

Abstract
The tuberous sclerosis complex (TSC) 1/2 is a negative regulator of the nutrient‐sensing kinase mechanistic target of rapamycin complex (mTORC1), and its function is generally associated with tumor suppression. Nevertheless, biallelic loss of function of TSC1 or TSC2 is rarely found in malignant tumors. Here, we show that TSC1/2 is highly expressed in Burkitt's lymphoma cell lines and patient samples of human Burkitt's lymphoma, a prototypical MYC‐driven cancer. Mechanistically, we show that MYC induces TSC1 expression by transcriptional activation of the TSC1 promoter and repression of miR‐15a. TSC1 knockdown results in elevated mTORC1‐dependent mitochondrial respiration enhanced ROS production and apoptosis. Moreover, TSC1 deficiency attenuates tumor growth in a xenograft mouse model. Our study reveals a novel role for TSC1 in securing homeostasis between MYC and mTORC1 that is required for cell survival and tumor maintenance in Burkitt's lymphoma. The study identifies TSC1/2 inhibition and/or mTORC1 hyperactivation as a novel therapeutic strategy for MYC‐driven cancers.
Keywords: Burkitt's lymphoma, cancer, mTORC1, MYC, TSC1/2
Subject Categories: Cancer, Signal Transduction
Introduction
TSC1/2 is a critical upstream regulator of the mechanistic target of rapamycin complex (mTORC) 1 kinase. TSC1 (hamartin) stabilizes TSC2 (tuberin), which is the GTPase‐activating protein (GAP) for Rheb (Ras homolog enriched in brain) in the regulation of mTORC1 (Mieulet & Lamb, 2010). Germline mutations in either TSC1 or TSC2 result in the development of benign tumors (hamartomas) due to hyperactive mTORC1 signaling, which, however, usually does not result in malignancy. Moreover, loss of function of either TSC1 or TSC2 is rarely found in malignant tumors (Mieulet & Lamb, 2010) with some known exceptions like somatic mutations of TSC1 in bladder cancer (Pymar et al, 2008) or TSC2 in hepatocellular carcinoma's (Huynh et al, 2015). Therefore, retaining functional TSC1/2‐mTORC1 regulation may be beneficial for certain cancer cells (Mieulet & Lamb, 2010). An important role of TSC1/2 in metabolic homeostasis was revealed in hematopoietic stem cells (HSCs) where deletion of TSC1 results in elevated mTORC1 activity and increased ROS production with detrimental effects on HCS function and survival (Chen et al, 2008). A database survey revealed that TSC1‐mRNA expression is the highest in Burkitt's lymphoma‐derived cell lines compared to 36 different tumor‐type cell lines in the Cancer Cell Line Encyclopedia (CCLE; http://www.broadinstitute.org/ccle) (Fig EV1A). Moreover, the COSMIC database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) lists no mutations for TSC1 or TSC2 in Burkitt's lymphoma and they are not under the recurrently mutated genes in Burkitt's lymphoma identified by genomic approaches in two studies (Richter et al, 2012; Schmitz et al, 2012). Burkitt's lymphoma is an aggressively growing malignancy characterized by a MYC translocation that induces very high expression levels of the proto‐oncogenic transcription factor MYC (Molyneux et al, 2012). MYC promotes cell proliferation by multiple mechanisms including stimulation of cell cycle progression, ribosome biogenesis, tRNA synthesis, translation and metabolic adjustments to increase provision of metabolic intermediates (Dang, 2012).
Figure EV1. TSC1 is highly expressed in Burkitt's lymphoma.
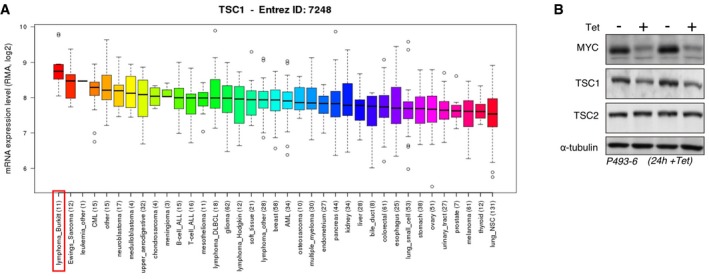
- Box plots showing the relative TSC1 mRNA expression levels across different cancer cell line types with the horizontal line showing the median, whiskers showing upper and lower non‐outlier limits, the box representing the first to the third quartiles, and open circles representing outliers. Data extracted from CCLE_Expression_Entrez_2012‐10‐18.res, with gene‐centric robust multi‐array analysis (RMA)‐normalized mRNA expression data (the number of different cell lines is indicated in parentheses).
- TSC1 protein reduction precedes TSC2 reduction following repression of MYC (+Tet, 24 h) in P493‐6 cells. Immunoblots showing expression levels of MYC, TSC1, TSC2, or α‐tubulin in low (+Tet) versus high MYC (−Tet) P493‐6 cells (in comparison with 72 h MYC repression shown in Fig 1B).
In this study, we reveal that MYC stimulates the expression of the mTORC1‐inhibitor TSC1 by a feed‐forward mechanism combining TSC1 transcriptional activation and alleviation of microRNA miR‐15a‐mediated repression. Loss of TSC1 function in Burkitt's lymphoma cells results in enhanced mitochondrial respiration and accumulation of toxic ROS levels. Our study is the first to provide evidence that TSC1 has tumor maintenance function designating the TSC1/2‐mTORC1 axis as a novel therapeutic target in MYC‐driven Burkitt's lymphoma.
Results
MYC controls mTORC1 through upregulation of TSC1/2 in Burkitt's lymphoma
To examine a potential MYC‐TSC1 regulation in Burkitt's lymphoma (BL), we analyzed TSC1/2 expression in human BL cell lines, which express high levels of MYC, in comparison with low MYC expressing Hodgkin lymphoma (HL) cell lines. Immunoblotting revealed that high expression of TSC1/2 correlates with high MYC expression in BL cells and that low TSC1/2 expression correlates with low MYC in HL cells (Fig 1A). To investigate MYC‐TSC1/2‐mTORC1 regulation, we used the EBV immortalized human B‐cell line P493‐6 that carries a conditional, tetracycline‐repressible MYC allele to study MYC‐induced B‐cell proliferation (Pajic et al, 2000). Also in this system, high MYC levels correlate with high TSC1/2 levels, and suppression of MYC (+Tet, 72 h) resulted in a reduction of TSC1 and TSC2 (Fig 1B). Quantitative real‐time PCR (qRT–PCR) analysis revealed a strong reduction of TSC1 mRNA versus a minor reduction of TSC2 mRNA following 24‐h repression of MYC (+Tet; Fig 1C). In addition, the decline in TSC1 protein occurred prior to the TSC2 reduction at the earlier 24‐h time point (Fig EV1B). Since TSC1 stabilizes TSC2, these data suggest that low MYC levels primarily affect TSC1 expression followed by destabilization of TSC2. TSC1/2 is the major inhibitor of mTORC1 signaling and accordingly expression of high levels of MYC (−Tet) in P493‐6 cells resulted in a strong reduction of phosphorylation of the mTORC1 substrate p70‐S6‐kinase1 (S6K) and its substrate ribosomal protein S6 measured over 24–72 h (Fig 1D). Knockdown of TSC1 in MYC expressing P493‐6 (−Tet) resulted in lower levels of TSC2 and in stimulation of mTORC1 signaling, revealing integral MYC‐TSC1/TSC2‐mTORC1 regulation (Fig 1E). The phosphorylation of S6K and S6 in the low MYC (+Tet) cells is abrogated by rapamycin showing that the observed effects are mTORC1 linked (Fig 1F).
Figure 1. MYC controls mTORC1 signaling through regulation of the TSC1.
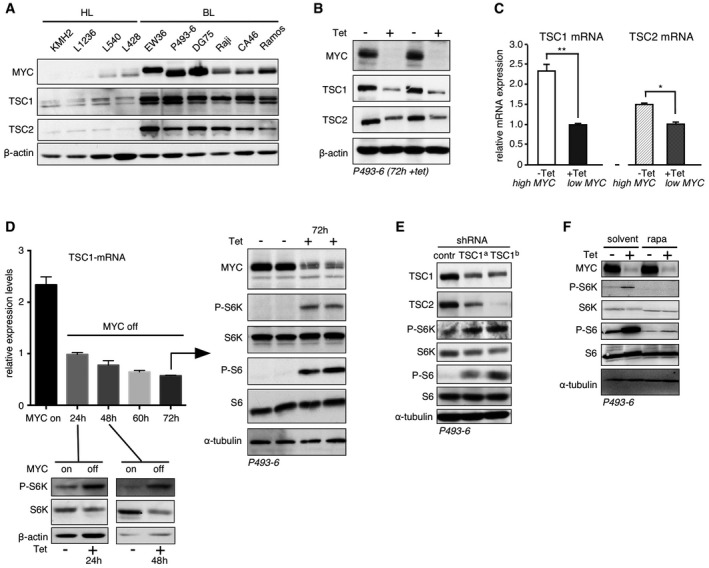
- Immunoblot of expression levels of MYC, TSC1, TSC2, and β‐actin loading control in high MYC Burkitt's lymphoma (BL) cells compared to low MYC Hodgkin lymphoma (HL) cells.
- Immunoblots showing expression levels of MYC, TSC1, TSC2, or β‐actin loading control in P493‐6 cells treated with tetracycline for 72 hours (+Tet) or in untreated cells (−Tet).
- Relative TSC1 and TSC2 mRNA expression levels determined by qRT–PCR for high MYC (−Tet) versus low MYC (+Tet) P493‐6 cells treated for 24 h with tetracycline (mean ± SD, n = 3 technical replicates). *P < 0.05; **P < 0.01; statistical relevance was determined by unpaired t‐test (two‐tailed).
- qRT–PCR analysis of TSC1 mRNA levels upon MYC suppression for 24 h–72 h (+Tet). Immunoblots for 24 h and 48 h (+Tet) show S6K and phosphorylation (P‐) of S6K as downstream mTORC1 target, and β‐actin loading control. For 72 h (+Tet), the immunoblots show expression of MYC and phosphorylation (P‐) of downstream mTORC1 targets S6K and S6, and α‐tubulin as loading control.
- Upper immunoblot shows the reduction in TSC1 levels upon expression of two different TSC1‐specific shRNAs compared to scrambled control shRNA in P493‐6 cells. Other blots show the expression levels of TSC2, S6K/P‐S6K, S6/P‐S6, and α‐tubulin for loading control.
- Immunoblots of indicated proteins in P493‐6 cells with high MYC (−Tet, 72 h) or low MYC (+Tet, 72 h) levels either treated with rapamycin or solvent.
Next, we analyzed the expression of MYC and TSC1 by immunohistochemistry of human BL tissue samples versus control reactive lymph node tissue samples. We found significantly higher expression of MYC and TSC1 protein levels in the BL samples compared to the B lymphocytes that reside in the germinal centers of control lymph nodes (Figs 2A and EV2A and E). In addition, in a second cohort of BL patient samples, we found significantly higher expression of TSC1 and TSC2 proteins by immunoblotting in BL compared to control tonsils and reactive lymph nodes (LN) (Figs 2B and EV2B). Thus, our results show that high TSC1/2 expression correlates with high MYC expression in BL and BL cell lines. Finally, S6K‐phosphorylation levels as determined by immunoblotting were lower for BL patient samples compared to reactive lymph node controls (Fig EV2C). S6‐phosphorylation was virtually absent in seven BL tissue samples and present in one BL sample. In the reactive control lymph nodes, S6‐phosphorylation staining was observed in a mosaic fashion and with different intensities (Fig EV2D and F).
Figure 2. Levels of TSC1 and MYC correlate in Burkitt's lymphoma.
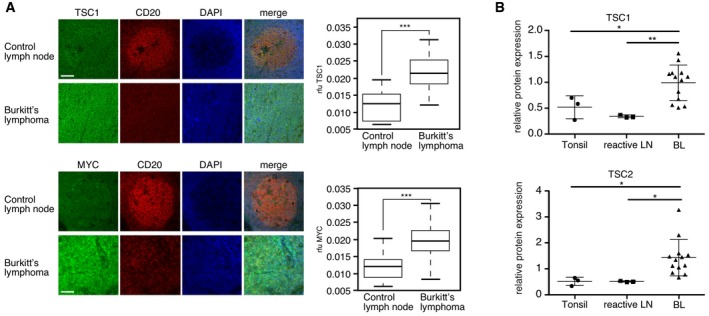
- Elevated TSC1 and MYC expression in BL (cohort 1). Example of immune staining of TSC1, MYC, the B‐cell marker CD20, and DAPI nuclear DNA‐staining in germinal centers of control lymph nodes (upper rows) and samples from BL patients (lower rows). Boxplots at the right show quantification of TSC1 or MYC staining from control germinal centers and BL samples (see materials and methods; the horizontal line shows the median, whiskers show maximum and minimum data points, and the box represents the first to the third quartiles, n = 56 fields for tumor samples and n = 21 fields for control germinal centers; scale bar = 100 μm).
- Quantification of immunoblot analysis of TSC expression in BL (cohort 2) (n = 13) compared to healthy tonsils (n = 3) or reactive lymph nodes (n = 3) (mean ± SD) shown in Fig EV2B.
Figure EV2. Reduced activity of the mTORC1 pathway in BL patient samples.
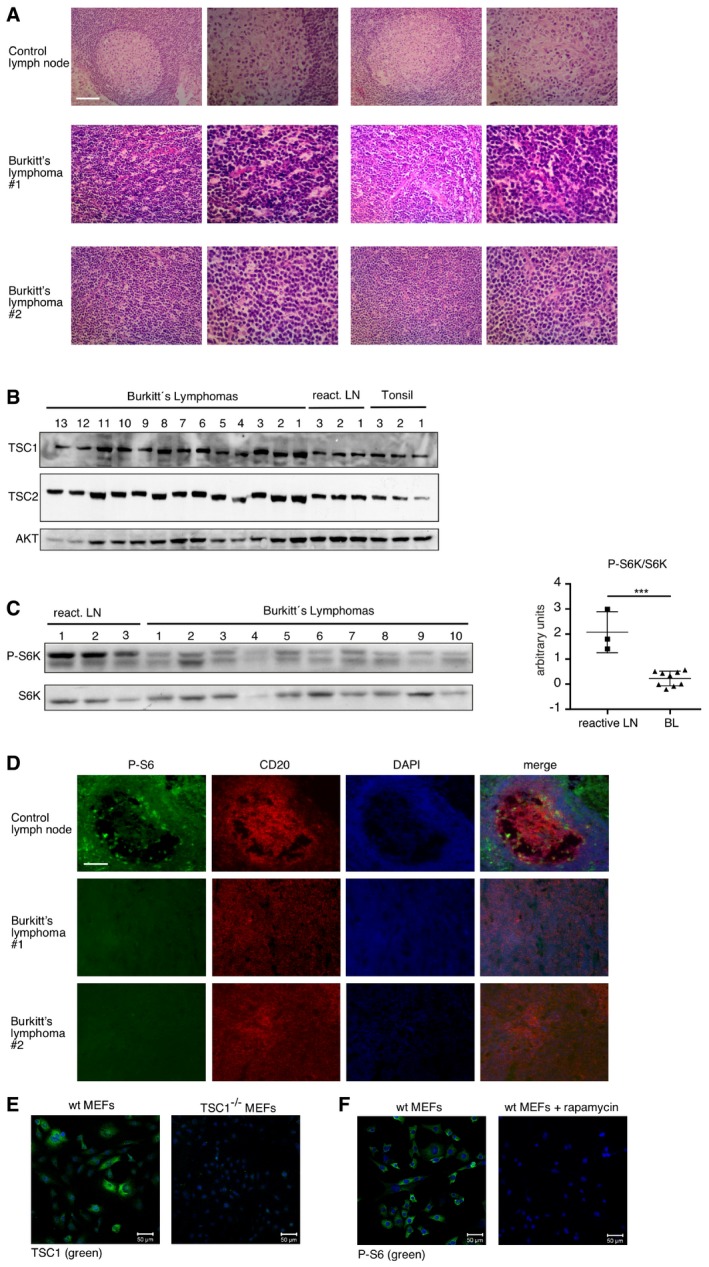
- Immunoblot of the same samples as in (B) with the indicated antibodies. Graph at the right shows the quantification of the P‐S685K/S6K ratio performed with ImageJ (mean ± SD, n = 3 for reactive LN, n = 10 for lymphomas). ***P < 0.001, statistical relevance was determined by unpaired t‐test (two‐tailed).
- Example of immune staining of P‐S6, the B‐cell marker CD20, and DAPI nuclear DNA‐staining in control lymph nodes (upper row) and Burkitt's lymphoma (lower rows). Of eight investigated lymphoma samples, we found seven with virtual absence of P‐S6 staining and one with positive P‐S6 staining. The samples belong to the same cohort used in Figs 2A and EV2A, scale bar = 100 μm.
- Wild‐type (wt) MEFs and TSC1‐deficient MEFs immunostained with anti‐TSC1 antibody to confirm its specificity. Scale bar, 50 μm.
- wt MEFs untreated and wt MEFs treated with 20 nM rapamycin for 12 h were immunostained with anti‐phosphorylated‐S6 antibody to confirm its specificity. Scale bar, 50 μm.
Altogether, our data show that TSC1/2 expression is remarkably high in MYC BL systems and suggesting that during oncogenesis MYC maintains control of mTORC1 signaling through stimulation of TSC function.
Loss of TSC1 function is lethal for MYC‐driven cancer cells
Given the anticipated role of TSC1 as a tumor suppressor, these rather unexpected findings led us to examine whether TSC1 upregulation is required for the oncogenic potential of MYC in the cellular BL model. Strikingly, TSC1 knockdown in high (−Tet) MYC P493‐6 cells resulted in a strong decrease in viable cell numbers (Fig 3A, left graph). AnnexinV/7AAD staining revealed that apoptosis was increased in TSC1 knockdown cells (Fig 3A, right graph), suggesting that the upregulation of TSC1 by MYC is required for cell survival. Notably, the decreased P493‐6 cell viability in response to TSC1 knockdown could be rescued by treatment with the mTORC1 inhibitor rapamycin, showing that enhanced mTORC1 activity is responsible for the increased apoptosis (Fig 3B). To further analyze a potential synthetic lethal interaction between MYC deregulation and mTORC1 hyperactivation, we made use of an U2OS cell line expressing a MYC‐ER fusion protein (Liu et al, 2012). In cells with physiological MYC activity (−OHT), the activation of mTORC1 through knockdown of TSC1 had little effect on cell viability. However, the combined activation of MYC (+OHT) and mTORC1 (TSC1 knockdown) synergistically increased apoptosis, which could be prevented with rapamycin treatment (Figs 3C and EV3A). This shows that cells with deregulated MYC require restriction of mTORC1 signaling for survival and that TSC inhibition is synthetic lethal with MYC overexpression.
Figure 3. TSC1 is crucial for survival of Burkitt's lymphoma (BL) cells.
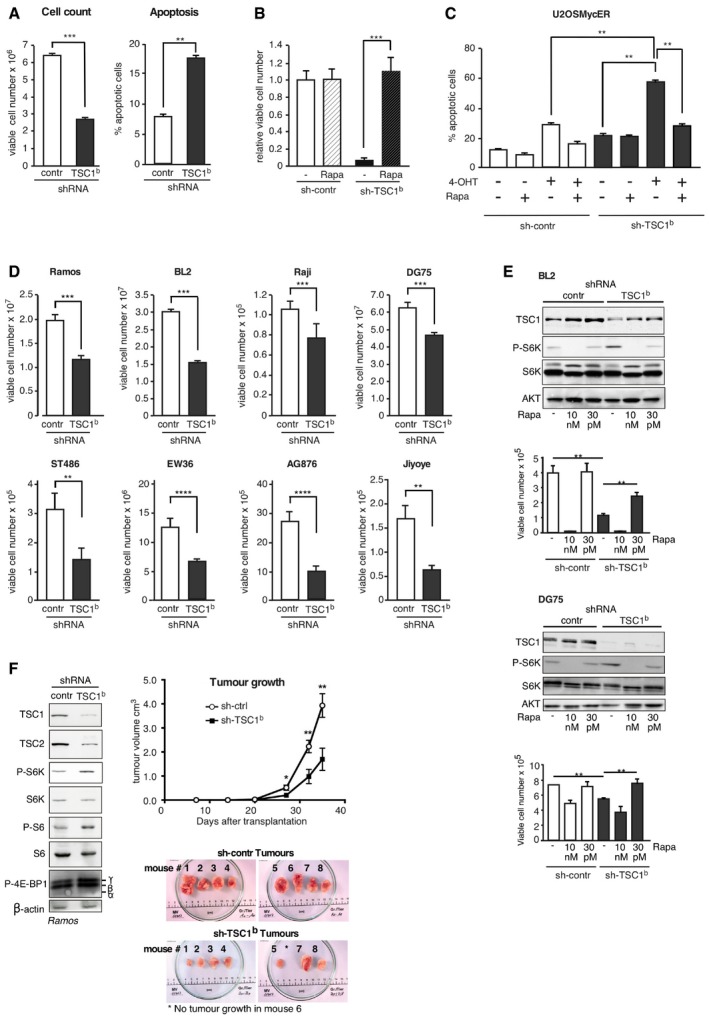
- Left graph shows the multiplication rate of P493‐6 (−Tet) cells expressing either a scrambled control shRNA or a TSC1‐specific shRNA determined by viable cell counting 3 days after seeding of equal number of viable cells (determined by Trypan blue exclusion; mean ± SD, n = 3 biological replicates). Right graph shows percentage of apoptotic P493‐6 (−Tet) cells expressing scrambled control shRNA or a TSC1‐specific shRNA determined by FACS analysis of AnnexinV/7AAD‐stained cells (mean ± SD, n = 3 biological replicates).
- Rapamycin treatment recovers survival of TSC1 knockdown in P493‐6 cells. Relative viable cell number counts of P493‐6 (−Tet) cells expressing scrambled control shRNA or TSC1‐specific shRNA 14 days after seeding equal number of viable cells (Trypan blue exclusion), in the presence of 30 pM rapamycin where indicated (mean ± SD, n = 3 biological replicates).
- TSC1 knockdown is synthetic lethal with MYC deregulation. U2OS‐MYC‐ER cells expressing either scrambled control shRNA or TSC1‐specific shRNA were treated with hydroxytamoxifen (4‐OHT) to induce MYC and rapamycin (100 nM) where indicated. Percentage of apoptotic cells was determined with Annexin/7AAD staining 4 days after MYC induction (mean ± SD, n = 3 biological replicates).
- Survival rate of different BL cell lines upon TSC1 knockdown. Graphs show numbers of viable cells expressing either a scrambled control shRNA or a TSC1‐specific shRNA 3 days after seeding of equal number of viable cells (determined by Trypan blue exclusion; mean ± SD, n = 3 biological replicates).
- Immunoblots of control‐ or TSC1‐shRNA expressing BL2 or DG75 cells treated with different concentrations of rapamycin to either completely inhibit mTORC1 activity (10 nM) or titrate the activity to control levels (30 pM), and survival rate of these cells over 7 days (mean ± SD, n = 3 biological replicates); (BL2 cells were selected for stable knockdown with puromycin, DG75 cells without selection).
- Ramos cells expressing either a TSC1‐specific or a control shRNA were inoculated into NOD/SCID mice, and tumor volume was measured regularly. The immunoblot shows the level of knockdown of TSC1 (sh‐TSC1b) compared to control (sh‐contr), and levels of TSC2 and phosphorylated mTORC1 target proteins before inoculation. Tumor growth curves show mean ± SEM (n = 8/group). Pictures of xenotransplanted human Ramos lymphoma tumors 35 days after inoculation. The top panels show tumors derived from Ramos cells stably expressing control shRNA (sh‐contr); the lower panels show tumors derived from Ramos cells stably expressing TSC1‐specific shRNA (sh‐TSC1b).
Figure EV3. TSC1 is required for survival of BL cells.
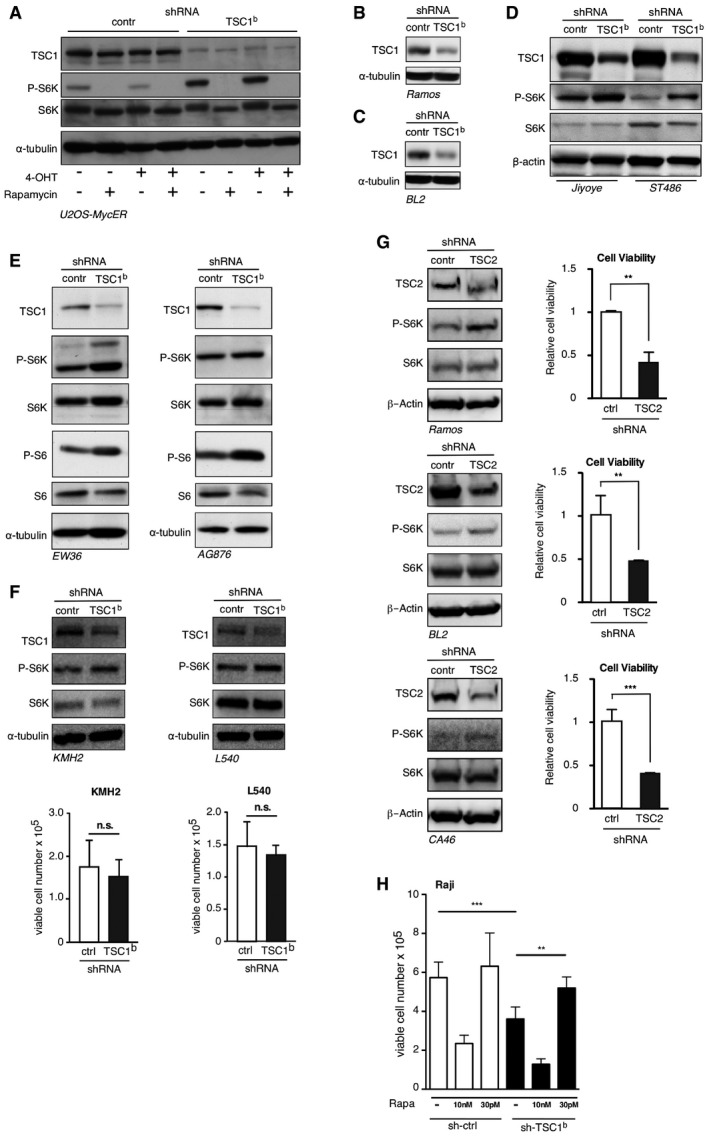
-
AImmunoblots of the experiment in Fig 3C. U2OS‐MYC‐ER cells expressing scrambled control shRNA or TSC1‐specific shRNA, treated with hydroxytamoxifen (4‐OHT) or rapamycin where indicated. Immunoblots showing expression of TSC1, S6K/P‐S6K, or α‐tubulin.
- B–E
-
FAt the top, immunoblots of indicated proteins in Hodgkin lymphoma (HL) cell lines KMH2 and L540 expressing either a TSC1‐specific shRNA or scrambled control shRNA. The bar graphs below show that TSC1 knockdown does not affect cell viability in KMH2 or L540 cells (mean ± SD, n = 3 biological replicates). See Fig 1A for MYC and TSC1 expression levels.
-
GImmunoblots show the reduction of TSC2 levels upon expression of a TSC2‐specific shRNA compared to scrambled control shRNA in the indicated BL cell lines and in addition the expression levels of S6K/PS6K and β‐actin as loading control. The bar graphs at the right show the relative cell viability of the same BL cell lines expressing either TSC2‐specific shRNA or scrambled control shRNA 2 days after seeding equal amounts of viable cells as determined by a cell viability assay (mean ± SD, biological replicates n = 3 for Ramos, n = 5 for BL2 and n = 6 for CA46).
-
HControl‐ or TSC1‐shRNA expressing Raji BL cells treated with different concentrations of rapamycin to either completely inhibit mTORC1 activity (10 nM) or titrate the activity to control levels (30 pM), and survival rate of these cells over 7 days (mean ± SD, n = 3 biological replicates).
In order to have a better representation of the human disease, we studied the effect of TSC1 knockdown on viability for BL cell lines. TSC1 knockdown reduced cell viability of all eight tested BL cell lines (Fig 3D; see Figs EV3B–E and EV4D–E for TSC1 knockdown). TSC1 knockdown does not reduce cell viability in the low MYC and low TSC1 Hodgkin lymphoma (HL) cell lines KMH2 and L540 (Fig EV3F). The data suggest that high expression of TSC1 is a specific requirement for BL cells to maintain cell survival. To investigate whether loss of TSC2 as the other part of the TSC1/2 complex similarly affects cell survival, we knocked down TSC2 in three selected BL cell lines. In all three cell lines, TSC2 knockdown resulted in increased S6K‐phosphorylation and reduced of cell viability (Fig EV3G). Our findings may seem to be at odds with studies showing that mTORC1 signaling is required for survival of lymphomas in the Eμ‐Myc mouse model (Wall et al, 2013) or in TCF3‐activated tonic BCR signaling that activates mTORC1 (Schmitz et al, 2012). Therefore, we hypothesized that maintaining control of mTORC1 to prevent hypo‐ as well as hyperactivity is important for Burkitt's lymphoma survival. To test this hypothesis, we performed a rapamycin titration experiment using the BL cell lines BL2, DG75, and Raji. As expected, strong inhibition of mTORC1 with a high dose of rapamycin (10 nM) severely reduced the cell viability in both control and TSC1 knockdown cells. In contrast, the decreased viability of the TSC1 knockdown cells was recovered by treatment with low dose of rapamycin (30 pM), which titrated the mTORC1 activity to comparable levels of that in control cells (Figs 3E and EV3H). Thus, a controlled level of mTORC1 activity that is in equilibrium with MYC expression levels is critical for the survival of BL cells and probably other MYC‐driven cancer cells.
Figure EV4. TSC1 knockdown increases mitochondrial function.
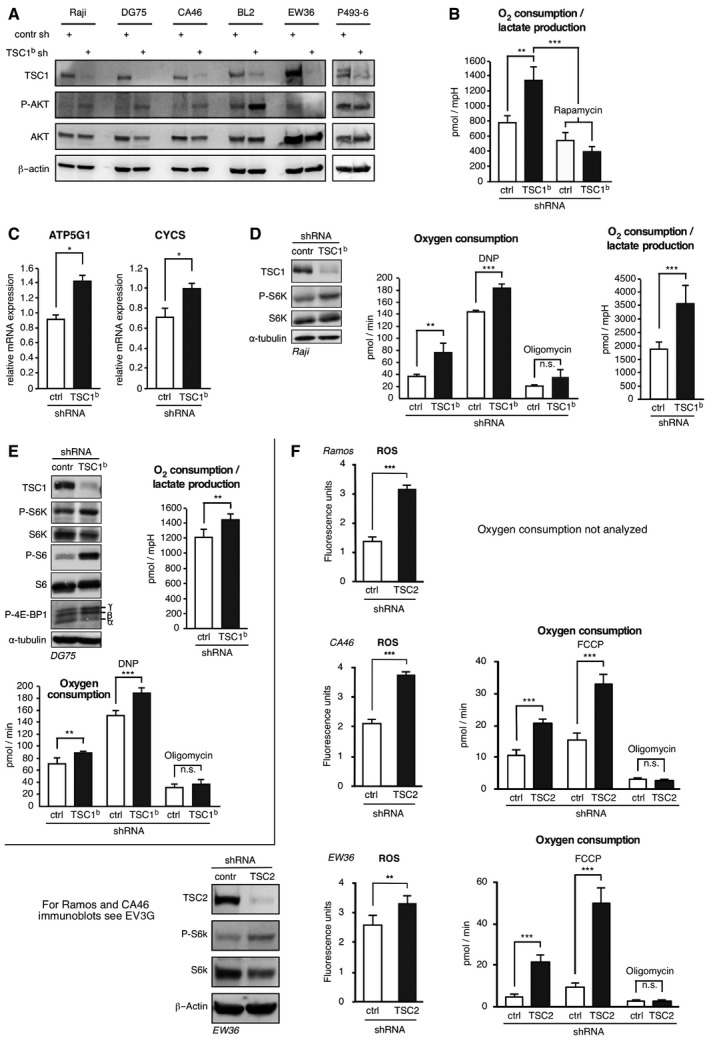
- Immunoblots of control‐ or TSC1‐shRNA expressing BL cells as indicated showing the effect of the TSC1 knockdown on phosphorylation of AKT (P‐Ser473). Antibodies specific for AKT and β‐actin served as controls. The incubation of the same immunoblots with an antibody specific for P‐Thr308 AKT did not produce any detectable signal.
- TSC1 knockdown increases ratio of oxygen consumption to lactate production (as measured by acidification) in a rapamycin‐dependent manner. Ratio of oxygen consumption to lactate production rates determined in the experiment of Figure 4A in high MYC P493‐6 (−Tet) cells expressing scrambled control shRNA or TSC1‐specific shRNA, treated with 20 nM rapamycin for 12 h where indicated (mean ± SD, n = 6 biological replicates).
- Relative mRNA expression determined by qRT–PCR of ATP5G1 (left graph) or cytochrome C (CYCS) (right graph) in Raji cells expressing scrambled control shRNA or TSC1‐specific shRNA (mean ± SD, n = 3).
- From left to right in Raji cells expressing scrambled control shRNA or TSC1‐specific shRNA: immunoblots showing expression of TSC1, S6K/P‐S6K, or α‐tubulin; rate of oxygen consumption, basal and in response to 10 μM DNP or 10 μM oligomycin where indicated; ratio of oxygen consumption to lactate production rates (mean ± SD, n = 8).
- From left to right in DG75 cells expressing scrambled control shRNA or TSC1‐specific shRNA: immunoblots showing expression of TSC1, S6K/P‐S6K, S6/P‐S6, P‐4E‐BP1, or α‐tubulin; rate of oxygen consumption, basal and in response to 10 μM DNP or 10 μM oligomycin where indicated; ratio of oxygen consumption to lactate production rates (mean ± SD, n = 8).
- Left graphs show increased ROS levels in BL cell lines as indicated expressing a TSC2‐specific shRNA compared to scrambled control shRNA expressing cells as determined by FACS analysis of CellRox‐stained cells (mean ± SD, n = 6 biological replicates). Right graphs show increased rate of basal and maximal (in response to 600 nM of the chemical uncoupler FCCP) oxygen consumption in TSC2 knockdown cells compared to scrambled control shRNA expressing cells. Oxygen consumption in response to the ATPase inhibitor oligomycin (10 μM) was comparable between TSC2 knockdown and control cells (mean ± SD, n = 8). Immunoblots at the bottom left show expression levels of TSC2, PS6K/S6K, and β‐actin as loading control for the EW36 cell line. Corresponding immunoblots for Ramos and CA46 cell lines are shown in Fig EV3G.
To determine whether TSC1 function is crucial for BL tumor growth in vivo, we established xenotransplant models of the aggressive human Ramos BL cell line in NOD/SCID mice. Ramos cells stably expressing either a control shRNA or a TSC1‐specific shRNA were injected subcutaneously into NOD/SCID mice and tumor growth was monitored. 35 days after cell injection, tumors derived from TSC1 knockdown cells showed a reduced tumor volume to 43% compared to tumors derived from the control shRNA expressing cells (Fig 3F). These data indicate that knockdown of TSC1 significantly slows BL tumor growth in vivo.
Loss of TSC1 results in toxic ROS production
Next, we addressed what causes the reduced cell viability by TSC1 knockdown. Hyperactivation of mTORC1 in certain cell types results in suppression of AKT kinase activity through negative feedback, which is reflected by the hypophosphorylation of Thr308 and Ser493 (Harrington et al, 2004; Manning, 2004; Rozengurt et al, 2014). Since AKT has a known anti‐apoptotic activity (Ahmed et al, 1997; Dudek et al, 1997; Kauffmann‐Zeh et al, 1997; Kennedy et al, 1997), a potential inhibition of AKT could be involved in the observed cell death. However, in a panel of BL cell lines, TSC1 knockdown either resulted in an increase in Ser493 phosphorylation or did not change Ser493 phosphorylation of AKT, while we were unable to detect any Thr308 phosphorylation in our assay (Fig EV4A). These data suggest that decreased AKT activity is not a cause for cell death following TSC1 knockdown. The enhanced Ser493 phosphorylation of AKT might reflect a compensatory response to counteract apoptosis.
Excessive mitochondrial respiration might result in toxic levels of reactive oxygen species (ROS) and apoptosis in cancer cells (DeNicola et al, 2011). Since both mTORC1 and MYC are known to increase mitochondrial respiration (Li et al, 2005; Cunningham et al, 2007), we examined mitochondrial respiration and ROS production in the cellular BL model. TSC1 knockdown in P493‐6 cells increased total oxygen consumption as well as the ratio of oxygen consumption over lactate production (as determined by acidification), which was measured with the Seahorse XF extracellular flux analyzer (Fig 4A, left graph and Fig EV4B). Treatment with rapamycin reverted these effects demonstrating the involvement of mTORC1 signaling. Suppression of MYC by tetracycline lowered oxygen consumption of both TSC1 knockdown and control cells revealing MYC's contribution in boosting mitochondrial function (Fig 4A, right graph). In the TSC1 knockdown cells, we detected a higher maximal respiratory capacity compared to control cells, which was determined by treatment of the cells with the decoupling drug 2,4‐dinitrophenol (DNP; Fig 4B). In response to the ATPase proton channel inhibitor oligomycin, oxygen consumption was reduced to a similar extent in both the TSC1‐shRNA and control shRNA expressing cells, demonstrating that the observed alterations in respiration are not due to proton leakage (Fig 4B). These data show that loss of TSC1 function and the resulting increased mTORC1 activity shifts metabolism to more mitochondrial respiration. In agreement with enhanced mitochondrial oxidative function, we found an increased ratio of mitochondrial to genomic DNA upon TSC1 knockdown (Fig 4C), indicating enhanced mitochondrial biogenesis. Moreover, mRNA expression of cytochrome C (CYCS) and the subunit ATP5G1 of the mitochondrial ATPase that are involved in oxidative phosphorylation were enhanced in TSC1 knockdown cells (Fig 4D). These alterations were reversed by rapamycin treatment showing their dependence on mTORC1 function. To expand our study from the P493‐6 model to other BL cell lines, we performed shRNA‐mediated knockdown of TSC1 in Raji (Fig EV4C and D) and DG75 (Fig EV4E) cells. This resulted in phenotypes similar to those observed in P493‐6 cells including enhanced S6K‐phosphorylation, increased oxygen consumption, and higher expression of CYCS and ATP5G1.
Figure 4. TSC1 function restricts oxidative phosphorylation and ROS production.
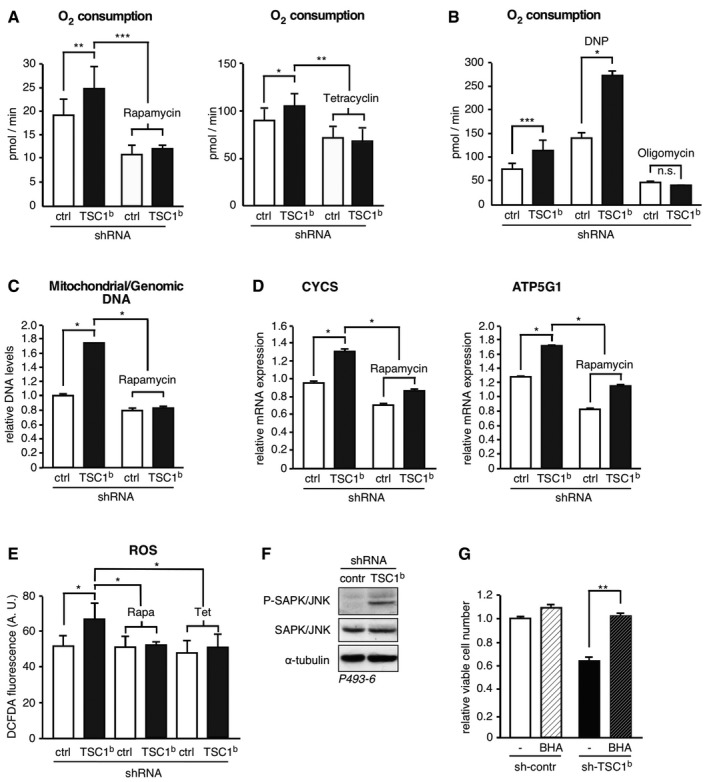
-
A, BTSC1 knockdown increases respiration in an mTORC1‐ and MYC‐dependent manner. Rate of basal oxygen consumption in P493‐6 (−Tet) cells expressing scrambled control shRNA or TSC‐specific shRNA, treated with 20 nM rapamycin for 12 h, or tetracycline to repress MYC for 48 h where indicated (A) or in response to 10 μM of the chemical uncoupler DNP to determine maximal respiration or 10 μM of the ATP synthase inhibitor oligomycin where indicated (B) (mean ± SD, n = 6 biological replicates).
-
C, DRatio of mitochondrial to genomic DNA (C) and expression of cytochrome C (CYCS) or ATP5G1 mRNAs (D) determined by qRT–PCR in P493‐6 (−Tet) cells expressing either scrambled control shRNA or a TSC1‐specific shRNA, and treated with 20 nM rapamycin for 12 h where indicated (mean ± SD, n = 3 technical replicates).
-
ETSC1 knockdown results in elevated ROS levels in a MYC‐ and mTORC1‐dependent manner. FACS analysis of DCF‐DA‐stained P493‐6 (−Tet) cells expressing scrambled control shRNA or a TSC1‐specific shRNA to evaluate ROS production and treated with 20 nM rapamycin for 12 h and tetracycline for 48 h where indicated (mean ± SD, n = 3 biological replicates).
-
FTSC1 knockdown results in increased phosphorylation of SAPK/JNK. Immunoblot of SAPK/JNK Thr183/Tyr185‐phosphorylation in P493‐6 (−Tet) cells expressing scrambled control shRNA or a TSC1‐specific shRNA. α‐tubulin expression serves as loading control.
-
GAntioxidant treatment rescues cells from death caused by TSC1 knockdown. Relative viable cell number counts of P493‐6 (−Tet) cells expressing scrambled control shRNA or TSC1‐specific shRNA 3 days after seeding equal number of viable cells (Trypan blue exclusion), in the presence of 10 μM of the antioxidant BHA where indicated (mean ± SD, n = 3 biological replicates).
To examine whether the increased mitochondrial respiration in response to mTORC1 activation in TSC1 knockdown cells is accompanied by increased intracellular ROS levels, we analyzed DCF‐DA‐stained cells by flow cytometry. Knockdown of TSC1 resulted in an increase in oxidized and fluorescent DCF‐DA compared to the control cells, indicating an increase in ROS production (Fig 4E). In agreement with enhanced oxidative stress, the ROS‐sensitive stress‐activated protein kinase/c‐Jun N‐terminal kinase (SAPK/JNK) was activated upon TSC1 knockdown (Fig 4F). Notably, the increase in ROS production in P496‐3 (−Tet) cells as a result of TSC1 knockdown could be normalized to control levels by mTORC1 inhibition through rapamycin treatment or by tetracycline‐mediated MYC repression (Fig 4E). Similarly, TSC2 knockdown resulted in increased mitochondrial respiration and increased ROS levels in BL cell lines (Fig EV4F). To examine whether elevated ROS levels are responsible for the increased lethality of TSC1 knockdown cells, we treated the cells with the antioxidant butylated hydroxyanisole (BHA). BHA treatment restored survival of high MYC expressing P493‐6 cells after knockdown of TSC1 (Fig 4G), showing that ROS production is responsible for the enhanced apoptosis.
Altogether, these data show that the combined activation of MYC and mTORC1 leads to synergistic enhancement of mitochondrial respiration, which increases ROS production to a level that induces apoptosis. To prevent cell death by metabolic overloading, MYC controls mTORC1 signaling in BL cancer cells through the upregulation of TSC1.
MYC induces TSC1 involving transcription and suppression of miR15a
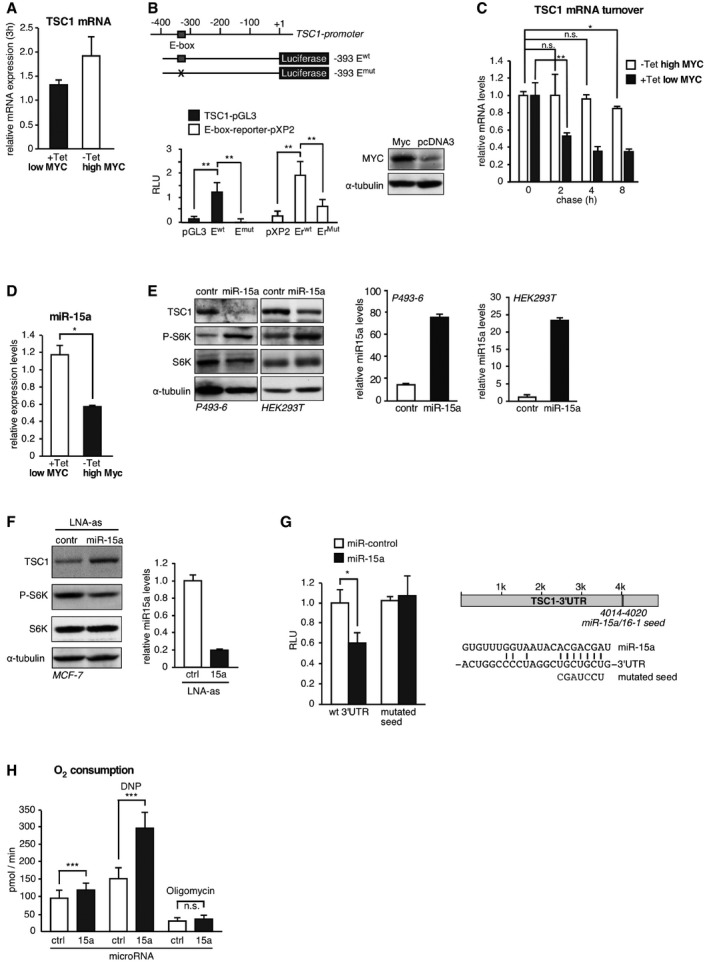
Finally, we set out to investigate the mechanism of TSC1 regulation by MYC. Steady‐state TSC1 mRNA levels were increased in high MYC (−Tet) P493‐6 cells compared to low MYC (+Tet) cells as determined with qRT–PCR (Fig 1C) suggesting a regulation at the level of either transcription or mRNA stability. However, pulse labeling revealed only a moderately enhanced accumulation of nascent TSC1 mRNA over 3 h in high (−Tet) MYC cells (Fig 5A). We used TFSEARCH software (Heinemeyer et al, 1998) to search for potential MYC binding sites and found a single site in the proximal TSC1‐promoter (E‐box: CACGTG, pos. −317/−322). Using TSC1‐promoter‐reporter constructs, we detected activation of the TSC1‐promoter upon coexpression of MYC in the presence of the wild‐type E‐box although to a lower extend as of a control E‐box reporter. Mutation of the E‐boxes resulted in loss of MYC‐induced activation (Fig 5B). These findings are in agreement with the ENCODE database that records a MYC‐associated DNA fragment of intermediate signal strength covering the same sequence (http://genome.ucsc.edu/ENCODE/). The rather modest transcriptional regulation of TSC1 suggested that it might not be the sole mechanism involved. Therefore, we performed a pulse‐chase labeling experiment, which revealed a strongly enhanced TSC1 mRNA turnover in low MYC (+Tet) compared to high MYC (−Tet) P493‐6 cells (Fig 5C), indicating that MYC function stabilizes the TSC1 mRNA. Since MYC is a known regulator of microRNA (miRNA) expression (Chang et al, 2008), we examined whether the observed increase in mRNA turnover in response to MYC repression may be caused by miRNA regulation, which generally results in deadenylation and decay of target mRNAs. In search of potentially involved miRNAs, we identified conserved binding sites for the MYC‐suppressed miRNAs miR‐15a/16‐1, ‐22, ‐23ab ‐26ab, ‐29abc, ‐30e‐5p, and ‐195 in the 3′untranslated region (3′UTR) of the TSC1 mRNA by using the TargetScan algorithm (http://www.targetscan.org). Importantly, those miRNAs are direct MYC targets and were found to be among the suppressed miRNAs in high MYC expressing BL patient samples compared to B cells from hyperplastic tonsils or samples from patients with MYC translocation negative non‐Hodgkin lymphomas (NHLs), including mantle cell lymphoma (MCL), follicular lymphoma (FL), and chronic lymphocytic leukemia (CLL) (Robertus et al, 2010; Table EV1). Overexpression of single miRNAs in HEK293T cells revealed that miR‐15a, miR‐22, and miR‐23a strongly suppressed TSC1 and increased P‐S6K with miR‐15a having the strongest effect on inducing phosphorylation of S6K (Fig EV5A). MiR‐15a and miR‐16‐1 have identical seed sequences and reside in the DLEU2/miR‐15a/16‐1 cluster in the chromosomal region 13q14 whose deletion is commonly associated with the B‐cell malignancy chronic lymphocytic leukemia (B‐CLL; Klein et al, 2010). It was shown by others that MYC represses pri‐microRNA 15a/16‐1 transcription in P493‐6 cells through direct interaction with the pri‐microRNA promoter (Chang et al, 2008). Accordingly, we found that induction of MYC reduced miR‐15a levels in P493‐6 cells (Fig 5D). Overexpression of miR‐15a in P493‐6 and HEK293T cells resulted in decreased TSC1 protein levels compared to the control microRNA with concomitant induction of S6K phosphorylation in both cell lines (Fig 5E). Moreover, knockdown of miR‐15a in MCF‐7 cells by transfection of LNA modified anti‐sense miR‐15a oligonucleotides resulted in elevated TSC1 expression and reduced S6K phosphorylation compared to cells that were transfected with control LNAs (Fig 5F). Next, we examined whether miR‐15a suppresses TSC1 mRNA through direct interaction with the TSC1‐3′UTR. Overexpression of miR‐15a reduced the luciferase expression from a TSC1‐3′UTR‐driven luciferase reporter construct and mutation of the miR‐15a seed sequence relieved the miR‐15a‐mediated repression (Fig 5G). Thus, the TSC1 mRNA is suppressed through direct interaction with miR‐15a. Although our experiments show that miR‐15a is sufficient for modulation of the TSC1‐mTORC1 axis, the other MYC‐suppressed miRNAs that target TSC1 may further solidify the regulation. Notably, oxygen consumption as well as maximal respiratory capacity was also increased in P493‐6 cells that overexpress miR‐15a (Fig 5H), revealing the crucial function of this miRNA in the MYC‐TSC1‐mTORC1 regulatory pathway and in the regulation of mitochondrial physiology. Furthermore, miR‐15a overexpression decreased cell viability in two BL cells (Fig EV5B). Taken together, the results indicate that MYC augments TSC1 expression by a feed‐forward mechanism—one of the most common motifs in transcriptional networks—combining the stimulation of TSC1 transcription with the alleviation of miR‐15a function and thereby increasing TSC1 mRNA stability.
Figure 5. MYC controls TSC1 expression through transcriptional and miR‐15a‐mediated regulation.
- Accumulation of EU‐labeled (Click‐It, Invitrogen) TSC1‐mRNA over 3 h in high MYC (‐Tet) versus low MYC (+Tet) P493‐6 cells (mean ± SD, n = 3 technical replicates).
- Drawing of the TSC1 promoter and luciferase reporter constructs with the predicted E‐box (CACGTG, pos. −317/−322) indicated. Relative luciferase units (RLU) of co‐transfection experiments using the indicated TSC1‐promoter‐reporters with MYC normalized to empty pcDNA3 expression vector in HeLa cells (mean ± SD, n = 3). Immunoblot shows MYC overexpression and α‐tubulin as loading control.
- TSC1 mRNA turnover determined by pulse and chase (Click‐It, Invitrogen) over 8 h in P493‐6 cells, either treated with tetracycline to repress MYC or left untreated (mean ± SD, n = 3 technical replicates).
- miR‐15a expression determined by qRT–PCR in P493‐6 cells treated with tetracycline for 3 days (+Tet) to suppress MYC expression or in untreated (−Tet) cells (mean ± SD, n = 3 technical replicates).
- Immunoblots showing expression levels of TSC1, S6K/P‐S6K, and α‐tubulin in P493‐6 and HEK293T cells overexpressing miR‐15a or a control miRNA. At the right, miR‐15a overexpression was determined by qRT–PCR in P493‐6 and HEK293T cells (mean ± SD, n = 3 technical replicates).
- Immunoblot showing TSC1 expression, S6K/P‐S6K, and α‐tubulin in MCF‐7 cells transfected with LNA modified anti‐sense miR‐15a oligos or control LNAs. At the right, successful miR‐15a knockdown determined by qRT–PCR in MCF‐7 cells is shown (mean ± SD, n = 3 technical replicates).
- Relative luciferase units (RLU) derived from reporter constructs containing TSC1‐3′UTR wild‐type sequences or with mutated miR‐15a seed sequence binding site on co‐transfection with miR‐15a in MCF‐7 cells (mean ± SD, n = 3 biological replicates). The drawing at the right shows the TSC1‐3′UTR with the position of the miR‐15a/16‐1 seed sequence and used mutation indicated.
- Rate of oxygen consumption in P493‐6 (−Tet) cells ectopically expressing miR‐15a or control miRNA vector, under basal conditions and in response to 10 μM DNP or 10 μM oligomycin where indicated (mean ± SD, n = 8 biological replicates).
Figure EV5. Effect of MYC‐suppressed microRNAs on mTORC1 activity and BL cell survival and NUAK mRNA expression levels in different types of cancer cell lines.
- MYC‐suppressed microRNAs regulate the TSC1‐mTORC1 axis. Immunoblot with the indicated antibodies of HEK293T cells transfected with the indicated microRNAs or a control miRNA.
- Reduced relative cell viability of the BL cell lines CA46 (left) and BL2 (right) expressing miR‐15a compared to control miR expressing cells (mean ± SD, n = 5 biological replicates). ***P < 0.001, statistical relevance was determined by unpaired t‐test (two‐tailed).
- Box plots showing the relative NUAK1/ARK5 mRNA expression levels across different cancer cell line types with the horizontal line showing the median, whiskers showing upper and lower non‐outlier limits, the box representing the first to the third quartiles, and open circles representing outliers. Data extracted from CCLE_Expression_Entrez_2012‐10‐18.res, with gene‐centric robust multi‐array analysis (RMA)‐normalized mRNA expression data. The number of cell lines used for each tumor type is indicated in parentheses.
Discussion
The proto‐oncogene MYC is often deregulated in human cancers yet therapeutic strategies to directly target MYC are currently not available. Identifying synthetic lethal interactions with oncogenic functions of MYC may lead to new therapeutic strategies. In this study, we present a connection between MYC and mTORC1 in Burkitt's lymphoma that may provide the foundation for novel therapeutic opportunities. MYC induces the expression of the mTORC1 inhibitor TSC1 through stimulation of TSC1 transcription and downregulation of miR‐15a that targets the TSC1‐mRNA. Other identified MYC‐repressed and TSC1‐targeting microRNAs that were not further investigated here may reinforce this regulation. The upregulation of TSC1 by MYC results in the attenuation of mTORC1 signaling in order to safeguard mitochondrial homeostasis and thereby preventing the accumulation of toxic ROS levels. In addition, knockdown of either TSC1 or TSC2 results in an upregulation mTORC1 signalling, accompanied by enhanced mitochondrial respiration, elevated ROS levels, and reduced cell viability, showing that the effects are not specific for TSC1 but related to TSC1/2 complex function. Since TSC1/2 is generally considered to function as a tumor suppressor (Mieulet & Lamb, 2010) and elevated mTORC1 signaling has been detected in a large proportion of human cancers including B‐cell malignancies (Dazert & Hall, 2011), our finding was surprising. However, we detected elevated TSC1/2 expression in tumors of two independent cohorts of BL patients and in several BL‐derived cell lines. Furthermore, our data show that TSC1/2 controls mTORC1 in BL and is crucial for survival in MYC overexpressing cells. Altogether, this argues against a role for TSC1/2 in tumor suppression in human BL, but rather for a role in tumor maintenance. We propose a model for BL in which a window of mTORC1 activity that supports cell survival is controlled by MYC through maintenance of TSC expression (Fig 6). In spontaneously immortalized rat fibroblast (Rat1A cell lines), Myc was shown to repress TSC2 transcription resulting in increased S6K activity and soft agar colony formation (Ravitz et al, 2007), which seems to be at odds with our findings. Notwithstanding, our data clearly reveal a positive correlation between high MYC and high TSC1/2 levels in BL cell lines as well as in patient material (Figs 1A and 2), and the positive transcriptional and post‐transcriptional regulation of TSC1 transcript by MYC (Figs 1C and D, and 5A–C). Possibly, these apparently opposing results are caused by different wiring of TSC2 regulation in fibroblasts and BL cells. In support of our finding is a study in the fly D. melanogaster where TSC was identified as an essential regulator of intestinal stem cells in the Drosophila midgut, using an RNAi‐based genetic screen. Loss of TSC results in excessive cell growth and halted cell division, which is rescued by reducing Myc levels or inhibition of TORC1 by treatment with rapamycin (Amcheslavsky et al, 2011). The latter study and the data presented here show that at least in certain context high MYC levels are incompatible with high mTORC1 activation.
Figure 6. Model of the MYC‐miR‐15a‐TSC1 axis controlling mTORC1 activity.
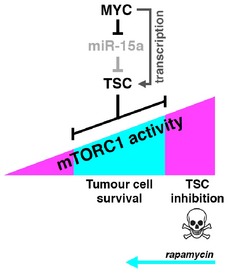
Hyperactivation of mTORC1 through loss of TSC1 function results in metabolic stress, ROS production, and apoptosis. Under this condition, low dose of rapamycin restores mTORC1 levels that are compatible with cell viability.
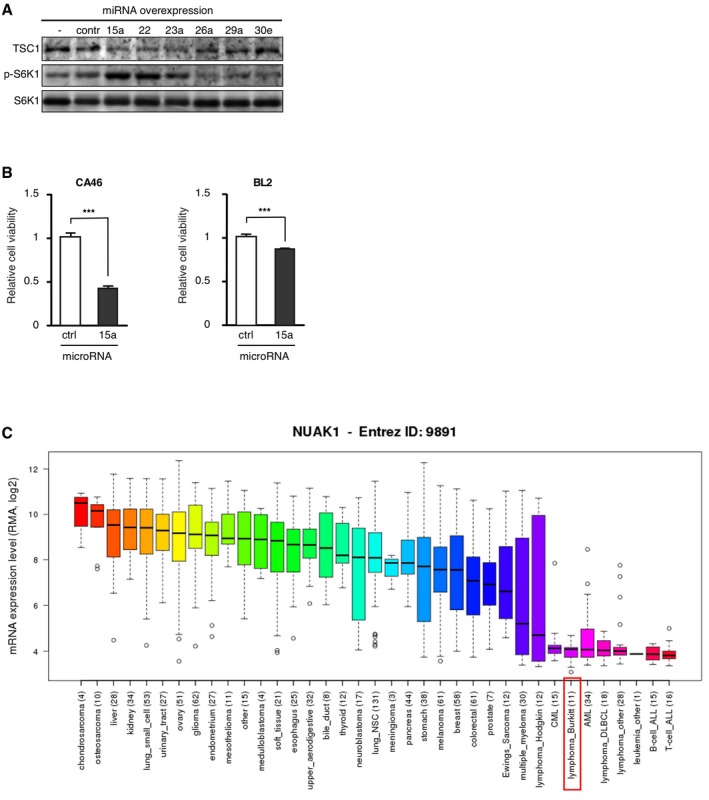
Activation of mTORC1 in cancer has been shown in numerous studies (Zoncu et al, 2011). mTORC1 and MYC have overlapping functions, including stimulating effects on cell growth and proliferation, ribosome biogenesis, and protein synthesis. In addition, mTORC1 induces oxidative phosphorylation by facilitating YY1/PGC1α complex formation (Cunningham et al, 2007) and supports aerobic glycolysis (Duvel et al, 2010). In the Eμ‐Myc mouse model, mTORC1 activation was shown to accelerate lymphomagenesis by inhibiting Myc‐induced apoptosis through translational upregulation of the anti‐apoptotic protein myeloid cell leukemia 1 (Mcl1; Mills et al, 2008), suggesting a collaboration of Myc and mTORC1 in tumor initiation. On the contrary, MYC amplification was shown to confer resistance to mTORC1 inhibition through transcriptional activation of the eukaryotic translation initiation factor 4E (eIF4E), suggesting that mTORC1 function is not critical in the context of high MYC expression (Ilic et al, 2011). Correspondingly, in a chemical screen study elevated MYC levels correlated with enhanced resistance to PI3K/mTORC1 inhibition (Muellner et al, 2011). In line with our study, others have shown that deregulation of MYC may result in energy (ATP) depletion and suppression of mTORC1 through the activation of NUAK1/ARK5‐AMPK, which is required for metabolic homeostasis and cell survival (Liu et al, 2012). However, this regulation may not be relevant for BL and other leukemia, since the CCLE data (http://www.broadinstitute.org/ccle) reveal that NUAK1/ARK5 mRNA expression is very low in most leukemia and lymphoma‐derived cell lines, compared to cell lines derived from solid tumors (Fig EV5C). Altogether, multiple studies suggest that MYC‐driven oncogenesis does not obligatory require mTORC1 activation and that time‐ and dose‐dependent variation in mTORC1 signaling may be required for different stages of tumor development.
Our study for the first time reveals a tumor maintenance function for TSC1/2 and thereby questions the general role of the TSC1/2 complex as a tumor suppressor. Although our results point to canonical mTORC1 inhibition as being responsible for the tumor maintenance activity, we cannot rule out the involvement of other TSC1/2 functions. So far, we do not know whether the requirement of TSC1/2 for cancer cell survival is restricted to BL or whether it might be a characteristic also for other MYC‐driven cancers. Nonetheless, in specific cancer (sub‐)types, the inhibition of TSC1/2 function or the activation of mTORC1 may be considered for a novel therapeutic strategy.
Material and Methods
Cell culture and plasmids
P493‐6, Raji, BL2, Ramos, DG75, CA46, ST486, Jiyoye, EW36, AG876, KMH2, L428, L450, L1236, and MCF‐7 cells were maintained in RPMI 1640. HEK293T, U2OS‐MYC‐ER cells (Liu et al, 2012), wt MEFs, and TSC1‐deficient MEFs (Hsieh et al, 2012) were maintained in DMEM. All media were supplemented with 10% FCS (Tet‐free FCS from Clontech was used for P493‐6 cells) or 15% (EW36, AG876, KMH2, L428, L450, and L1236), non‐essential amino acids, and penicillin/streptomycin. For repression of MYC in P493‐6 cells, 0.1 μg/ml tetracycline was added to the culture medium, and for activation of MYC in U2OS‐MYC‐ER cells, the culture medium was supplemented with 100 μM hydroxytamoxifen (4‐OHT) for 4 days. Rapamycin was added in a concentration range from 30 pM to 100 nM for different time periods. For antioxidant treatment, cells were grown for 3 days in the presence of 1 μM butylated hydroxyanisole (BHA, Sigma‐Aldrich).
Transfection and transduction
HEK293T cells were seeded to a density of 2 × 106 cells in 10‐cm culture dishes. 24 h later, transfection was carried out using the calcium phosphate method. pcDNA‐6.2‐GW/EmGFP‐miRNA (Invitrogen)‐based microRNA expression vectors were transfected in P493‐6 cells using the Amaxa Nucleofector (Lonza) following the manufacturer's instructions, and stable microRNA expressing cell lines were obtained by selecting on blasticidin. Locked nucleic acids (LNA) anti‐miR‐15a and control LNAs (Exiqon) were transfected using HiPerfect (Qiagen) in 6‐well plates following the manufacturer's instructions. Cells were harvested after 72 h. Transfection of P493‐6 cells with self‐delivery Accell siRNAs (Dharmacon) was performed according to the provider's instructions in 96‐well plates. MicroRNA expression vectors were generated using the Block‐it system (Invitrogen) using the pcDNA‐6.2‐GW/EmGFP‐miR‐ vector. Cells were infected following a standard protocol with pLKO.1 lentiviral constructs containing shRNAs against human TSC1: sh‐a 5′‐CCG GGC ACT CTT TCA TCG CCT TTA TCT CGA GAT AAA GGC GAT GAA AGA GTG CTT TTT G‐3′; sh‐b 5′‐CCG GGC CAA GAA AGA CCA CCT TCT TCT CGA GAA GAA GGT GGT CTT TCT TGG CTT TTT G‐3′, TSC2: 5′‐CCG GGC TCA TCA ACA GGC AGT TCT ACT CGA GTA GAA CTG CCT GTT GAT GAG CTT TTT G‐3′ and MYC: 5′‐CCG GCC TGA GAC AGA TCA GCA ACA ACT CGA GTT GTT GCT GAT CTG TCT CAG GTT TTT G‐3′ or non‐target shRNA control (Sigma‐Aldrich). Stable shRNA expressing cells were obtained by selection on puromycin.
MicroRNA expression plasmids
MicroRNA expression vectors were generated using the Block‐it system (Invitrogen). Briefly, oligos were phosphorylated, annealed, and ligated with the pcDNA‐6.2‐GW/EmGFP‐miR‐ vector following the manufacturer's instructions. The following oligos were used for ligation: miR‐15a 5′‐TGC TGT AGC AGC ACA TAA TGG TTT GTG GTT TTG GCC‐3′ (top strand) and 5′‐ACT GAC TGA CCA CAA ACC TAT GTG CTG CTA‐3′ (bottom strand), miR‐22 5′‐TGC TGA AGC TGC CAG TTG AAG AAC TGT GTT TTG GCC ACT GAC TGA CAC AGT TCT AAC TGG CAG CTT‐3′ (top strand) and 5′‐CCT GAA GCT GCC AGT TAG AAC TGT GTC AGT CAG TGG CCA AAA CAC AGT TCT TCA ACT GGC AGC TTC‐3′ (bottom strand), miR‐23a 5′‐TGC TGA TCA CAT TGC CAG GGA TTT CCG TTT TGG CCA CTG ACT GAC GGA AAT CCG GCA ATG TGA T‐3′ (top strand) and 5′‐CCT GAT CAC ATT GCC GGA TTT CCG TCA GTC AGT GGC CAA AAC GGA AAT CCC TGG CAA TGT GAT C‐3′ (bottom strand), miR‐26a 5′‐TGC TGT TCA AGT AAT CCA GGA TAG GCT GTT TTG GCC ACT GAC TGA CAG CCT ATC GGA TTA CTT GAA‐3′ (top strand) and 5′‐CCT GTT CAA GTA ATC CGA TAG GCT GTC AGT CAG TGG CCA AAA CAG CCT ATC CTG GAT TAC TTG AAC‐3′ (bottom strand), miR‐29a 5′‐TGC TGT AGC ACC ATC TGA AAT CGG TTA GTT TTG GCC ACT GAC TGA CTA ACC GAT CAG ATG GTG CTA‐3′ (top strand) and 5′‐CCT GTA GCA CCA TCT GAT CGG TTA GTC AGT CAG TGG CCA AAA CTA ACC GAT TTC AGA TGG TGC TAC‐3′ (bottom strand), miR‐30e 5′‐TGC TGT GTA AAC ATC CTT GAC TGG AAG GTT TTG GCC ACT GAC TGA CCT TCC AGT AGG ATG TTT ACA‐3′ (top strand) and 5′‐CCT GTG TAA ACA TCC TAC TGG AAG GTC AGT CAG TGG CCA AAA CCT TCC AGT CAA GGA TGT TTA CAC‐3′ (bottom strand), and a non‐targeting miRNA was used as a control (Invitrogen).
Immunoblotting
Immunoblotting was performed as described earlier (Zidek et al, 2015) with the following antibodies: from Cell Signaling Technology, anti‐TSC1 (#4906), anti‐TSC2 (#4308); anti‐pS6K (Thr389) (#9206) and (Thr389) (#9234), anti‐S6K (#9202), anti‐pS6 (Ser235/236) (#2211), anti‐S6 (#2317), anti‐pSAPK/JNK (Thr183/Tyr185) (#9251); anti‐p‐4E‐BP1 (T37/46) (#9459); anti‐p‐AKT (Ser473) (#4060); anti‐AKT (#9272); anti‐MYC (#5605); from Sigma‐Aldrich, anti‐β‐actin (A3853); from Santa Cruz Biotechnology, anti‐α‐tubulin (sc‐8035), anti‐TSC2 (sc‐893), anti‐MYC (sc‐40), anti‐SAPK/JNK (sc‐571); and from MP Biomedicals, anti‐β‐actin (#69100). Bands were visualized by chemoluminescence (Western Lightning Plus‐ECL, Perkin Elmer).
qRT–PCR
RNA was isolated using the RNAeasy kit (Qiagen), and cDNA synthesis was done using the first strand cDNA kit (ROCHE). qRT–PCR was performed on a LC480 Light Cycler (Roche) using SYBR Green (Roche) and the following primers: TSC1 5′‐CAA ACT CCA GGC AAG AGG AC‐3′ (forward) and 5′‐CCA ATT CAA ACA CCT GGG TTA‐3′ (reverse), TSC2 5′‐TGC AAG CCG TCT TCC ACA T‐3′ (forward) and 5′‐ATG GAC ACA AAG TCG TTG C‐3′, β‐actin 5′‐AGA GGG AAA TCG TGC GTG AC‐3′ (forward) and 5′‐CAA TAG TGA TGA CCT GGC CGT‐3′ (reverse), ATP5G1 5′‐AGG GCT AAA GCT GGG AGA CTG AA‐3′ (forward) and 5′‐GTC TGG CCA CCT GGA GTG GGA‐3′ (reverse), CYCS 5′‐AGG GAC AGA ATT TAA ATA TGG GTG A‐3′ (forward) and 5′‐AGA TTT GGC CCA GTC TTG TG‐3′ (reverse), HPRT (for normalization) 5′‐TGA CAC TGG CAA AAC AAT GCA‐3′ (forward) and 5′‐GGT CCT TTT CAC CAG CAA GCT‐3′ (reverse). For microRNA qRT–PCR, RNA was isolated using the miRNeasy kit (Qiagen), and primers obtained from the miScript System (Qiagen) were used for expression analysis with a primer for U6 RNA used for normalization. Primers for the analysis of mitochondrial DNA to genomic DNA ratio: genomic glucagon‐intron 5′‐TGA CAA AGA CGG ACT TGA CG‐3′ (forward) and 5′‐CCC TGT GTC ACA AGC AGA TG‐3′ (reverse); mitochondrial COX2 5′‐TTC ATG ATC ACG CCC TCA TA‐3′ (forward) and 5′‐TAA AGG ATG CGT AGG GAT GG‐3′ (reverse).
mRNA labeling experiments
TSC1 nascent transcription or mRNA stability was determined using the Click‐It Nascent RNA Capture Kit (Invitrogen) according to the protocol of the manufacturer. P493‐6 cells were grown for 3 days in the absence or presence of tetracycline. Cells were seeded in a concentration of 1 × 106 cells/ml growth medium (RPMI), and mRNA was labeled with 5‐ethynyl uridine (EU) in a final concentration of 200 μM in the absence or presence of tetracycline. Cells were harvested after 3‐h labeling for analysis of initial transcription rates. For pulse chase, cells were labeled o.n., washed twice with PBS, and chased for different time points with normal growth medium in the absence or presence of tetracycline. Non‐labeled cells were used as control (for each time point, 1.5 × 107 cells were used). Total RNA was prepared using the RNeasy kit from Qiagen, and 2 μg RNA was biotinylated using 0.5 mM biotin azide. After RNA precipitation, the biotinylated RNA was isolated using Streptavidin T1 magnetic beads and used for cDNA synthesis using the SuperScript Reverse Transcriptase (Roche). The amount of biotinylated hTSC1 mRNA was determined by qRT–PCR on a LC480 Light Cycler (Roche) and normalized to β‐actin mRNA levels.
Luciferase assay
Luciferase constructs were obtained by cloning TSC1‐3′UTR sequence containing miR‐15a binding site into pGL3 Vector (Promega). Briefly, 3′UTR sequence was PCR amplified using the following primers: 5′‐ATG CTC TAG ATC TGC TGC ACC TTC ACT CTC‐3′ (forward) and 5′‐ATG CTC TAG AGC AAA ATC TGT TCC TCC GTA A‐3′ (reverse), XbaI digested and ligated into the XbaI restriction site of the pGL3‐promoter vector. For site‐directed mutagenesis of the miR‐15a binding site by PCR, the following primers were used: 5′‐GAC AGG AGG TAT GGC GAT CCT CTG TGT AGC A‐3′ (forward) and 5′‐TGC TAC ACA GAG GAT CGC CAT ACC CTC CTG TC‐3′ (reverse). For the luciferase assay, 2 × 104 MCF‐7 cells per well were seeded into 96‐well plates. After 24 h, cells were co‐transfected with TSC1‐3′UTR‐pGL3 luciferase reporter, Renilla reporter, and either miR‐15a or control pcDNA‐6.2‐GW/EmGFP‐miRNA expression vectors. After additional 24 h, luciferase activity was measured using the Stop and Glow kit (Promega) following the manufacturer's protocol with a Mithras LB 940 (Berthold technologies).
For generating the TSC1‐promoter luciferase reporters, TSC1 promoter fragments were amplified by PCR from genomic DNA using following forward primers: E1wt 5′‐GCG GGT ACC GGG CGC TCC AAC CAC ACC CAG‐3′, in combination with the reverse primer 5′‐GCG CTC GAG CTT GGA CGT ACA GCA CCT CCC‐3′. The mutation of E‐box (E1mut) was performed by PCR using the forward primer for E1wt and the reverse primer as outer primers, and the primers: 5′‐CCG CCC GCC AGT CAC CCG ACC GCC CGC CCC‐3′ and 5′‐GGG GCG GGC GGT CGG GTG ACT GGC GGG CGG‐3′ carrying the mutation. PRC fragments were digested with KpnI and XhoI and cloned into the pGL3‐Basic Firefly luciferase vector (Promega). The pTK‐Ap2/E (renamed in E‐boxRep) and pTK‐AP2/mut4 (renamed in E‐boxRepMut) E‐box luciferase reporter constructs were obtained from Martin Eilers (Gaubatz et al, 1995). 20 h before transfection, HeLa cells were seeded in a 96‐well plate (5,000 cells per well). Transfection was performed in triplicates using 100 ng pGL4 Renilla vector, 300 ng Firefly reporter, and either 600 ng pcDNA3, 600 ng pCMV‐Myc, and 3 μl Fugene (Promega) according to the protocol of the manufacturer. 5 h after transfection, medium was exchanged, and luciferase activity was analyzed 48 h after transfection by using the Stop and Glow kit (Promega) with a Mithras LB 940 (Berthold Technologies).
Measurement of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR)
To measure the rate change of dissolved oxygen (oxygen consumption rate) in the culture medium, a Seahorse XF96 was used. To calculate the ratio of oxygen consumption over lactate production, the pH change in the medium (acidification rate as indirect method for lactate production) was measured simultaneously. Between 4 × 104 and 8 × 104 cells were seeded in unbuffered DMEM in poly‐lysine treated XF96 plates 1 h prior to the assay, and the measurement was made over 2 min. Rapamycin (20 nM) treatment was done for 12 h prior to the measurement. Concentrations of the injected drugs were as follows: oligomycin (10 μM), 2,4‐dinitrophenol (DNP) (10 μM), FCCP (600 nM), all from Sigma‐Aldrich.
Apoptosis assay and ROS measurement
To determine apoptosis, the AnnexinV/7AAD Apoptosis detection kit from BD Biosciences was used. Cells were treated according to the manufacturer's protocol and analyzed by FACS using a BD FACSCanto II. For cell multiplication, equal number of viable cells (Trypan blue exclusion) were seeded and assayed for viability 3 days after seeding, using a cell viability assay from Promega (CellTiter Glo). For ROS measurements, cells were incubated with 10 μM DCF‐DA (Sigma‐Aldrich) or 5 μM CellRox (Invitrogen) for 30 min under normal culture conditions, washed and subjected to FACS analysis using a BD FACSCanto II.
Tumor xenotransplant mouse studies
4.5 × 106 Ramos cells, expressing either a scrambled shRNA or a shRNA against TSC1 (titer 1 × 108, MISSION shRNAs, Sigma), were inoculated into immunodeficient NOD/SCID mice (n = 8/group) subcutaneously, and tumor growth was monitored over time. Tumor volume was calculated in all experiments according to V = (length × width2)/2, and animals were sacrificed when tumor size exceeded 2 cm3. Mice experiments were performed by EPO GmBH (Berlin, Germany) according to the German Animal Protection Law with permission of the responsible authorities. Statistical significance was determined by unpaired t‐test (two‐tailed). The experiment was not randomized. The investigators were not blinded to allocation during the experiment.
Tissue samples
Individual diagnoses were reviewed by an experienced hematopathologist for consistent morphology and immunophenotype according to the 2008 WHO classification (Swerdlow et al, 2008). Pediatric BL presented with abdominal mass and was CD10+, BCL2−, and MYC/8q24 breakpoint positive. All protocols for obtaining human tissue samples were performed in accordance to the guidelines from the Institutional Review Board or Medical Ethical Committee of the University Medical Center Groningen and University Hospital Jena.
Histological analysis
Lymph node and Burkitt's lymphoma (BL) tissue sections were deparaffinized; heat‐mediated antigen retrieval was done in citrate buffer for 15 min at the sub‐boiling point. Sections were cooled down to room temperature, washed three times with PBS, and were then blocked with 2% BSA in PBS for 1 h at room temperature. Primary antibody was incubated in 1% BSA, in a humidified chamber o.n. at 4°C with the following dilutions: TSC1 (Abcam, ab 40872) 1:75, CD20 (Abcam, ab 9475) 1:20, MYC (Abcam, ab 39688) 1:50, P‐S6 (Ser235/236) (Cell Signaling Technology, #4857) 1:75. Sections were washed with PBS and incubated with the fluorescence‐conjugated secondary antibody (Alexa Fluor 488 goat anti‐rabbit or Alexa Fluor 568 goat anti‐mouse, Invitrogen) in 1% BSA for 2 h at room temperature. After washing with PBS, sections were sealed with coverslips in diamine phenylindole (DAPI) containing mounting medium (Fluoroshield, ImmunoBioScience Corp.). Microscopic analysis was performed with the Zeiss AxioImager Z1 microscope. For the quantification of TSC1 and MYC staining in CD20‐positive B cells, images (200× magnified) of seven follicles of the superficial cortex of three control lymph nodes each (21 images) were compared with seven images taken from eight BL samples derived from different sites, lymph node (4×), cerebellum (1×), bone marrow (1×), oropharynx (1×), nasopharynx (1×). Analysis was performed using CellProfiler (Carpenter et al, 2006). Nuclei were visualized by the DAPI staining, and the cell outline was identified from the TSC1 staining. The TSC1‐positive cytoplasm was identified by subtracting the nuclear area from the cell outline, and TSC1 fluorescence intensities were measured in the cytoplasm of an average of 1,500 cells per image. Statistical significance of mean fluorescence intensities was tested with the t‐test (unpaired, two‐tailed) using R (R Development Core Team (2008). “R: A Language and Environment for Statistical Computing”. from http://www.R-project.org).
Author contributions
CFC supervised the project. GH and CFC conceived and designed the study. GH, CM, AK, LMZ, CD, RW, SE, and GK performed experiments. HS, JK, IP, and AB provided patient materials and advised on Burkitt's lymphoma. CK and Z‐QW provided recourses for and advised on mouse experiments. GH, CM, JK, AB, and CFC were involved in writing the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgments
We thank Dirk Eick from the Helmholtz Centre Munich for the P493‐6 cells, Daniel Murphy from the University of Glasgow for the U2OS‐MYC‐ER cells and helpful discussions, Marieke von Lindern from Sanquin in Amsterdam for the TSC1, TSC2, MYC, and control shRNA vectors, Martin Eilers from the Theodor Boveri Institute of the University of Würzburg for MYC expression vector, the pTK‐Ap2/E and pTK‐AP2/mut4 E‐box‐containing reporter constructs and advise on the related experiments. From the Friedrich Schiller University of Jena, we thank Berit Jungnickel for the BL2, Ramos, and DG75 cells, and Kim Zarse and Michael Ristow for advise on the experiments with antioxidants. At the FLI, we thank Annemarie Carlstedt for technical assistance, Simone Tänzer, Anne Gompf, and Ulrike Baschant for help with FACS, and Kristin Dreffke for providing the apoptosis assay protocol. J.K. and A vd B. were supported by the Pediatric Oncology Foundation Groningen (#SKOG 11‐001). G.H. was supported by the Deutsche Krebshilfe e.V. through a grant (#110193) to C.F.C. G.H. and A.K. were supported by the Leibniz Graduate School on Ageing and Age‐Related Diseases (LGSA; http://www.fli-leibniz.de/phd/), and L.M.Z. was supported by the Deutsche Forschungsgemeinschaft through a grant (CA 283/1‐1) to C.F.C. C.M. and G.K. are supported by the KWF kankerbestrijding (Dutch Cancer Society) to a grant to C.F.C (KWF 11010).
The EMBO Journal (2018) 37: e98589
References
- Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN (1997) Transduction of interleukin‐2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci USA 94: 3627–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amcheslavsky A, Ito N, Jiang J, Ip YT (2011) Tuberous sclerosis complex and Myc coordinate the growth and division of Drosophila intestinal stem cells. J Cell Biol 193: 695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas‐Tikhonenko A, Mendell JT (2008) Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 40: 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Zheng P (2008) TSC‐mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 205: 2397–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P (2007) mTOR controls mitochondrial oxidative function through a YY1‐PGC‐1alpha transcriptional complex. Nature 450: 736–740 [DOI] [PubMed] [Google Scholar]
- Dang CV (2012) MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dazert E, Hall MN (2011) mTOR signaling in disease. Curr Opin Cell Biol 23: 744–755 [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio‐Donahue C, Kern SE, Blair IA, Tuveson DA (2011) Oncogene‐induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475: 106–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine‐threonine protein kinase Akt. Science 275: 661–665 [DOI] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39: 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaubatz S, Imhof A, Dosch R, Werner O, Mitchell P, Buettner R, Eilers M (1995) Transcriptional activation by Myc is under negative control by the transcription factor AP‐2. EMBO J 14: 1508–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF (2004) The TSC1‐2 tumor suppressor controls insulin‐PI3K signaling via regulation of IRS proteins. J Cell Biol 166: 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA (1998) Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res 26: 362–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh H, Hao HX, Chan SL, Chen D, Ong R, Soo KC, Pochanard P, Yang D, Ruddy D, Liu M, Derti A, Balak MN, Palmer MR, Wang Y, Lee BH, Sellami D, Zhu AX, Schlegel R, Huang A (2015) Loss of tuberous sclerosis complex 2 (TSC2) is frequent in hepatocellular carcinoma and predicts response to mTORC1 inhibitor everolimus. Mol Cancer Ther 14: 1224–1235 [DOI] [PubMed] [Google Scholar]
- Ilic N, Utermark T, Widlund HR, Roberts TM (2011) PI3K‐targeted therapy can be evaded by gene amplification along the MYC‐eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci USA 108: E699–E708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffmann‐Zeh A, Rodriguez‐Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G (1997) Suppression of c‐Myc‐induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 385: 544–548 [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N (1997) The PI 3‐kinase/Akt signaling pathway delivers an anti‐apoptotic signal. Genes Dev 11: 701–713 [DOI] [PubMed] [Google Scholar]
- Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi‐Impiombato A, Califano A, Migliazza A, Bhagat G, Dalla‐Favera R (2010) The DLEU2/miR‐15a/16‐1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17: 28–40 [DOI] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV (2005) Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 25: 6225–6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, Muthalagu N, Rycak L, Rudalska R, Moll R, Kempa S, Zender L, Eilers M, Murphy DJ (2012) Deregulated MYC expression induces dependence upon AMPK‐related kinase 5. Nature 483: 608–612 [DOI] [PubMed] [Google Scholar]
- Manning BD (2004) Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol 167: 399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieulet V, Lamb RF (2010) Tuberous sclerosis complex: linking cancer to metabolism. Trends Mol Med 16: 329–335 [DOI] [PubMed] [Google Scholar]
- Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, Lowe SW, Pelletier J (2008) mTORC1 promotes survival through translational control of Mcl‐1. Proc Natl Acad Sci USA 105: 10853–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneux EM, Rochford R, Griffin B, Newton R, Jackson G, Menon G, Harrison CJ, Israels T, Bailey S (2012) Burkitt's lymphoma. Lancet 379: 1234–1244 [DOI] [PubMed] [Google Scholar]
- Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig‐Mueller N, Colinge J, Duernberger G, Nijman SM (2011) A chemical‐genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol 7: 787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajic A, Spitkovsky D, Christoph B, Kempkes B, Schuhmacher M, Staege MS, Brielmeier M, Ellwart J, Kohlhuber F, Bornkamm GW, Polack A, Eick D (2000) Cell cycle activation by c‐myc in a burkitt lymphoma model cell line. Int J Cancer 87: 787–793 [DOI] [PubMed] [Google Scholar]
- Pymar LS, Platt FM, Askham JM, Morrison EE, Knowles MA (2008) Bladder tumour‐derived somatic TSC1 missense mutations cause loss of function via distinct mechanisms. Hum Mol Genet 17: 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravitz MJ, Chen L, Lynch M, Schmidt EV (2007) c‐myc Repression of TSC2 contributes to control of translation initiation and Myc‐induced transformation. Cancer Res 67: 11209–11217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, Rosolowski M, Ammerpohl O, Wagener R, Bernhart SH, Lenze D, Szczepanowski M, Paulsen M, Lipinski S, Russell RB, Adam‐Klages S, Apic G, Claviez A, Hasenclever D, Hovestadt V et al (2012) Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet 44: 1316–1320 [DOI] [PubMed] [Google Scholar]
- Robertus JL, Kluiver J, Weggemans C, Harms G, Reijmers RM, Swart Y, Kok K, Rosati S, Schuuring E, van Imhoff G, Pals ST, Kluin P, van den Berg A (2010) MiRNA profiling in B non‐Hodgkin lymphoma: a MYC‐related miRNA profile characterizes Burkitt lymphoma. Br J Haematol 149: 896–899 [DOI] [PubMed] [Google Scholar]
- Rozengurt E, Soares HP, Sinnet‐Smith J (2014) Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther 13: 2477–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, Liu X, Powell J, Yang Y, Xu W, Zhao H, Kohlhammer H, Rosenwald A, Kluin P, Muller‐Hermelink HK, Ott G et al (2012) Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490: 116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J (2008) WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC; [Google Scholar]
- Wall M, Poortinga G, Stanley KL, Lindemann RK, Bots M, Chan CJ, Bywater MJ, Kinross KM, Astle MV, Waldeck K, Hannan KM, Shortt J, Smyth MJ, Lowe SW, Hannan RD, Pearson RB, Johnstone RW, McArthur GA (2013) The mTORC1 inhibitor everolimus prevents and treats Emu‐Myc lymphoma by restoring oncogene‐induced senescence. Cancer Discov 3: 82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidek LM, Ackermann T, Hartleben G, Eichwald S, Kortman G, Kiehntopf M, Leutz A, Sonenberg N, Wang Z‐Q, von Maltzahn J, Müller C, Calkhoven CF (2015) Deficiency in mTORC1‐controlled C/EBPβ‐mRNA translation improves metabolic health in mice. EMBO Reports 16: 1022–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File