

Key Points
Pre–B-cell ALL induces T-cell dysfunction in vivo, mediated in part by a non–T-cell receptor–linked mechanism.
Prior exposure of T cells to pre–B-cell ALL in vivo impairs subsequent functionality of CAR-expressing T cells.
Abstract
Adoptive transfer of patient-derived T cells modified to express chimeric antigen receptors (CARTs) has demonstrated dramatic success in relapsed/refractory pre–B-cell acute lymphoblastic leukemia (ALL), but response and durability of remission requires exponential CART expansion and persistence. Tumors are known to affect T-cell function, but this has not been well studied in ALL and in the context of chimeric antigen receptor (CAR) expression. Using TCF3/PBX1 and MLL-AF4–driven murine ALL models, we assessed the impact of progressive ALL on T-cell function in vivo. Vaccines protect against TCF3/PBX1.3 but were ineffective when administered after leukemia injection, suggesting immunosuppression induced early during ALL progression. T cells from leukemia-bearing mice exhibited increased expression of inhibitory receptors, including PD1, Tim3, and LAG3, and were dysfunctional following adoptive transfer in a model of T-cell receptor (TCR)–dependent leukemia clearance. Although expression of inhibitory receptors has been linked to TCR signaling, pre–B-cell ALL induced inhibitory receptor expression, at least in part, in a TCR-independent manner. Finally, introduction of a CAR into T cells generated from leukemia-bearing mice failed to fully reverse poor in vivo function.
Visual Abstract
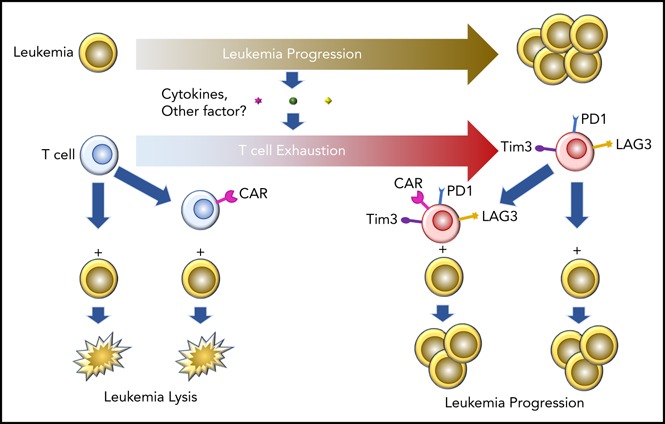
Introduction
Pre–B-cell acute lymphoblastic leukemia (ALL) is the most common oncologic diagnosis in children. Modern risk-adapted multiagent regimens, which incorporate prolonged maintenance in combination with central nervous system prophylaxis, has led to cure rates for pediatric pre–B-cell ALL reaching ∼90%. Nonetheless, leukemia remains a leading cause of cancer-related death in children, and outcomes for patients with relapsed or chemotherapy refractory ALL have not changed substantially over recent decades despite maximization of the intensity of cytotoxic regimens in such patients.1-4 Furthermore, adolescents and adults with pre–B-cell ALL have worse outcomes than younger children.5 Allogeneic hematopoietic stem cell transplantation offers a curative option for high-risk patients with a clear contribution from the graft-versus-leukemia (GVL) effect.6,7 However, in some,8-10 but not all,11 studies, the potency of GVL for ALL is inferior to that seen in myeloid malignancies. This has been attributed in part to suboptimal antigen presentation by ALL blasts in combination with diminished T-cell function secondary to impaired priming or direct tolerization of T cells by ALL blasts12-14 resulting in inherent resistance to T-cell receptor (TCR)–mediated therapy. There has been dramatic success with immunotherapeutic targeting of ALL using patient-derived T cells modified to express chimeric antigen receptors (CARTs) that redirects specificity toward the B-cell antigen CD19.15-19 However poor CART expansion and relapses in a substantial number of patients suggests that adoptive T-cell therapy for acute B precursor ALL could be enhanced by the identification of pathways that contribute to suboptimal of T-cell function.
T-cell function can be negatively regulated by interactions between ligands expressed on antigen-presenting cells or target cells and inhibitory receptors expressed on the T-cell surface.20 The prototypic negative regulatory receptors are CTLA4 (CD152), which binds B71 and B72, and the programmed death receptor 1 (PD1), which binds either PDL1 or PDL2. Checkpoint inhibitors, which block either the CTLA4 or PD1 axis, have induced objective tumor responses in humans, illustrating the importance of these negative regulators of immunity in cancer biology.21 However, T-cell exhaustion is a complex, progressive phenomena, and PD1+ T cells are not inherently dysfunctional.22 T-cell dysfunction and exhaustion have been well described in the setting of many solid tumors and some types of hematologic malignancies, including chronic lymphocytic leukemia,23 multiple myeloma,24 and AML25,26 but has been poorly studied in the context of pre–B-cell ALL. Furthermore, whether cancer-induced T-cell dysfunction can be overcome through introduction of a synthetic CAR generating a non-TCR signal is not known.
The majority of the preclinical studies of adoptive cell therapy for hematologic malignancies use xenograft systems in which human T cells are infused into highly immunodeficient mice bearing human leukemia. These models have major limitations in terms of studying in vivo immunobiology due to the lack of a complete immune system in the murine recipient and the development of xenogeneic graft-versus-host disease. We established a syngeneic murine pre–B-cell ALL model in which PD1 is rapidly upregulated on bone marrow T cells in the presence of ALL and is associated with poor T-cell functionality. We further demonstrate that ALL-induced T-cell dysfunction can occur in a TCR-independent manner, is not reversed by blockade of the PD1 axis, and persists despite in vitro T-cell expansion and redirection of specificity by a synthetic CD19 CAR. These findings have important implications for the optimization of immunotherapy for ALL, especially with regards to adoptive cell therapies utilizing CARTs generated from patients with leukemia.
Materials and methods
Mice
C57BL/6(H-2b)(B6) and B6/Ly5.2 (CD45.1) were purchased from the Animal Production Unit, National Cancer Institute (NCI). B6.129S7-Rag1 < tm1Mom>/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). OT1/Rag2−/− mice were purchased from Taconic. All mice were housed in a pathogen-free animal facility at the National Institutes of Health. Animal protocols were approved by the NCI Animal Care and Use Committee.
Cell lines
TCF3/PBX1 cell lines were derived from the spleen of a leukemic TCF3/PBX1 × CD3ε−/− (E2a/PBX1) leukemic mice and were generous gift from Janet Bijl.27 Leukemia cells were first injected IV into sublethally irradiated (250 cGy) B6 mice. By 3 weeks, all of these mice developed splenomegaly, hepatomegaly, and lymphadenopathy consistent with leukemia. Fresh splenocytes from these leukemic mice were then used for in vitro culture to establish a stable cell line we called TCF3/PBX1.3.3. Initially, TCF3/PBX1.3.3 was cultured in StemSpan SFEM (Vancouver, BC) media, containing recombinant human interleukin-7 (rhIL-7; 50 U/mL), rhIL2 (20 U/mL), recombinant murine IL-3 (20 ng/mL), stem cell factor (50 ng/mL), Flt3-L (25 ng/mL), β-mercaptoethanol, and penicillin/streptomycin/glutamine. After initial culture in stem cell media supplemented with cytokines, murine leukemia cells were passaged at least 15 times in 10% complete mouse media (RPMI 1640 with 10% heat-inactivated fetal calf serum, 1% nonessential amino acids, 1% sodium pyruvate, 1% penicillin/streptomycin, 1% l-glutamine [Invitrogen, Carlsbad, CA], and 1% N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer [Sigma-Aldrich, St. Louis, MO]) without additional cytokines to ensure a stable cell line. Reverse-transcription polymerase chain reaction confirmed preservation of the TCF3/PBX1 fusion gene (data not show). In culture, TCF3/PBX1.3.3 cells are spherical and have lymphoid blast morphology. TCF3/PBX1 was genotyped with 96 microsatellite markers to scan the 19 mouse autosomes. The Genome Scan scores indicated TCF3/PBX1 is the equivalent of a N6 conventional backcross of C57BL/6J. C57/Bl6 MLL-AF4 ALL cell lines were kindly provided by Scott Armstrong.28 Mice engrafted with MLL-AF4 received 250 cGy prior to injection. TCF3/PBX1-OVA cells were generated by transduction the ovalbumin gene using a retroviral vector into TCF3/PBX1 cells, and TCF3/PBX1-OVA-GFP cells were generated by transducing the Lenti-GFP into TCF3/PBX1-OVA cells. The transduced cells were further sorted with a fluorescence activated cell sorter for the desired expression of ovalbumin or GFP.
Human leukemia samples
Human ALL samples were collected and stored after informed consent to a NCI institutional review board–approved tissue-acquisition protocol. Bone marrow specimens were collected only at the time of a clinically indicated bone marrow evaluation that was performed as part of routine restaging either prior to or after implementation of treatment (either standard chemotherapy or experimental). Peripheral blood specimens were generally obtained when patients had known circulating blasts and volume did not exceed prespecified guidelines for research blood draws. All research specimens from human subjects were obtained with informed consent in accordance with the Declaration of Helsinki.
Adoptive T-cell transfer, vaccines, CART production, and in vivo cell depletion
See supplemental Methods (available on the Blood Web site) for more information.
In vitro colony-forming assays
Colony-forming assays were performed as described in the manufacturer’s protocol (Stem Cell Technology catalog number 04100). Briefly, TCF3/PBX1 cells were serially diluted (24 300 cells/100 μL, 8100 cells/100 μL, 2700 cells/100 μL, 900 cells/100 μL, 300 cells/100 μL, 100 cells/100 μL) and incubated in triplicate in 1% MethoCult H4100 plus RPMI (containing 10 ng/mL rhIL-7) for 6 to 7 days at 37°C and 5% CO2. The number of colonies was then counted and recorded. The frequency of tumor-initiating cells was calculated based on the slope of the linear range of the plot formed by the average of colonies at each dilution.
Flow cytometry analysis
Fluorochrome-conjugated anti-CD45R (B220), anti-CD127 (IL-7Rα), anti-BP1, anti-CD43, anti–immunoglobulin M (anti-IgM), anti-IgD, anti-CD8, anti-CD19, anti-μ chain, anti-CD43, anti-BP1, anti-PDL1, anti-Tim3, anti-LAG3, anti-CD45.2, and streptavidin–fluorescein isothiocyanate were purchased from Becton Dickinson (BD), eBioscience, or Jackson Immunoresearch. Via-Probe was purchased from Becton Dickinson. Anti-human CD45RO, CD8a, CD3, PDL1, PDL2, Tim3, and LAG3 were purchased from BioLegend. Labeled cells were analyzed with a dual-laser flow cytometer (FACSCalibur, BD) or 3-laser flow cytometer (LSR Fortessa, BD) and data analyzed using FlowJo software (Tree Star, Ashland, OR)
Flow sorting to isolate PD1+ and PD1− T cells
Single-cell suspensions were made from freshly harvested mouse spleen and enriched for T cells with a Miltenyi Pan T Cell Isolation Kit II (catalog number 130-095-130) following the manufacturer’s protocol. T cells were stained and sorted into PD1+ and PD1− populations using the BD FACSAria II.
Immunohistopathology
TCF3/PBX1 cells were stained on a glass slide with Wright-Giemsa stain. Spleen and liver from leukemic mice were fixed with 10% formalin and transferred to 70% ethanol. Specimens were sent for paraffin block generation, sectioned at 5 µm, and stained with hematoxylin and eosin (American Histo Labs). Femurs were harvested from naive or tumor-bearing mice, fixed in 4% paraformaldehyde overnight at 4°C, and decalcified in 0.1 M Tris/0.26 M EDTA (8% EDTA) pH 7.4 buffer with constant agitation at room temperature for 5 days. Slides were then washed with EDTA in 1× phosphate-buffered saline and resuspended in 30% vol/vol glucose (phosphate-buffered saline) at 4°C and sent to Histoserv (Germantown, MD) for sectioning and hematoxylin and eosin staining.
Serum cytokines
Mice were bled into Microtainer tubes (BD reference 365967), and serum was recovered from after centrifugation for 15 minutes at maximum speed in a Sorvall Legend Micro 17R centrifuge. Serum cytokine levels were detected using a BD Cytometric Bead Array Mouse Th1/Th2/Th17 Cytokine Kit (catalog 560485) following the manufacturer’s procedure.
Checkpoint inhibitor blockade
Anti-PD1 (clone RMP1-14), anti-Tim3 (clone RMT3-23), and anti-PDL1 (clone 10F.9G2) were purchased from BioXCell. Anti-PD1 and/or anti-Tim3 were given every 3 to 4 days at a dose of 200 μg/mouse. All antibodies were administered intraperitoneally after tumor challenge and continued until the mice became terminally ill.
Microarray
Untouched B-cell progenitors from bone marrow were isolated using CD43 (Ly-48) MicroBeads (Miltenyi Biotec, catalog number 130-049-801). CARTs were made from either naive or tumor-bearing mice as described above. RNA was extracted for microarray hybridization and global gene expression on Affimatrix chips (mouse Gene ST 1.0). The array results were further analyzed for differential gene expression between samples and compared against data a tumor database (https://pob.abcc.ncifcrf.gov/cgi-bin/JK).
Statistical analysis
Statistical tests were performed using GraphPad Prism 7 Version 7.0a for Mac OS X (GraphPad Software, San Diego, CA). Significant differences comparing the survival curve of 2 groups were determined by a log-rank (Mantel-Cox) test. A P value < .05 was considered significant. Comparisons between 2 groups were performed using a Mann-Whitney U test. Comparisons between multiple groups were performed using a 1-way analysis of variance.
Results
Characterization and validation of a syngeneic pre–B-cell ALL model
To comprehensively evaluate the impact of progressive pre–B-cell ALL on an intact immune system, we generated a stable, transplantable murine leukemia cell line (TCF3/PBX1.3) from TCF3/PBX1 transgene–expressing mice.27 TCF3/PBX1.3 expresses immunophenotypic markers (CD19+B220+CD127+ with cytoplasmic μ-chain but no surface IgM expression) consistent with an early B-cell phenotype (Figure 1A; supplemental Figure 1A) representative of pro–B and pre–B-1-cell stages.29 Morphologically, TCF3/PBX1.3 appears lymphoblastic (Figure 1B). Gene expression confirmed this early B-cell phenotype, demonstrating a profile that was similar to human pre–B-cell ALL based on unsupervised hierarchical clustering (supplemental Figure 1B).
Figure 1.

TCF3/PBX1.3 exhibits an immunophenotype and in vivo behavior consistent with pre–B-cell ALL. (A) Cultured TCF3/PBX1.3 cells (analyzed by flow cytometry) demonstrating expression of B-lineage receptors CD19+ and B220+ as well as the immature B-cell marker CD127+ (IL-7Rα) with cytoplasmic μ-chain present but without surface IgM expression. (B) Wright-Giemsa stain of TCF3/PBX1.3 demonstrating typical lymphoblastic morphology. (C) Spleens of C57BL/6 mice 2 weeks after injection of 1 × 105 TCF3/PBX1.3 cells IV (top) or saline (bottom). (D) Histological sections of tissues from mice in panel C stained with hematoxylin and eosin. (E) CD45.1 congenic mice received 1 × 105 TCF3/PBX1.3 cells (CD45.2) IV. At the time points indicated, bone marrow was collected analyzed by flow cytometry for CD45.2+B220+ TCF3/PBX1.3 cells. (F) TCF3/PBX1.3 cells were injected IV at decreasing doses into female C57BL/6 mice. Lethality was confirmed to be leukemia induced at autopsy. (G) Rag1−/−, irradiated (250 cGy) C57BL/6, or unirradiated, immune-competent C57BL/6 mice received decreasing doses of leukemia (P < .004, Mantel-Cox test; irradiated vs healthy) or mice (P < .003, Mantel-Cox test; Rag1−/− vs healthy).
Following injection into immunocompetent mice, TCF3/PBX1.3 rapidly infiltrates lymphoid tissues, resulting in loss of primary tissue architecture (Figure 1C-D). Distribution of TCF3/PBX1.3 to the bone marrow can be detected as early as 5 days after injection (Figure 1E) with subsequent leukemic infiltration of the meninges (data not shown) resulting in hindlimb paralysis. TCF3/PBX1.3 is lethal following injection of as few as 1000 cells into immune-competent mice (Figure 1F).
Immune resistance attenuates ALL progression
To establish the role of adaptive immune cells in delaying progression of TCF3/PBX1.3, time to lethality was compared in immunodeficient and immunocompetent mice. TCF3/PBX1.3 cells (105) were injected into unconditioned C57BL/6 mice, sublethally irradiated (250 cGy) C57BL/6 mice, and T-cell/B-cell–deficient Rag1−/− mice. Disease progression was more rapid in irradiated and Rag1−/− mice than in immunocompetent recipients (Figure 1G) with consistent leukemic engraftment and lethality occurring in irradiated recipients following injection of 100 cells (supplemental Figure 1C). Colony-forming assays in semisolid methylcellulose estimated a colony-initiating cell frequency of ∼4% (supplemental Figure 1D), consistent with engraftment of 100 cells in immunodeficient mice.
Vaccination mediates T-cell–dependent protection against TCF3/PBX1.3 but is therapeutically ineffective
Based on the accelerated progression of TCF/E2aPBX1.3 in lympho-deficient RAG−/− and irradiated recipients, we next tested whether vaccination could protect against leukemia progression. C57BL/6 mice were primed (2 weeks prior to leukemia challenge) and boosted (1 week prior) with 1 of 3 types of vaccines: dendritic cells (DCs) pulsed with apoptotic (irradiated 15 000 cGy) TCF3/PBX1.3, unpulsed DCs, or apoptotic TCF3/PBX1.3 alone (Figure 2A-B). Both apoptotic cell-loaded mature DCs and apoptotic TCF3/PBX1.3 alone provided comparable protection. We next investigated the contribution of T cells to vaccine-mediated leukemic resistance. CD4+ T cells and/or CD8+ T cells were depleted from C57BL/6 mice following vaccination and 3 days before tumor challenge. Depleting either CD4+ or CD8+ T cells had a minimal impact on vaccine-mediated protection; however, depleting both subsets fully abrogated the protective effects of vaccination (Figure 2C). Finally, we sought to determine whether a tumor vaccine was therapeutic when given to mice already bearing leukemia. Despite the robust protection provided by vaccination prior to leukemia challenge, there was no effect on survival when administered 3 days following TCF3/PBX1.3 challenge (∼0.1% bone marrow infiltration; Figure 1E) followed by a boost at day 10 (Figure 2D). Thus, even minimal leukemic burden prevents therapeutic efficacy of vaccination.
Figure 2.

Vaccination induces T-cell–mediated protection against leukemia but is unable to prevent progression of established disease. C57BL/6 mice were primed and boosted with a vaccine as indicated and then challenged with 1 × 105 TCF3/PBX1.3 cells 1 week later. (A) Survival. (B) Flow cytometry or bone marrow of representative mice receiving unpulsed DCs (left) or irradiated TCF3/PBX1.3 25 days after leukemia injection. (C) Immune cell depletion with anti-CD4, anti-CD8, or both anti-CD4 and CD8 antibodies initiated 3 days before tumor challenge, given 3 times weekly, and continued for 3 weeks. (D) Vaccines were prepared as described in panel A and injected 7 and 21 days after 1 × 105 TCF3/PBX1.3.
Leukemia progression promotes phenotypic T-cell exhaustion and functional impairment
We hypothesized that therapeutic vaccine failure could be due to the impact of preexistent ALL on T-cell function. CD8+ T cells from leukemia-vaccinated and live leukemia-bearing C57BL/6 mice were analyzed by flow cytometry 12 days after tumor challenge or vaccination. As expected, the majority of CD8+ T cells from healthy mice demonstrated a naive phenotype (CD44−CD62L+). In contrast, the bone marrow of leukemia-bearing mice contained a majority of effector memory T cells (CD44+CD62L−; Figure 3A) that was more pronounced than in the bone marrow of vaccinated mice that contained substantial numbers of central memory CD8+ T cells (CD44+CD62L+). We next investigated whether phenotypic markers of T-cell exhaustion were upregulated during leukemia progression. Indeed, there was a significant increase in the proportion of PD1-expressing CD4+ and CD8+ T cells in the bone marrow 1 week after leukemia challenge compared with non–leukemia-bearing mice (Figure 3B). Further characterization of PD1+ T cells in leukemia-bearing mice demonstrated coexpression of Tim3 and Lag3 (Figure 3C-D). We next confirmed that the phenotypic exhaustion associated with progression of the TCF3/PBX1 cell line was also observed in the setting of primary murine leukemia (supplemental Figure 2) and in the bone marrow and peripheral blood of patients with ALL (supplemental Figure 3).
Figure 3.

Progressive leukemia induces phenotypic changes consistent with T-cell dysfunction. (A) Bone marrow from healthy C57BL/6 mice was analyzed by flow cytometry 12 days after challenge with 1 × 105 viable or irradiated TCF3/PBX1.3. (B) Percentage of bone marrow CD4+ and CD8+ T cells expressing PD1 at the time points indicated. Line represents the mean, and error bars represent standard error of the mean. (C) Representative dot plots of bone marrow cells at day 12 following injection of TCF3/PBX1.3 compared with non–leukemia-bearing mice. Bottom panels represent gating on PD1+ CD4+ and CD8+ T cells. (D) Scatter plots showing percentage of PD1+ CD4+ and PD1+CD8+ bone marrow T cells expressing Tim3 and LAG3 at day 12 following injection of TCF3/PBX1.3 compared with non–leukemia-bearing mice. (P < .001, unpaired Student t test). (E) Leukemia-bearing mice were given 5 × 106 T cells from mice bearing leukemia or vaccinated with irradiated TCF3/PBX1.3 (as in panel A). Mice received 105 TCF3/PBX1.3 on day 0, 500 cGy irradiation on day 2 and adoptive transfer of T cells on day 2. Mice receiving T cells from vaccinated donors demonstrated survival benefit compared with leukemia-bearing donors (P < .0001, Gehan-Breslow-Wilcoxon test). (F) Percentage of CD8+ T cells expressing PD1, Tim3, and LAG3 in mice bearing MLL-AF4 ALL at day 23 following leukemia injection compared with non–leukemia-bearing irradiated mice. Control mice received 250 cGy irradiation. Line represents the mean, and error bars represent standard error of the mean.
TCF3/PBX1.3 expresses PDL1 at higher levels than bone-marrow derived DCs pulsed with TCF3/PBX1.3 (supplemental Figure 4A) suggesting that the presence of leukemia could induce functional impairment of PD1-expressing T cells. Thus, we next evaluated the ability of T cells from vaccinated and leukemia-bearing donors to clear leukemia following adoptive transfer into secondary recipients. T cells from apoptotic cell–vaccinated mice mediated a significant survival benefit compared with naive T cells (Figure 3E; P < .0001, Mantel-Cox test). However, T cells from mice bearing viable leukemia (harvested at 12 days following leukemia injection) provided no therapeutic benefit (P < .0001, Mantel-Cox test), further indicating that leukemia rapidly induces impairment in T-cell functionality. Finally, to confirm that the upregulation of exhaustion markers was not unique to TCF3/PBX1.3, we also analyzed T cells in the bone marrow of mice bearing a syngeneic ALL driven by overexpression of MLL-AF4, which also demonstrated increased expression of PD1, Tim3, and Lag3 (Figure 3F).
Phenotypic T-cell exhaustion occurs in the absence of cognate antigen
We next used a TCR transgenic model in which all T cells express a TCR that recognizes an antigen not expressed on TCF3/PBX1.3 to test whether antigen recognition is required for upregulation of PD1 in the presence of leukemia. CD8+ T cells from leukemia-bearing OT-1/Rag−/− transgenic mice in which T cells express a TCR specific for the ovalbumin antigen also upregulate PD1 in the presence of pre–B-cell ALL not expressing ovalbumin (Figure 4A) as well as Tim3 and Lag3 (Figure 4B). We next adoptively transfer polyclonal CD8+ T cells from CD45.1 congenic mice and CD45.2 OT-1 T cells into lymphocyte-deficient recipients and compared PD1 induction in the presence of TCF3/PBX1.3 or TCF3/PBX1.3 expressing ovalbumin. As expected, there was a marked increase in the level of PD1 expression on OT-1 T cells in the presence of ovalbumin-expressing leukemia consistent with TCR-induced PD1 upregulation (supplemental Figure 4B). However, there was also a significant increase in PD1 expression on both OT-1 T cells in the presence of leukemia not expressing cognate antigen. Taken, together these results suggest that TCR-independent mechanisms contribute to leukemia-induced T-cell dysfunction occurring in the context of pre–B-cell ALL. T cells from mice bearing either TCF3/PBX1.3 or MLL-AF4 were adoptively transferred into secondary recipients 2 days after injection with TCF3/PBX1.3 transduced with ovalbumin. As shown in Figure 4C-D, OT-1 T cells from TCF3/PBX1.3- or MLL-AF4–bearing donors were less effective at clearing leukemia at day 14. Finally, OT1 cells from leukemia-bearing donors demonstrated reduced proliferation (supplemental Figure 5A) and cytotoxicity (supplemental Figure 5B) upon stimulation by ovalbumin-expressing TCF3/PBX1.3.
Figure 4.

PD1 expression induced by progressive ALL is partially TCR independent and does not define functional impairment, nor does blockade of inhibitory receptors prevent or reverse dysfunction. (A) CD8+ T cells from the bone marrow and spleens of OT1/RAG−/− TCR transgenic mice were analyzed by flow cytometry for PD1 expression 10 days after injection of 1 × 105 TCF3/PBX1.3 challenge. (P < .01, unpaired Student t test). (B) Representative zebra plots of mice represented in panel A. (C) CD8+ T cells from the bone marrow and spleens of OT1/RAG−/− TCR transgenic mice harvested and enriched using a T-cell selection column 14 days after injection with TCF3/E2aPBX1.3 and transferred into secondary recipients (1 × 106/recipient) 2 days after injection with ovalbumin-transduced TCF3/E2aPBX1.3. Fourteen days later, bone marrow analyzed by flow cytometry for ovalbumin-expressing CD19+ B220+ cells. (D) Experimental design as in panel D with the exception that OT1 donors were irradiated (250 cGy) and injected with MLL-AF4 and T cells were harvested 21 days later. Non–leukemia-bearing donors also received irradiation. (E) Leukemia-bearing mice were given a subcurative dose (1 × 106) of sorted PD1+ or PD1− T cells from mice bearing leukemia or vaccinated with irradiated TCF3/PBX1.3. RAG1 mice received 105 TCF3/PBX1.3 on day 0 and adoptive transfer of T cells on day 2. (F) Anti-PD1 (200 µg/dose) and/or anti-PDL1 (200 µg /dose) was administered intraperitoneally every 3 days beginning 1 day after 105 TCF3/PBX1.3 challenge. (G) Anti-TIM3 (250 μg/dose) and/or anti-PD1 was administered as in panel D. (H) Splenic CD8+ T cells were collected from irradiated TCF3/PBX1.3 vaccinated mice (as in Figure 3G) and administered with or without PD1 and/or TIM3 blockade. Antibody administration was initiated 1 day prior to T-cell transfer and continued for up to 5 weeks (P < .0001, Mantel-Cox test).
Leukemia-induced phenotypic changes on T cells is not reversible by blockade of PD1 or Tim3
To determine whether ALL-induced T cell dysfunction segregated with PD1-expression, we adoptively transferred T cells sorted based on PD1 from vaccinated or leukemia-bearing donors into Rag1−/− mice (supplemental Figure 6). PD1+ and PD1− T cells from mice primed with irradiated leukemia were equivalent in delaying leukemia progression (Figure 4E). In contrast, both PD1+ and PD1− T cells from mice with viable leukemia were less effective at delaying leukemia progression in secondary recipients, suggesting that T-cell dysfunction in the presence of pre–B-cell ALL is not associated with PD1 expression. Indeed, blockade of PD1 did not reverse leukemia-induced T-cell dysfunction (Figure 4F). Combined PD1 or Tim3 blockade also failed to reduce time to lethality (Figure 4G). Finally, adoptive transfer of vaccine-primed T cells with immune checkpoint blockade with anti-PD1 and anti-Tim3 was also unable to improve functionality of primed T cells into leukemia-bearing mice (Figure 4H).
Exhaustion markers can be induced by cytokines present in leukemia-bearing mice
Cytokines may contribute to TCR-independent induction markers of T-cell exhaustion.30,31 Multiple cytokines, including IL-6, IL-10, and tumor necrosis factor α (TNF-α), are elevated in the serum of mice bearing both TCF3/PBX1.3 and MLL-AF4 (Figure 5A-B; supplemental Figure 7). Furthermore, exposure of CD4 and CD8 T cells to IL-6, IL-10, and TNF-α in vitro results in increased expression of Tim3 (Figure 5C). Exposure of T cells to all 3 cytokines further increases Tim3 expression, suggesting that cytokines may contribute TCR-independent induction of exhaustion markers induced in the presence of pre–B-cell ALL.
Figure 5.

Leukemia-derived cytokines may contribute to phenotypic T cell exhaustion. (A) Serum was collected from leukemia-bearing mice 12 days after TCF3/PBX1.3 injection. Cytokines were measured by multiplex assay. Unirradiated C57Bl/6 mice were used as controls. (B) Serum was collected from MLL-AF4–bearing mice (conditioned with 250 cGy irradiation prior to injection) as in panel A and serum analyzed for cytokines. Irradiated mice were used as controls. (C) TCF3/PBX1.3.3 was cultured in the presence of IL-6 (200 µg /mL), IL-10 (100 µg /mL), TNF-α (100 µg/mL), or all 3 cytokines combined. Tim3 expression was evaluated by flow cytometry after 2 or 3 days of culture.
CAR expression does not fully reverse leukemia-induced T-cell dysfunction
We have previously shown that a murine CD19-targeted CAR can mediate complete regression of TCF3/PBX1.3 when generated from leukemia-naive donors.32 We next tested whether progressive ALL impacts functionality of CARTs generated from leukemia-bearing donors. Activation and expansion of T cells in the presence of anti-CD3/CD28 beads transiently induces expression of PD1 on T cells during the process of CART production from leukemia-naive donors. However, PD1, Tim3, and LAG3 expression persists on CARTs generated from leukemia-bearing donors (Figure 6A-C). Finally, CARTs from healthy donors completely eradicated leukemia, resulting in 100% survival. In contrast, while CARTs from leukemia-bearing donors demonstrated activity, leukemia-induced lethality occurred in 60% of treated mice (Figure 6D). Finally, CARTs generated from MLL-AF4–bearing donors were also less effective at clearing TCF3/PBX1.3 (supplemental Figure 8). Thus, leukemia-induced T-cell dysfunction persists following the introduction of a CAR and has implications for the efficacy of adoptive cell therapies for ALL.
Figure 6.

CARTs derived from leukemia-bearing donors remain dysfunctional following in vitro activation, expansion and transduction. (A) Splenocytes from mice were enriched for CD3+ cells, which were used to generate mCD19 CARTs as described in methods. Representative dot plots showing PD1, Tim3, and LAG3 expression on after 4 days in culture. (B-C) Geometric mean of PD1 expression (B) and TIM3 expression (C) on mCD19 CAR-expressing T cells generated from healthy and leukemia-bearing donors. P < .05 at day 4 (Mann-Whitney U test). (D) C57BL/6 mice received 500 cGy radiation followed by tumor challenge with 105 TCF3/PBX1.3. CARTs or mock (GFP+) T cells were generated from healthy and leukemia-bearing donors. CARs were administered IV at a dose of 3 × 105 CARTs 3 days after tumor challenge, 500 cGy irradiation on day 3. Mock CAR provided no survival benefit compared with untreated mice, but mCD19 CAR resulted in 100% survival (P = .02, Mantel-Cox test). (E) Microarray was performed on mCD19 CARTs generated as in panel A on day 5 of culture. Heat map of representative gene expression associated with TCR signaling and T cell exhaustion is shown.
Distinct gene expression profile on CARTs generated from leukemia-bearing donors vs healthy donors
Since PD1 blockade does not reverse the impaired functionality of CARTs generated from leukemia-bearing donors, we performed gene expression profiling to identify alternative pathways that could contribute to leukemia-induced T-cell dysfunction. Consistent with phenotypic data, CARTs produced from leukemia-bearing donors expressed more PD1, Tim3, and LAG3 in addition to other genes known to be associated with T-cell dysfunction (Figure 6E).22 Finally, unbiased pathway analysis revealed differential expression of genes regulating a number of other cellular processes (carbohydrate and cholesterol metabolism, chromatin remodeling, and hypoxia) that could serve as targets to improve functionality of T cells used for the generation of adoptive cell therapies (supplemental Figure 9).
Discussion
There has been tremendous success in the treatment of pediatric B precursor ALL over the past 3 decades, with the majority of patients being cured using standard chemotherapy. However, there remains a subset of children who will relapse following upfront cytotoxic chemotherapy despite. Furthermore, adolescents and adults with ALL fare less well, with lower remission induction to upfront chemotherapy and higher rates of relapse.5 Allogeneic hematopoietic stem cell transplantation (allo-HSCT) offers the chance of cure following high-risk relapse if patients can be rendered minimal residual disease negative,33 but the contribution of GVL to success of allo-HSCT for ALL is limited compared with other hematologic malignancies.10 Because T cells are a major contributor to the GVL effect, this finding suggests that pre–B-cell ALL may be inherently less responsive to unmodified T-cell immunotherapy than other hematopoietic malignancies. Malignancy-induced T-cell dysfunction has been well described in hematologic malignancies, including acute myelogenous leukemia,34 but it has not been well studied in the context of ALL. One of the challenges in studying ALL immune biology has been the relatively limited availability of murine models for B-cell precursor leukemia, as the majority of syngeneic murine models of lymphoid malignancies are phenotypically mature.35 Evaluation of immunotherapy in syngeneic models of mature B-cell malignancies may not be predictive in lymphoblastic leukemia, as interactions between adaptive immune cells and other hematopoietic cells in the bone marrow are likely to have significant immunobiological consequences.
Using syngeneic murine models of pre–B-cell ALL in immune competent mice, we demonstrate that T-cell dysfunction is induced by progressive leukemia and that this can impact the efficacy of immunotherapeutic approaches that rely on T cells residing in the leukemic host. T-cell depletion has been associated with loss of protection in a syngeneic murine36 model, and our data extend this observation to the context of vaccines and CARTs. Furthermore, we establish that T-cell dysfunction is initiated early in the course of leukemia progression, precluding efficacy of a therapeutic vaccine. Although the data presented here were generated using 2 separate leukemia models, one limitation is that we used a transplantable model (rather than a primary leukemia model) to achieve consistent onset and allow for systematic analysis of T-cell function. Nonetheless, these findings have important implications for the successful generation of immunotherapy for ALL in general and suggest that in contrast to AML and mature B-cell malignancies, where checkpoint inhibitors have achieved success, immune checkpoint blockade may not be effective as an isolated modality in ALL.
Our results may also shed light on the low potency of the GVL following allo-HSCT. Rather than inherent resistance of ALL blasts to T cell killing, the data presented here are consistent with a model wherein ALL blasts are not only poor antigen-presenting cells12,37 but also actively suppress the function of host-resident T cells. Interestingly, the potency of irradiated leukemia cells at inducing an immune response protective against leukemia challenge is also consistent with prior studies showing that ALL blasts can be manipulated to improve antigen-presenting cell function.14 Indeed, vaccines administered during immune reconstitution following an allo-HSCT and prior to relapse are therapeutically effective and may be a strategy to improve outcomes following allo-HSCT.38 However, our finding that even minimal levels of ALL present early during leukemia progression inhibit T-cell responses to a vaccine are consistent with recent data that MRD levels of ALL impair GVL and result in rapid posttransplant relapse.33 T-cell dysfunction induced early during solid tumor initiation has also been described.39
Dramatic success in ALL has been achieved with the ex-vivo expansion of T cells genetically modified to express a CAR targeting a leukemia-associated protein. In most trials, CARTs are derived from T cells harvested from peripheral blood of patients harboring leukemia at the time of collection or prior to collection. Despite potent activity at inducing remission, a CAR product cannot be generated for all patients, the marked expansion required for remission induction is not universal, and the persistence of CARTs appears to improve the likelihood of durable remissions. Thus, T cell quality is an important component of CART efficacy. The data presented here indicate that the presence of leukemia in the host may impact the potency of a CART product despite in vitro expansion and genetic manipulation to redirect specificity. Generation of CARTs from a third-party donor or an allogeneic donor in the case of transplanted patients obviates the impact of ALL on T-cell quality. There is also enthusiasm for combining immune checkpoint inhibition with CARTs in the setting of poor expansion or relapse. The data presented here would suggest that inhibiting the PD1-PDL1 access may not be sufficient, but clinical trials will be required.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Javed Khan and Qingrong Chen for Metagene analysis of TCF3/PBX1.3 and marrow-derived pre–B cells with multiple types of murine and human tumor lines, John Buckley for expert assistance in performing animal experiments, and Vishal Koparde and Ajeet Mandal for help with array data submission to the public database.
This work was supported by the Intramural Research Program of the NCI.
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: H.Q. and T.J.F. designed experiments; H.Q., K.I., S.N., P.P.S., C.R.B., B.B.D., N.N.S., and S.T. performed experiments and analyzed data; B.-H.K., M.E.K., and T.J.F. analyzed data; and H.Q., B.B.D., N.N.S., and T.J.F. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for T.J.F. is Department of Pediatrics, University of Colorado Denver and Children’s Hospital Colorado, Aurora, CO.
Correspondence: Terry J. Fry, 13123 East 16th Ave, Box 115, Aurora, CO 80045; e-mail: terry.fry@ucdenver.edu.
REFERENCES
- 1.Hunger SP, Loh ML, Whitlock JA, et al. ; COG Acute Lymphoblastic Leukemia Committee. Children’s Oncology Group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer. 2013;60(6):957-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14(6):e205-e217. [DOI] [PubMed] [Google Scholar]
- 3.Ko RH, Ji L, Barnette P, et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol. 2010;28(4):648-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tallen G, Ratei R, Mann G, et al. Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol. 2010;28(14):2339-2347. [DOI] [PubMed] [Google Scholar]
- 5.Curran E, Stock W. How I treat acute lymphoblastic leukemia in older adolescents and young adults [published correction appears in Blood. 2015;126(15):1868]. Blood. 2015;125(24):3702-3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pulsipher MA, Langholz B, Wall DA, et al. The addition of sirolimus to tacrolimus/methotrexate GVHD prophylaxis in children with ALL: a phase 3 Children’s Oncology Group/Pediatric Blood and Marrow Transplant Consortium trial. Blood. 2014;123(13):2017-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn T, Wall D, Camitta B, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in children: an evidence-based review. Biol Blood Marrow Transplant. 2005;11(11):823-861. [DOI] [PubMed] [Google Scholar]
- 8.Collins RH Jr, Goldstein S, Giralt S, et al. Donor leukocyte infusions in acute lymphocytic leukemia. Bone Marrow Transplant. 2000;26(5):511-516. [DOI] [PubMed] [Google Scholar]
- 9.Choi SJ, Lee JH, Lee JH, et al. Treatment of relapsed acute lymphoblastic leukemia after allogeneic bone marrow transplantation with chemotherapy followed by G-CSF-primed donor leukocyte infusion: a prospective study. Bone Marrow Transplant. 2005;36(2):163-169. [DOI] [PubMed] [Google Scholar]
- 10.Fry TJ, Willasch A, Bader P. The graft-versus-tumor effect in pediatric malignancy. Pediatr Clin North Am. 2010;57(1):67-81. [DOI] [PubMed] [Google Scholar]
- 11.Stern M, de Wreede LC, Brand R, et al. Sensitivity of hematological malignancies to graft-versus-host effects: an EBMT megafile analysis. Leukemia. 2014;28(11):2235-2240. [DOI] [PubMed] [Google Scholar]
- 12.Cardoso AA, Schultze JL, Boussiotis VA, et al. Pre-B acute lymphoblastic leukemia cells may induce T-cell anergy to alloantigen. Blood. 1996;88(1):41-48. [PubMed] [Google Scholar]
- 13.D’Amico G, Vulcano M, Bugarin C, et al. CD40 activation of BCP-ALL cells generates IL-10-producing, IL-12-defective APCs that induce allogeneic T-cell anergy. Blood. 2004;104(3):744-751. [DOI] [PubMed] [Google Scholar]
- 14.Reid GS, She K, Terrett L, Food MR, Trudeau JD, Schultz KR. CpG stimulation of precursor B-lineage acute lymphoblastic leukemia induces a distinct change in costimulatory molecule expression and shifts allogeneic T cells toward a Th1 response. Blood. 2005;105(9):3641-3647. [DOI] [PubMed] [Google Scholar]
- 15.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125(26):4017-4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709-2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56-61. [DOI] [PubMed] [Google Scholar]
- 22.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492-499. [DOI] [PubMed] [Google Scholar]
- 23.Brusa D, Serra S, Coscia M, et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica. 2013;98(6):953-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atanackovic D, Luetkens T, Kröger N. Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia. 2014;28(5):993-1000. [DOI] [PubMed] [Google Scholar]
- 25.Le Dieu R, Taussig DC, Ramsay AG, et al. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood. 2009;114(18):3909-3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. 2011;118(19):5084-5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bijl J, Sauvageau M, Thompson A, Sauvageau G. High incidence of proviral integrations in the Hoxa locus in a new model of E2a-PBX1-induced B-cell leukemia. Genes Dev. 2005;19(2):224-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krivtsov AV, Feng Z, Lemieux ME, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14(5):355-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardy RR, Kincade PW, Dorshkind K. The protean nature of cells in the B lymphocyte lineage. Immunity. 2007;26(6):703-714. [DOI] [PubMed] [Google Scholar]
- 30.Kato T, Nishida T, Ito Y, Murase M, Murata M, Naoe T. Correlations of programmed death 1 expression and serum IL-6 level with exhaustion of cytomegalovirus-specific T cells after allogeneic hematopoietic stem cell transplantation. Cell Immunol. 2014;288(1-2):53-59. [DOI] [PubMed] [Google Scholar]
- 31.Kinter AL, Godbout EJ, McNally JP, et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008;181(10):6738-6746. [DOI] [PubMed] [Google Scholar]
- 32.Jacoby E, Yang Y, Qin H, Chien CD, Kochenderfer JN, Fry TJ. Murine allogeneic CD19 CAR T cells harbor potent antileukemic activity but have the potential to mediate lethal GVHD. Blood. 2016;127(10):1361-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pulsipher MA, Carlson C, Langholz B, et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood. 2015;125(22):3501-3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Q, Munger ME, Highfill SL, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116(14):2484-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacoby E, Chien CD, Fry TJ. Murine models of acute leukemia: important tools in current pediatric leukemia research. Front Oncol. 2014;4:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jo S, Lee JH, Mattei JJ, et al. Generation of a multi-antigen-directed immune response for durable control of acute lymphoblastic leukemia. Leukemia. 2018;32(2):539-542. [DOI] [PubMed] [Google Scholar]
- 37.Haining WN, Cardoso AA, Keczkemethy HL, et al. Failure to define window of time for autologous tumor vaccination in patients with newly diagnosed or relapsed acute lymphoblastic leukemia. Exp Hematol. 2005;33(3):286-294. [DOI] [PubMed] [Google Scholar]
- 38.Oka Y, Tsuboi A, Nakata J, et al. Wilms’ tumor gene 1 (WT1) peptide vaccine therapy for hematological malignancies: from CTL epitope identification to recent progress in clinical studies including a cure-oriented strategy. Oncol Res Treat. 2017;40(11):682-690. [DOI] [PubMed] [Google Scholar]
- 39.Schietinger A, Philip M, Krisnawan VE, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. 2016;45(2):389-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.