

Abstract
Although lysosomes perform a number of essential cellular functions, damaged lysosomes represent a potential hazard to the cell. Such lysosomes are therefore engulfed by autophagic membranes in the process known as lysophagy, which is initiated by recognition of luminal glycoprotein domains by cytosolic lectins such as Galectin‐3. Here, we show that, under various conditions that cause injury to the lysosome membrane, components of the endosomal sorting complex required for transport (ESCRT)‐I, ESCRT‐II, and ESCRT‐III are recruited. This recruitment occurs before that of Galectin‐3 and the lysophagy machinery. Subunits of the ESCRT‐III complex show a particularly prominent recruitment, which depends on the ESCRT‐I component TSG101 and the TSG101‐ and ESCRT‐III‐binding protein ALIX. Interference with ESCRT recruitment abolishes lysosome repair and causes otherwise reversible lysosome damage to become cell lethal. Vacuoles containing the intracellular pathogen Coxiella burnetii show reversible ESCRT recruitment, and interference with this recruitment reduces intravacuolar bacterial replication. We conclude that the cell is equipped with an endogenous mechanism for lysosome repair which protects against lysosomal damage‐induced cell death but which also provides a potential advantage for intracellular pathogens.
Keywords: autophagy, endosome, lysophagy, lysosome, membrane repair
Subject Categories: Autophagy & Cell Death; Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction
Introduction
Lysosomes are essential organelles that carry out numerous cellular functions, including degradation of macromolecules, pathogen killing, and signaling functions. On the other hand, because of their low intraluminal pH and high content of Ca2+ and enzymes that can potentially trigger cell death, lysosome damage caused by pathogens, sharp crystals, amphiphilic drugs, or other membrane‐disrupting agents impose a serious threat to cell viability (Kroemer & Jaattela, 2005; Papadopoulos & Meyer, 2017). Previous work has uncovered a lysosome‐protective function of heat‐shock protein 70 (Kirkegaard et al, 2010) and the existence of an autophagic pathway that sequesters and degrades damaged lysosomes (Hung et al, 2013; Maejima et al, 2013). This pathway, termed lysophagy, is triggered by sensing of lysosomal membrane lesions by cytosolic lectins such as Galectin‐3, which recognize exposed intraluminal carbohydrate chains of lysosomal glycoproteins (Maejima et al, 2013; Papadopoulos & Meyer, 2017). This is followed by ubiquitination of lysosomal membrane proteins, processing by the ATPase p97, and mobilization of LC3‐containing autophagic membranes (Papadopoulos & Meyer, 2017).
The existence of pathways that protect lysosomes from damage and incapacitate damaged lysosomes has begged the question whether repair mechanisms for damaged lysosomes also exist (Papadopoulos & Meyer, 2017). Here, we have tested the hypothesis that the endosomal sorting complex required for transport (ESCRT) machinery (Henne et al, 2013) might play a role in lysosome repair and thereby have a cytoprotective function. Originally identified for its function in protein sorting to the yeast lysosome equivalent, the vacuole (Katzmann et al, 2002), the ESCRT machinery has recently been shown to have a more general function in membrane involution and scission processes that occur in the direction “away” from cytosol, such as virus budding from the plasma membrane, daughter cell separation during cytokinesis, and sealing of the nuclear envelope during mitotic exit (Christ et al, 2017; Schoneberg et al, 2017). Interestingly, the ESCRT machinery has also been shown to mediate repair of both the plasma membrane and the nuclear envelope (Jimenez et al, 2014; Scheffer et al, 2014; Denais et al, 2016; Raab et al, 2016), raising the possibility that it might also function in repair of other membranes.
Based on biochemical and genetic evidence, the ESCRT machinery can be divided into four subcomplexes termed ESCRT‐0, ESCRT‐I, ESCRT‐II, and ESCRT‐III, of which ESCRT‐III is thought to mediate membrane sealing/scission through formation of membrane‐active oligomeric filaments (Christ et al, 2017). Even though ESCRT‐0 and ESCRT‐II are important during endosomal protein sorting into intraluminal vesicles of endosomes (Katzmann et al, 2001, 2003; Babst et al, 2002b; Raiborg et al, 2002; Bache et al, 2003), these subcomplexes appear to be dispensable for most known ESCRT functions (Christ et al, 2017). On the other hand, ESCRT‐I is frequently involved in recruitment of ESCRT‐III, and so is ALIX, a Bro1 domain‐containing protein which can potentially form a physical link between ESCRT‐I and ESCRT‐III (Bissig & Gruenberg, 2014; Christ et al, 2017).
Here, we show that ESCRT‐III is indeed recruited to damaged lysosomes and that this requires ESCRT‐I and ALIX. Interference with this mechanism abolishes the cell's ability to repair damaged lysosomes and causes otherwise reversible lysosome damage to become cell lethal. Surprisingly, we also find that ESCRTs are not only recruited to vacuoles containing the replicating form of the intracellular bacterium Coxiella burnetii, but they also provide the bacterium with an advantage by maintaining a niche for its intracellular replication.
Results
The ESCRT‐III subunit CHMP4B is recruited to damaged lysosomes
In order to achieve permeabilization of lysosomes in an acute manner, we used L‐leucyl‐L‐leucine methyl ester (LLOMe), which is converted into a membranolytic polymeric form in the lysosome lumen by lysosomal hydrolases (Thiele & Lipsky, 1990). Using an antibody against Galectin‐3 as marker for lysosome permeabilization (Paz et al, 2010), we observed by fluorescence microscopy that Galectin‐3 translocated to vesicular structures in HeLa cells treated with LLOMe, and its co‐occurrence with the lysosome marker LAMP1 confirmed that these structures are lysosomes (Fig 1A). We generated a stable HeLa line expressing low levels of mCherry‐tagged Galectin‐3 and the major ESCRT‐III subunit CHMP4B tagged with eGFP and monitored the effect of LLOMe on CHMP4B distribution. Interestingly, whereas both mCherry‐Galectin‐3 and CHMP4B‐eGFP displayed mainly cytosolic staining in untreated cells, treatment with a low (250 μM) concentration of LLOMe for 1 h caused a strong redistribution of both molecules to vesicles positive for the late endosome/lysosome marker CD63 (Fig 1B). Likewise, antibodies against CHMP4B stained CD63‐, Galectin‐3‐, and LAMP1‐positive lysosomes after LLOMe incubation of HeLa, RPE‐1, and H‐460 cells, in contrast to untreated cells (Fig EV1A–C). We conclude that LLOMe‐induced membrane damage causes recruitment of CHMP4B to lysosomes in various cell types.
Figure 1. The ESCRT‐III subunit CHMP4B is recruited to Galectin‐3‐positive damaged endolysosomal membranes.
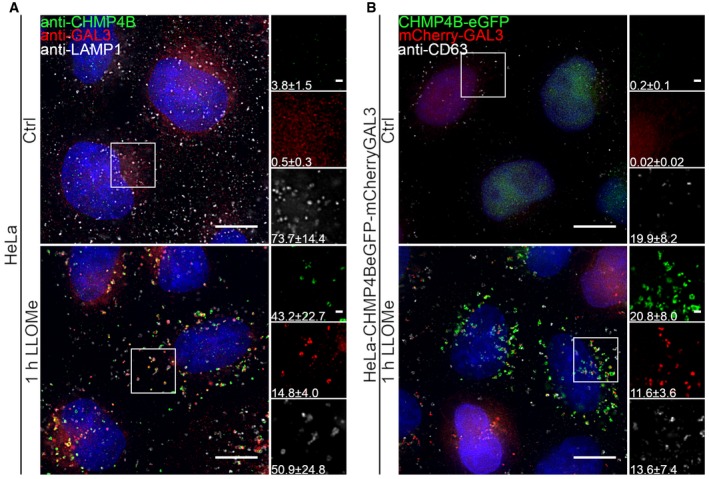
- Representative fluorescence micrographs of HeLa cells treated with 250 μM lysosomotropic drug LLOMe or equal volume of DMSO (Ctrl) for 1 h before fixation and immunostained with Hoechst (blue), anti‐CHMP4B (green), anti‐GAL3 (red), and anti‐LAMP1 (white) are presented. Cells treated with LLOMe show increased recruitment of ESCRT‐III protein to lysosomes when compared to Ctrl cells. Number of foci per cell was quantified and is indicated as mean ± SD. Data are quantified from >86 cells per condition from three independent experiments. CHMP4B, GAL3, LAMP1 foci per cell (Ctrl versus LLOMe treatment): P = 0.0398, P = 0.0033, P = 0.2400 (Student's t‐test), respectively.
- HeLa cells stably expressing CHMP4B‐eGFP and mCherry‐Galectin‐3 treated as in (A) and stained with CD63 antibody are shown. Number of foci per cell was quantified and is indicated as mean ± SD. Data are quantified from >87 cells per condition from three independent experiments. CHMP4B, GAL3, CD63 foci per cell (Ctrl versus LLOMe treatment): P = 0.0111, P = 0.0050, P = 0.3793 (Student's t‐test), respectively.
Figure EV1. CHMP4B is recruited to Galectin‐3‐positive damaged endolysosomal membranes in various cell lines.
- Representative fluorescence images of a HeLa‐mCherry‐Galectin‐3 stable cell line treated with 250 μM lysosomotropic drug LLOMe for 1 h, fixed and labeled with CD63 antibody. To determine the statistical significance, the number of CHMP4B, GAL3, and CD63 foci per cell observed after LLOMe treatment was compared to Ctrl using Student's t‐test. P‐values for CHMP4B, GAL3, and CD63 are as follows: P = 0.000001, P = 0.0017, P = 0.4160, respectively.
- Experimental setup as in (A) where RPE‐1 cells are labeled with CHMP4B, Galectin‐3, and LAMP1 antibodies. P‐values for CHMP4B, GAL3, and LAMP1 foci per cell (Ctrl versus LLOMe treatment): P = 0.00001, P = 0.00018, P = 0.1368 (Student's t‐test), respectively.
- Representative images of fixed H‐460 cells after 1‐h treatment with 250 μM LLOMe. Cells were stained for endogenous CHMP4B, GAL3, and LAMP1. CHMP4B, GAL3, and LAMP1 foci per cell (Ctrl versus LLOMe treatment): P = 0.0192, P = 0.0493, P = 0.6333 (Student's t‐test), respectively.
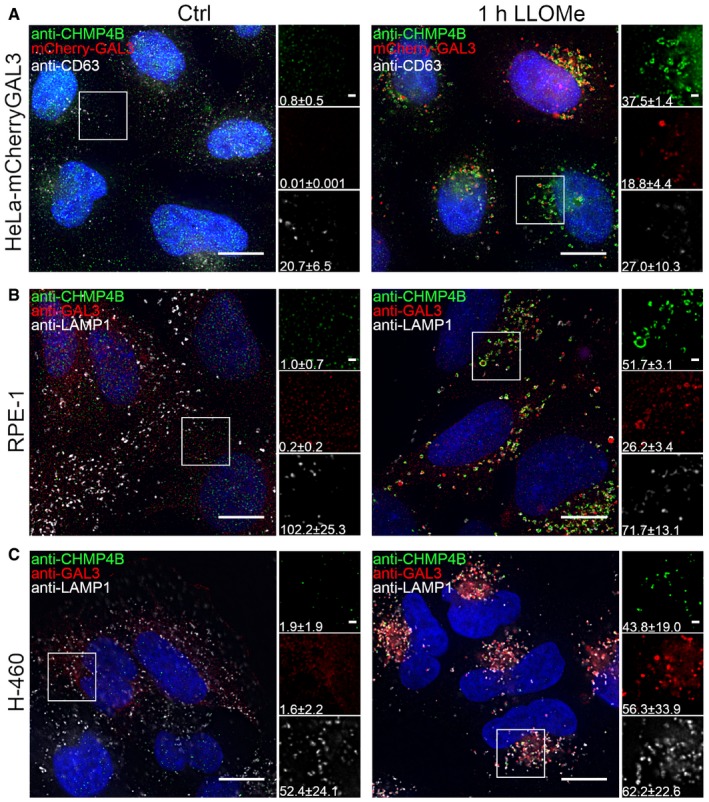
We next investigated whether other agents that cause lysosomal membrane damage also induce CHMP4B recruitment. Glycyl‐L‐phenylalanine‐beta‐naphthylamide (GPN) is known to cause lysosomal membrane permeabilization via osmotic swelling (Berg et al, 1994), and we indeed observed that this compound caused a profound recruitment of both mCherry‐Galectin‐3 and CHMP4B‐eGFP to LAMP1‐containing lysosomes (Fig EV2A). Amphiphilic antihistamines have been shown to induce permeabilization of the lysosome membrane (Ellegaard et al, 2016), and the antihistamine terfenadine induced recruitment of Galectin‐3 and CHMP4B to LAMP1‐positive lysosomes in RPE‐1 cells, as detected with antibodies against the endogenous proteins (Fig EV2B). Collectively, these results show that CHMP4B, like Galectin‐3, is recruited to lysosomes upon various types of membrane damage.
Figure EV2. CHMP4B is recruited to damaged endolysosomal membranes after treatment with different agents.
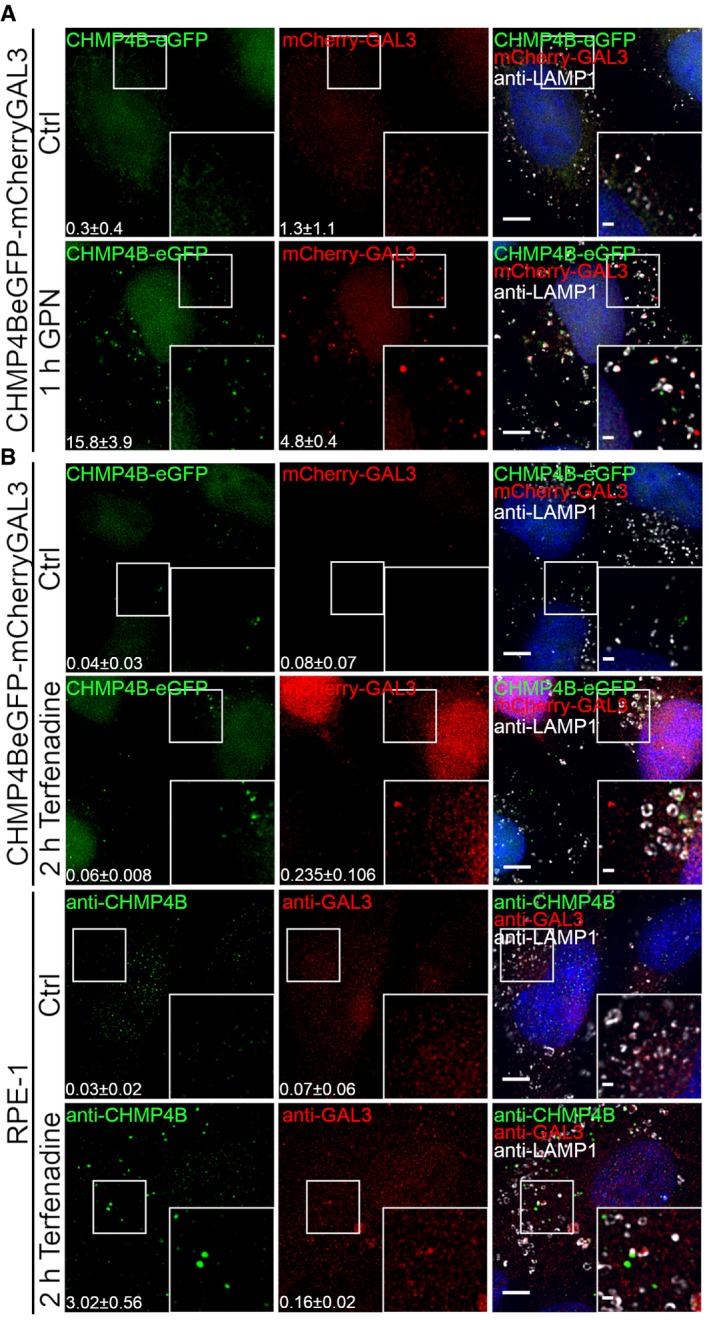
- HeLa cells stably expressing CHMP4B‐eGFP and mCherry‐Galectin‐3 were treated with 250 μM lysosomotropic drug GPN for 1 h, fixed and imaged. Representative images showing recruitment of CHMP4B to damaged endolysosomal membranes are presented. Number of foci per cell (>90 cells per condition from four independent experiments) was quantified and is indicated as mean ± SD. P‐values for comparisons CHMP4B, GAL3, and LAMP1 foci per cell (Ctrl versus GPN treatment) are as follows: P = 0.0002, P = 0.0008, P = 0.1113 (Student's t‐test), respectively.
- Representative fluorescence images showing recruitment of CHMP4B to damaged membranes in HeLa cells stably expressing CHMP4B‐eGFP and mCherry‐Galectin‐3 and RPE‐1 cells after 2‐h treatment with 7 μm cationic amphiphilic drug terfenadine. In HeLa cells, there is a slight but not significant increase in CHMP4B‐positive foci. On the other hand, RPE‐1 cells labeled with CHMP4B, GAL3, and LAMP1 antibodies have elevated number of CHMP4 and GAL3 foci per cell. For HeLa‐CHMP4BeGFP‐mCherry‐Galectin‐3: CHMP4B, GAL3 foci per cell (Ctrl versus terfenadine treatment): P = 0.2679, P = 0.2266 (Student's t‐test), respectively. For RPE‐1 cell line: CHMP4B, GAL3 foci per cell (Ctrl versus terfenadine treatment): P = 0.0169, P = 0.1806 (Student's t‐test), respectively. Number of foci per cell was quantified from >75 cells per condition from two independent experiments and is indicated in the figure as mean ± SD.
ESCRT‐I, ESCRT‐II, ESCRT‐III, ALIX, and VPS4A are recruited to damaged lysosomes
The observed recruitment of CHMP4B to damaged lysosomes begged the question of which other ESCRT subunits are recruited. Specifically, because ESCRT‐0, ESCRT‐I, and ESCRT‐II are upstream of ESCRT‐III in endosomal sorting (Babst et al, 2002a; Bache et al, 2003), we wondered if this might be the case with recruitment to damaged lysosomes as well. In HeLa cells treated with LLOMe for 30 min, we found no evidence for recruitment of the ESCRT‐0 component HRS. In contrast, the ESCRT‐I subunit TSG101 and the ESCRT‐II subunit EAP30 were clearly recruited, as was VPS4A (Schoneberg et al, 2017), an ATPase that controls ESCRT‐III dynamics (Fig 2). As expected, not only CHMP4B, but also another ESCRT‐III subunit, CHMP2A was found to be recruited, as was the ESCRT‐III‐related protein IST1 (Figs 2 and EV3). Interestingly, ALIX, a Bro1 domain‐containing protein that can bridge ESCRT‐I with ESCRT‐III (Bissig & Gruenberg, 2014), was also recruited, whereas we were unable to detect recruitment of another Bro1 domain protein, HD‐PTP (Fig 2). We were also unable to detect recruitment of the VPS4 isoform VPS4B (Fig EV3). From these studies, we conclude that ESCRT‐I, ESCRT‐II, ESCRT‐III, ALIX, and VPS4A are recruited to damaged lysosomes.
Figure 2. ESCRT‐I, ESCRT‐II, ESCRT‐III, ALIX, and VPS4A are recruited to endolysosomes upon LLOMe treatment.
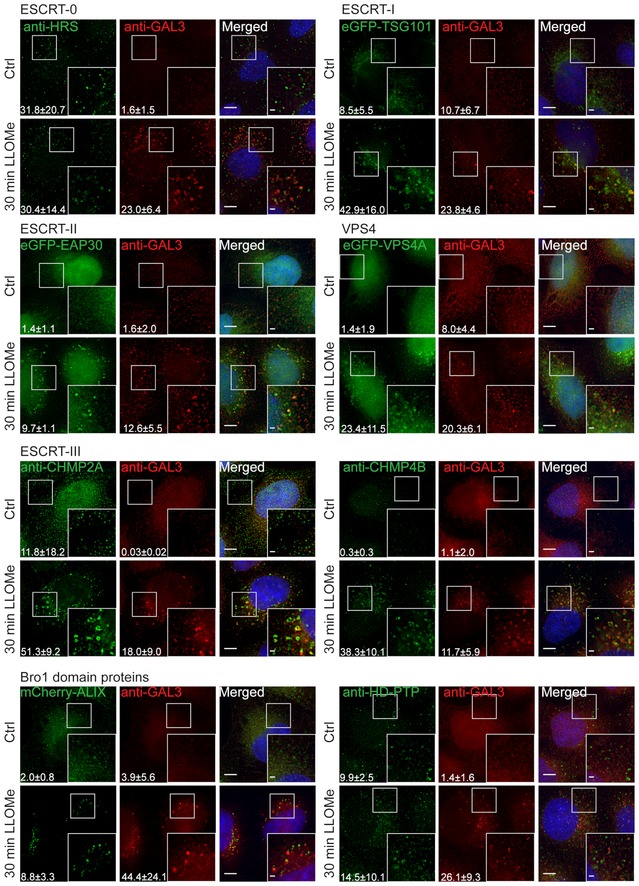
To screen for ESCRT proteins that are involved in the endolysosomal repair process, cells were incubated with 250 μM LLOMe for 30 min and processed for immunofluorescence. TSG101, a component of the ESCRT‐I complex, and EAP30, an ESCRT‐II protein, are clearly recruited to the sites of damage compared to the DMSO control (Ctrl). As shown, the ESCRT‐III complex together with ALIX is recruited to damaged endolysosomes. In contrast, HRS, a component of the ESCRT‐0 complex, and HD‐PTP show no recruitment to damaged endomembranes. Number of foci per cell was quantified from >85 cells per condition from three independent experiments and are presented as mean ± SD. Statistical significance for number of foci per cell in Ctrl versus LLOMe treatment was determined using Student's t‐test, the P‐values for which are as follows: HRS P = 0.9257, GAL3 P = 0.0050; TSG101 P = 0.0243, GAL3 P = 0.0489; EAP30 P = 0.0006, GAL3 P = 0.0307; CHMP2A P = 0.0284, GAL3 P = 0.0260; CHMP4B P = 0.0028, GAL3 P = 0.0414; VPS4A P = 0.0309, GAL3 P = 0.0461; ALIX P = 0.0249, GAL3 P = 0.0471; HD‐PTP P = 0.4898, GAL3 P = 0.0105. Scale bars: 5 μm and 1 μm (inset).
Figure EV3. Screening for additional ESCRT proteins that are involved in the lysosomal repair process.
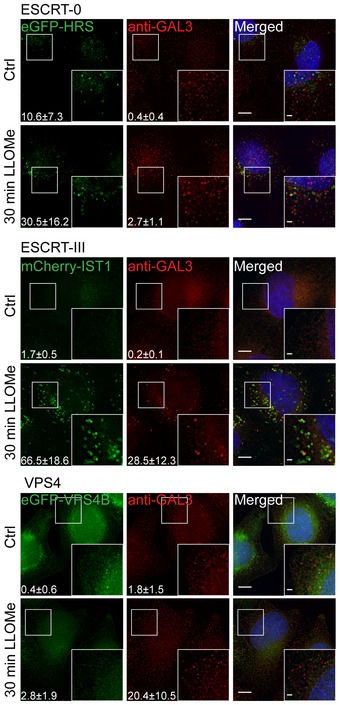
ESCRT‐0: To clarify the role of HRS, a cell line stably expressing eGFP‐HRS was incubated with 250 μm LLOMe for 30 min, pre‐permeabilized with PEM buffer (for details see Materials and Methods), and fixed. HRS shows no significant changes upon lysosomal damage when compared to untreated cells. ESCRT‐III: After treatment with 250 μm LLOMe for 30 min, HeLa cells stably expressing mCherry‐IST1 or eGFP‐VPS4B were fixed and imaged. As shown in the representative fluorescence images, IST1 is recruited to damaged lysosomes whereas VPS4B shows no change in localization upon endolysosomal membrane damage. Number of foci per cell was quantified and is indicated as mean ± SD. Data were obtained from >71 cells per condition from three independent experiments. Number of foci per cell (Ctrl versus LLOMe treatment): HRS P = 0.1241, GAL3 P = 0.0309 (Student's t‐test); IST1 P = 0.0038, GAL3 P = 0.0164 (Student's t‐test); VPS4B P = 0.1102, GAL3 P = 0.0387 (Student's t‐test). Scale bars: 5 μm and 1 μm (inset).
TSG101 depletion inhibits CHMP4B recruitment on damaged lysosomes, whereas CHMP2A knockdown stabilizes it
The above results suggested that TSG101 and ALIX might have a role in ESCRT‐III recruitment, and we tested this further using siRNA to deplete various ESCRT proteins in HeLa cells expressing CHMP4B‐eGFP and monitoring CHMP4B‐eGFP recruitment by live‐cell microscopy. Knockdown of the ESCRT‐0 subunit HRS was without effect on CHMP4B‐eGFP recruitment to lysosomes upon LLOMe treatment of the cells (Fig 3A, Movie EV1), consistent with our finding that this ESCRT subunit is not recruited itself. On the other hand, depletion of TSG101 caused a strong delay in CHMP4B‐eGFP recruitment (Fig 3B, Movie EV2). Conversely, depletion of CHMP2A caused increased accumulation of CHMP4B‐eGFP on LLOMe‐damaged lysosomes (Fig 3C, Movie EV3), in agreement with previous studies on ESCRT recruitment to other membranes, suggesting that CHMP2A limits the extent of CHMP4B recruitment (Vietri et al, 2015). Whereas depletion of ALIX was without detectable effect on CHM4B‐eGFP recruitment (Fig 3D, Movie EV4), co‐depletion of TSG101 and ALIX led to an almost complete lack of CHMP4B‐eGFP recruitment (Fig 3E, Movie EV5). These results confirm that TSG101 in ESCRT‐I is upstream of ESCRT‐III in recruitment to damaged lysosomes and also reveal a role for ALIX.
Figure 3. Dynamics of CHMP4B accumulation at the damaged endolysosomal membranes depend on other ESCRT components.
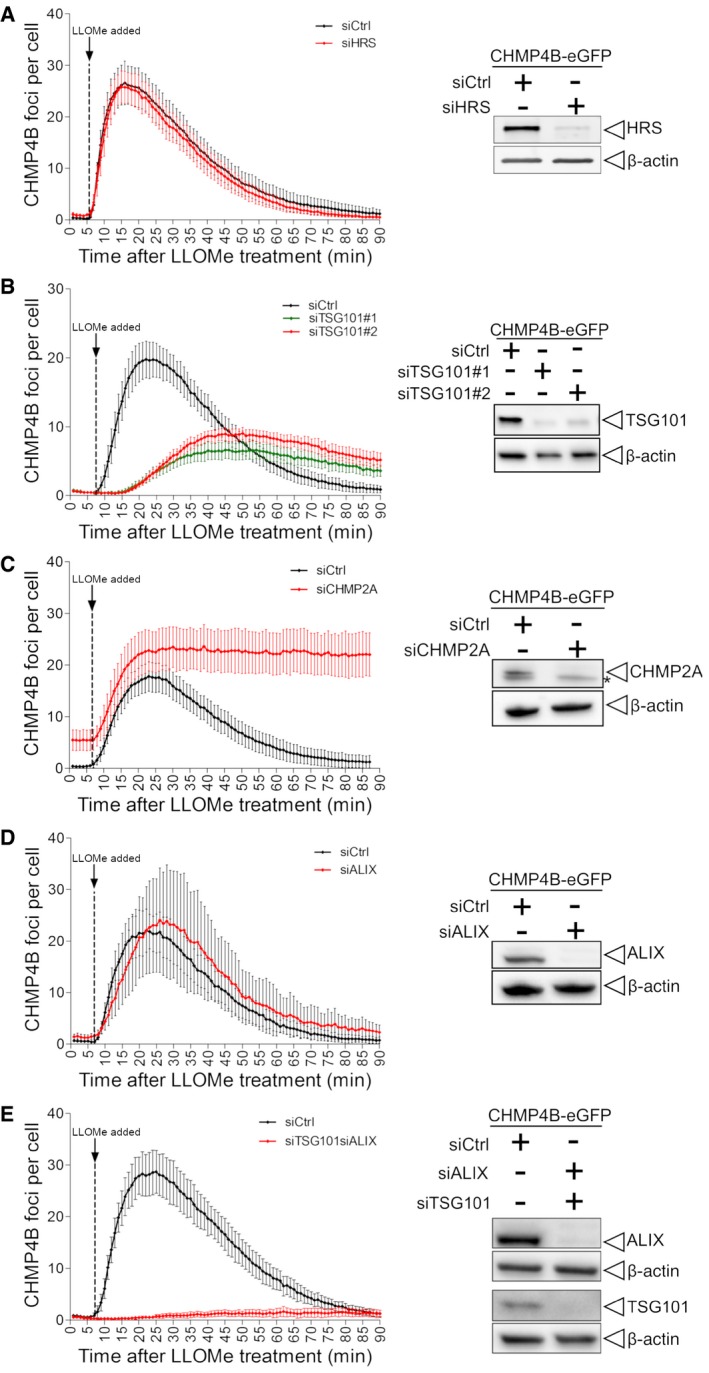
- Depletion of HRS does not alter the dynamics of CHMP4B‐eGFP recruitment as compared to siCtrl during treatment with LLOMe.
- Downregulation of TSG101, with two independent siRNAs, causes a delay in CHMP4B‐eGFP recruitment as compared to siCtrl, indicating an important role of the ESCRT‐I complex in recruiting the downstream components.
- CHMP2A depletion accumulates and stabilizes CHMP4B‐eGFP‐positive foci.
- siRNA‐mediated depletion of ALIX shows no significant change on dynamics of CHMP4B recruitment.
- Simultaneous depletion of TSG101 and ALIX caused almost no recruitment of CHMP4B upon induction of endolysosomal damage.
ESCRT is required for repair of the lysosome membrane
We next asked whether the ESCRT machinery is involved in repair of damaged lysosome membranes. As an assay for membrane permeability, we used the ability of lysosomes to retain Lysotracker, a weak base which accumulates in acidic lysosomes and is fluorescent at low pH (Chazotte, 2011). As expected, incubation of HeLa cells with LLOMe at 250 μM caused reduced Lysotracker staining of lysosomes (Fig 4A). Interestingly, however, at this relatively low LLOMe concentration, Lysotracker staining fully recovered within 30–60 min, indicating that the LLOMe‐induced membrane damage was repaired (Fig 4A and B). Importantly, whereas Lysotracker staining recovered after LLOMe treatment of control cells, lysosomes in cells depleted for TSG101 and ALIX failed to recover Lysotracker fluorescence (Fig 4C). This indicates that the ESCRT machinery mediates repair of damaged lysosomes.
Figure 4. ESCRTs are essential for the repair after endolysosomal damage.
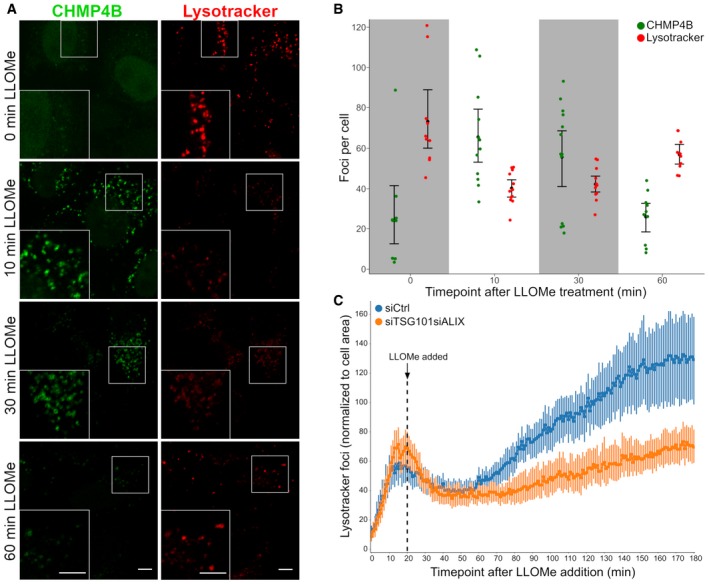
- HeLa cells stably expressing CHMP4B‐eGFP were treated with 250 μM LLOMe and 75 nM Lysotracker DND‐99 and fixed at different time points as indicated. After 10 min of LLOMe treatment, CHMP4B is recruited whereas the number of Lysotracker‐positive foci is reduced. After 30 min, lysosomes gain back functionality (judging by the increased number of Lysotracker foci) and appear recovered after 1 h indicating that the ESCRT complex is able to seal the damaged endolysosomal membranes. Representative confocal images for each time point are shown. Scale bars: 5 μm.
- Quantification graph (>250 cells per condition from four independent experiments) showing CHMP4B and Lysotracker‐positive foci per cell at different time points. Error bars correspond to 95% confidence intervals.
- Quantification graph showing dynamics of Lysotracker recovery in control (siCtrl) and both TSG101‐ and ALIX‐depleted cells. HeLa cells stably expressing CHMP4B‐eGFP were co‐transfected with siRNAs against TSG101 and ALIX. Forty‐eight hours post‐transfection, cells were pre‐treated for 20 min with 75 nM Lysotracker Deep Red, which was used as a read‐out. While the decrease in the number of Lysotracker spots is quickly recovered in siCtrl, simultaneous depletion of ALIX and TSG101 leads to a severe impairment in lysosomal repair. Data from >40 cells per condition from four independent live‐cell imaging experiments are shown. Graph is normalized to the area occupied by the cells. Error bars correspond to 95% confidence intervals.
ESCRT‐III shows much faster recruitment kinetics than Galectin‐3 to damaged lysosomes
In order to monitor the dynamics of ESCRT recruitment to damaged lysosomes, we used live microscopy to study HeLa cells stably expressing low levels of CHMP4B‐eGFP and mCherry‐Galectin‐3. Upon LLOMe addition, mCherry‐Galectin‐3 accumulated slowly on the lysosomes as expected (Fig 5). Remarkably, CHMP4B‐eGFP accumulated much faster, with detectable levels already after 1 min and reaching a peak level at about 10 min (Fig 5 and Movie EV6). This indicates that the ESCRT machinery detects a subtler membrane damage than the exposure of intraluminal β‐galactosides detected by Galectin‐3.
Figure 5. CHMP4B is recruited before Galectin‐3 to the damaged endolysosomal membranes.
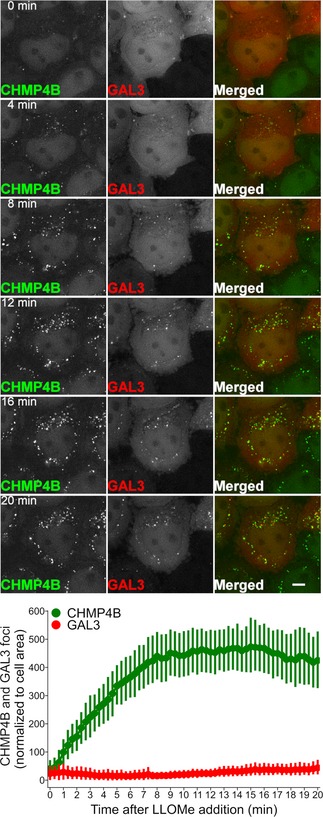
Representative movie montage of a live‐cell imaging experiment illustrating the dynamics of CHMP4B‐eGFP and mCherry‐Galectin‐3 recruitment at different time points before and after LLOMe treatment. Using the perfusion system, 250 μM LLOMe was added to HeLa cells stably expressing CHMP4B‐eGFP and mCherry‐Galectin‐3 and images were acquired every 20 s for a period of 20 min. As shown in the representative movie montage and the quantification graph, CHMP4B recruitment precedes Galectin‐3, which is an established marker for detection of damaged endolysosomal membranes. The quantification graph is normalized to the area occupied by the cells. Data were obtained from >40 cells per condition from three independent experiments. Error bars correspond to 95% confidence intervals. Scale bars: 5 μm.
ESCRT‐III recruitment to damaged lysosomes precedes lysophagy
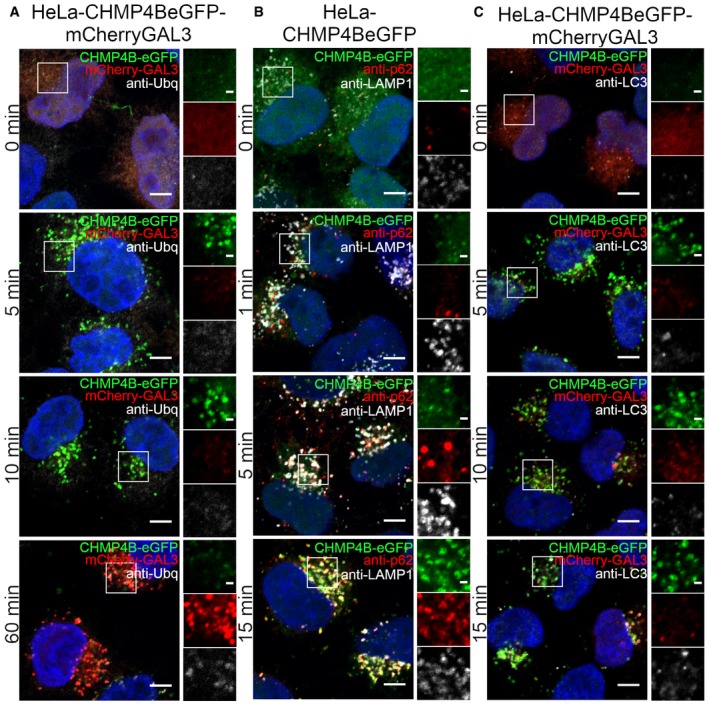
Because Galectin‐3 recruitment to lysosomes triggers lysophagy, we next compared the dynamics of CHMP4B recruitment with the dynamics of various molecules involved in lysophagy by confocal microscopy of HeLa cells fixed at various time points after LLOMe addition. Ubiquitination of lysosomal membrane proteins is known to occur early during lysophagy (Maejima et al, 2013), and we therefore monitored the kinetics of ubiquitin acquisition on lysosomes using an antibody against conjugated ubiquitin. Like with Galectin‐3 recruitment, ubiquitin appeared on the damaged lysosomes much slower than CHMP4B (Figs 6A and EV4A). We also studied recruitment of the autophagy adaptor p62/SQSTM1, a protein known to connect ubiquitinated autophagic cargoes to autophagic membranes (Bjørkøy et al, 2005; Maejima et al, 2013), and a major constituent of the autophagy machinery, LC3 (Kabeya et al, 2000; Maejima et al, 2013). Both p62 and LC3 appeared on damaged lysosomes with much slower kinetics than CHMP4B (Figs 6B and C, and EV4B and C). The autophagy inhibitor SAR405 (Ronan et al, 2014) was without effect on CHMP4B‐eGFP recruitment (Movie EV7). We conclude from these studies that the ESCRT machinery is recruited much prior to the autophagy machinery to damaged endolysosomes.
Figure 6. ESCRT‐III recruitment to damaged lysosomes precedes lysophagy.
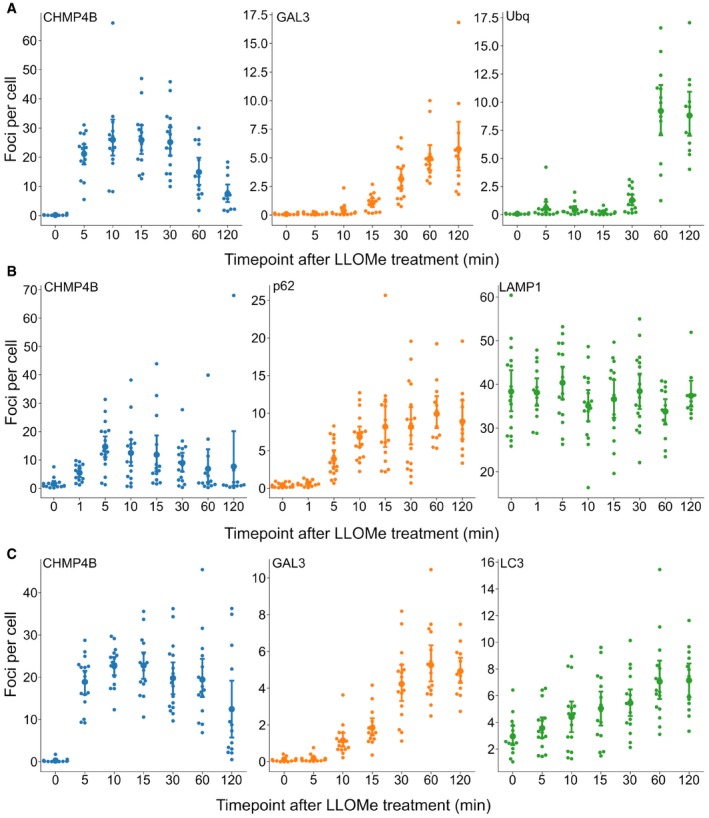
- HeLa cells stably expressing CHMP4B‐eGFP and mCherry‐Galectin‐3 were incubated with 250 μM LLOMe, fixed at different time points as indicated and stained for ubiquitin. As presented, CHMP4B is recruited before ubiquitin and Galectin‐3 upon lysosomal membrane damage.
- HeLa cells stably expressing CHMP4B‐eGFP were treated as in (A) and stained for p62. CHMP4B is recruited before p62 on the damaged lysosomes. The number of LAMP1‐positive foci appears stable independently of LLOMe treatment.
- Experimental setup as in (A and B) where HeLa cells stably expressing CHMP4B‐eGFP and mCherry‐Galectin‐3 were stained for LC3. As shown, CHMP4B is recruited prior to LC3.
Figure EV4. Representative confocal images corresponding to Fig 6 .
-
ARecruitment of CHMP4B as early as 5 min following LLOMe treatment whereas ubiquitin appears to be recruited later.
-
B, CRecruitment of CHMP4B to damaged endolysosomal membranes precedes p62 and LC3. In addition, p62 is recruited before LC3.
ESCRT recruitment protects cells against cell death caused by lysosome damage
Having found that ESCRT recruitment mediates lysosome repair and occurs prior to lysophagy, we next addressed the biological importance of this mechanism. By live‐cell imaging of TSG101‐ALIX‐depleted cells treated with LLOMe, we had observed that some cells started dying upon prolonged incubation (Movie EV5), and we therefore hypothesized that ESCRT‐mediated lysosome repair might provide a cell survival advantage. To test this hypothesis, we depleted cells of TSG101, ALIX, or both and used flow cytometry to monitor cell death 3, 6, and 10 h after LLOMe addition. Strikingly, whereas LLOMe at 250 μM had minimal effect on cell viability, siRNA‐mediated depletion of TSG101 led to a dramatic rise in cell death in the presence of LLOMe. While depletion of ALIX had only a minor effect on cell death, the combined depletion of both TSG101 and ALIX led to death of a large fraction of the cells (Figs 7 and EV5A and B, Movie EV8). This indicates that ESCRT‐mediated lysosome repair plays an important role in promoting cell viability after lysosome injury.
Figure 7. ESCRTs are essential for cell viability after endolysosomal damage.
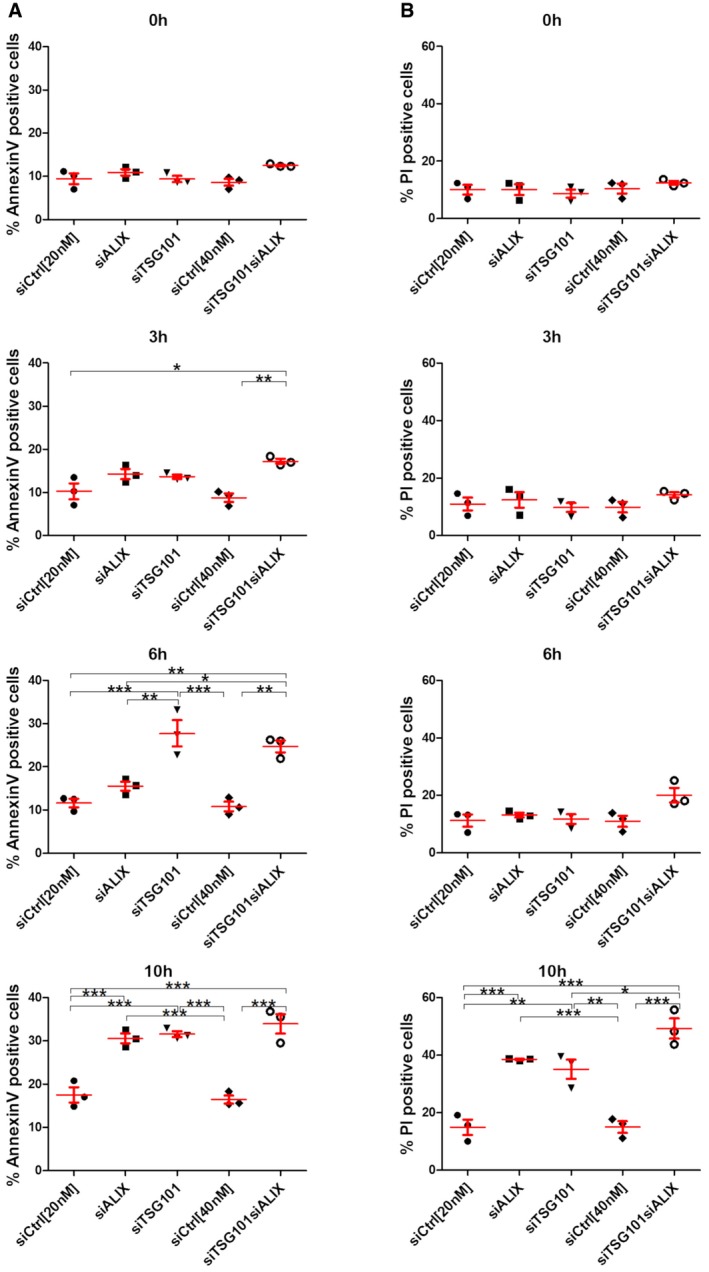
- HeLa cells were transfected with different siRNAs as indicated. Forty‐eight hours post‐transfection, cells were treated with 250 μM LLOMe for 3, 6, and 10 h harvested and stained for Annexin V. Cells were immediately processed for flow cytometry. Already after 3 h of treatment with LLOMe, cells co‐transfected with TSG101+ALIX siRNA showed an increase in apoptotic cell death, whereas TSG101 siRNA‐transfected cells showed elevated Annexin V staining after 6 h of treatment.
- Experimental set up as in (A) with additional propidium iodide (PI) staining of transfected HeLa cells.
Figure EV5. ESCRTs are essential for cell viability after endolysosomal damage.
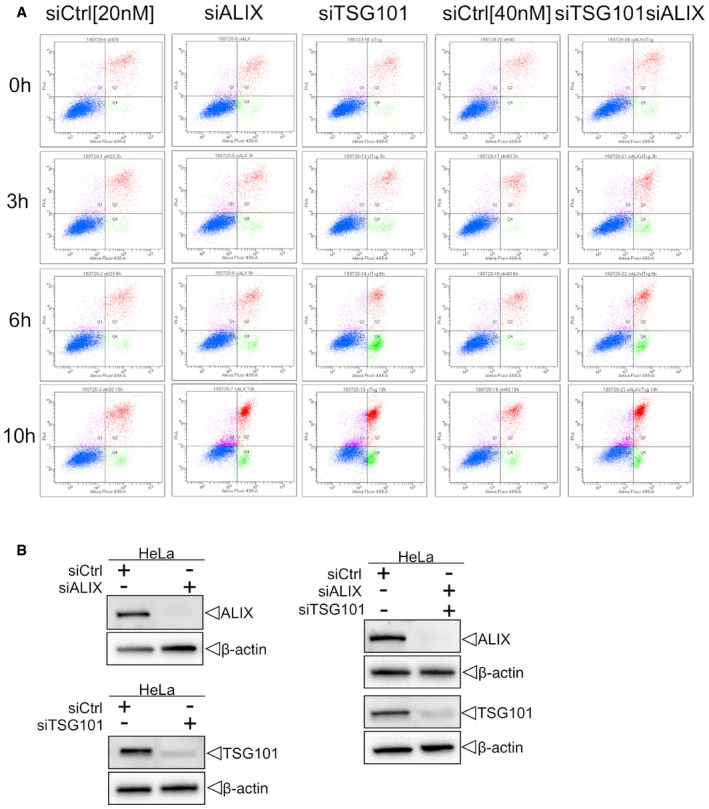
- Cells were transfected with control (siCtrl), TSG101, ALIX, or both siRNAs. Forty‐eight hours after transfection, cells were harvested and profiled with flow cytometry. Dot plots and gating strategy of one out of three experiments are shown.
- HeLa cells were transfected with control (siCtrl), ALIX, TSG101, or both siRNA. Efficient depletion was confirmed by Western blot analysis. β‐actin was used as loading control.
Source data are available online for this figure.
ESCRT recruitment to Coxiella burnetii vacuoles promotes bacterial replication
Many intracellular pathogens are able to survive and even replicate within modified phagosomes of host cells (Hybiske & Stephens, 2008). One very interesting example is the small Gram‐negative bacterium Coxiella burnetii, the causative agent of Q fever (Pechstein et al, 2017). Coxiella burnetii is an obligate intracellular pathogen which has the remarkable property of replicating inside an acidic lysosome‐like vacuole. A recent study has revealed that Galectin‐3 is recruited to C. burnetii vacuoles (Mansilla Pareja et al, 2017), and we therefore asked whether the ESCRT machinery might be recruited as well. To address this question, we infected HeLa cells expressing mCherry‐Galectin‐3 and CHMP4B‐eGFP with C. burnetii and used long‐time live microscopy to monitor protein recruitment. Interestingly, after a lag time of several hours, bacterium‐containing vacuoles became positive for both Galectin‐3 and CHMP4B before they quickly turned negative again. This was repeated several times (Fig 8A and Movie EV9), and the vacuole expansion that occurred after CHMP4B and Galectin‐3 recruitment indicated that the replicative niche was kept intact (Movie EV10). We interpret this as sporadic ruptures of the vacuole membrane which were repeatedly repaired by the ESCRT machinery.
Figure 8. ESCRT‐III and Galectin‐3 are recruited to the Coxiella burnetii vacuole.
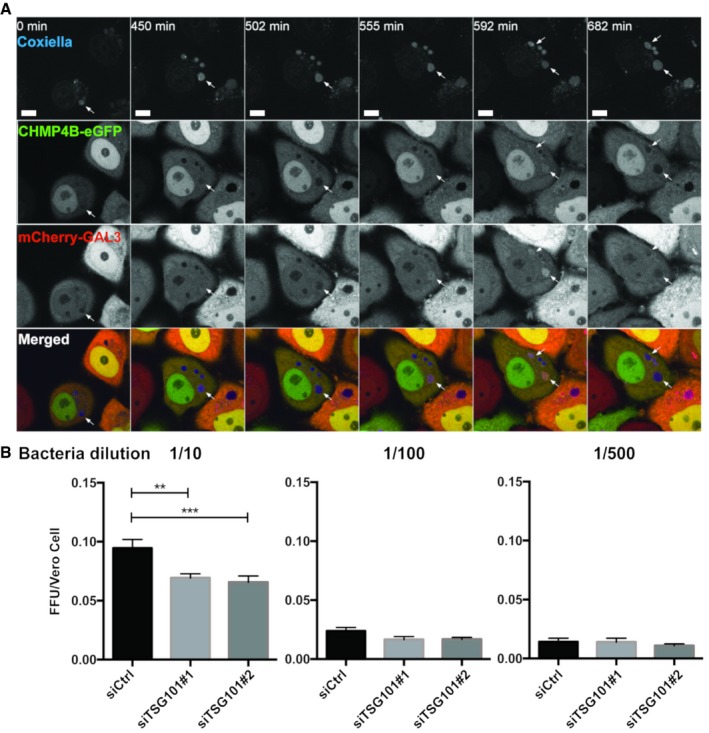
- HeLa cells stably expressing CHMP4B‐eGFP were transfected with the mCherry‐Galectin‐3 plasmid. Twenty‐four hours later, cells were infected with WT Coxiella burnetii. Live‐cell imaging started 24 h post‐infection, and several time points are indicated during the next 24 h of observation (time indicated in minutes, see Movie EV9). Arrows indicate Coxiella‐containing vacuole (DRAQ5 labeling) becoming positive for CHMP4B‐eGFP and mCherry‐Galectin‐3. Scale bars: 50 μm.
- Bacterial viability and replication assay upon TSG101 KD. HeLa cells were treated with either siCtrl or with two different siTSG101 for 48 h. Then, cells were infected with mCherry‐Coxiella burnetii for 48 h before lysis and serial dilutions were made to infect Vero cells. Seventy‐two hours later, infected cells were fixed, DAPI stained, and processed for quantitative image analysis. Between 30 and 40 fields representing more than 9,000 cells per condition of three independent experiments were analyzed. Error bars represent SD. Statistical significance was determined using one‐way ANOVA test. **P ≤ 0.01 and ***P ≤ 0.001.
We hypothesized that failure of ESCRT‐mediated vacuole repair might interfere with bacterial infection. To test this hypothesis, we performed a fluorescent foci unit (FFU) assay in which we used lysates from C. burnetii‐containing vacuoles from HeLa cells with or without depletion of TSG101 to infect Vero cells at different dilutions. Interestingly, the bacterial viability and replication FFU assay showed that depletion of TSG101 decreased C. burnetii replication (Fig 8B). We conclude that the ESCRT machinery provides the bacterium a replicative advantage which can probably be explained by the requirement of an intact vacuole for efficient C. burnetii replication to proceed.
Discussion
Here, we have shown that ESCRT‐mediated repair of damaged lysosomes occurs independently of lysophagy, which is in excellent agreement with results reported in a very recent paper (Skowyra et al, 2018). In addition, we show that this mechanism is crucial for promoting cell viability after lysosome injury and that ESCRT‐mediated membrane repair can also be exploited for maintaining an intact replicative niche for an intracellular pathogen. Thus, ESCRT‐mediated repair of lysosomal membranes can have both beneficial and harmful consequences for the host cell.
Whereas lysophagy is triggered by holes in the lysosome membrane large enough for detection of intraluminal β‐galactosides by cytosolic lectins such as Galectin‐3, the ESCRT machinery must be recruited by subtler cues. Because knockdown of the ESCRT‐I subunit TSG101 resulted in a strong delay in ESCRT‐III recruitment, ESCRT‐I is likely to mediate recruitment of ESCRT‐III to sites of injury in the lysosome membrane. Even though depletion of ALIX by itself had no detectable effect on ESCRT‐III recruitment, simultaneous depletion of ALIX and TSG101 resulted in a complete block of ESCRT‐III recruitment, suggesting that ALIX and TSG101 cooperate to recruit ESCRT‐III. This resembles the two‐pronged recruitment of ESCRT‐III by ESCRT‐I and Bro1/ALIX during endosomal sorting and cytokinetic abscission (Christ et al, 2016; Tang et al, 2016).
Previous work has uncovered cellular mechanisms that protect the lysosome membrane from damage (Kirkegaard et al, 2010) and that lead to elimination of damaged lysosomes (Hung et al, 2013; Maejima et al, 2013). We have here shown the existence of an ESCRT‐mediated lysosomal repair pathway that kicks in before engagement of the lysophagy machinery. Thus, the cell is equipped with several strategies for preventing leakage of lysosomal contents into the cytosol. Our finding that otherwise reversible lysosomal membrane damage becomes cell lethal in the absence of ESCRT recruitment illustrates the importance of the lysosomal repair pathway.
Many intracellular pathogens are able to replicate inside host cells, and in some cases, replication can take place in lysosome‐like bacterial vacuoles. C. burnetii is a good example of this (Pechstein et al, 2017), and it was interesting to observe the reversible recruitment of ESCRT proteins to the C. burnetii vacuole. Galectin‐3 was also recruited, although our time‐lapse movies had too low temporal resolution to determine whether Galectin‐3 was recruited after ESCRT‐III, as found with damaged lysosomes. The fact that both ESCRT‐III and Galectin‐3 recruitment was reversible suggests that the C. burnetii vacuoles undergo sporadic membrane damage that can be repaired by the ESCRT machinery. Upon recruitment and repair, Galectin‐3 might be degraded in the lysosome‐like lumen of the vacuole, whereas ESCRT‐III would dissociate upon fulfilling its function in membrane repair. Our finding that ESCRT depletion inhibits C. burnetii replication indicates that ESCRT‐mediated repair of the sporadically injured vacuole membrane is required for keeping the vacuole intact over the several hours required for optimal replication.
Our findings raise several questions and perspectives. First, how is lysosomal membrane repair achieved by ESCRT proteins? This has to be clarified by future experiments, but our working hypothesis is that the membrane patch containing the lesion becomes internalized into intraluminal vesicles by the same mechanism as that used in ESCRT‐mediated endosomal protein sorting and biogenesis of multivesicular endosomes (Raiborg & Stenmark, 2009), even though the kinetics appear to be different (Wenzel et al, 2018). Another important question concerns the cues that trigger ESCRT recruitment to damaged lysosomes. Ca2+ leakage out of the injured lysosome, detected by ALIX and its Ca2+ binding partner ALG2, has been put forward as a mechanism (Skowyra et al, 2018), but we note that TSG101 appears to be more important than ALIX for mediating ESCRT‐III recruitment. We therefore hypothesize that additional mechanisms may exist for ESCRT recruitment to damaged lysosomes. Lysosome damage has been shown to induce cell death by several alternative mechanisms (Papadopoulos & Meyer, 2017), and it will be interesting to learn which principal mechanism causes cell death upon lysosome damage in ESCRT‐depleted cells. Our observation that the ESCRT machinery facilitates intracellular replication of C. burnetii raises the question whether other intracellular pathogens also take advantage of the ESCRTs. If so, pharmacological ESCRT inhibition might offer a potential strategy for future treatment of infections caused by intracellular pathogens.
Materials and Methods
Reagents, cell culture, and stable cell lines
Lysosomotropic drugs used in this study, L‐leucyl‐L‐leucine methyl ester (cat. no. 16008) and Gly‐Phe‐β‐naphthylamide (cat. no. 14634), were purchased from Cayman Chemical and used at 250 μM. The antihistamine terfenadine (cat. no. 9652) was purchased from Sigma‐Aldrich. All compounds were dissolved in dimethyl sulfoxide (DMSO) and stored at −80°C. Other reagents used in this study: Dead Cell Apoptosis Kit with Annexin V Alexa Fluor™ 488 and Propidium Iodide (PI) (cat. no. V13241) was purchased from Thermo Fisher Scientific; LysoTracker DND‐99 (cat. no. 7528) for immunofluorescence and LysoTracker Deep Red (cat. no. 12492) for live‐cell imaging were purchased from Molecular Probes and were used at 75 nM. Propidium iodide solution (cat. no. P3566) was purchased from Sigma‐Aldrich and used at 1 μg/ml final concentration.
Human HeLa “Kyoto” and hTERT‐RPE‐1 cells (human retinal pigment epithelial cells immortalized with telomerase) were maintained in DMEM (Gibco) and F12:DMEM medium, respectively, supplemented with 10% fetal bovine serum (FBS), 5 U/ml penicillin, and 50 μg/ml streptomycin. The human large‐cell lung carcinoma cell line H‐460 was maintained in medium recommended by ATCC. All cells were maintained at 37°C supplemented with 5% CO2. A stable HeLa cell line expressing CHMP4B‐eGFP was obtained from Anthony A. Hyman (Max Planck Institute for Molecular Cell Biology and Genetics, Dresden, Germany; Poser et al, 2008).
All other stable cell lines used in this study were lentivirus‐generated pools, using plasmids pCDH‐PGK‐IRES‐Blast‐mCherry‐Galectin‐3 and pCDH‐PGK‐IRES‐Blast‐mCherry‐ALIX for stable expression of mCherry‐Galectin‐3 and mCherry‐ALIX, respectively. The weak PGK promoter was used for transgene expression in order to achieve low expression levels. Third‐generation lentivirus was generated as previously published in Campeau et al (2009). Briefly, mCherry or eGFP fusions were generated as Gateway Entry plasmids using standard molecular biology techniques. From these vectors, lentiviral transfer vectors were generated by recombination into pLenti Destination vectors using a Gateway LR reaction. VSV‐G‐pseudotyped lentiviral particles were packaged using a third‐generation packaging system (Dull et al, 1998). Cells were then transduced with low virus titers, and stable expressing populations were generated by antibiotic selection. Other cell lines stably expressing tagged versions of HRS, TSG101, VPS4A, VPS4B, EAP30, HD‐PTP, and IST1 were kindly provided by Eva M. Wenzel (Oslo University Hospital) and Coen Campsteijn (University of Oslo, Norway). RPE‐1 cell line stably expressing CHMP4B‐eGFP was kindly provided by Yan Zhen (Oslo University Hospital).
siRNA transfections
Silencer Select siRNAs against HRS (5′‐CGACAAGAACCCACACGTC‐3′), TSG101 (#1, 5′‐CCGUUUAGAUCAAGAAGUA‐3′; #2, 5′‐CCUCCAGUCUUCUCUCGUC‐3′), siCHMP2A (5′‐AAGAUGAAGAGGAGAGUGA‐3′), ALIX (#1, 5′‐GCAGUGAGGUUGUAAAUGU‐3′; #2, 5′‐CCUGGAUAAUGAUGAAGGA‐3′), and nontargeting control siRNA (predesigned, cat. no. 4390844) were purchased from Ambion. Cells at 50% confluency were transfected with 20–50 nM final siRNA concentration using Lipofectamine RNAiMax transfection reagent (Life Technologies) according to the manufacturer's instructions and harvested after 48 or 96 h.
Immunofluorescence staining and antibodies
Cells seeded on coverslips were fixed in 4% EM‐grade paraformaldehyde for 10 min and permeabilized with 0.05% saponin in phosphate‐buffered saline (PBS). For HRS localization experiments, cells were permeabilized with 0.05% saponin in PEM buffer (80 mM K‐Pipes, pH 6.8, 5 mM EGTA, and 1 mM MgCl2) for 5 min on ice to decrease the fluorescence signal from the cytosolic pool of proteins before fixation. Cells were washed twice in PBS and once in PBS containing 0.05% saponin before staining with the indicated primary antibodies for 1 h. Prior staining with the secondary antibodies for 1 h, cells were washed three times in PBS containing 0.05% saponin. Cells were mounted in Mowiol containing 2 mg/ml Hoechst 33342 (Sigma‐Aldrich). Rabbit anti‐ALIX, rabbit anti‐CHMP4B, and rabbit anti‐HRS antibodies were described previously (Christ et al, 2016). Goat anti‐Galectin‐3 antibody (cat. no. AF1154) and human Galectin‐3 Alexa Fluor 488‐conjugated antibody (cat. no. IC1154G) were purchased from R&D Systems, and rabbit anti‐CHMP2A (cat. no. 10477‐1‐AP) and rabbit anti‐HD‐PTP (cat. no. 10472‐1‐AP) were purchased from Proteintech. Mouse anti‐LAMP1 (cat. no. H4A3) was purchased from Developmental Studies Hybridoma Bank, rabbit anti‐LC3 (cat. no. PM036) from MBL International Corporation, mouse anti‐Ubq (cat. no. PW8810) from Enzo Life Sciences, mouse anti‐β‐actin (cat. no. A5316) from Sigma‐Aldrich, and mouse anti‐TSG101 (cat. no. 612697) from BD Transduction Laboratories. All secondary antibodies used for immunofluorescence studies and Western blotting were obtained from Jacksons ImmunoResearch Laboratories or from Molecular Probes (Life Technologies).
Immunoblotting
Cells were washed with cold PBS and lysed in 2× sample buffer (125 mM Tris–HCl, pH 6.8, 4% SDS, 20% glycerol, 200 mM DTT, and 0.004% bromophenol blue). Whole‐cell lysates were subjected to SDS–PAGE on 4–20% gradient gels (Mini‐PROTEAN TGX; Bio‐Rad). Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Trans‐Blot® Turbo™ LF PVDF, Bio‐Rad) followed by blocking in 5% fat‐free milk powder and antibody incubation in 5% fat‐free milk powder in Tris‐buffered saline with 0.1% Tween‐20. Membranes incubated with horseradish peroxidase‐conjugated antibodies were developed using Clarity Western ECL Substrate Solutions (Bio‐Rad) with a ChemiDoc XRS+ imaging system (Bio‐Rad).
Confocal fluorescence microscopy
Stained coverslips were examined with a Zeiss LSM 780 confocal microscope (Carl Zeiss) equipped with an Ar laser multiline (458/488/514 nm), a DPSS‐561 10 (561 nm), a laser diode 405‐30 CW (405 nm), and a HeNe laser (633 nm). The objective used was a Zeiss Plan‐Apochromat 63×/1.40 Oil DIC M27 (Carl Zeiss). Image processing was performed with ImageJ software (National Institutes of Health). Intensity settings for the relevant channels were kept constant during imaging. Images shown in figures are representative of at least three independent experiments.
Live‐cell imaging
Cells seeded in MatTek 35‐mm petri dish, 20‐mm Microwell No. 1.5 coverglass were imaged on a DeltaVision microscope (Applied Precision) equipped with Elite TruLight Illumination System, a CoolSNAP HQ2 camera, and a 60× Plan‐Apochromat (1.42 NA) lens. For temperature control during live observation, the microscope stage was kept at 37°C by a temperature‐controlled incubation chamber. Time‐lapse images (6 z‐sections 2,2 μm apart) were acquired every 1–3 min over a total time period of 4–6 h and deconvolved using the softWoRx software (Applied Precision). In addition, DeltaVision OMX V4 microscope equipped with three PCO.edge sCMOS cameras, a solid‐state light source, a 60× 1.42 NA objective, and a laser‐based autofocus was used. Environmental control was provided by a heated stage and an objective heater (20‐20 Technologies). Images were deconvolved using softWoRx software and processed in ImageJ/FIJI.
Flow cytometry
Forty‐eight hours post‐transfection, cells were treated with 250 μM LLOMe for 3, 6, and 10 h. Cells were harvested using accutase, and detection of apoptosis was performed using Dead Cell Apoptosis Kit with Annexin V Alexa Fluor™ 488 and Propidium Iodide according to the manufacturer's instructions. Cells were analyzed by flow cytometry on a LSRII flow cytometer (BD Biosciences) using FACS Diva (BD Biosciences) software.
Coxiella infection
Propagation of mCherry‐Coxiella burnetii (generously provided by Dr. Robert Heinzen, Rocky Mountain Laboratories, NIAID, NIH, Hamilton, MT, USA) was performed in Vero cells as previously described (Romano et al, 2007). A fluorescent foci unit (FFU) infection assay was used to quantify the replication and viability of C. burnetii in HeLa cells in 24‐well plates treated with control or TSG101 siRNA for 48 h (with silencing extent verified by Western blotting). In brief, after 48 h treatment with siRNAs, cells were infected as previously described (Campoy et al, 2013) for 48 h and lysed. Samples were serially diluted and used to infect Vero cells in a 24‐well plate. After 72 h of infection, Vero cells were fixed, DAPI stained, and processed for fluorescent microscopy. Several thousands of cells were automatically scored per condition to determine an average number of FFU for each sample, and three independent experiments were pooled.
Statistical analysis
The number of individual experiments and the number of cells analyzed are indicated in the figure legends. We tested our datasets for normal distribution and chose an appropriate test accordingly using GraphPad Prism version 5.01. The unpaired, two‐tailed Student's t‐test was used to calculate the significance of number of foci per cell in Figs 1 and 2, and EV1, EV2, EV3. The statistical tests performed for each experiment are specified in the figure legends. P‐values are indicated for each experiment, and P‐values below 0.05 were regarded as significant. In instances of multiple means comparisons, we used one‐way analysis of variance (ANOVA) followed by the Bonferroni post hoc test to determine statistical significance.
Image analysis and post‐processing
Images were analyzed in Fiji/ImageJ using custom python scripts. Briefly, images were filtered to remove noise and foci of ESCRTs and/or associated proteins were segmented by the “Find maxima” function and scored (Figs 4 and 5). Alternatively, foci were segmented by manual thresholding and objects were then identified and scored by the “Analyze Particles” function of ImageJ (Figs 1, 2, 3 and 6, and EV1, EV2, EV3). Manual thresholds were chosen to detect prominent LLOMe‐induced structures; therefore, only spots above a certain intensity were scored. All post‐processing was performed using Python and the “pandas” package, plots were generated using Seaborn. For live‐cell movies, foci counts were normalized to the area occupied by cells within a given field of view. For fixed cell analysis, nuclei were segmented and counted, and all foci counts were normalized to the number of nuclei within a given field of view. Analysis scripts have been deposited at https://github.com/koschink/Radulovic_et_al.
Author contributions
MR performed transient and stable transfections, microscopy and image quantifications, prepared movies and figures, and wrote the manuscript together with HS with input from all co‐authors. KOS developed image analysis and data processing software, and performed live‐cell imaging, image processing, and data analysis for Fig 5. EMW prepared stable transfectants, performed microscopy and Western blot analyses for Fig 3A, and prepared figures. VN performed flow cytometry and analyzed flow cytometry data. AB and FL performed and analyzed all experiments with C. burnetii. HS conceived and supervised the study and wrote the manuscript together with MR with input from all co‐authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Acknowledgements
We thank Coen Campsteijn for kindly providing plasmid constructs, and M.E. Mansilla and M.‐I. Colombo for sharing protocols and reagents. We thank the Flow Cytometry Core Facility at Oslo University Hospital for help with cell death measurements and the Core Facility for Advanced Light Microscopy at Oslo University Hospital for providing access to microscopes. M.R. is a post‐doctoral fellow of the South‐Eastern Norway Regional Health Authority. K.O.S. holds a career development fellowship and V.N. a post‐doctoral fellowship from the Norwegian Cancer Society. E.M.W. is a senior research fellow of the South‐Eastern Norway Regional Health Authority (grant number 2015014). H.S. is supported by project grants from the Norwegian Cancer Society (grant number 182698) and the South‐Eastern Norway Regional Health Authority (grant number 2016087). F.L. is supported by the ANR, project number 15‐CE15‐0017. This work was partly supported by the Research Council of Norway through its Centres of Excellence funding scheme, project number 262652.
The EMBO Journal (2018) 37: e99753
References
- Babst M, Katzmann DJ, Estepa‐Sabal EJ, Meerloo T, Emr SD (2002a) Escrt‐III: an endosome‐associated heterooligomeric protein complex required for mvb sorting. Dev Cell 3: 271–282 [DOI] [PubMed] [Google Scholar]
- Babst M, Katzmann DJ, Snyder WB, Wendland B, Emr SD (2002b) Endosome‐associated complex, ESCRT‐II, recruits transport machinery for protein sorting at the multivesicular body. Dev Cell 3: 283–289 [DOI] [PubMed] [Google Scholar]
- Bache KG, Brech A, Mehlum A, Stenmark H (2003) Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol 162: 435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg TO, Stromhaug E, Lovdal T, Seglen O, Berg T (1994) Use of glycyl‐L‐phenylalanine 2‐naphthylamide, a lysosome‐disrupting cathepsin C substrate, to distinguish between lysosomes and prelysosomal endocytic vacuoles. Biochem J 300(Pt 1): 229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig C, Gruenberg J (2014) ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol 24: 19–25 [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4: e6529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campoy EM, Mansilla ME, Colombo MI (2013) Endocytic SNAREs are involved in optimal Coxiella burnetii vacuole development. Cell Microbiol 15: 922–941 [DOI] [PubMed] [Google Scholar]
- Chazotte B (2011) Labeling lysosomes in live cells with LysoTracker. Cold Spring Harb Protoc 2011: pdb prot5571 [DOI] [PubMed] [Google Scholar]
- Christ L, Wenzel EM, Liestol K, Raiborg C, Campsteijn C, Stenmark H (2016) ALIX and ESCRT‐I/II function as parallel ESCRT‐III recruiters in cytokinetic abscission. J Cell Biol 212: 499–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ L, Raiborg C, Wenzel EM, Campsteijn C, Stenmark H (2017) Cellular functions and molecular mechanisms of the ESCRT membrane‐scission machinery. Trends Biochem Sci 42: 42–56 [DOI] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J (2016) Nuclear envelope rupture and repair during cancer cell migration. Science 352: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L (1998) A third‐generation lentivirus vector with a conditional packaging system. J Virol 72: 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegaard AM, Dehlendorff C, Vind AC, Anand A, Cederkvist L, Petersen NHT, Nylandsted J, Stenvang J, Mellemgaard A, Osterlind K, Friis S, Jaattela M (2016) Repurposing cationic amphiphilic antihistamines for cancer treatment. EBioMedicine 9: 130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Stenmark H, Emr SD (2013) Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol 5: a016766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YH, Chen LM, Yang JY, Yang WY (2013) Spatiotemporally controlled induction of autophagy‐mediated lysosome turnover. Nat Commun 4: 2111 [DOI] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS (2008) Exit strategies of intracellular pathogens. Nat Rev Microbiol 6: 99–110 [DOI] [PubMed] [Google Scholar]
- Jimenez AJ, Maiuri P, Lafaurie‐Janvore J, Divoux S, Piel M, Perez F (2014) ESCRT machinery is required for plasma membrane repair. Science 343: 1247136 [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Babst M, Emr SD (2001) Ubiquitin‐dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT‐I. Cell 106: 145–155 [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002) Receptor downregulation and multivesicular‐body sorting. Nat Rev Mol Cell Biol 3: 893–905 [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Stefan CJ, Babst M, Emr SD (2003) Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol 162: 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C, Kinnunen PK, Nylandsted J, Jaattela M (2010) Hsp70 stabilizes lysosomes and reverts Niemann‐Pick disease‐associated lysosomal pathology. Nature 463: 549–553 [DOI] [PubMed] [Google Scholar]
- Kroemer G, Jaattela M (2005) Lysosomes and autophagy in cell death control. Nat Rev Cancer 5: 886–897 [DOI] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, Yoshimori T (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansilla Pareja ME, Bongiovanni A, Lafont F, Colombo MI (2017) Alterations of the Coxiella burnetii replicative vacuole membrane integrity and interplay with the autophagy pathway. Front Cell Infect Microbiol 7: 112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Meyer H (2017) Detection and clearance of damaged lysosomes by the endo‐lysosomal damage response and lysophagy. Curr Biol 27: R1330–R1341 [DOI] [PubMed] [Google Scholar]
- Paz I, Sachse M, Dupont N, Mounier J, Cederfur C, Enninga J, Leffler H, Poirier F, Prevost MC, Lafont F, Sansonetti P (2010) Galectin‐3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol 12: 530–544 [DOI] [PubMed] [Google Scholar]
- Pechstein J, Schulze‐Luehrmann J, Luhrmann A (2017) Coxiella burnetii as a useful tool to investigate bacteria‐friendly host cell compartments. Int J Med Microbiol 308: 77–83 [DOI] [PubMed] [Google Scholar]
- Poser I, Sarov M, Hutchins JR, Heriche JK, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW, Pelletier L, Kittler R, Hua S, Naumann R, Augsburg M, Sykora MM, Hofemeister H, Zhang Y, Nasmyth K, White KP et al (2008) BAC TransgeneOmics: a high‐throughput method for exploration of protein function in mammals. Nat Methods 5: 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon‐Dumenil AM, Manel N, Piel M (2016) ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352: 359–362 [DOI] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H (2002) Hrs sorts ubiquitinated proteins into clathrin‐coated microdomains of early endosomes. Nat Cell Biol 4: 394–398 [DOI] [PubMed] [Google Scholar]
- Raiborg C, Stenmark H (2009) The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458: 445–452 [DOI] [PubMed] [Google Scholar]
- Romano PS, Gutierrez MG, Beron W, Rabinovitch M, Colombo MI (2007) The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol 9: 891–909 [DOI] [PubMed] [Google Scholar]
- Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot MF, Lamberton A, Mathieu M, Bertrand T, Marquette JP, El‐Ahmad Y, Filoche‐Romme B, Schio L, Garcia‐Echeverria C, Goulaouic H, Pasquier B (2014) A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol 10: 1013–1019 [DOI] [PubMed] [Google Scholar]
- Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, Jaiswal JK (2014) Mechanism of Ca(2)(+)‐triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun 5: 5646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoneberg J, Lee IH, Iwasa JH, Hurley JH (2017) Reverse‐topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol 18: 5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI (2018) Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360: eaar5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Buchkovich NJ, Henne WM, Banjade S, Kim YJ, Emr SD (2016) ESCRT‐III activation by parallel action of ESCRT‐I/II and ESCRT‐0/Bro1 during MVB biogenesis. Elife 5: e15507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele DL, Lipsky PE (1990) Mechanism of L‐leucyl‐L‐leucine methyl ester‐mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci USA 87: 83–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C, Stenmark H (2015) Spastin and ESCRT‐III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522: 231–235 [DOI] [PubMed] [Google Scholar]
- Wenzel EM, Schultz SW, Schink KO, Pedersen NM, Nahse V, Carlson A, Brech A, Stenmark H, Raiborg C (2018) Concerted ESCRT and clathrin recruitment waves define the timing and morphology of intraluminal vesicle formation. Nat Commun 9: 2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Source Data for Expanded View
Review Process File
Source Data for Figure 3