:557–566. doi: 10.1016/j.tips.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hashimoto K. Sigma-1 receptor chaperone and brain-derived neurotrophic factor: Emerging links between cardiovascular disease and depression. Prog. Neurobiol. 2013;100:15–29. doi: 10.1016/j.pneurobio.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Su T.P., Su T.C., Nakamura Y., Tsai S.Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016;37:262–278. doi: 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanner M., Moebius F.F., Flandorfer A., Knaus H.G., Striessnig J., Kempner E., Glossmann H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci USA. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt H.R., Zheng S., Gurpinar E., Koehl A., Manglik A., Kruse A.C. Crystal structure of the human sigma1 receptor. Nature. 2016;532:527–530. doi: 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maurice T. Improving Alzheimer’s Disease-Related Cognitive Deficits with sigma1 Receptor Agonists. Drug News Perspect. 2002;15:617–625. doi: 10.1358/dnp.2002.15.10.740241. [DOI] [PubMed] [Google Scholar]
- 9.Mesangeau C., Amata E., Alsharif W., Seminerio M.J., Robson M.J., Matsumoto R.R., Poupaert J.H., McCurdy C.R. Synthesis and pharmacological evaluation of indole-based sigma receptor ligands. Eur. J. Med. Chem. 2011;46:5154–5161. doi: 10.1016/j.ejmech.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olivieri M., Amata E., Vinciguerra S., Fiorito J., Giurdanella G., Drago F., Caporarello N., Prezzavento O., Arena E., Salerno L., et al. Antiangiogenic Effect of (+/−)-Haloperidol Metabolite II Valproate Ester [(+/−)-MRJF22] in Human Microvascular Retinal Endothelial Cells. J. Med. Chem. 2016;59:9960–9966. doi: 10.1021/acs.jmedchem.6b01039. [DOI] [PubMed] [Google Scholar]
- 11.Chu U.B., Ruoho A.E. Biochemical Pharmacology of the Sigma-1 Receptor. Mol. Pharmacol. 2016;89:142–153. doi: 10.1124/mol.115.101170. [DOI] [PubMed] [Google Scholar]
- 12.Prezzavento O., Arena E., Sanchez-Fernandez C., Turnaturi R., Parenti C., Marrazzo A., Catalano R., Amata E., Pasquinucci L., Cobos E.J. (+)-and (−)-Phenazocine enantiomers: Evaluation of their dual opioid agonist/sigma1 antagonist properties and antinociceptive effects. Eur. J. Med. Chem. 2017;125:603–610. doi: 10.1016/j.ejmech.2016.09.077. [DOI] [PubMed] [Google Scholar]
- 13.Amata E., Dichiara M., Arena E., Pittalà V., Pistarà V., Cardile V., Graziano A.C.E., Fraix A., Marrazzo A., Sortino S., et al. Novel Sigma Receptor Ligand-Nitric Oxide Photodonors: Molecular Hybrids for Double-Targeted Antiproliferative Effect. J. Med. Chem. 2017;60:9531–9544. doi: 10.1021/acs.jmedchem.7b00791. [DOI] [PubMed] [Google Scholar]
- 14.Amata E., Rescifina A., Prezzavento O., Arena E., Dichiara M., Pittalà V., Montilla-García Á., Punzo F., Merino P., Cobos E.J., et al. (+)-Methyl (1R,2S)-2-{[4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl]methyl}-1-phenylcyclopropanecarboxylate [(+)-MR200] Derivatives as Potent and Selective Sigma Receptor Ligands: Stereochemistry and Pharmacological Properties. J. Med. Chem. 2018;61:372–384. doi: 10.1021/acs.jmedchem.7b01584. [DOI] [PubMed] [Google Scholar]
- 15.Kabe Y., Nakane T., Koike I., Yamamoto T., Sugiura Y., Harada E., Sugase K., Shimamura T., Ohmura M., Muraoka K., et al. Haem-dependent dimerization of PGRMC1/Sigma-2 receptor facilitates cancer proliferation and chemoresistance. Nat. Commun. 2016;7:11030. doi: 10.1038/ncomms11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu U.B., Mavlyutov T.A., Chu M.L., Yang H., Schulman A., Mesangeau C., McCurdy C.R., Guo L.W., Ruoho A.E. The sigma-2 receptor and progesterone receptor membrane component 1 are different binding sites derived from independent genes. Ebiomedicine. 2015;2:1806–1813. doi: 10.1016/j.ebiom.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto R.R., Bown W.D., Su T.P. Sigma Receptors: Chemistry, Cell Biology and Clinical Implications. Springer; Berlin, Germany: 2007. [Google Scholar]
- 18.Yi B., Sahn J.J., Ardestani P.M., Evans A.K., Scott L.L., Chan J.Z., Iyer S., Crisp A., Zuniga G., Pierce J.T., et al. Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer’s disease. J. Neurochem. 2017;140:561–575. doi: 10.1111/jnc.13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei Z., Mousseau D.D., Dai Y., Cao X., Li X.M. Haloperidol induces apoptosis via the sigma2 receptor system and Bcl-XS. Pharmacogenom. J. 2006;6:279–288. doi: 10.1038/sj.tpj.6500373. [DOI] [PubMed] [Google Scholar]
- 20.Ostenfeld M.S., Fehrenbacher N., Hoyer-Hansen M., Thomsen C., Farkas T., Jaattela M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005;65:8975–8983. doi: 10.1158/0008-5472.CAN-05-0269. [DOI] [PubMed] [Google Scholar]
- 21.Zeng C., Rothfuss J., Zhang J., Chu W., Vangveravong S., Tu Z., Pan F., Chang K.C., Hotchkiss R., Mach R.H. Sigma-2 ligands induce tumour cell death by multiple signalling pathways. Br. J. Cancer. 2012;106:693–701. doi: 10.1038/bjc.2011.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonhede S., Petersen A., Zetterberg M., Karlsson J.O. Acute effects of the sigma-2 receptor agonist siramesine on lysosomal and extra-lysosomal proteolytic systems in lens epithelial cells. Mol. Vis. 2010;16:819–827. [PMC free article] [PubMed] [Google Scholar]
- 23.Dehdashti F., Laforest R., Gao F., Shoghi K.I., Aft R.L., Nussenbaum B., Kreisel F.H., Bartlett N.L., Cashen A., Wagner-Johnston N., et al. Assessment of cellular proliferation in tumors by PET using 18F-ISO-1. J. Nucl. Med. 2013;54:350–357. doi: 10.2967/jnumed.112.111948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ClinicalTrials.gov. [(accessed on 11 October 2018)];[18F]ISO-1 PET/CT in Breast Cancer. 2014 Available online: https://clinicaltrials.gov/ct2/show/NCT02284919.
- 25.ClinicalTrials.gov. [(accessed on 11 October 2018)];Imaging of In Vivo Sigma-2 Receptor Expression with 18F-ISO-1 Positron Emission Tomography in Metastatic Breast Cancer. 2016 Available online: https://clinicaltrials.gov/ct2/show/NCT03057743.
- 26.Mach R.H., Zeng C., Hawkins W.G. The sigma2 receptor: A novel protein for the imaging and treatment of cancer. J. Med. Chem. 2013;56:7137–7160. doi: 10.1021/jm301545c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schininà B., Martorana A., Colabufo N.A., Contino M., Niso M., Perrone M.G., De Guidi G., Catalfo A., Rappazzo G., Zuccarello E., et al. 4-Nitro-2,1,3-benzoxadiazole derivatives as potential fluorescent sigma receptor probes. RSC Adv. 2015;5:47108–47116. doi: 10.1039/C5RA08639F. [DOI] [Google Scholar]
- 28.Srinivasarao M., Galliford C.V., Low P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discov. 2015;14:203–219. doi: 10.1038/nrd4519. [DOI] [PubMed] [Google Scholar]
- 29.Leelananda S.P., Lindert S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016;12:2694–2718. doi: 10.3762/bjoc.12.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rescifina A., Floresta G., Marrazzo A., Parenti C., Prezzavento O., Nastasi G., Dichiara M., Amata E. Development of a Sigma-2 Receptor affinity filter through a Monte Carlo based QSAR analysis. Eur. J. Pharm. Sci. 2017;106:94–101. doi: 10.1016/j.ejps.2017.05.061. [DOI] [PubMed] [Google Scholar]
- 31.Rescifina A., Floresta G., Marrazzo A., Parenti C., Prezzavento O., Nastasi G., Dichiara M., Amata E. Sigma-2 receptor ligands QSAR model dataset. Data Brief. 2017;13:514–535. doi: 10.1016/j.dib.2017.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Floresta G., Rescifina A., Marrazzo A., Dichiara M., Pistarà V., Pittalà V., Prezzavento O., Amata E. Hyphenated 3D-QSAR statistical model-scaffold hopping analysis for the identification of potentially potent and selective sigma-2 receptor ligands. Eur. J. Med. Chem. 2017;139:884–891. doi: 10.1016/j.ejmech.2017.08.053. [DOI] [PubMed] [Google Scholar]
- 33.Nastasi G., Miceli C., Pittala V., Modica M.N., Prezzavento O., Romeo G., Rescifina A., Marrazzo A., Amata E. S2RSLDB: A comprehensive manually curated, internet-accessible database of the sigma-2 receptor selective ligands. J. Cheminform. 2017;9:3. doi: 10.1186/s13321-017-0191-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salerno L., Amata E., Romeo G., Marrazzo A., Prezzavento O., Floresta G., Sorrenti V., Barbagallo I., Rescifina A., Pittalà V. Potholing of the hydrophobic heme oxygenase-1 western region for the search of potent and selective imidazole-based inhibitors. Eur. J. Med. Chem. 2018;148:54–62. doi: 10.1016/j.ejmech.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 35.Floresta G., Amata E., Dichiara M., Marrazzo A., Salerno L., Romeo G., Prezzavento O., Pittala V., Rescifina A. Identification of Potentially Potent Heme Oxygenase1 Inhibitors through 3D-QSAR Coupled to Scaffold-Hopping Analysis. ChemMedChem. 2018;13:1336–1342. doi: 10.1002/cmdc.201800176. [DOI] [PubMed] [Google Scholar]
- 36.Greish K.F., Salerno L., Al Zahrani R., Amata E., Modica M.N., Romeo G., Marrazzo A., Prezzavento O., Sorrenti V., Rescifina A., et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules. 2018;23:1209. doi: 10.3390/molecules23051209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Floresta G., Pistara V., Amata E., Dichiara M., Damigella A., Marrazzo A., Prezzavento O., Punzo F., Rescifina A. Molecular modeling studies of pseudouridine isoxazolidinyl nucleoside analogues as potential inhibitors of the pseudouridine 5’-monophosphate glycosidase. Chem. Biol. Drug Des. 2018;91:519–525. doi: 10.1111/cbdd.13113. [DOI] [PubMed] [Google Scholar]
- 38.Floresta G., Apirakkan O., Rescifina A., Abbate V. Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis. Molecules. 2018;23:2183. doi: 10.3390/molecules23092183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Floresta G., Pittala V., Sorrenti V., Romeo G., Salerno L., Rescifina A. Development of new HO-1 inhibitors by a thorough scaffold-hopping analysis. Bioorg. Chem. 2018;81:334–339. doi: 10.1016/j.bioorg.2018.08.023. [DOI] [PubMed] [Google Scholar]
- 40.Davis G.D., Vasanthi A.H. Seaweed metabolite database (SWMD): A database of natural compounds from marine algae. Bioinformation. 2011;5:361–364. doi: 10.6026/97320630005361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Degtyarenko K., de Matos P., Ennis M., Hastings J., Zbinden M., McNaught A., Alcantara R., Darsow M., Guedj M., Ashburner M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008;36:D344–D350. doi: 10.1093/nar/gkm791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pereira F., Latino D.A., Gaudencio S.P. QSAR-assisted virtual screening of lead-like molecules from marine and microbial natural sources for antitumor and antibiotic drug discovery. Molecules. 2015;20:4848–4873. doi: 10.3390/molecules20034848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amata E., Marrazzo A., Dichiara M., Modica M.N., Salerno L., Prezzavento O., Nastasi G., Rescifina A., Romeo G., Pittala V. Heme Oxygenase Database (HemeOxDB) and QSAR analysis of isoform 1 inhibitors. ChemMedChem. 2017 doi: 10.1002/cmdc.201700321. [DOI] [PubMed] [Google Scholar]
- 44.Amata E., Marrazzo A., Dichiara M., Modica M.N., Salerno L., Prezzavento O., Nastasi G., Rescifina A., Romeo G., Pittala V. Comprehensive data on a 2D-QSAR model for Heme Oxygenase isoform 1 inhibitors. Data Brief. 2017;15:281–299. doi: 10.1016/j.dib.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toropova M.A., Toropov A.A., Raska I., Jr., Raskova M. Searching therapeutic agents for treatment of Alzheimer disease using the Monte Carlo method. Comput. Biol. Med. 2015;64:148–154. doi: 10.1016/j.compbiomed.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 46.Toropova A.P., Toropov A.A., Martyanov S.E., Benfenati E., Gini G., Leszczynska D., Leszczynski J. CORAL: Monte Carlo Method as a Tool for the Prediction of the Bioconcentration Factor of Industrial Pollutants. Mol. Inform. 2013;32:145–154. doi: 10.1002/minf.201200069. [DOI] [PubMed] [Google Scholar]
- 47.Cheeseright T., Mackey M., Rose S., Vinter A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006;46:665–676. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- 48.Vyas V.K., Ukawala R.D., Ghate M., Chintha C. Homology Modeling a Fast Tool for Drug Discovery: Current Perspectives. Indian J. Pharm. Sci. 2012;74:1–17. doi: 10.4103/0250-474X.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hopf T.A., Scharfe C.P.I., Rodrigues J.P.G.L.M., Green A.G., Kohlbacher O., Sander C., Bonvin A.M.J.J., Marks D.S. Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife. 2014;3:e03430. doi: 10.7554/eLife.03430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laskowski R.A., Macarthur M.W., Moss D.S., Thornton J.M. Procheck—A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 51.Alon A., Schmidt H.R., Wood M.D., Sahn J.J., Martin S.F., Kruse A.C. Identification of the gene that codes for the σ2 receptor. Proc. Natl. Acad. Sci. USA. 2017;114:7160–7165. doi: 10.1073/pnas.1705154114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheu J.H., Huang S.Y., Wang G.H., Duh C.Y. Study on cytotoxic oxygenated desmosterols isolated from the red alga Galaxaura marginata. J. Nat. Prod. 1997;60:900–903. doi: 10.1021/np9701844. [DOI] [PubMed] [Google Scholar]
- 53.Qi X., Liu G., Qiu L., Lin X., Liu M. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, represses angiogenesis in HUVEC cells and in zebrafish embryos via inhibiting the VEGF signal systems. Biomed. Pharmacother. 2015;75:58–66. doi: 10.1016/j.biopha.2015.08.033. [DOI] [PubMed] [Google Scholar]
- 54.Liu M., Zhang W., Wei J., Qiu L., Lin X. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, induces mitochondrial apoptosis in K562 cells and inhibits topoisomerase I in vitro. Toxicol. Lett. 2012;211:126–134. doi: 10.1016/j.toxlet.2012.03.771. [DOI] [PubMed] [Google Scholar]
- 55.Shi D.Y., Li J., Guo S.J., Su H., Fan X. The antitumor effect of bromophenol derivatives in vitro and Leathesia nana extract in vivo. Chin. J. Oceanol. Limnol. 2009;27:277–282. doi: 10.1007/s00343-009-9119-x. [DOI] [Google Scholar]
- 56.Fernandez J.J., Souto M.L., Norte M. Evaluation of the cytotoxic activity of polyethers isolated from Laurencia. Bioorg. Med. Chem. 1998;6:2237–2243. doi: 10.1016/S0968-0896(98)80004-7. [DOI] [PubMed] [Google Scholar]
- 57.Norte M., Fernandez J.J., Souto M.L., GarciaGravalos M.D. Two new antitumoral polyether squalene derivatives. Tetrahedron Lett. 1996;37:2671–2674. doi: 10.1016/0040-4039(96)00357-7. [DOI] [Google Scholar]
- 58.Pacheco F.C., Villa-Pulgarin J.A., Mollinedo F., Martin M.N., Fernandez J.J., Daranas A.H. New Polyether Triterpenoids from Laurencia viridis and Their Biological Evaluation. Mar. Drugs. 2011;9:2220–2235. doi: 10.3390/md9112220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pati M.L., Niso M., Ferorelli S., Abate C., Berardi F. Novel metal chelators thiosemicarbazones with activity at the sigma(2) receptors and P-glycoprotein: An innovative strategy for resistant tumor treatment. RSC Adv. 2015;5:103131–103146. doi: 10.1039/C5RA19857G. [DOI] [Google Scholar]
- 60.Niso M., Abate C., Contino M., Ferorelli S., Azzariti A., Perrone R., Colabufo N.A., Berardi F. Sigma-2 Receptor Agonists as Possible Antitumor Agents in Resistant Tumors: Hints for Collateral Sensitivity. ChemMedChem. 2013;8:2026–2035. doi: 10.1002/cmdc.201300291. [DOI] [PubMed] [Google Scholar]
- 61.Azzariti A., Colabufo N.A., Berardi F., Porcelli L., Niso M., Simone G.M., Perrone R., Paradiso A. Cyclohexylpiperazine derivative PB28, a sigma(2) agonist and sigma(1) antagonist receptor, inhibits cell growth, modulates P-glycoprotein, and synergizes with anthracyclines in breast cancer. Mol Cancer Ther. 2006;5:1807–1816. doi: 10.1158/1535-7163.MCT-05-0402. [DOI] [PubMed] [Google Scholar]
- 62.Al-Nabulsi I., Mach R.H., Wang L.M., Wallen C.A., Keng P.C., Sten K., Childers S.R., Wheeler K.T. Effect of ploidy, recruitment, environmental factors, and tamoxifen treatment on the expression of sigma-2 receptors in proliferating and quiescent tumour cells. Br. J. Cancer. 1999;81:925–933. doi: 10.1038/sj.bjc.6690789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johannessen M., Fontanilla D., Mavlyutov T., Ruoho A.E., Jackson M.B. Antagonist action of progesterone at sigma-receptors in the modulation of voltage-gated sodium channels. Am. J. Physiol.-Cell Physiol. 2011;300:C328–C337. doi: 10.1152/ajpcell.00383.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barf T., Lehmann F., Hammer K., Haile S., Axen E., Medina C., Uppenberg J., Svensson S., Rondahl L., Lundback T. N-Benzyl-indolo carboxylic acids: Design and synthesis of potent and selective adipocyte fatty-acid binding protein (A-FABP) inhibitors. Bioorg. Med. Chem. Lett. 2009;19:1745–1748. doi: 10.1016/j.bmcl.2009.01.084. [DOI] [PubMed] [Google Scholar]
- 65.Stewart J.J. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004;10:155–164. doi: 10.1007/s00894-004-0183-z. [DOI] [PubMed] [Google Scholar]
- 66.Alemán C., Luque F.J., Orozco M. Suitability of the PM3-derived molecular electrostatic potentials. J. Comput. Chem. 1993;14:799–808. doi: 10.1002/jcc.540140706. [DOI] [Google Scholar]
- 67.Qiao F., Luo L., Peng H., Luo S., Huang W., Cui J., Li X., Kong L., Jiang D., Chitwood D.J., et al. Characterization of Three Novel Fatty Acid- and Retinoid-Binding Protein Genes (Ha-far-1, Ha-far-2 and Hf-far-1) from the Cereal Cyst Nematodes Heterodera avenae and H. filipjevi. PLoS ONE. 2016;11:e0160003. doi: 10.1371/journal.pone.0160003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Meer G., Voelker D.R., Feigenson G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.