

Abstract
Nonsense‐mediated mRNA decay (NMD) is a translation‐dependent RNA degradation pathway involved in many cellular pathways and crucial for telomere maintenance and embryo development. Core NMD factors Upf1, Upf2 and Upf3 are conserved from yeast to mammals, but a universal NMD model is lacking. We used affinity purification coupled with mass spectrometry and an improved data analysis protocol to characterize the composition and dynamics of yeast NMD complexes in yeast (112 experiments). Unexpectedly, we identified two distinct complexes associated with Upf1: Upf1‐23 (Upf1, Upf2, Upf3) and Upf1‐decapping. Upf1‐decapping contained the mRNA decapping enzyme, together with Nmd4 and Ebs1, two proteins that globally affected NMD and were critical for RNA degradation mediated by the Upf1 C‐terminal helicase region. The fact that Nmd4 association with RNA was partially dependent on Upf1‐23 components and the similarity between Nmd4/Ebs1 and mammalian Smg5‐7 proteins suggest that NMD operates through conserved, successive Upf1‐23 and Upf1‐decapping complexes. This model can be extended to accommodate steps that are missing in yeast, to serve for further mechanistic studies of NMD in eukaryotes.
Keywords: affinity purification, NMD, quantitative mass spectrometry, RNA decay, Saccharomyces cerevisiae
Subject Categories: Protein Biosynthesis & Quality Control, RNA Biology
Introduction
Nonsense‐mediated mRNA decay (NMD) is a major surveillance pathway defined by its importance for the fast decay of mRNAs with premature termination codons (PTC). Rapid translation‐dependent RNA degradation prevents the production of truncated proteins (Hall & Thein, 1994) and reduces the loss of ribosomes “locked” on RNA at aberrant termination sites (Ghosh et al, 2010; Serdar et al, 2016). NMD factors are essential for the development of metazoans (Medghalchi et al, 2001) and participate to telomere maintenance and telomerase activity regulation in yeast (Addinall et al, 2011) and human cells (Azzalin et al, 2007). As a quality control pathway, NMD shapes the transcriptome of eukaryotic cells, both under normal and pathologic conditions, including cancer (Lindeboom et al, 2016). While NMD substrates were initially thought to be rather rare, single‐nucleotide resolution RNA sequencing in yeast revealed that genome‐wide transcription generates thousands of RNAs that are degraded by NMD (Malabat et al, 2015).
Large‐scale results confirmed and validated previous studies on individual NMD reporters that described two classes of NMD substrates: one in which a long 3′ untranslated region (UTR) extension follows a short coding sequence and another that includes an exon–exon junction, bound by the exon–exon junction complex (EJC), downstream a stop codon. While long 3′‐UTRs destabilize RNAs in all the eukaryotes studied to date, EJC‐enhanced NMD is absent from Saccharomyces cerevisiae, Schizosaccharomyces pombe (Wen & Brogna, 2010) or Caenorhabditis elegans (Gatfield et al, 2003). Most of mRNAs physiologically regulated by NMD belong to the long 3′‐UTR category (Yepiskoposyan et al, 2011), but many mRNAs generated by point mutation in neoplastic cells belong to the EJC class (Lindeboom et al, 2016). Degradation of both types of NMD substrates requires three universally conserved “core” factors: Upf1, Upf2 and Upf3 (Leeds et al, 1991, 1992; Pulak & Anderson, 1993; Cui et al, 1995; Perlick et al, 1996). Independent of the initial triggering feature, RNA degradation in NMD occurs through activation of general RNA degradation pathways: decapping and endonucleolytic cleavage. Final degradation of RNA depends on the major 5′ to 3′ exonuclease Xrn1 (He & Jacobson, 2001) and the cytoplasmic exosome 3′ to 5′ exonucleolytic activity (Eberle et al, 2009).
How the NMD machinery differentiates normal termination codons from PTC has been a long‐standing question (He & Jacobson, 2015). For long 3′‐UTR NMD substrates, the prevalent model proposes that detection of a stop codon as premature occurs when the stop is far from the poly(A) tail (“faux 3′‐UTR” model, Amrani et al, 2004). In this model, the polyA‐binding protein, Pab1 in yeast and PABPC1 in mammals, affects translation termination efficiency. Slow termination allows recruitment of Upf1, Upf2, Upf3, which leads to rapid decapping and degradation of NMD substrates (Muhlrad & Parker, 1994, 1999). A long distance from the PTC to PABPC1 triggers NMD in all studied species (Bühler et al, 2006; Eberle et al, 2008; Singh et al, 2008; Huang et al, 2011).
Exon–exon junction complex‐enhanced NMD does not require a long 3′‐UTR and depends on an exon–exon junction located at more than 50 nucleotides downstream the PTC (Holbrook et al, 2004; Maquat, 2004). EJC positioning is an important element of the prevalent model for the first steps in mammalian NMD, called SURF/DECID. This model involves the formation of an initial complex that contains a ribosome stalled at a termination codon, the translation termination factors eRF1 and eRF3, Upf1 and the protein kinase Smg1 (SURF). Recruitment of the Upf2 and Upf3 proteins to SURF via a proximal EJC leads to formation of the DECID complex in which Upf1 N‐ and C‐terminal regions are phosphorylated by Smg1 (Kashima et al, 2006). Phosphorylated Upf1 binds the Smg6 endonuclease and the Smg5‐7 heterodimer, which, in turn, activates RNA deadenylation and decapping. Recruitment of a phosphatase ensures the return of Upf1 to its initial state for another NMD cycle (Ohnishi et al, 2003).
While the SURF/DECID model offers an explanation for EJC‐enhanced NMD, it does not apply to organisms that do not have EJC components, such as the budding yeast, and does not explain how NMD works on RNAs with long 3′‐UTRs. Understanding how such substrates are degraded through NMD is relevant for mammalian cells since several physiologically important NMD substrates do not depend on an EJC downstream the termination codon. A salient example is the mRNA of the GADD45A gene (Chapin et al, 2014; Lykke‐Andersen et al, 2014), a transcript whose destabilization through NMD is essential for the development of fly and mammal embryos (Nelson et al, 2016). It is possible that EJC‐enhanced and EJC‐independent NMD are two versions of the same, not yet understood, generic NMD mechanisms. Such a mechanism is unlikely to rely on a SURF/DECID‐like sequence of events for several reasons. First, the SURF/DECID model proposes crucial roles for factors or events that are not conserved in all eukaryotes, even if the key NMD proteins are present from yeast to humans. For example, the protein kinase Smg1, a central component of the SURF complex, has no known equivalent in S. cerevisiae and its absence has little impact on NMD in Drosophila melanogaster embryos (Chen et al, 2005). Second, a major assumption of the SURF/DECID model, the role of EJC in bringing Upf3 in the proximity of Upf1 (Le Hir et al, 2001; Chamieh et al, 2008), was challenged by very recent biochemical analyses that show interactions of Upf3 with translation termination complexes independent of EJC components (Neu‐Yilik et al, 2017). Third, while all the RNA degradation mechanisms can play a role in NMD, the model leaves open the question of the mechanisms of recruitment for the RNA degradation factors. Major roles have been attributed to the decapping machinery (Muhlrad & Parker, 1994; Lykke‐Andersen, 2002), endonucleolytic cleavage (Huntzinger et al, 2008; Eberle et al, 2009) or deadenylation (Loh et al, 2013), depending on the reporter RNA used. The molecular mechanisms responsible for the redundancy and interplay between these different degradation pathways (Metze et al, 2013; Colombo et al, 2016) remain unclear. Hence, a re‐evaluation of the available data and new biochemical results on the involved complexes are critical to uncover a universally conserved mechanism of NMD in eukaryotes. Such a mechanism could then serve as the basis to understand the evolution of NMD, its variations across species and additional levels of complexity.
The enumerated weaknesses of current NMD models originate, at least in part, from the lack of a global view of the biochemistry of the NMD process. We show here that fast affinity purification of yeast NMD complexes coupled with high‐resolution quantitative mass spectrometry (LC‐MS/MS) allows a global and unprecedented view of NMD complexes. Our results uncover the existence of two distinct and successive protein complexes that both contain the essential NMD factor Upf1. We identified two yeast accessory NMD factors that interact with Upf1 and become essential for NMD under limiting conditions. Their similarity with Smg6 and Smg5/7 implies that comparable molecular mechanisms for NMD have been conserved from yeast to humans. Furthermore, our results suggest that Upf1 might suffer major conformation changes to accommodate a switch from a “Upf1‐23” complex to an “Upf1‐decapping” complex, that could trigger RNA decapping, the first step in NMD substrates degradation.
Results
Quantitative mass spectrometry reveals the global composition of Upf1‐associated complexes
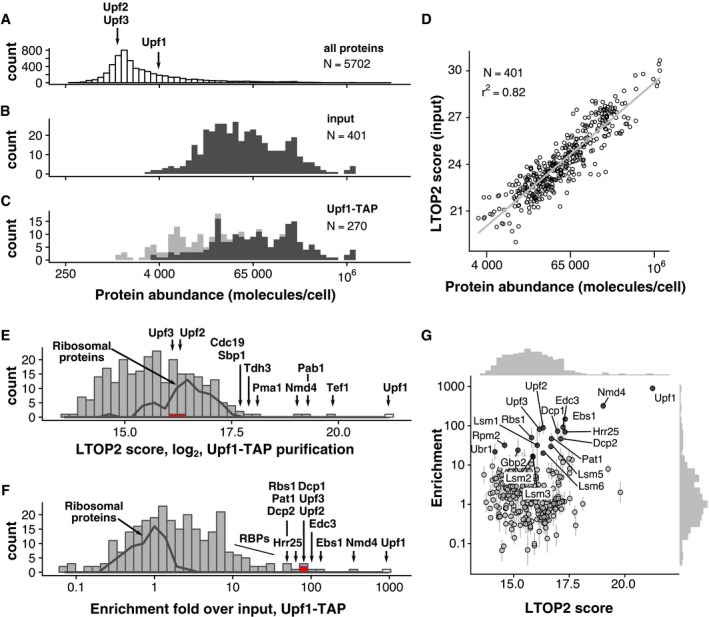
Previous large‐scale attempts to describe the composition of Upf1‐associated complexes in yeast failed to identify known Upf1 partners, such as Upf2 or Upf3 (Gavin et al, 2006; Krogan et al, 2006; Collins et al, 2007) even if these studies provided high‐quality interaction data on other proteins. This negative result could be due to the relatively low abundance of Upf2 and Upf3, estimated to 1,470 and 1,439 molecules per cell (Ho et al, 2018), which places them in the category of the 25% least abundant proteins in yeast (Fig 1A). The instability of the complexes during the purification procedure could also play a role. To overcome these problems, we used a combination of fast affinity purification (Oeffinger et al, 2007) with the current high sensitivity of mass spectrometry methods for protein identification. TAP‐tagged (Rigaut et al, 1999) Upf1 was isolated from cell extracts obtained from 6 l of yeast culture grown in exponential phase. The total cell extract was used as a reference and an untagged strain as a negative control. Four hundred proteins were reliably identified in the total extract and corresponded, as expected, to abundant yeast proteins (the frequency distribution of the abundance for the identified proteins is shown as dark grey bars in Fig 1B). Many abundant proteins were also identified in the Upf1‐TAP‐purified fraction (distribution in dark grey, Fig 1C). In contrast to the input fraction, less abundant proteins were also identified in the purified fraction (distribution in light grey, Fig 1C).
Figure 1. Enrichment analysis accurately describes Upf1‐associated proteins.
-
A–CPositioning of Upf1, Upf2 and Upf3 in the abundance distribution of all yeast proteins, based on the data compiled by Ho et al (2018) (A). The distribution of abundance for the proteins identified by mass spectrometry in input (B), and purified samples (C), shows the expected bias for proteins present in high copy numbers. Light grey colour highlights the positions of proteins that were only identified in the Upf1‐TAP samples (six replicates) and not in the total extract (three replicates).
-
DCorrelation of LTOP2‐based protein level estimates in a total yeast protein extract with published protein abundance data (Ho et al, 2018).
-
EDistribution of the intensity for the proteins quantified in association with Upf1 (LTOP2 score, log2 scale). Positioning of Upf1 is highlighted as a white rectangle while Upf2 and Upf3 are highlighted in red. The dark grey line indicates the region of intensities corresponding to ribosomal proteins.
-
FDistribution of the protein enrichment values for Upf1‐associated proteins based on LTOP2 scores and known protein abundance in a total yeast extract. Upf1, Upf2 and Upf3 positions are also highlighted as white and red rectangles.
-
GA combination of the data presented in (E) and (F) as a scatter plot, to see both the amount and enrichment of proteins in Upf1‐TAP‐purified samples. The horizontal axis represents the LTOP2 score, with the vertical axis showing enrichment over input values. Both axes use log2 transformed values.
Source data are available online for this figure.
To assess to what extent the identified proteins were specific to the Upf1 purification, we used a label‐free quantification method based on the intensity of the most abundant peptides for each of the identified proteins (Silva et al, 2006), a method that accurately estimates protein levels (Ahrné et al, 2013). For each identified protein, we derived a peptide intensity‐based score that we called LTOP2—“L”, because it includes a log2 transformation, and “TOP2” for the minimum number of required peptides (see Materials and Methods). The LTOP2 score of proteins identified in the total protein extract was well correlated with protein abundance estimates (Ho et al, 2018), with a Pearson determination coefficient of 0.82 (Fig 1D).
Upf1 had the highest LTOP2 score in the Upf1‐TAP purification, as expected, validating both the use of this MS score for protein quantitation and the efficiency of the affinity purification. Most of the other proteins with high LTOP2 scores in the purification, such as the plasma membrane proton pump Pma1, or the glyceraldehyde‐3‐phosphate dehydrogenase Tdh3 (Fig 1E), were unrelated to Upf1 or NMD, but are among the 10% most abundant proteins in yeast. NMD factors Upf2 and Upf3, the best‐known partners of Upf1, ranked low in the list of 270 LTOP2 scores, with position 80 and 91, respectively (Dataset EV1). Thus, many of the proteins present in the affinity‐purified sample were contaminants, known to pollute affinity purifications and complicate the conclusions of such experiments (Mellacheruvu et al, 2013). To filter this type of contamination, we chose to calculate an enrichment score, which reflects the ratio between the proportion of a protein in a sample and its proportion in a total extract (Ho et al, 2018). Top enrichment scores corresponded to the tagged protein Upf1, Upf2 (rank 5) and Upf3 (rank 6), validating enrichment as an excellent noise removal strategy in the identification of specific partners in affinity‐purified samples (Fig 1F and Dataset EV1). To simultaneously visualize both the amount of protein found in a purified sample and its enrichment score, we combined LTOP2 and enrichment in a graphical representation that is similar to the intensity versus fold change “MA” plots used for the analysis of differential gene expression (Fig 1G). The workflow for mass spectrometry data analysis is presented in Appendix Fig S1.
In addition to the canonical Upf1‐Upf2‐Upf3 interactions (He et al, 1996, 1997), our enrichment strategy identified the RNA decapping enzyme Dcp2, its co‐factor Dcp1 and decapping activators, Edc3, Lsm1‐7 and Pat1 as Upf1 partners. This result was correlated with the requirement for decapping for the degradation of NMD substrates in yeast (Muhlrad & Parker, 1994). Direct or indirect interactions of Upf1 with these different factors have been previously reported, mostly through yeast two‐hybrid, or affinity purification coupled with immunoblots (He & Jacobson, 2015). Unexpectedly, two poorly characterized proteins, Nmd4 and Ebs1, ranked second and third in the Upf1 purification. Both proteins were previously linked with NMD. Nmd4 was identified in a yeast Upf1 two‐hybrid screen but was not further studied (He & Jacobson, 1995). The deletion of the NMD4 gene shows a weak positive genetic interaction with the deletion of UPF1 and modifies the growth phenotype of several mutant strains expressing essential gene alleles for which the corresponding mRNAs are sensitive to NMD (Wilmes et al, 2008). Ebs1, identified through sequence similarity as a potential yeast equivalent of human Smg7, a component of mammalian NMD, was shown to co‐purify with Upf1 and to affect yeast NMD (Luke et al, 2007). Other proteins enriched in the Upf1‐associated fraction by a factor of 10 or more (Rbs1, Hrr25, Gbp2, Gar1, Cbf5, Sbp1, Ssd1, Lsm12, Lhp1 and Rbg1) have functions related to RNA metabolism and could be indirectly associated with the Upf1 RNA‐containing complexes. None of the proteins found enriched with Upf1 by a factor of 8 or higher were detected in the control purification (Dataset EV1). These results indicated that Upf1 has more protein partners than previously thought and validated quantitative mass spectrometry of purified complexes as a powerful strategy to characterize yeast NMD complexes.
Upf1‐associated proteins are grouped in two mutually exclusive complexes
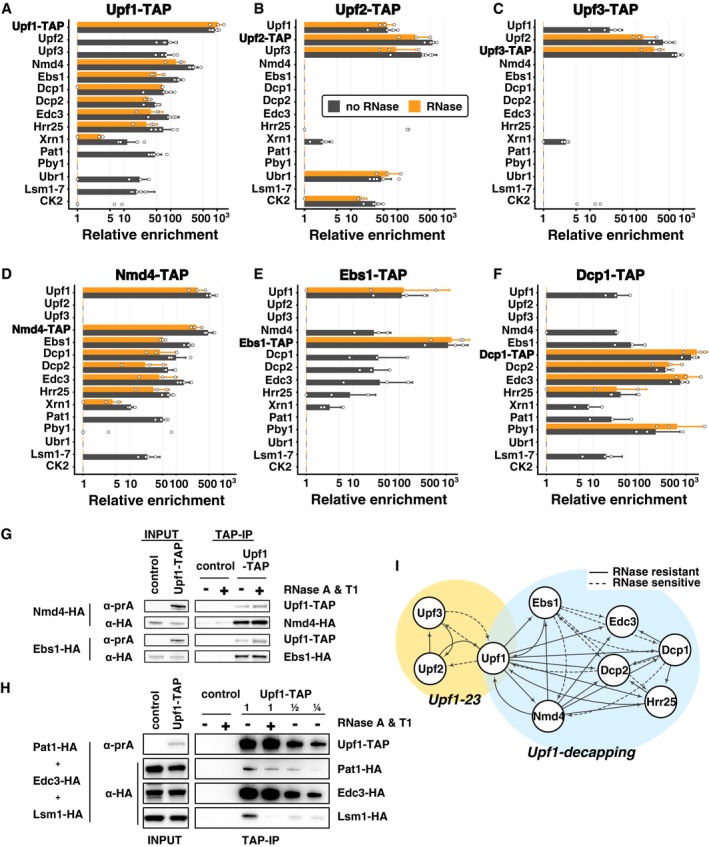
Since our data indicated that Upf1 is associated with a relatively large number of factors, and since Upf1 directly binds RNA (Weng et al, 1998) in large polysomal complexes (Atkin et al, 1997), some identified interactions could be mediated by RNA. To establish which interactions were RNA dependent, we repeated the Upf1‐TAP purification, in a protocol including two RNase A and RNase T1 treatments. Ribosome constituents and RNA binding proteins were lost following this harsh RNase treatment (P‐values associated with GO term enrichment analysis between 10−7 and 10−30 for the corresponding terms, Appendix Table S1). RNase treatment was efficient, suggesting that the proteins that remained specifically enriched were bound to the Upf1 complex through direct protein–protein interactions. Among these proteins, we identified the decapping enzyme Dcp2 and its co‐factors Dcp1 and Edc3, Nmd4, Ebs1 and the protein kinase Hrr25, a homologue of mammalian casein kinase delta, CSNK1D (Fig 2A). Upf2 and Upf3 interactions with Upf1‐TAP were sensitive to RNase. In view of these results, and to obtain a global view of NMD complexes in yeast, we affinity‐purified TAP‐tagged versions of the components of Upf1‐associated complexes identified here with or without RNase treatment: Upf2, Upf3, Nmd4, Ebs1, Dcp1 and Hrr25 (Dataset EV1). All the used tagged strains showed close to wild‐type levels of the RPL28 unspliced pre‐mRNA, a well‐known NMD substrate in yeast (Fig EV1A and B), indicating that the TAP tag had a minimal impact on the function of the corresponding proteins.
Figure 2. Purification of NMD factors reveals two distinct complexes containing Upf1.
-
A–FEnrichment values for proteins identified in tagged Upf1 (A), Upf2 (B), Upf3 (C), Nmd4 (D), Ebs1 (E) and Dcp1 (F) purifications. Tagged proteins are indicated in bold for each experiment. Black bars correspond to average enrichment values obtained in purifications done without RNase and orange bars with an RNase A and RNase T1 treatment. Error bars represent SD. Dots represent individual enrichment values for each protein in the six replicates for Upf1‐TAP, 3 for Upf1‐TAP and RNase, 6 for Upf2‐TAP, 3 for Upf2‐TAP and RNase, 5 for Upf3‐TAP, 3 for Upf3‐TAP and RNase, 4 for Nmd4‐TAP, 3 for Nmd4‐TAP and RNase, 3 for Ebs1‐TAP, with and without RNase and 3 for Dcp1‐TAP with or without RNase. Only proteins enriched by a factor of 16 or more in one of the purifications presented here are shown. For clarity, groups of related proteins were combined (the group of Lsm1 to Lsm7 is marked as “Lsm1‐7” and the values correspond to the mean of all the values found in purifications; CK2 group corresponds to Cka1, Cka2, Ckb1 and Ckb2).
-
GImmunoblot validation of Upf1‐TAP interactions with HA‐tagged Nmd4 and Ebs1. Control strains did not express Upf1‐TAP.
-
HImmunoblot validation of Upf1‐TAP interaction with Pat1‐HA, Lsm1‐HA and Edc3‐HA. The purification was performed with a mix of three strains, expressing Upf1‐TAP or not (control).
-
IRepresentation of binary interactions identified by our experiments: dashed lines correspond to interactions that are sensitive to RNase treatment; the arrows start at the tagged protein and indicate the enriched factor.
Source data are available online for this figure.
Figure EV1. Tagged proteins are functional for NMD .
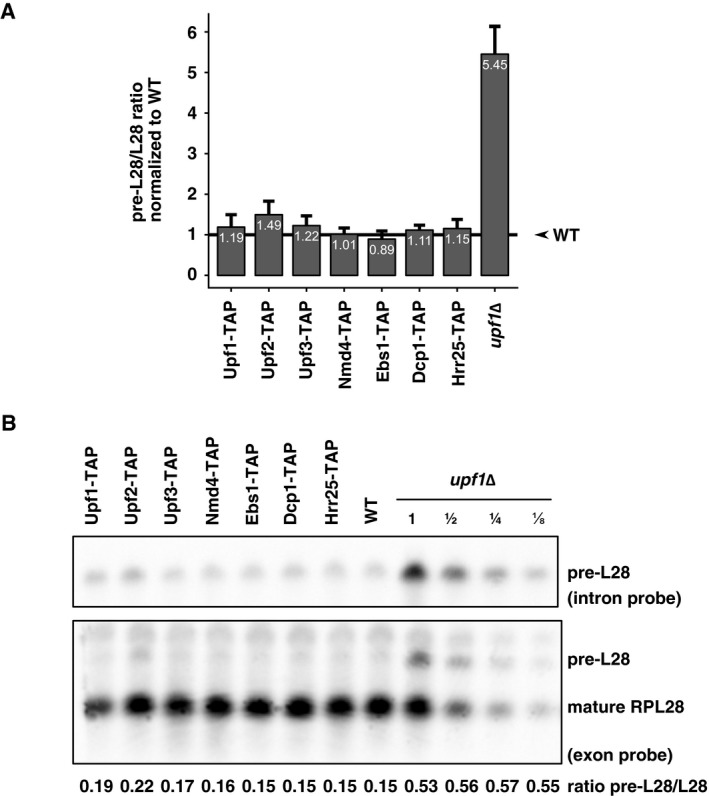
- RNA from the indicated tagged strains and upf1Δ, as control, was quantified by reverse transcription followed by quantitative PCR for the unspliced precursor of RPL28 mRNA and for a normalization RNA, insensitive for NMD (RIM1). Mean values and standard deviations are depicted.
- The stabilization of the endogenous NMD target pre‐RPL28 was tested in comparison with the mature form (RPL28) by northern blot using dsDNA probes and chemiluminescent detection.
One of the most surprising results of the extensive purification data was the difference between the sets of proteins enriched with Upf2‐TAP (Fig 2B) or Upf3‐TAP (Fig 2C) in comparison with Upf1‐TAP (Figs 1G and 2A). Many of the proteins present in the Upf1 complex, such as the decapping factors, Ebs1, and Nmd4, were absent in the Upf2‐TAP and Upf3‐TAP purifications. Conversely, the purifications done with Nmd4‐TAP, Ebs1‐TAP, Dcp1‐TAP or Hrr25‐TAP did not co‐purify Upf2 or Upf3, even if Upf1 was specifically enriched with or without RNase treatment (Fig 2D–F and Dataset EV1). Nmd4‐TAP shared the same purification profile with Upf1‐TAP except for Upf2 and Upf3 (Fig 2A compared with D). Ebs1‐TAP, also purified the same set of proteins, but the enrichment of most of the factors, except Upf1, was dependent on RNA (Fig 2E). Dcp1 co‐purified with its known partners Dcp2 and Edc3, but also with Upf1, Nmd4, Ebs1 and Hrr25 (Fig 2F). In addition to Upf1‐associated proteins, purifications with Dcp1‐TAP and Hrr25‐TAP revealed their presence in other protein complexes. For example, Dcp1 purification also showed the enrichment of another decapping‐associated factor, Pby1 (Fig 2F). The results, including the variability of biological replicates, are presented in Dataset EV1 and can be explored at hub05.hosting.pasteur.fr/NMD_complexes.
To confirm the mass spectrometry quantitative results by a different detection method, we purified Upf1‐TAP in strains in which Nmd4, Ebs1, Pat1, Edc3 and Lsm1 were tagged with three repeats of the HA epitope at the C‐termini. Immunoblots on purified fractions showed that Nmd4‐HA and Ebs1‐HA were enriched with Upf1 with or without RNase treatment, as expected (Fig 2G). RNase treatment reduced the amounts of Pat1‐HA co‐purified with Upf1 by a factor of two and reduced the amounts of Lsm1‐HA more than fourfold, while Edc3‐HA was not affected, in an experiment in which a mix of the three strains was used as the input fraction (Fig 2H).
The obtained results suggest that Upf1 is part of two mutually exclusive complexes, one composed of Upf1, Upf2 and Upf3 and the other containing Upf1, Nmd4, Ebs1 and decapping factors (Fig 2I). Since the second complex contains the decapping machinery, known to trigger the degradation of NMD substrates (Muhlrad & Parker, 1994), we refer to this complex as the “Upf1‐decapping”. It is likely that the action of Upf2 and Upf3 precedes decapping and ensures specificity for NMD substrates, so we decided to call the Upf1‐Upf2‐Upf3 complex, the “Upf1‐23”. Altogether, these data suggest that Upf1 coordinates the recognition and degradation of NMD by binding to two mutually exclusive sets of proteins.
The Upf1 cysteine/histidine‐rich N‐terminal domain is an interaction “hub” in vivo
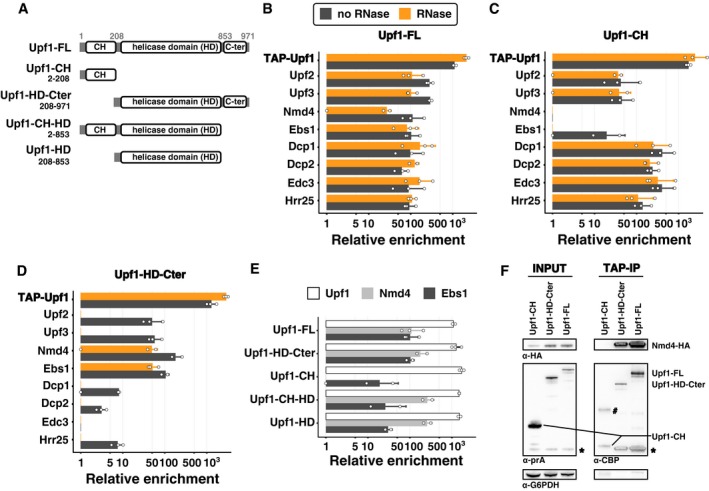
To find out how the NMD complexes are organized and to investigate why the Upf1‐binding proteins in Upf1‐23 and Upf1‐decapping were mutually exclusive, we tested the importance of Upf1 domains in the interaction with the associated factors. We built plasmids for the expression of tagged Upf1 fragments (Fig 3A) to test the cysteine/histidine‐rich N‐terminal domain (Upf1‐CH, 2–208), the C‐terminal helicase domain (Upf1‐HD‐Cter, 208–971), as well as variants of these constructs, comprising either the helicase domain alone (Upf1‐HD, 208–853) or a truncated Upf1 lacking the C‐terminal extension (Upf1‐CH‐HD, 2–853). Strains deleted for endogenous UPF1 were transformed with plasmids expressing N‐terminal TAP‐tagged full‐length Upf1 (Upf1‐FL) or the various fragments. TAP‐Upf1‐FL was expressed to levels 10‐fold to 50‐fold higher than the C‐terminal tagged Upf1 expressed from the chromosomal locus, as estimated by immunoblots on total protein cell extracts. The various fragments were also stably expressed to high levels at the expected size (Fig EV2A).
Figure 3. Nmd4 and Ebs1 are the only Upf1‐decapping components that interact with Upf1 independent of the N‐terminal CH domain.
-
AUpf1 fragments tested: FL is for the full‐length protein; HD‐Cter for the region containing the helicase domain and the C‐terminal part of Upf1 (208–971); CH for the N‐terminal domain (2–208) that contains the N‐terminal unstructured region and the cysteine/histidine‐rich domain; CH‐HD for a version a full‐length Upf1 lacking the C‐terminal region 854–971; and HD for the helicase domain of Upf1 alone (not to scale). N‐terminal TAP‐tagged Upf1 versions were expressed from a single‐copy vector under an inducible tetO7 promoter.
-
B–DResults of purifications using Upf1‐FL (B), Upf1‐CH (C) and Upf1‐Cter (D) as bait with the x‐axis showing the average enrichment value (log2 scale) for Upf1‐decapping and Upf1‐23 components. The colour of the bars illustrates the treatment used during the purification, black without RNase and orange with an RNase treatment. Error bars represent SD. White dots correspond to enrichment values obtained in individual experiments.
-
EComparison of the enrichment of Upf1, Nmd4 and Ebs1 in the purification of the Upf1 fragments. The Upf1 C‐terminal region (854–971) affected the association with Nmd4 or Ebs1 (Student t‐test with a one‐sided alternative hypothesis). We compared the enrichment of each of Ebs1 and Nmd4 between purifications of Upf1 fragments having this region (Upf1‐FL and Upf1‐HD‐Cter, six experiments) and purifications of Upf1 fragments lacking the extension (Upf1‐CH‐HD and Upf1‐HD, four experiments). The C‐terminal region had no effect on Nmd4 enrichment (P‐value ≈ 0.98), whereas there was significantly less associated Ebs1 on this small region (P‐value ≈ 0.0013).
-
FWestern blot of input and eluates of Upf1 domains purification in a Nmd4‐HA strain. The band with the # might correspond to a dimer of Upf1‐CH, bands marked with a star correspond to residual signal with the anti‐HA antibodies (Nmd4). Fragments in the eluate have a smaller size because the protein A part of the tag was removed by digestion with the TEV protease. G6PDH served as a loading control in the input samples.
Source data are available online for this figure.
Figure EV2. N‐terminal and C‐terminal tagged Upf1 enrich similar sets of specific proteins.
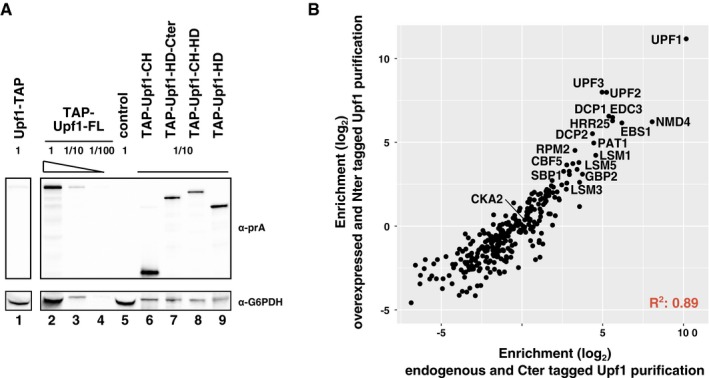
- Estimation of the levels of overexpression for N‐terminal tagged Upf1 fragments, in comparison with chromosomally C‐terminal tagged protein. G6PDH was used as a loading control. Serial dilutions were used to test the ability of the immunoblot signal to estimate protein levels.
- Average enrichment values for purifications done with chromosomally C‐terminal tagged Upf1 (x‐axis) and N‐terminally TAP‐tagged Upf1.
Enrichment of the various proteins in the purification done with overexpressed TAP‐Upf1‐FL was well correlated with the enrichment of the same proteins in the previous purifications done with the protein tagged at its C‐terminus and expressed from the endogenous UPF1 locus (Fig EV2B and Dataset EV1), confirming that neither overexpression nor tag position had major effects on Upf1‐23 and Upf1‐decapping composition (see Fig 2A compared with Fig 3B). The only marked difference between the results with the overexpressed N‐terminal Upf1 and the C‐terminal tagged Upf1 was the persistence of Upf2 and Upf3 among the enriched proteins after RNase treatment (Fig 3B, orange bars). Since the Upf1 interaction was also preserved when the Upf1‐23 complex was purified using tagged Upf2 (Fig 2B), but not when using tagged Upf3 (Fig 2C), this variability in Upf1‐23 sensitivity to RNase indicates the importance of RNA for the stability of the Upf1‐Upf2‐Upf3 complex. In contrast, the interactions of Upf1 with the Upf1‐decapping complex proteins, and especially with Nmd4, were insensitive to RNase in all the studied situations.
Tagged Upf1‐CH recovered a majority of the proteins associated with the full‐length protein, with the notable exception of Nmd4 (not detected) and of Ebs1, which was detected only in the sample not treated with RNase, albeit to a lower intensity than in the full‐length Upf1 purification (Fig 3B and C). The observation that the other proteins of the Upf1‐decapping and Upf1‐23 complexes were highly enriched in this purification and the fact that the interactions were not sensitive to RNase (Fig 3C), raise the possibility that Upf2 and Upf3 compete with the decapping factors for binding to Upf1‐CH domain. Thus, the CH domain seems to be crucial for both Upf1‐23 and Upf1‐decapping organization.
Nmd4 and Ebs1 are tightly associated with Upf1‐HD‐Cter
Nmd4 and Ebs1 were the only proteins interacting specifically with the HD‐Cter domain of Upf1 independently of RNA (Fig 3D). To establish whether the C‐terminal domain of Upf1 could play a role in these interactions, we purified Upf1‐CH‐HD and Upf1‐HD and evaluated the relative enrichment levels for Ebs1 and Nmd4 in these conditions. While Nmd4 was recovered with both Upf1 fragments, Ebs1 levels were substantially lower in the absence of the C‐terminal extension (Fig 3E, t‐test P‐value 0.0013, n = 4). These results suggest that the C‐terminal extension of Upf1 has a crucial effect on binding Ebs1, and does not affect binding of Nmd4.
To confirm the observed strong Nmd4‐Upf1 interactions, we built Nmd4‐HA strains and tested the presence of the tagged protein in fractions enriched with overexpressed tagged Upf1‐FL, Upf1‐CH and Upf1‐HD‐Cter. The purification of Upf1‐FL and Upf1‐HD‐Cter co‐enriched Nmd4‐HA, while the Upf1‐CH region alone did not. Unexpectedly, overexpression of Upf1‐FL or Upf1‐HD‐Cter also led to increased levels of Nmd4‐HA in the total extracts (Fig 3F), suggesting that Nmd4‐HA protein stability can be affected by its interaction with Upf1. To test the importance of the observed interaction between Nmd4 and Upf1 for the association with other factors of the Upf1‐decapping complex, we purified Upf1‐TAP in the absence of NMD4. We observed no major effects on the various associated proteins (Fig 4A, Appendix Fig S2 and Dataset EV1). In the reciprocal experiment, in which we purified the Nmd4‐associated complexes in the absence of UPF1, all the specific components of the Upf1‐decapping complex were lost (Fig 4B).
Figure 4. Nmd4 interaction with the Upf1‐decapping complex is direct and mediated by Upf1.
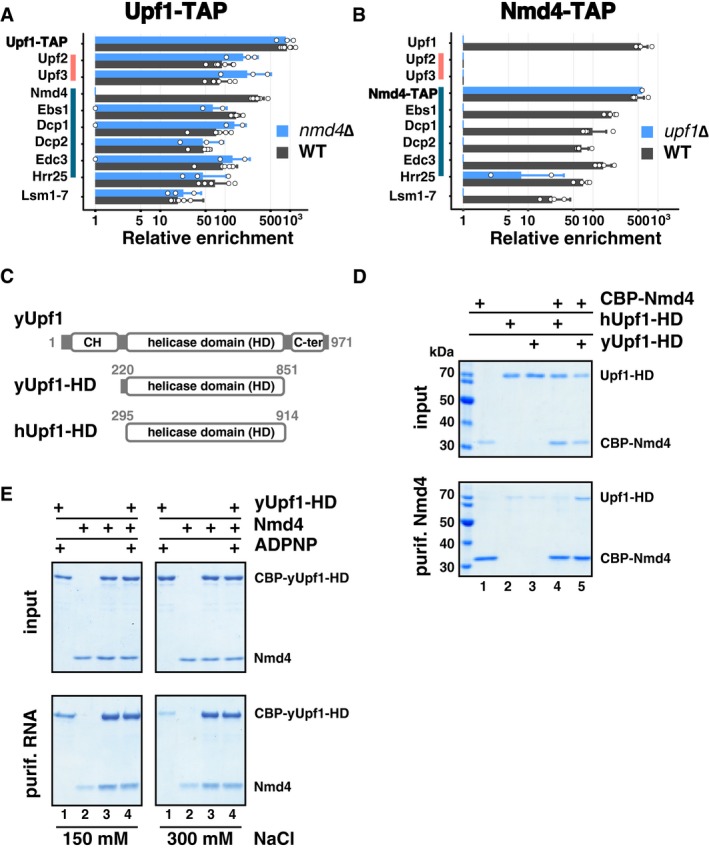
- Average enrichment of Upf1‐23 and Upf1‐decapping components in purified Upf1‐TAP in the presence (black bars) and absence (blue bars) of NMD4. Pink and dark blue vertical lines highlight proteins of the Upf1‐23 and Upf1‐decapping complexes, respectively. White dots correspond to individual enrichment values obtained in replicate experiments, 6 for Upf1‐TAP and 3 for the nmd4Δ condition. Error bars correspond to SD.
- Average enrichment of Upf1‐decapping components in Nmd4‐TAP in the presence (black bars) and absence (blue bars) of UPF1. White dots correspond to individual enrichment values obtained in replicate experiments, 4 for Nmd4‐TAP and 2 for the upf1Δ condition. Error bars correspond to SD.
- Schematics of Upf1 fragments used for the in vitro interaction assay; yUpf1‐HD is the helicase domain (220–851) of the yeast Upf1 protein, hUpf1‐HD is the helicase domain (295–914) of human Upf1 (not to scale).
- CBP‐Nmd4 was mixed with hUPF1‐HD or yUPF1‐HD (all the proteins overexpressed in Escherichia coli and purified). Protein mixtures before (input, 20% of total) or after purification on calmodulin affinity beads were separated on 10% SDS–PAGE (w/v) acrylamide gels.
- Upf1 interaction with a 30‐mer 3′ biotinylated RNA fragment was tested by mixing purified Upf1 helicase domain with Nmd4 and testing the fraction of recovered protein on streptavidin beads after washes with 150 and 300 mM NaCl containing buffer solution.
Source data are available online for this figure.
These results, together with the strong and RNase insensitive enrichment of Nmd4 in Upf1 complexes, suggested that Upf1 directly interacts with Nmd4. To validate this hypothesis, we expressed CBP‐Nmd4 as a recombinant protein in Escherichia coli and tested its association in vitro with recombinant purified Upf1 helicase domains of yeast and human origin (yUpf1‐HD 220–851 and hUpf1‐HD 295–914, Fig 4C). Yeast Upf1, but not human Upf1, co‐purified with CBP‐Nmd4, showing that the interaction is direct and specific (Fig 4D). To test whether this interaction could affect Upf1 function, we used a short 3′ biotinylated RNA fragment to purify Upf1, as previously described (Fiorini et al, 2012). The concomitant presence of Nmd4 led to a stronger binding of Upf1 to RNA, especially visible at higher ionic strength (Fig 4E). These results designate Nmd4 as a tightly bound Upf1 co‐factor, specific to the conformation of Upf1 present in the Upf1‐decapping complex, and affecting the affinity of Upf1 for RNA.
Potential Smg5‐6‐7 homologues can be essential for NMD efficiency under limiting conditions
The association of Nmd4 and Ebs1 with Upf1 support the hypothesis that they are the yeast functional equivalents of human Smg6 and Smg5/7. In line with this idea, Nmd4 contains an endonuclease‐like region from the PIN domain family (Clissold & Ponting, 2000), like Smg6 and Smg5 (Fig 5A). Ebs1 has strong similarities with the N‐terminal 14‐3‐3 domains of Smg5, Smg6 and Smg7 (Luke et al, 2007), with similar percentages of identity in the aligned sequences for this domain (Fig 5A, Appendix Fig S3). The interactions that we described here indicate that Nmd4 and Ebs1 are components of the Upf1‐decapping complex and co‐factors of Upf1. While a role for Ebs1 in the degradation of an NMD reporter RNA has been previously reported (Luke et al, 2007), no data were available about a potential global role of NMD4 and EBS1 in NMD. To investigate the impact of NMD4 and EBS1 on NMD on a large scale, we performed strand‐specific RNA‐Seq experiments in strains deleted for each of the two genes.
Figure 5. Nmd4 and Ebs1 are essential for NMD elicited by the overexpression of the helicase domain of Upf1.
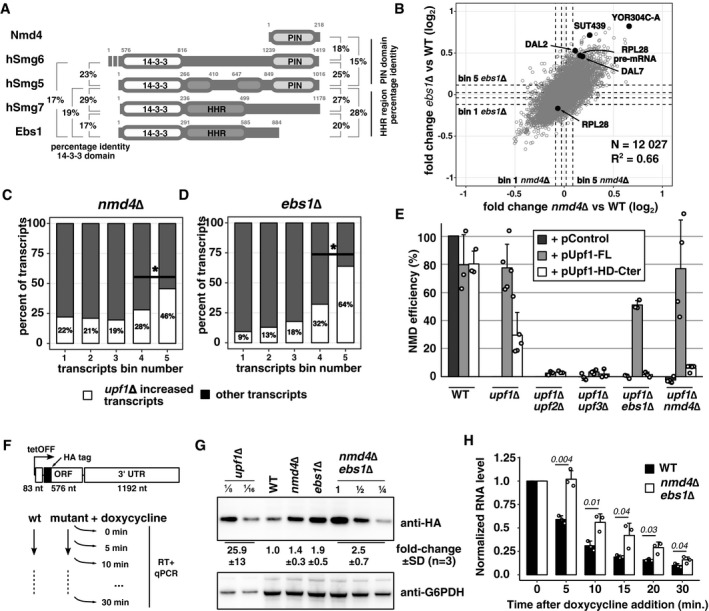
- Schematic representation of the domain structure of Nmd4 and Ebs1 from Saccharomyces cerevisiae compared with human (h) Smg6, Smg5 and Smg7. 14‐3‐3, HHR and PIN domains were defined based on literature data (Fukuhara et al, 2005; Luke et al, 2007). Boundaries of the PIN domains were chosen based on a Mafft alignment of Smg5/6 of several species, while for Nmd4 the entire protein sequence was used. Drawing is not to scale. Identity percentages among the different domains of Smg proteins, Nmd4 and Ebs1, are indicated. Values represent per cent of identical residues as a fraction of all the aligned residues.
- Scatter plot of the mean fold change of transcripts in nmd4∆ RNA‐Seq experiment against transcript mean fold change in ebs1∆ (Pearson correlation, r = 0.66, P‐value < 2 × 10−16). Black dots correspond to NMD substrate examples for which individual traces are shown in Fig EV3. Dashed lines represent boundaries of five bins of equal numbers of transcripts (2,046 transcripts or notable features per bin), with bin 5 containing the transcripts that were most increased in the mutant condition compared with wild type.
- Percentage of transcripts affected by UPF1 deletion (increase by at least 1.4‐fold) among nmd4∆ bins, as defined in (B). Differences between percentages of upf1∆ affected transcripts in bin 4 and 5 and bin 3 and 4 were significant (binomial test, P < 10−24).
- Same as in (C), for ebs1∆. Differences between distribution of transcripts in bin n and bin n + 1 were significant (binomial test, P < 10−9).
- NMD efficiency of WT, upf1∆ or double‐mutant strains complemented with Upf1‐FL, Upf1‐HD‐Cter or an empty plasmid. The NMD efficiency for each strain is based on reverse transcription followed by quantitative PCR for RPL28 pre‐mRNA. A wild‐type strain has 100% NMD efficiency and a upf1∆ strain has 0% NMD efficiency, by definition. Each value is an average of replicate experiments; 3 for the WT condition; 5 for upf1Δ; 3 for ufp1Δ/upf2Δ, upf1Δ/ufp3Δ and upf1Δ/ebs1Δ; and 4 for the upf1Δ/nmd4Δ condition. Error bars represent SD. Dots represent values obtained in the various replicate experiments.
- Schematic drawing of an NMD reporter whose transcription can be repressed by doxycycline addition to the culture.
- Changes in the steady‐state level of the reporter encoded HA‐tagged protein in upf1Δ, nmd4Δ, ebs1Δ and the nmd4Δ/ebs1Δ strains were estimated by immunoblot from three independent experiments, with one example shown.
- RNA decay in a wild‐type and nmd4Δ/ebs1Δ strains was tested by reverse transcription quantitative PCR specific to the 5′ end of the reporter RNA at different times after transcription shut‐off. The quantifications represent mean RNA amounts and standard deviation of the average (three independent experiments).
Source data are available online for this figure.
The obtained RNA‐Seq data allowed us to quantify variations of RNA levels for 12,027 transcripts (mapping statistics available in Appendix Table S2 and quantifications in Dataset EV2). DESeq2 (Love et al, 2014) was used to adjust, normalize and compute the changes in expression levels in ebs1∆ and nmd4∆ strains in comparison with a wild‐type strain. Even if the amplitude of change in transcript levels was modest (examples shown in Fig EV3A–E), the observed variations were highly correlated between the two strains (Fig 5B, N = 12,027; Pearson correlation coefficient = 0.66; P‐value < 2.2 × 10‐16). To see to what extent the transcripts enriched in these mutant strains were related to transcripts stabilized in the absence of UPF1, we performed a similar RNA‐Seq experiment by comparing upf1∆ to a wild‐type strain. 3,271 transcripts showed an increase of more than 1.4‐fold in the absence of UPF1 and were used as a reference set for NMD‐sensitive RNAs (Fig EV3F). To estimate the presence of such RNAs in the population of transcripts affected by the absence of NMD4 or EBS1, we divided the set of obtained values in five bins of equal size (N = 2,406 per bin, Fig 5B). We found a strong enrichment of UPF1‐affected RNA in bin 5, which contains the RNAs most increased in the absence of NMD4 (Fig 5C) or EBS1 (Fig 5D; P‐value < 10−74, binomial test performed with one‐sided alternative null hypothesis, N = 2,406).
Figure EV3. Deletion of NMD4 and EBS1 stabilize a set of transcripts that is also stabilized in the absence of UPF1 .
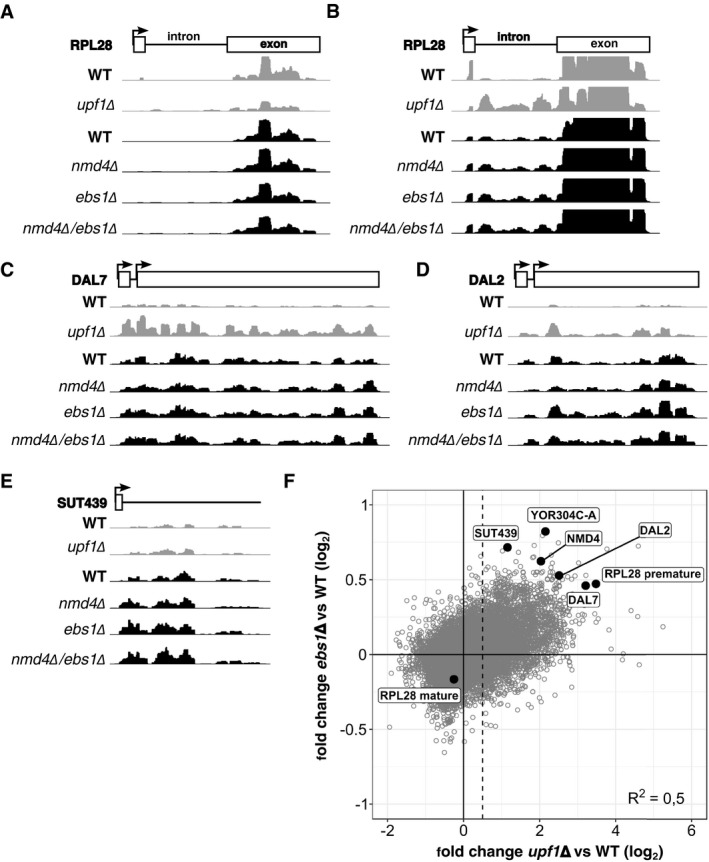
-
A–EExamples of NMD substrates sequencing profiles in WT, upf1∆, nmd4∆, ebs1∆ and nmd4∆/ebs1∆ experiments. NMD substrates belong to different classes, intron containing (RPL28; A and B, different scaling to show intron signal), uORF (DAL7, DAL2; C and D) and non‐coding RNA (SUT439; E). Profiles were normalized using the samples median counts.
-
FScatter plot of transcript log2 fold change in upf1∆ against ebs1∆. The dashed line represents the limit over which RNA was considered as stabilized, with a log2 value of 0.5 (1.4 fold change).
To establish how NMD4 and EBS1 could affect NMD substrates, we searched for conditions that would simplify our phenotypic analysis. We chose to focus on the helicase and C‐terminal domain of Upf1, important for binding of both Nmd4 and Ebs1 (Fig 3D). It has been previously observed that overexpression of this region of Upf1 can complement the deletion of the UPF1 gene (Weng et al, 1996). We thus tested whether NMD4 and EBS1 were required for this complementation. First, we validated the effect of overexpressing full‐length Upf1 or its different domains (depicted in Fig 3A) on the levels of RPL28 (also known as CYH2) pre‐mRNA, a well‐studied NMD substrate. To facilitate the visualization of the complementation levels, we devised an NMD efficiency measure that takes into account RPL28 pre‐mRNA levels in WT (100% NMD) and upf1Δ (0% NMD) strains (Materials and Methods and Appendix). While overexpression of full‐length UPF1 led to the destabilization of RPL28 pre‐mRNA, with an estimated reconstituted NMD efficiency of 80%, overexpression of Upf1‐HD‐Cter allowed recovery of 30% of NMD in a upf1∆ strain (Figs 5E and EV4A). This partial complementation of NMD required classical NMD factors since it was abolished in strains lacking UPF2 and UPF3, in strains lacking the 5′ to 3′ exonuclease XRN1 and in a strain in which the decapping enzyme Dcp2, required for yeast NMD, was depleted (Figs 5E, and EV4A and B). Deletion of either EBS1 or NMD4 abolished the partial complementation effect (Fig 5E, Appendix Fig S4), suggesting that under limiting conditions, EBS1 and NMD4 become essential for NMD. These results correlated with the inability of the Upf1‐HD fragment, which showed decreased binding of Ebs1 (Fig 3E), to complement NMD (Fig EV4A). In line with these observations, EBS1 deletion also decreased the NMD efficiency of Upf1‐FL (Fig 5E). Altogether, these results indicate that the mechanism by which Upf1‐HD‐Cter destabilizes NMD substrates depends on the presence of Nmd4 and Ebs1, the only factors that specifically interact with this region of Upf1 independent on RNA (Fig 3D).
Figure EV4. The helicase domain of Upf1 alone can destabilize RPL28 pre‐mRNA, an NMD substrate.
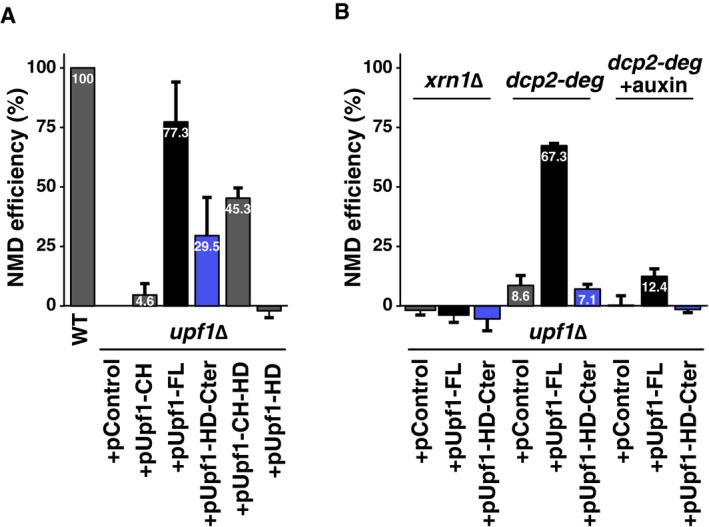
- Total RNA from wild‐type or a upf1Δ strain transformed with an empty plasmid (pControl) or plasmids expressing various Upf1 fragments (see Fig 3A) was tested by reverse transcription and quantitative PCR. The levels of RPL28 pre‐mRNA were normalized using an NMD‐insensitive transcript (RIM1), and an NMD efficiency score was calculated based on the difference between a wild‐type (100%) and the upf1Δ (0% NMD) strain.
- Complementation of UPF1 deletion was tested in combination with the deletion of XRN1, and in strains with a degron‐regulated Dcp2 protein, when protein degradation was not induced (dcp2‐deg) or was induced by addition of auxin (dcp2‐deg + auxin) and incubation for 1 h. Values represent average and standard deviation for at least three independent experiments.
While the biochemical association, RNA‐Seq data and the NMD complementation assay strongly suggested a specific role for Nmd4 and Ebs1 in NMD, we wanted to test the impact of the corresponding genes in a reporter system that allows measuring RNA half‐life changes. To this end, we used an NMD reporter that codes for a HA‐tagged protein fragment and whose transcription can be shut off by the addition of doxycycline (Fig 5F). Immunoblots with anti‐HA antibodies showed a consistent increase in the amounts of produced protein in strains lacking either NMD4, EBS1 or both genes (Fig 5G). Since the strongest effect was seen in the double‐mutant strain, we tested the decrease in the reporter NMD RNA levels at different times after transcription shut‐off in this condition. Compared with the wild type, the nmd4Δ/ebs1Δ strain showed a delay in the decrease of RNA levels, best seen at 5 min after doxycycline addition (Fig 5H). These data suggest that Ebs1 and Nmd4 are NMD factors that affect the degradation of NMD substrates through their specific interactions with the Upf1 helicase and C‐terminal domain.
Upf1‐23 forms by binding of a Upf2/Upf3 heterodimer to Upf1 in vivo
While the results presented in the previous paragraphs identified a role for Ebs1 and Nmd4 linked with the helicase domain of Upf1, the CH domain of the protein binds the best‐known NMD factors Upf2 and Upf3. Among the results that stood out from the purifications of Upf1‐23 complex components, one of the most striking was the loss of Upf2 and Upf3 in the Upf1 complex after RNase treatment (Fig 2A). This result was unexpected, since a complex can be reconstituted with domains of the three proteins in the absence of RNA (Chamieh et al, 2008). The reciprocal experiments using Upf2‐TAP and Upf3‐TAP as baits showed that Upf1 could be efficiently recovered in the presence or absence of RNase with Upf2‐TAP (Fig 2B), but its interaction with Upf3‐TAP was sensitive to RNase (Fig 2C). Thus, the role played by RNA in the stability of the purified Upf1‐Upf2‐Upf3 complex suggested that Upf1‐23 is assembled on RNA. To investigate the assembly process, we analysed the composition of complexes purified with Upf1‐TAP, Upf2‐TAP and Upf3‐TAP in the absence of UPF1, UPF2 or UPF3. Deletion of either UPF2 or UPF3 had a major impact on the enrichment of Upf3 and Upf2 with Upf1 (Fig 6A and B). In the absence of UPF2, we could no longer detect Upf3 (Fig 6A), which correlates with previously published data that described human Upf2 as bridging Upf1 and Upf3 (Chamieh et al, 2008). Unexpectedly, in the absence of Upf3, we could not detect Upf2 in the Upf1‐associated complex (Fig 6B). Thus, in the absence of UPF2, Upf3 no longer stably associated with Upf1 and in the absence of UPF3, Upf2 was lost from the Upf1‐associated factors. We verified that these changes were not due to changes in the total levels of Upf3‐TAP and Upf2‐TAP in the absence of UPF2 (Fig 6C) and UPF3 (Fig 6D). The absence of Upf1 had no impact on the formation of the Upf2‐Upf3 complex, independent of which of the two proteins was tagged (Fig 6E and Dataset EV1). Altogether, these results suggest that Upf2 and Upf3 form a heterodimer independent of Upf1 and that both proteins are required for a stable association with Upf1 in the Upf1‐23 complex.
Figure 6. Upf2 and Upf3 function as a heterodimer in the formation of Upf1‐23.
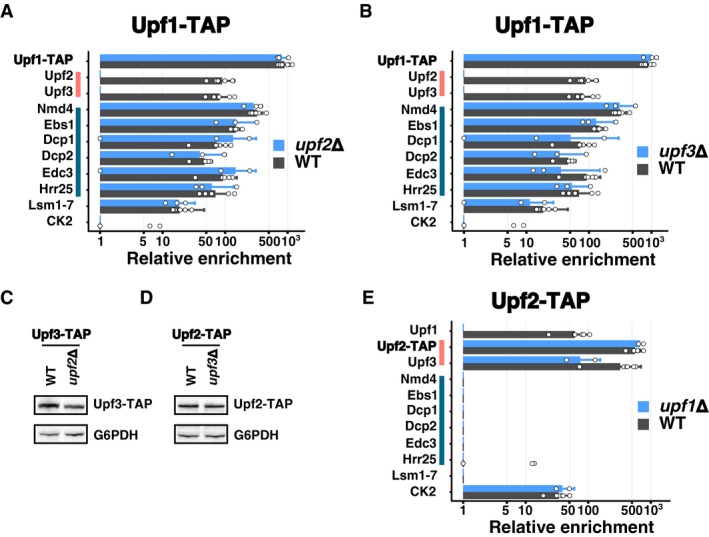
-
A, BComparison between average enrichment values for Upf1‐decapping and Upf1‐23 components, Lsm proteins and CK subunits in Upf1‐TAP‐purified samples in the presence (grey bars) or the absence (blue bars) of UPF2 (A) or UPF3 (B). Error bars represent SD. White dots represent enrichment values for individual replicates, 6 for Upf1‐TAP, 3 for the upf2Δ and upf3Δ conditions.
-
C, DEvaluation of the effects of UPF2 deletion on Upf3‐TAP levels (C) and of UPF3 deletion on Upf2‐TAP levels (D) in total extracts, in comparison with a loading control (G6PDH), was done by immunoblot.
-
EComparison between average enrichment values for Upf1‐decapping and Upf1‐23 components, Lsm proteins and CK subunits in Upf2‐TAP‐purified samples in the presence (grey bars, six experiments) or the absence (blue bars, two experiments) of UPF1. Error bars represent SD. The levels of expression of Upf2‐TAP and Upf3‐TAP proteins have been verified in this condition (Appendix Fig S2).
Source data are available online for this figure.
Binding of Upf1‐decapping to NMD substrates depends on Upf2
The formation of Upf1‐23 on NMD substrates could be followed by a switch to Upf1‐decapping, for RNA decapping and initiation of degradation. Surprisingly, the absence of Upf2 or Upf3 did not alter the protein composition of the purified Upf1‐decapping complex (Fig 6A and B). This puzzling result made us wonder if Upf1‐decapping exists in two forms, one that associates with NMD substrates, and another that would correspond to Upf1 and the decapping machinery in the process of being recycled as RNA‐free complexes. To explore this hypothesis, we took advantage of the fact that Upf1 binds preferentially to RNAs that are degraded through NMD (Johansson et al, 2007). To distinguish between the Upf1‐decapping‐bound and Upf1‐23‐bound Upf1, we purified Upf1‐decapping via Nmd4‐TAP (Fig 2D). The sixfold enrichment of RPL28 pre‐mRNA as compared with total RNA in the input fraction (Fig 7A) indicated that a fraction of the Upf1‐decapping complex binds NMD substrates.
Figure 7. Association of Nmd4 with NMD substrates depends on Upf1‐23.

-
AUnspliced RPL28, an NMD substrate, was enriched in Nmd4‐TAP purification in comparison with a control untagged strain, as measured by reverse transcription followed by quantitative PCR. Bars correspond to the mean of pre‐RPL28 enrichment for three independent experiments, as compared with RIM1, a non‐NMD mRNA. Error bars correspond to SD. The indicated P‐value was computed using the Welch t‐test, single‐sided comparison.
-
B–DDistribution of Nmd4‐TAP, Nog1 (marker of free 60S subunits) and Rps8 (marker of 40S, 80S and polysome fractions) in wild‐type (B), upf1∆ (C) and upf2∆ (D) strains, tested by immunoblot. Fractions 1, 2, 3 are light fractions; fractions 4, 5 and 6 correspond, respectively, to ribosomal 40S, 60S and 80S positions; fractions 7–12 correspond to polysomes. The per cent of total signal for Nmd4‐TAP in three independent replicates along with standard deviation of the values are indicated for each fraction.
-
EQuantified relative changes in Nmd4‐TAP average levels in light and monosome/polysome fractions in upf1∆ and upf2∆ strains (100% correspond to levels in the wild‐type strain). Indicated P‐values correspond to the Welch t‐test, single‐sided comparison of four replicate experiments (individual values are indicated as dots). Error bars correspond to SD.
-
FSummary of the Upf1 domains interaction with Upf1‐23 and Upf1‐decapping components, based on the data presented in this work.
-
GProposed Upf1‐23/Upf1‐decapping sequence of events for yeast NMD.
Source data are available online for this figure.
We next wanted to know whether Upf1‐decapping binding to RNA was dependent on Upf1‐23 components, Upf2 and Upf3, which would be suggestive of a mandatory sequence of association of the NMD complexes with the RNA substrates. We could not test this hypothesis directly, because deletion of UPF2 or UPF3 leads to a massive stabilization of NMD‐sensitive RNAs and renders RNA enrichment calculations and comparisons with the wild‐type situation unreliable. To overcome this problem, we looked at the association of the Upf1‐decapping complex with RNA by measuring the distribution of Nmd4‐TAP in a polysome gradient in the absence or presence of UPF2. While most of Nmd4 sedimented in the upper part of the gradient, a fraction of the protein was found in the polysomal and monosomal fractions (Fig 7B). The monosomal region concentrates the majority of NMD substrates (Heyer & Moore, 2016). In the absence of UPF1, which mediates all the interactions between Nmd4 and the other components of the Upf1‐decapping complex (Fig 4B), Nmd4 was lost from the monosome and polysome fractions (Fig 7C). In the absence of UPF2, an essential component of the Upf1‐23 complex, we also observed a similar change in the distribution of Nmd4 (Fig 7D). Compared with the wild‐type situation, the absence of Upf1 or Upf2 led to an increase in Nmd4‐TAP levels in the lower molecular weight fractions and a decrease in the polysome fractions (Fig 7E). These changes were specific for Nmd4, since the gradient distribution of Rps8, a component of the 40S ribosomal subunits used as a control, was not altered, neither in the upf1 (Fig 7C) nor in the upf2∆ strain (Fig 7D).
The association of Nmd4, a specific component of the Upf1‐decapping complex, with NMD substrates, and its redistribution into lower molecular weight complexes in the absence of the Upf1‐23 formation, suggests that Upf1‐23 and Upf1‐decapping are obligatory successive complexes on RNA, and allows us to propose a succession of events compatible with our observations for yeast NMD (Fig 7F and G). This model includes two forms of Upf1‐decapping: one that is RNA‐bound and depends on Upf1‐23 components Upf2 and Upf3, and another that is RNA‐free.
The fact that Nmd4 was bound to a region of Upf1 not required for other interactions and still could only be enriched in Upf1‐decapping complexes suggests that Upf1 has a different conformation in Upf1‐23 and Upf1‐decapping. A switch from the Upf1‐23 to the Upf1‐decapping conformation, described here, would permit recruitment of decapping factors, required to degrade NMD substrates (Muhlrad & Parker, 1994), and could be an essential step of the NMD mechanism.
Discussion
The current study addresses the composition and dynamics of NMD complexes in yeast. We identified two distinct, mutually exclusive (Fig 2) and potentially successive (Fig 7) complexes containing Upf1. We propose that the Upf1‐23 complex, composed of Upf1, Upf2 and Upf3, is recruited on NMD substrates. A complete re‐organization of the initial complex leads to the replacement of the Upf2/3 heterodimer with Nmd4, Ebs1 and the decapping factors Dcp1, Dcp2 and Edc3. Nmd4 and Ebs1 are accessory factors for NMD in yeast and could be functional homologues of human Smg6 and Smg5/7, respectively (Figs 4 and 5). Both the decapping machinery and Upf2/3 interacted with the CH domain of Upf1 (Fig 3), in support of the hypothesis that decapping competes with Upf2‐3 to form the Upf1‐decapping. This competition is likely to occur on RNA, since Nmd4 was depleted from large RNA‐associated complexes in the absence of UPF2 (Fig 7). Thus, recruitment of decapping on NMD substrates is likely to depend on this switch on RNA, which could be a crucial, albeit not yet explored step in NMD.
How exactly the switch from the “Upf1‐23” to an “Upf1‐decapping” complex occurs is unclear, but similar events could be also important for recruiting RNA degradation factors for NMD in other organisms. Since all the factors that we describe in yeast NMD complexes have mammalian equivalents, and since our results are compatible with a large body of experimental evidence in various other organisms, we propose an extension of the canonical model of NMD (Appendix Fig S5). We discuss how a revised NMD model fits the available experimental data in yeast and other species and to what extent it can serve as a basis to build NMD models that incorporate RNA splicing and Upf1 phosphorylation, two molecular events that do not affect NMD in S. cerevisiae.
Affinity purification strategies for NMD
Before entering into the description of the proposed model, it is important to see how the results presented here extend and improve those obtained in previous studies. Affinity purification followed by mass spectrometry is the most effective way to find functional association of proteins and surpasses in precision all the other large‐scale interaction methods in yeast (Benschop et al, 2010). However, despite the excellent results obtained with this method on hundreds of different complexes, large‐scale affinity purification failed to assign specific interactions of Upf1, Upf2 or Upf3 with other yeast proteins (Gavin et al, 2006). As a result, most of what was known on the composition of yeast NMD complexes was based on two‐hybrid experiments and the use of co‐purification and immunoblotting for previously identified factors (e.g. He & Jacobson, 1995; Czaplinski et al, 1998; Swisher & Parker, 2011). The situation is similar in other species and can be illustrated by the results obtained with the recently published SONAR approach (Brannan et al, 2016), which analysed the proteins co‐purified with tagged human Upf1 (hUpf1). Human Upf2 was found as the 112th most enriched factor in the list of Upf1‐associated proteins and only reached position 28 when the sample was treated with RNase A. Except for Upf3b, ranked second, no other specific NMD factors were identified in the first 100 proteins associated with hUpf1. Thus, without previous knowledge of the identity of NMD factors, the number of false‐positive results precludes the use of these data to define NMD complexes in human cells.
To obtain a global and high‐resolution view of the composition and dynamics of NMD complexes, we used a combination of affinity isolation with an innovative computational workflow. Fast affinity purification preserves transient interactions (Oeffinger et al, 2007), and the use of surface‐coated magnetic beads gives access to the large RNA–protein complexes in which NMD takes place (Zhang et al, 1997) without the bias induced by porous chromatography media (Halbeisen et al, 2009). To get rid of abundant contaminants, we used the relative enrichment of proteins, based on mass spectrometry quantitations (see Materials and Methods), which efficiently highlighted specific interactions.
These robust quantitative data identified novel interactions, including two protein kinases associated with Upf1 (Hrr25; Figs 1F and G, and 2A, Dataset EV1) and Upf2 (Casein Kinase 2 complex; Fig 2B and Dataset EV1). Besides the ability to show the presence of interactions, quantitative mass spectrometry provided a way to define lost or absent proteins in a given complex. This led to the identification of mutually exclusive complexes, illustrated by the comparison of Upf1‐23 components purified via Upf2‐TAP with Upf1‐decapping complexes associated with Nmd4‐TAP (Fig 2B and D). The existence of these two distinct complexes in yeast NMD was not known but is compatible with previously published results, like the two‐hybrid interactions of Upf1 with Upf2 and Upf3 (He et al, 1997), the co‐purification of human Dcp1 and Dcp2 with Upf1 (Lykke‐Andersen, 2002) or the two‐hybrid interactions between yeast Edc3 and Upf1 (Swisher & Parker, 2011). Our results, showing the importance of the CH domain in the interactions between Upf1 and other components of Upf1‐23 and Upf1‐decapping (Fig 3), are also compatible with previous two‐hybrid data showing the importance of this domain in the interactions with Upf2 and Upf3 (He et al, 1997) and with Dcp2 (Swisher & Parker, 2011; He & Jacobson, 2015).
Altogether, our enrichment approach and the combination of RNase treated and native complexes analyses drastically reduced the number of NMD‐associated candidate proteins to a short list that only contains highly specific top hits and is devoid of factors that are not related to NMD (Fig 2I and Dataset EV1). The extensive measurements of the dynamics of NMD complexes in different mutant conditions bound or not to RNA support a new hierarchy of the molecular events required for yeast NMD (Fig 7G).
Upf1‐23 formation is both similar and distinct from SURF/DECID
The proposed yeast mechanism (Fig 7G, Appendix Fig S5) can be extended to other species to include elements of the canonical SURF/DECID model that was, until now, restricted to mammalian EJC‐enhanced NMD. Similar unified NMD models that include EJC‐enhanced and EJC‐independent NMD as two aspects of the same molecular mechanism have been proposed earlier (Stalder & Mühlemann, 2008), but differ from the extended Upf1‐23/Upf1‐decapping model in terms of the order of interactions during NMD substrate detection and of the importance of Smg1 in the process.
The first step in the model proposed here involves the recognition of an aberrant translation termination event (Amrani et al, 2004; Behm‐Ansmant et al, 2007) and the formation of a complex containing Upf1, Upf2 and Upf3. Its formation depends on an aberrant translation termination event and could occur through an interaction of Upf2/3 with the translation termination factors eRF1/eRF3 or a terminating ribosome. This type of mechanism has been recently proposed as an alternative to SURF formation in recruiting Upf1 to NMD substrates in human cells (Neu‐Yilik et al, 2017). A less detailed, but similar mechanism, has been proposed previously, based on experiments on human cells in culture (Lykke‐Andersen, 2002).
Our data support the formation of a Upf1‐Upf2‐Upf3 complex through the interaction of a Upf2‐3 heterodimer (Fig 6) with Upf1 on RNA (Fig 2A and C). The importance of heterodimerization for the function of Upf2‐3 was also observed in C. elegans, where co‐purification of Upf2/Smg3 with Upf1/Smg2 is strongly reduced in the absence of Upf3/Smg4 (Johns et al, 2007). The hypothesis of a recruitment of Upf1 through a Upf2‐3 heterodimer is also compatible with the fact that Upf2 and Upf3 co‐sediment in yeast polysomal fractions independent on the presence of UPF1, while Upf2 loses its association with heavy complexes in the absence of UPF3 (Atkin et al, 1997).
The importance of the C‐terminal Upf1 domain in yeast and mammalian NMD
We show here that the presence of the C‐terminal region of Upf1 is important for NMD in yeast, as demonstrated by its effect on the ability of Upf1 fragments to complement the deletion of the gene (Figs 5E and EV4A) and by its role in binding Ebs1 (Fig 3E). Thus, the C‐terminal extension of yeast Upf1 might function like the SQ C‐terminal extension in other species, even in the absence of the characteristic SQ/TQ motifs and phosphorylation. The C‐terminal extension of yeast Upf1 can thus be the functional equivalent of the C‐terminal Upf1 extension in other species. It is notable that Smg1, the protein kinase involved in the phosphorylation of Upf1 in many species, is absent from red and brown algae, excavates such as Trypanosoma brucei and Giardia lamblia (Lloyd & Davies, 2013) and ciliates, such as Tetrahymena thermophila (Tian et al, 2017). However, these species present active NMD mechanisms that depend on equivalents of Upf1, Upf2 and Upf3, suggesting that phosphorylation of the C‐terminal region is not an absolute requirement for NMD. In line with this observation, no phosphorylated residues have been reported in yeast Upf1 C‐terminal region, even if phosphorylated residues have been detected in Upf1 (Lasalde et al, 2013). These observations indicate that the C‐terminal domain per se is important for NMD and that the main role of the C‐terminal extension, with or without phosphorylation, is the recruitment of later factors and the formation of Upf1‐decapping‐like complexes. In support to this idea, a chimeric Upf1 where the helicase domain of the yeast protein was replaced with the similar region of human Upf1 partially complements the deletion of UPF1 in yeast as long as it preserves the fungal‐specific N and C‐terminal extensions (Perlick et al, 1996).
The similarity between the binding of Ebs1 to the C‐terminal domain of yeast Upf1 and the recruitment of Smg5/7 to phosphorylated SQ residues in metazoan NMD also extends to Nmd4. Nmd4 is a specific marker of the Upf1‐decapping complex that directly binds the helicase domain of Upf1 (Fig 4) and consists of a PIN domain similar to the PIN domain of mammalian Smg6 (Fig 5A). Nmd4 binding to the helicase domain of yeast Upf1 correlates with the known phosphorylation‐independent binding of Smg6 to a region of Upf1 helicase domain (Chakrabarti et al, 2014; Nicholson et al, 2014). Thus, the architecture and function of Upf1 and the associated factors seem to be more conserved than previously thought.
A molecular switch around Upf1 for the Upf1‐23 to Upf1‐decapping transition
The defining feature of the proposed NMD model is the major change in composition of Upf1‐bound complexes from the Upf1‐23 to the Upf1‐decapping. Upf1 conformation is likely to be dramatically different between the two complexes since Upf2 and Upf3 were completely absent from complexes purified using Nmd4‐TAP (Fig 2D); Nmd4 was bound to a region of Upf1 that is different from the N‐terminal CH domain, to which decapping factors and Upf2‐3 bind (Fig 3C and D); structures of Upf1 with and without a Upf2 C‐terminal fragment show a different organization of the Upf1 helicase sub‐domains 1B and 1C (Clerici et al, 2009); all the interactions of Nmd4 with Upf1‐decapping components depend on Upf1 (Fig 4B). This change was not apparent in previous studies that lacked quantitative estimations of all the components of purified complexes at the same time.
The main result of the switch is the recruitment of the decapping enzyme, specifically the core decapping proteins Dcp1, Dcp2 and Edc3 (Fig 2A, D and H). Decapping is the first and major step in the degradation of NMD substrates in yeast (Muhlrad & Parker, 1994) and is responsible for the degradation of about a third of NMD substrates in human cells (Lykke‐Andersen et al, 2014). The other two‐thirds of NMD substrates in human cells are likely to be degraded through a pathway that depends on the presence of catalytically active Smg6 (Huntzinger et al, 2008; Eberle et al, 2009). In view of the modest increase in NMD substrates levels in a nmd4Δ mutant (Figs 5 and EV3), it is unlikely that the Nmd4 PIN domain has an endonucleolytic activity in yeast NMD. However, this hypothesis cannot be completely ruled out, since the sequence of Nmd4 contains a conserved Asp residue in a position equivalent with Asp1392 of hSmg6, replaced by a Gly residue in the inactive PIN of hSmg5 (Appendix Fig S6). The tight association of Nmd4 with Upf1 (Figs 2A and D, and 4D), its impact on Upf1 binding to RNA (Fig 4E), and its effects on NMD triggered by overexpression of a truncated Upf1 (Fig 5E) and on a reporter RNA (Fig 5G and H) suggest that Nmd4 is a Upf1 co‐factor that assists the helicase in yeast NMD. The importance of Nmd4 in fungal NMD is supported by the presence of orthologs in many species, including Yarrowia lipolytica (Pryszcz et al, 2011), a yeast that is as distant from S. cerevisiae as the urochordate Ciona intestinalis is from humans (Dujon et al, 2004). Thus, it is likely that different organisms use the same basic molecular machineries to degrade NMD substrates, but the balance between decapping, endonucleolysis or other alternative degradation processes is variable depending on each species.
Conclusion
Our data and the extended NMD model provide a solution to the unclear situation of the conservation of NMD molecular mechanisms among eukaryotes and will allow future work on yet unsolved issues: how aberrant termination is recognized and leads to Upf1‐23 formation, where on RNA are positioned Upf1‐23 and Upf1‐decapping complexes, how Upf1‐decapping and Upf1‐23 components are recycled and at what steps ATP binding and hydrolysis by Upf1 play crucial roles.
Materials and Methods
Yeast and bacterial strains
Saccharomyces cerevisiae strains were derived from BY4741 (mat a) and BY4742 (mat alpha) strains. Deletion strains were part of the systematic yeast gene deletion collection (Giaever et al, 2002) distributed by EuroScarf (http://www.euroscarf.de/) or were built by transformation of BY4741 strain with a cassette containing a selection marker cassette flanked by long recombination arms located upstream and downstream the open reading frame of the gene. Deletions were tested by PCR amplification of the modified locus. The list of used strains is provided in Appendix Table S3.
Escherichia coli strain NEB 10‐beta (New England Biolabs) was used for construction of plasmids by Gibson assembly and their multiplication. Escherichia coli BL21(DE3) cells were used for the expression of recombinant Nmd4 and Upf1 proteins.
Plasmids construction for yeast experiments
To build single‐copy plasmids for the expression of N‐terminal TAP‐tagged Upf1 fragments in yeast, we used pCM189 (Garí et al, 1997) as a start, since it provides a Tet‐OFF promoter regulation system. Cloning of Upf1 fragments into plasmids was done by a modified version of Gibson assembly (Fu et al, 2014) using NotI digested pCM189‐NTAP (plasmid 1233, Saveanu et al, 2007) and the PCR product obtained with oligonucleotides CS1359 and CS1364 (UPF1‐FL), CS1359 and CS1361 (UPF1‐CH), CS1362 and CS1364 (UPF1‐Cter), CS1359 and CS1393 (UPF1‐NoCex) or CS1362 and CS1393 (UPF1‐Cter‐NoCex) on template pDEST14‐NAM7 (pl. 1350) or pDONR201‐NAM7 (pl. 1330).
The NMD reporter plasmid pTG189‐HA‐ALA1‐KAN was generated from a short‐lived NMD reporter used previously (Decourty et al, 2014) and which consists of a fragment of the yeast ALA1 coding sequence (576 nt), tagged by a HA tag at its N‐terminus and followed by the KanMX6 cassette ending with a TEF' terminator. The RNA has a long 3′ UTR of about 1,192 nucleotides, which induces degradation of the transcript through NMD, and its transcription is repressed by addition of doxycycline in the culture medium.
The list of plasmids and oligonucleotides used in this study is provided in Appendix Tables S4 and S5.
Plasmids construction for in vitro interaction studies
Coding sequences of yeast full‐length NMD4, yeast UPF1 helicase domain and human UPF1 helicase domains (Uniprot accession codes Q12129, P30771 and Q92900‐2, respectively) were cloned between NheI/NotI, NheI/XhoI and NdeI/XhoI in variants of pET28a (Novagen) for the in vitro interaction tests.
TAP affinity purifications
TAP‐tagged proteins were purified using a one‐step purification method. Briefly, frozen cell pellets of 2–8 l culture were resuspended in lysis buffer (20 mM HEPES K pH 7.4, 145 mM KCl, Protease inhibitor, 1X Vanadyl‐Ribonucleoside Complex, 40 units ml−1 RNasin) and lysed using a French press (two passages at 1200 PSI). The lysate was cleared at 4°C for 20 min in a JA‐25.50 rotor (Beckman) at 18,500 g. Magnetic beads (Dynabeads M‐270 epoxy) coupled to IgG were added to the protein extract and incubated for 1 h at 4°C (Oeffinger et al, 2007). Beads were collected with a magnet and extensively washed three times with a first buffer (HKI: 20 mM HEPES K pH 7.4, 145 mM KCl, 40 units ml−1 RNasin, 0.1% Igepal) and twice with a second buffer (HK: 20 mM HEPES K pH 7.4, 145 mM KCl, 40 units ml−1RNasin, 1 mM DTT). The lack of detergent in the second buffer ensured compatibility with mass spectrometry. After washing, complexes were eluted by incubation in elution buffer (HK + recombinant purified AcTEV, Thermo Fisher Scientific) for 1 h at 16°C on a rotating wheel. Supernatant that contained purified complexes was collected and precipitated using the methanol/chloroform method (Wessel & Flügge, 1984) before further analysis by silver staining, Western blot or mass spectrometry.
In case of RNase treatment, lysed cells were divided into two equal parts, one part was treated with RNase A and RNase T1 and the other part was not. Purifications were done in parallel. For treated samples, clarified lysate was treated first with 500 units ml−1 of RNase T1 (Thermo scientific) and 25 units of RNase A (Thermo scientific), 20 min on ice. The sample was also treated a second time, after adsorption of proteins on beads and washing, using a first wash with HK buffer with RNase T1 at 3000 units ml−1 and RNase A at 750 units ml−1 for 20 min at 25°C. When purified complexes were tested by immunoblot, the TEV elution was replaced with elution in 1% SDS (incubated 10 min at 65°C).
Mass spectrometry, sample preparation, acquisition and data analysis
Protein samples were treated with Endoprotease Lys‐C (Wako) and Trypsin (Trypsin Gold Mass Spec Grade; Promega). Peptide samples were desalted using OMIX C18 pipette tips (Agilent Technologies). The peptides mixtures were analysed by nano‐LC‐MS/MS using an Ultimate 3000 system (Thermo Fisher Scientific) coupled to an LTQ‐Orbitrap Velos mass spectrometer or LTQ‐Orbitrap XL (Thermo Fisher Scientific). Peptides were desalted on‐line using a trap column (C18 Pepmap100, 5 μm, 300 μm × 5 mm, Thermo Scientific) and then separated using 120 min RP gradient (5–45% acetonitrile/0.1% formic acid) on an Acclaim PepMap100 analytical column (C18, 3 μm, 100 Å, 75 μm id × 150 mm, Thermo Scientific) with a flow rate of 0.340 μl min−1. The mass spectrometer was operated in standard data‐dependent acquisition mode controlled by Xcalibur 2.2. The instrument was operated with a cycle of one MS (in the Orbitrap) acquired at a resolution of 60,000 at m/z 400, with the top 20 most abundant multiply‐charged (2+ and higher) ions subjected to CID fragmentation in the linear ion trap. An FTMS target value of 1e6 and an ion trap MSn target value of 10,000 were used. Dynamic exclusion was enabled with a repeat duration of 30 s with an exclusion list of 500 and exclusion duration of 60 s. Lock mass of 445.12002 was enabled for all experiments.
Raw data files from MS were used for spectra matching with the MaxQuant/Andromeda search engine (version 1.5.5.1 Cox & Mann, 2008; Cox et al, 2011) against the Uniprot S. cerevisiae proteome database (downloaded 12/02/2016; total 6,721 entries) completed with the 22 virus protein sequences from yeast, and a list of commonly observed contaminants supplied by MaxQuant. Searches allowed either trypsin specificity with two missed cleavages. Cysteine carbamidomethylation was selected as fixed modification, and oxidation of methionine and N‐terminal acetylation was searched as variable modifications. Peptide identification was performed using 6 ppm as mass tolerance at the MS level for FT mass analyser and 0.5 Da at the MS/MS level for IT analyser. MaxQuant/Andromeda used seven amino acid minimum for peptide length, with 0.01 false discovery rate for both protein and peptide identification. The protein identification was considered valid only when at least one unique or “razor” peptide was present. Following protein identification, the intensity for each identified protein was calculated using peptide signal intensities. Identification transfer protocol (“match‐between‐runs” feature in MaxQuant) was applied.
Only protein identifications based on a minimum of two peptides were selected for further quantitative studies. Bioinformatics analysis of the MaxQuant/Andromeda workflow output and the analysis of the abundances of the identified proteins were performed using R (workflow in Appendix Fig S1). A score called LTOP2 was computed for each protein group in the different experiments. This score is similar to the “top three” average described (Silva et al, 2006), with several differences. First, we built meta‐peptide intensities, based on the intensity of overlapping peptides with missed cleavages (minimum 4 residues overlapping). Next, we took the three most intense peptides (top 3), or the top 2 if only two peptide intensities were available, and calculated the average of log2‐transformed intensities. Finally, LTOP2 scores between experiments were normalized using the TEV protease as a spike in, as this protease is added in each sample during the purification process in equivalent quantities. To overcome some limitations of the score, and specifically the contamination by abundant yeast proteins, we calculated enrichments for each protein. Enrichment values were obtained by dividing the relative ratio of a purified protein with the ratio of the protein in a total extract.
RNA decay assay
Yeast strains were transformed with a single‐copy plasmid expressing an NMD reporter RNA that uses the TET‐off transcriptional repression system (Garí et al, 1997). Cells were grown overnight in a medium lacking uracil to maintain plasmid selection, diluted in YPD medium, grown for 2–4 h at 30°C and shifted for 1 h at 20°C, then split into several flasks in preparation for the time course experiment (20°C). Doxycycline was added to the wild‐type and mutant strain cultures at a final concentration of 10 μg ml−1 to ensure the presence of the drug in the cultures for 0–40 min. After repression, the flasks were incubated at the same time with agitation for 5 min in an ice‐containing water bath, the cultures were next centrifuged at 4°C, and the cell pellets kept at −80°C until RNA extraction. Reporter RNA levels were tested by reverse transcription followed by quantitative PCR with oligonucleotides that are specific to positions close to the 5′ end of the mRNA (CS1127 and CS1128, amplified region between 40 and 120 nucleotides downstream the potential transcription start site) and normalized to RIM1, an endogenous mRNA.
Reverse transcription and quantitative PCR
To measure the enrichment of NMD substrates such as the pre‐messenger RPL28 or the DAL7 mRNA in different experiments, we performed reverse transcription followed by qPCR. After RNA extraction and DNase treatment, 500 ng of RNA was reverse‐transcribed using Superscript III (Invitrogen) and a mix of reverse qPCR oligonucleotides, CS888 (RPL28‐premature), CS889 (RPL28), CS1077 (RIM1), CS1430 (DAL7); for RNA immuno‐precipitation, the purified and input samples were used for reverse transcription with transcript‐specific primers. Eightfold dilutions of cDNA were tested by quantitative PCR. The amplification was done in a Stratagene MX3005P and corresponding software (MXpro quantitative PCR system), with step 1 (95°C for 5 min) and step 2 (40 cycles of 95°C for 20 s and 60°C for 1 min).
NMD efficiency calculation
Complementation of an NMD‐deficient strain leads to a decrease in the levels of an NMD‐sensitive transcript like the unspliced precursor of the RPL28 mRNA. To be able to judge how the efficiency of NMD affects the level of a transcript and to simplify the interpretation of complementation experiments, we computed NMD efficiency using the maximum (no NMD) and minimum (NMD fully active) steady‐state levels of a given transcript. We assumed that RNA synthesis is constant and that the degradation of RNA occurs by two competitive pathways, with different first‐order rate constants.
Polysome fractionation
Polysome extracts were obtained from 150 ml of mid‐log phase (A 600 ≈ 0.6) yeast cells culture treated with 50 μg ml−1 cycloheximide for 5 min at 4°C and broken with glass beads using a MagNA lyser (three times for 60 s at 3,000 rpm) in breaking buffer (20 mM Tris–HCl, pH 7.4, 50 mM KCl, 10 mM MgCl2, 50 μg ml−1 cycloheximide, 1 mM DTT, 0.1 mg ml−1 Heparin and EDTA‐free protease inhibitors from Roche). Ten to 15 A 260 of clarified cell lysates were loaded on 10–50% sucrose gradients. Gradients were centrifuged for 3 h 15 min at 160,000 g with slow acceleration in a SW41‐Ti rotor (Beckman) in a Optima L‐80 ultracentrifuge. Half of collected fractions were precipitated with TCA, and the precipitate was resuspended in 50 μl of sample buffer. Ten microliter was loaded on polyacrylamide NuPAGE Novex 4–12% Bis‐Tris gels (Life technologies). After transfer to nitrocellulose membrane with a semi‐dry system, proteins were detected by hybridization with appropriate antibodies.
For polysome analysis, Western blot signals were quantified using Image Lab software (version 5.2.1 build 11—Bio‐Rad). We used the square volume tool to define boxes of identical size at the expected position of the protein band we wanted to quantify. We also defined a series of blank boxes for each lane for background definition. To normalize the results, we calculated the percentage of signal over all the fractions of a run for both Nmd4 and Rps8 (control). Statistical significance of the signal differences between conditions was assessed by the paired Student t‐test.
Data availability
The accession number for the sequencing data reported in this paper is GEO: GSE102099 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102099).
The MS proteomics data that support the findings of this study have been deposited in the ProteomeXchange Consortium via the PRIDE repository (Vizcaíno et al, 2016) with the dataset identifier PXD007159 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD007159).
In addition to the access to raw data, enrichment and intensity data are available as an interactive web application at: https://hub05.hosting.pasteur.fr/NMD_complexes.
Author contributions
MD, AN and CS conceived the project. MD, LD, AN, CP, JK, HLH, AJ and CS designed experiments. MD, LD, AN, JK and CS performed experiments. AN, LD, MD, HLH and CS analysed the data. MD and CS wrote the manuscript. All authors discussed results and participated in the corrections of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Magalie Duchateau, Julia Chamot‐Rooke and Mariette Matondo of the Mass Spectrometry for Biology UtechS at the Pasteur Institute for access to the mass spectrometry facility, Jean‐Yves Coppée, from the Transcriptome and Epigenome, Biomics platform for access to the sequencing facility. We thank Giorgio Dieci, University of Parma, Italy, for the Rps8 antibodies. We thank members of the Genetics of Macromolecular Interactions Unit: Gwenael Badis for providing RNA sequencing results for the upf1Δ strain, helpful discussions and critical reading of the manuscript; Antonia Doyen for performing initial affinity purification experiments and for building strains; Frank Feuerbach for providing strains, reagents, critical suggestions, advice and support in writing the manuscript; and Micheline Fromont‐Racine for discussions and critical reading of the manuscript. We thank Christophe Malabat from the Bioinformatics and Biostatistics hub, Pasteur Institute, for help with data analysis and support in making the results available through a web interface. The study was supported by the ANR CLEANMD grant (ANR‐14‐CE10‐0014) from the French Agence Nationale de la Recherche to C.S., A.J. and H.L‐H. and by the Ministère de l'Enseignement Supérieur et de la Recherche and the “Fondation ARC pour la recherche sur le cancer” PhD fellowships to M.D. and J.K., and by continuous financial support from the Institut Pasteur and Centre National de Recherche Scientifique, France.
The EMBO Journal (2018) 37: e99278
References
- Addinall SG, Holstein E‐M, Lawless C, Yu M, Chapman K, Banks AP, Ngo H‐P, Maringele L, Taschuk M, Young A, Ciesiolka A, Lister AL, Wipat A, Wilkinson DJ, Lydall D (2011) Quantitative fitness analysis shows that NMD proteins and many other protein complexes suppress or enhance distinct telomere cap defects. PLoS Genet 7: e1001362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrné E, Molzahn L, Glatter T, Schmidt A (2013) Critical assessment of proteome‐wide label‐free absolute abundance estimation strategies. Proteomics 13: 2567–2578 [DOI] [PubMed] [Google Scholar]
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A (2004) A faux 3′‐UTR promotes aberrant termination and triggers nonsense‐mediated mRNA decay. Nature 432: 112–118 [DOI] [PubMed] [Google Scholar]
- Atkin AL, Schenkman LR, Eastham M, Dahlseid JN, Lelivelt MJ, Culbertson MR (1997) Relationship between yeast polyribosomes and Upf proteins required for nonsense mRNA decay. J Biol Chem 272: 22163–22172 [DOI] [PubMed] [Google Scholar]
- Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J (2007) Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318: 798–801 [DOI] [PubMed] [Google Scholar]
- Behm‐Ansmant I, Gatfield D, Rehwinkel J, Hilgers V, Izaurralde E (2007) A conserved role for cytoplasmic poly(A)‐binding protein 1 (PABPC1) in nonsense‐mediated mRNA decay. EMBO J 26: 1591–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop JJ, Brabers N, van Leenen D, Bakker LV, van Deutekom HWM, van Berkum NL, Apweiler E, Lijnzaad P, Holstege FCP, Kemmeren P (2010) A consensus of core protein complex compositions for Saccharomyces cerevisiae . Mol Cell 38: 916–928 [DOI] [PubMed] [Google Scholar]
- Brannan KW, Jin W, Huelga SC, Banks CAS, Gilmore JM, Florens L, Washburn MP, Van Nostrand EL, Pratt GA, Schwinn MK, Daniels DL, Yeo GW (2016) SONAR discovers RNA‐binding proteins from analysis of large‐scale protein‐protein interactomes. Mol Cell 64: 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bühler M, Steiner S, Mohn F, Paillusson A, Mühlemann O (2006) EJC‐independent degradation of nonsense immunoglobulin‐μ mRNA depends on 3′ UTR length. Nat Struct Mol Biol 13: 462–464 [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Bonneau F, Schüssler S, Eppinger E, Conti E (2014) Phospho‐dependent and phospho‐independent interactions of the helicase UPF1 with the NMD factors SMG5–SMG7 and SMG6. Nucleic Acids Res 42: 9447–9460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamieh H, Ballut L, Bonneau F, Le Hir H (2008) NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol 15: 85–93 [DOI] [PubMed] [Google Scholar]
- Chapin A, Hu H, Rynearson SG, Hollien J, Yandell M, Metzstein MM (2014) In vivo determination of direct targets of the nonsense‐mediated decay pathway in Drosophila . G3 (Bethesda) 4: 485–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Smith KR, Batterham P, Robin C (2005) Smg1 nonsense mutations do not abolish nonsense‐mediated mRNA decay in Drosophila melanogaster . Genetics 171: 403–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mourao A, Gutsche I, Gehring NH, Hentze MW, Kulozik A, Kadlec J, Sattler M, Cusack S (2009) Unusual bipartite mode of interaction between the nonsense‐mediated decay factors, UPF1 and UPF2. EMBO J 28: 2293–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clissold PM, Ponting CP (2000) PIN domains in nonsense‐mediated mRNA decay and RNAi. Curr Biol 10: R888–R890 [DOI] [PubMed] [Google Scholar]
- Collins SR, Kemmeren P, Zhao X‐C, Greenblatt JF, Spencer F, Holstege FCP, Weissman JS, Krogan NJ (2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae . Mol Cell Proteomics 6: 439–450 [DOI] [PubMed] [Google Scholar]
- Colombo M, Karousis ED, Bourquin J, Bruggmann R, Mühlemann O (2016) Transcriptome‐wide identification of NMD‐targeted human mRNAs reveals extensive redundancy between SMG6‐ and SMG7‐mediated degradation pathways. RNA 23: 189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Cui Y, Hagan KW, Zhang S, Peltz SW (1995) Identification and characterization of genes that are required for the accelerated degradation of mRNAs containing a premature translational termination codon. Genes Dev 9: 423–436 [DOI] [PubMed] [Google Scholar]
- Czaplinski K, Ruiz‐Echevarria MJ, Paushkin SV, Han X, Weng Y, Perlick HA, Dietz HC, Ter‐Avanesyan MD, Peltz SW (1998) The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev 12: 1665–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decourty L, Doyen A, Malabat C, Frachon E, Rispal D, Séraphin B, Feuerbach F, Jacquier A, Saveanu C (2014) Long open reading frame transcripts escape nonsense‐mediated mRNA decay in yeast. Cell Rep 6: 593–598 [DOI] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuvéglise C, Talla E, Goffard N, Frangeul L, Aigle M, Anthouard V, Babour A, Barbe V, Barnay S, Blanchin S, Beckerich J‐M, Beyne E et al (2004) Genome evolution in yeasts. Nature 430: 35–44 [DOI] [PubMed] [Google Scholar]
- Eberle AB, Stalder L, Mathys H, Orozco RZ, Mühlemann O (2008) Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol 6: e92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AB, Lykke‐Andersen S, Mühlemann O, Jensen TH (2009) SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16: 49–55 [DOI] [PubMed] [Google Scholar]
- Fiorini F, Bonneau F, Le Hir H (2012) Biochemical characterization of the RNA helicase UPF1 involved in nonsense‐mediated mRNA decay. Methods Enzymol 511: 255–274 [DOI] [PubMed] [Google Scholar]
- Fu C, Donovan WP, Shikapwashya‐Hasser O, Ye X, Cole RH (2014) Hot fusion: an efficient method to clone multiple DNA fragments as well as inverted repeats without ligase. PLoS One 9: e115318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara N, Ebert J, Unterholzner L, Lindner D, Izaurralde E, Conti E (2005) SMG7 is a 14‐3‐3‐like adaptor in the nonsense‐mediated mRNA decay pathway. Mol Cell 17: 537–547 [DOI] [PubMed] [Google Scholar]
- Garí E, Piedrafita L, Aldea M, Herrero E (1997) A set of vectors with a tetracycline‐regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae . Yeast 13: 837–848 [DOI] [PubMed] [Google Scholar]
- Gatfield D, Unterholzner L, Ciccarelli FD, Bork P, Izaurralde E (2003) Nonsense‐mediated mRNA decay in Drosophila: at the intersection of the yeast and mammalian pathways. EMBO J 22: 3960–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A‐C, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dümpelfeld B, Edelmann A, Heurtier M‐A, Hoffman V, Hoefert C, Klein K, Hudak M, Michon A‐M, Schelder M, Schirle M, Remor M et al (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440: 631–636 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Ganesan R, Amrani N, Jacobson A (2010) Translational competence of ribosomes released from a premature termination codon is modulated by NMD factors. RNA 16: 1832–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau‐Danila A, Anderson K, André B, Arkin AP, Astromoff A, El‐Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A et al (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391 [DOI] [PubMed] [Google Scholar]
- Halbeisen RE, Scherrer T, Gerber AP (2009) Affinity purification of ribosomes to access the translatome. Methods 48: 306–310 [DOI] [PubMed] [Google Scholar]
- Hall GW, Thein S (1994) Nonsense codon mutations in the terminal exon of the beta‐globin gene are not associated with a reduction in beta‐mRNA accumulation: a mechanism for the phenotype of dominant beta‐thalassemia. Blood 83: 2031–2037 [PubMed] [Google Scholar]
- He F, Jacobson A (1995) Identification of a novel component of the nonsense‐mediated mRNA decay pathway by use of an interacting protein screen. Genes Dev 9: 437–454 [DOI] [PubMed] [Google Scholar]
- He F, Brown AH, Jacobson A (1996) Interaction between Nmd2p and Upf1p is required for activity but not for dominant‐negative inhibition of the nonsense‐mediated mRNA decay pathway in yeast. RNA 2: 153–170 [PMC free article] [PubMed] [Google Scholar]
- He F, Brown AH, Jacobson A (1997) Upf1p, Nmd2p, and Upf3p are interacting components of the yeast nonsense‐mediated mRNA decay pathway. Mol Cell Biol 17: 1580–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A (2001) Upf1p, Nmd2p, and Upf3p regulate the decapping and exonucleolytic degradation of both nonsense‐containing mRNAs and wild‐type mRNAs. Mol Cell Biol 21: 1515–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A (2015) Nonsense‐mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu Rev Genet 49: 339–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer EE, Moore MJ (2016) Redefining the translational status of 80S monosomes. Cell 164: 757–769 [DOI] [PubMed] [Google Scholar]
- Ho B, Baryshnikova A, Brown GW (2018) Unification of protein abundance datasets yields a quantitative Saccharomyces cerevisiae proteome. Cell Syst 6: 192–205 e3 [DOI] [PubMed] [Google Scholar]
- Holbrook JA, Neu‐Yilik G, Hentze MW, Kulozik AE (2004) Nonsense‐mediated decay approaches the clinic. Nat Genet 36: 801–808 [DOI] [PubMed] [Google Scholar]
- Huang L, Lou C‐H, Chan W, Shum EY, Shao A, Stone E, Karam R, Song H‐W, Wilkinson MF (2011) RNA homeostasis governed by cell type‐specific and branched feedback loops acting on NMD. Mol Cell 43: 950–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E (2008) SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14: 2609–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson MJO, He F, Spatrick P, Li C, Jacobson A (2007) Association of yeast Upf1p with direct substrates of the NMD pathway. Proc Natl Acad Sci USA 104: 20872–20877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns L, Grimson A, Kuchma SL, Newman CL, Anderson P (2007) Caenorhabditis elegans SMG‐2 selectively marks mRNAs containing premature translation termination codons. Mol Cell Biol 27: 5630–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S (2006) Binding of a novel SMG‐1–Upf1–eRF1–eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense‐mediated mRNA decay. Genes Dev 20: 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrín‐Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B et al (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae . Nature 440: 637–643 [DOI] [PubMed] [Google Scholar]
- Lasalde C, Rivera AV, León AJ, González‐Feliciano JA, Estrella LA, Rodríguez‐Cruz EN, Correa ME, Cajigas IJ, Bracho DP, Vega IE, Wilkinson MF, González CI (2013) Identification and functional analysis of novel phosphorylation sites in the RNA surveillance protein Upf1. Nucleic Acids Res 42: 1916–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Gatfield D, Izaurralde E, Moore MJ (2001) The exon–exon junction complex provides a binding platform for factors involved in mRNA export and nonsense‐mediated mRNA decay. EMBO J 20: 4987–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeds P, Peltz SW, Jacobson A, Culbertson MR (1991) The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev 5: 2303–2314 [DOI] [PubMed] [Google Scholar]
- Leeds P, Wood JM, Lee BS, Culbertson MR (1992) Gene products that promote mRNA turnover in Saccharomyces cerevisiae . Mol Cell Biol 12: 2165–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom RGH, Supek F, Lehner B (2016) The rules and impact of nonsense‐mediated mRNA decay in human cancers. Nat Genet 48: 1112–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd JPB, Davies B (2013) SMG1 is an ancient nonsense‐mediated mRNA decay effector. Plant J 76: 800–810 [DOI] [PubMed] [Google Scholar]
- Loh B, Jonas S, Izaurralde E (2013) The SMG5–SMG7 heterodimer directly recruits the CCR4–NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev 27: 2125–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke B, Azzalin CM, Hug N, Deplazes A, Peter M, Lingner J (2007) Saccharomyces cerevisiae Ebs1p is a putative ortholog of human Smg7 and promotes nonsense‐mediated mRNA decay. Nucleic Acids Res 35: 7688–7697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke‐Andersen J (2002) Identification of a human decapping complex associated with hUpf proteins in nonsense‐mediated decay. Mol Cell Biol 22: 8114–8121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke‐Andersen S, Chen Y, Ardal BR, Lilje B, Waage J, Sandelin A, Jensen TH (2014) Human nonsense‐mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev 28: 2498–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabat C, Feuerbach F, Ma L, Saveanu C, Jacquier A (2015) Quality control of transcription start site selection by nonsense‐mediated‐mRNA decay. eLife 4: e06722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE (2004) Nonsense‐mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5: 89–99 [DOI] [PubMed] [Google Scholar]
- Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, Dietz HC (2001) Rent1, a trans‐effector of nonsense‐mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet 10: 99–105 [DOI] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert J‐P, St‐Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, Halim VA, Bagshaw RD, Hubner NC, al‐Hakim A, Bouchard A, Faubert D, Fermin D, Dunham WH, Goudreault M, Lin Z‐Y et al (2013) The CRAPome: a contaminant repository for affinity purification‐mass spectrometry data. Nat Methods 10: 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metze S, Herzog VA, Ruepp M‐D, Mühlemann O (2013) Comparison of EJC‐enhanced and EJC‐independent NMD in human cells reveals two partially redundant degradation pathways. RNA 19: 1432–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Parker R (1994) Premature translational termination triggers mRNA decapping. Nature 370: 578–581 [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Parker R (1999) Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 5: 1299–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JO, Moore KA, Chapin A, Hollien J, Metzstein MM (2016) Degradation of Gadd45 mRNA by nonsense‐mediated decay is essential for viability. eLife 5: e12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu‐Yilik G, Raimondeau E, Eliseev B, Yeramala L, Amthor B, Deniaud A, Huard K, Kerschgens K, Hentze MW, Schaffitzel C, Kulozik AE (2017) Dual function of UPF3B in early and late translation termination. EMBO J 36: 2968–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P, Josi C, Kurosawa H, Yamashita A, Mühlemann O (2014) A novel phosphorylation‐independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res 42: 9217–9235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP (2007) Comprehensive analysis of diverse ribonucleoprotein complexes. Nat Methods 4: 951–956 [DOI] [PubMed] [Google Scholar]
- Ohnishi T, Yamashita A, Kashima I, Schell T, Anders KR, Grimson A, Hachiya T, Hentze MW, Anderson P, Ohno S (2003) Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG‐5 and hSMG‐7. Mol Cell 12: 1187–1200 [DOI] [PubMed] [Google Scholar]
- Perlick HA, Medghalchi SM, Spencer FA, Kendzior RJ, Dietz HC (1996) Mammalian orthologues of a yeast regulator of nonsense transcript stability. Proc Natl Acad Sci USA 93: 10928–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz LP, Huerta‐Cepas J, Gabaldón T (2011) MetaPhOrs: orthology and paralogy predictions from multiple phylogenetic evidence using a consistency‐based confidence score. Nucleic Acids Res 39: e32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulak R, Anderson P (1993) mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev 7: 1885–1897 [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Saveanu C, Rousselle J‐C, Lenormand P, Namane A, Jacquier A, Fromont‐Racine M (2007) The p21‐activated protein kinase inhibitor Skb15 and its budding yeast homologue are 60S ribosome assembly factors. Mol Cell Biol 27: 2897–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serdar LD, Whiteside DL, Baker KE (2016) ATP hydrolysis by UPF1 is required for efficient translation termination at premature stop codons. Nat Commun 7: 14021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JC, Gorenstein MV, Li G‐Z, Vissers JPC, Geromanos SJ (2006) Absolute quantification of proteins by LCMSE a virtue of parallel ms acquisition. Mol Cell Proteomics 5: 144–156 [DOI] [PubMed] [Google Scholar]
- Singh G, Rebbapragada I, Lykke‐Andersen J (2008) A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense‐mediated mRNA decay. PLoS Biol 6: e111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder L, Mühlemann O (2008) The meaning of nonsense. Trends Cell Biol 18: 315–321 [DOI] [PubMed] [Google Scholar]
- Swisher KD, Parker R (2011) Interactions between Upf1 and the decapping factors Edc3 and Pat1 in Saccharomyces cerevisiae . PLoS One 6: e26547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Yang W, Zhang J, Dang H, Lu X, Fu C, Miao W (2017) Nonsense‐mediated mRNA decay in Tetrahymena is EJC independent and requires a protozoa‐specific nuclease. Nucleic Acids Res 45: 6848–6863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno JA, Csordas A, del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T, Xu Q‐W, Wang R, Hermjakob H (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Brogna S (2010) Splicing‐dependent NMD does not require the EJC in Schizosaccharomyces pombe . EMBO J 29: 1537–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y, Czaplinski K, Peltz SW (1996) Identification and characterization of mutations in the UPF1 gene that affect nonsense suppression and the formation of the Upf protein complex but not mRNA turnover. Mol Cell Biol 16: 5491–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y, Czaplinski K, Peltz SW (1998) ATP is a cofactor of the Upf1 protein that modulates its translation termination and RNA binding activities. RNA 4: 205–214 [PMC free article] [PubMed] [Google Scholar]
- Wessel D, Flügge UI (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138: 141–143 [DOI] [PubMed] [Google Scholar]
- Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS, Ideker T, Guthrie C, Krogan NJ (2008) A genetic interaction map of RNA‐processing factors reveals links between Sem1/Dss1‐containing complexes and mRNA export and splicing. Mol Cell 32: 735–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Mühlemann O (2011) Autoregulation of the nonsense‐mediated mRNA decay pathway in human cells. RNA 17: 2108–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Welch EM, Hogan K, Brown AH, Peltz SW, Jacobson A (1997) Polysome‐associated mRNAs are substrates for the nonsense‐mediated mRNA decay pathway in Saccharomyces cerevisiae . RNA 3: 234–244 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The accession number for the sequencing data reported in this paper is GEO: GSE102099 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102099).
The MS proteomics data that support the findings of this study have been deposited in the ProteomeXchange Consortium via the PRIDE repository (Vizcaíno et al, 2016) with the dataset identifier PXD007159 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD007159).
In addition to the access to raw data, enrichment and intensity data are available as an interactive web application at: https://hub05.hosting.pasteur.fr/NMD_complexes.