

Abstract
The structures and physicochemical properties of chemicals are important for determining their potential toxicological effects, toxicokinetics, and route(s) of exposure. These data are needed to prioritize the risk for thousands of environmental chemicals, but experimental values are often lacking. In an attempt to efficiently fill data gaps in physicochemical property information, we generated new data for 200 structurally diverse compounds, which were rigorously selected from the USEPA Distributed Structure-Searchable Toxicity Database (DSSTox). This pilot study evaluated rapid experimental methods to determine five physicochemical properties including the log of the octanol:water partition coefficient (known as log(Kow) or logP), vapor pressure, water solubility, Henry’s law constant, and the acid dissociation constant (pKa). For most compounds experiments were successful for at least one property; log(Kow) yielded the largest return (176 values). It was determined that the presence of 21 structural features may have played an overall role in rapid measurement method failures. To gauge consistency with traditional measurement methods, the new measurements were compared with previous measurements (where available). Since quantitative structure-activity/property relationship (QSAR/QSPR) models are used to fill gaps in physicochemical property information, 5 suites of QSPRs were evaluated for their predictive ability and chemical coverage or applicability domain of new experimental measurements. The ability to have accurate measurements of these properties will facilitate better exposure predictions in two ways: 1) direct input of these experimental measurements into exposure models; and 2) construction of QSPRs with a wider applicability domain, as their predicted physicochemical values can be used to parameterize exposure models in the absence of experimental data.
Keywords: physicochemical properties, environmental chemicals, predictive modeling, QSAR, chemical features
GRAPHICAL ABSTRACT
1. INTRODUCTION
Physicochemical properties such as the log of the octanol:water partition coefficient (log(Kow) or logP), and vapor pressure (VP) play a critical role in addressing many aspects of a chemical’s behavior including in drug discovery1–5, migration through the environment and body6–13, and potential impact on human health and the environment14–17. In order to estimate the environmental risk posed by such chemicals, cheminformatics tools and predictive models rely on physicochemical properties to predict important aspects such as toxicity14–17, toxicokinetics10,12,23, and exposure6,7,11. However, there are tens of thousands of manufactured chemicals, that may find their way into living organisms and the environment18–22 but also have few or no physicochemical property data,. This lack of data is particularly problematic as physicochemical properties govern how chemicals 1) affect the biosphere (i.e., physiological and pathological effects) and 2) emit from or pass through the lithosphere (soil), hydrosphere (water), and atmosphere (air) to arrive at biological sites of exposure. Advancements in computational toxicology methods support on-going efforts to develop rapid toxicity information to inform the anthroposphere (human evaluation and regulation) in a decision-making context24–31. The USEPA’s Exposure Forecasting (ExpoCast) project32 relies upon robust physicochemical property information in order to develop high-throughput exposure and toxicokinetic models. In lieu of measured data, property prediction models can potentially be used, but the validity and relevance of these models needs to be assessed as new chemistries are developed. One of the goals of the ExpoCast project is to assess the utility and reliability31 of available existing physicochemical property models. This requires new experimental physicochemical data to be generated, but given the thousands of chemicals of potential interest, higher-throughput methods are attractive if they can be shown to be reliable.
Physicochemical properties have been found to be useful descriptors in predicting a wide range of properties33, including absorption34, distribution10,12,34, clearance34, permeability35,36, membrane (lipid bilayer) affinity37,38, plasma protein binding39,40, in vitro assay concentration41,42, and predictive ability of in vitro toxicokinetics (TK) assays23. Models derived from the use of physicochemical properties as well as molecular structure descriptors also enable the prediction of chemical functional use in products (e.g., emulsifiers and dyes)2,4,5. Thus, any resulting model’s accuracy will be affected by uncertainty in the properties used, which subsequently impacts their utility for chemical risk assessment.
Physicochemical properties can be predicted from chemical structure via quantitative structure-property relationships (QSPRs)43–49. A QSPR expresses, in mathematical form, the quantitative relationship that may exist between the chemical structure of a series of chemicals and their measured properties. Many QSPRs are derived using machine learning algorithms which seek out statistically relevant correspondence between specific structural features and property values for a training set of chemicals43–49. Applicability domains (AD) are typically defined for QSPRs to facilitate reliable use. The AD for a QSPR is defined as the response and chemical structure space in which the model makes predictions with a given acceptable reliability. There are many different types of ADs that can be defined for QSPR models; for statistically based QSPR models relying on structural features, interpolation methods are often used43–46,50. Chemicals within the AD are associated with model-specific prediction uncertainty based on approximations to experimental measurements51. In addition, it is worth noting that prediction uncertainty is assumed to increase for chemicals determined to be outside of the AD or for models with unknown AD boundaries43,45,52. Since predicted physicochemical properties are often used as inputs to derive QSAR models, whether they be for toxicity, environmental fate or toxicokinetic parameters, any prediction uncertainty will ultimately cascade to these model predictions also.52 Unfortunately, for many chemicals of interest, the relevant physicochemical properties of interest have not been measured and/or are out of the any predictive models’ interpolation spaces6,53,54.
There are a number of QSPRs for physicochemical properties that have been incorporated into software tools for ease of use. Some of these tools are open-source and free, such as OPEn (quantitative) structure-activity Relationship Application (OPERA)55,56, whilst others are proprietary but free, such as Estimation Program Interface (EPI Suite)53 and Online Chemical Database (OCHEM)57 . There are also a number of tools that are proprietary and commercial such as Simulations Plus58 and Advanced Chemistry Development, Inc. (ACD/Labs)59 or a mix of different categories such as ChemAxon60 products that provide QSPR models. Some of these models are not transparent in terms of providing end-users with the information needed to assess their reliability, specifically details such as the AD, underlying training set and details of the model algorithms. For example, EPI Suite53 does not provide an AD for any of its physicochemical QSPR models.
Here we describe a pilot study where we rapidly generated experimental data for 5 physicochemical properties of 200 structurally diverse chemicals using high-throughput approaches. The selected parameters were log(Kow), VP, water solubility (WS), Henry’s law constant (HLC), and the acid dissociation constant (pKa). The chemicals were identified to incorporate: 1) those with existing values measured using traditional, lower-throughput protocols (Note: There was no accessible database of measured values for pKa); 2) those that are structurally similar to chemicals with existing data; and 3) those that are structurally diverse from chemicals with existing property data. New experimental measurements were thus compared with their respective previously measured values that resulted from traditional measurement protocols (e.g., one compound at a time). Structural features were identified that lend themselves to failed measurements. Furthermore, it was determined whether successful experiments might be added to the already existing measured data in publicly accessible databases. We compared the experimental measurements to model predictions from five models that were either available publicly or licensed for use (ACD/Labs59,61, Chemical Properties Estimation Software (ChemProp)62, EPI Suite53,61, National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM)61,63 and OPERA55,56,61).
2. METHODS
2.1. Test Chemical Selection
Multiple factors were considered in the selection of the set of test compounds. First, resources allowed for only 200 compounds to be tested. Then, selection criteria were used to gauge experimental reproducibility and the ability to retrieve new experimental data for chemical moieties that have substantial data gaps. Figure 1 illustrates the workflow which was used to filter chemicals in the Distributed Structure-Searchable Toxicity (DSSTox) database64 based on the following: 1) whether enough chemical sample was readily in stock (i.e., greater than 20 mg), 2) whether chemical properties had been previously measured, 3) limits of detection (LOD) for each property, which are the bounds within which experimental measurements are most reliable (as determined by the analytical methods to be used), and 4) computed similarity coefficients for chemicals within and outside the LOD. This workflow was then implemented in KNIME65 (V2.9), a free and open-source environment for data mining with a multitude of integrated cheminformatics tools. Starting with 2,553 DSSTox compounds (Figure 1) that were in stock, 60 of the compounds had measured properties for log(Kow), VP, WS, and HLC as provided in a curated version of the PHYSPROP56,66 database, a publicly accessible resource for existing physicochemical property measurements. The remaining 2,493 chemicals were filtered based on two approaches to maximize chemical diversity: consideration of the LOD of the experiments and identification of various patterns of structural similarity. 782 chemicals were within the range of new measurement LOD based on EPI Suite predictions: Kow (0<log(Kow)<6)67 and VP (10−7<VP<102 Pa at 25°C)68. While HLC and WS had previous experimental data in PHYSPROP, neither one had a pre-specified LOD range prior to experimentation. Although there were no previous experimental pKa data in PHYSPROP, new measurement values were based on a specific pH range (3<pH<12)69.
Figure 1.
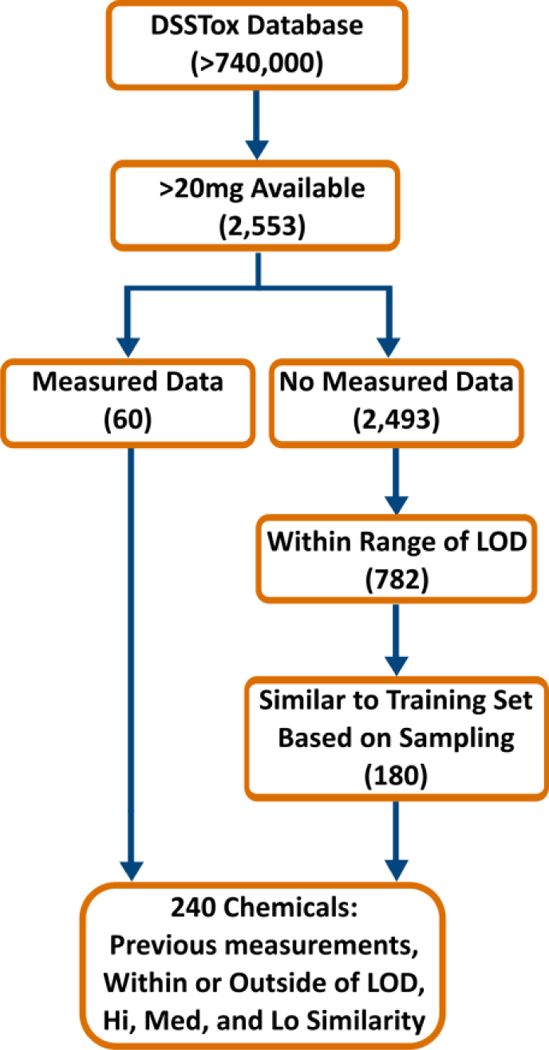
Simplified workflow for selecting chemicals for testing. An excess of 20% of the compounds were filtered and 200 compounds were submitted for experimental measurements of five physicochemical properties: octanol:water partition coefficient (log(Kow)), vapor pressure (VP), water solubility (WS), Henry’s law constant (HLC), and acid dissociation constant (pKa).
In order to include the most diverse chemicals from the set of 782 chemicals, Tanimoto70 similarity indices (S) were computed based on extended CDK fingerprints71 (~4000 bits), such that if any two compounds achieved a similarity score of greater than 0.6, then one of two compounds was randomly removed from consideration in this pilot study. The remaining 385 compounds (with S ≤ 0.6) were further filtered based on their similarity to chemicals in PHYSPROP datasets that were used to train the EPI Suite models for log(Kow), VP, and HLC. Tanimoto indices were calculated between the 385 remaining compounds and the compounds in PHYSPROP with a measured value for at least one of 3 properties: log(Kow), VP, and HLC. Sixty compounds were then selected from each of three similarity index ranges: high (S > 0.7); medium (0.5 ≤ S < 0.7) and low (S < 0.5) similarity, for the purpose of providing chemicals inside and outside of the chemical space covered by EPI Suite for the three models. This led to 180 compounds for which there were no measured values and therefore resulted in a total of 240 compounds for measurement analysis (60 already measured and 180 with no measurement). We aimed for 200 chemicals and the addition of 20% (40 chemicals) in excess of the goal as a precaution for any problems with the measurement methods. Out of the 240 computationally selected chemicals, the 200 most abundant in stock were submitted to be tested where 22% (44) had previously measured values for log(Kow), VP, and HLC, while there were 62 compounds where WS had been previously measured. For the remaining chemicals, the measured values were mostly within or near the pre-specified LODs. The KNIME65 workflow with a detailed description, a screenshot, and summary statistics are provided in Supplemental Code S1. Results of experimental measurements for all 5 properties are available in Supplemental Table S1.
2.2. Determination of Octanol:Water Partition Coefficient
For the high-throughput experimental measurements of log(Kow), the procedures in the Estimation by Liquid Chromatography and OECD Test Guideline 11767 and the EPA OPPTS 830.7570 Partition Coefficient (n-Octanol/Water)72 were adapted and modified. In reverse phase high-performance liquid chromatography (HPLC), the chemicals are retained in proportion to their hydrocarbon:water partition coefficient. Hydrophilic chemicals elute first and lipophilic chemicals elute last. This enables the relationship between the retention time on a reverse phase column and the octanol:water partition coefficient to be established. The HPLC method is applicable to compounds with log(Kow) in the range of 0 – 6. The HPLC method described in the EPA and OECD methods was converted to high-throughput with the use of ultra-high pressure liquid chromatography (UHPLC) with sub-2 μm particles column. A Waters ACQUITY system equipped with a 2996 photodiode array detector was used for analysis. The analytical column used was Waters BEH C18 (2.1 × 50 mm, 1.8 μm). An isocratic mobile phase system using 55% water and 45% acetonitrile maintained at 25oC was used to elute the chemicals. The flow rate was maintained at 0.3 mL/min. By plotting the capacity factor (k) generated from the retention times of the reference compounds versus log(Kow), a linear regression curve was generated to estimate the log(Kow) of the compound in question by its retention time using Equations (1) and (2).
| Eq. 1 |
| Eq. 2 |
where tR is the retention time of the test compound, and t0 is the dead-time of the analytical system. Three reference compounds: nitrobenzene, toluene, and benzyl benzoate with their log(Kow) values of 1.85, 2.73, and 3.97, respectively62, were used in the initial screening process. Test compounds with retention time results within range of these three compounds were considered valid. Test compounds that eluted after benzyl benzoate were reanalyzed a second time using a different mobile phase system with 15% water and 85% acetonitrile. For this system, benzyl benzoate, fluoranthene, and 4,4’-DDT were used as the reference compounds, whereby the reference log(Kow) values used were 3.97, 5.1, and 6.5, respectively62.
2.3. Determination of Vapor Pressure
Several methods developed by the ASTM International73 organization were available for use in VP determination but these methods were primarily intended for liquid petroleum products. Here, capillary gas chromatography (GC) using the relative retention time method was evaluated as an option to estimate VP of the compounds68. This GC method was an indirect measurement and involved establishing the retention times of compounds using a capillary GC column with a non-polar phase such as 100% dimethyl-polysiloxane. The GC technique was fast, easily automated, and required minimal amounts of sample. The disadvantage of using this method was selecting the appropriate reference chemicals to relate to the VP of the test substance. The chemical structures, functional groups and polarities of chemicals in the DSSTox library are very diverse which made the data measurement and interpretation difficult. Two compounds di-n-propyl phthalate (DnPP) and di-2-ethylhexyl phthalate (DEHP) were selected as the reference compounds for the VP measurement. The reference VP values used were 1.76 × 10−2 and 8.27 × 10−6 Pa for DnPP and DEHP, respectively63.
An Agilent 6890 gas chromatograph coupled to an Agilent 5973 mass spectrometer was used for the measurement of VP. The instrument was equipped with a Restek Rtx-1MS column (15 m; 0.25 mm id; 0.25 μm film thickness). Helium was used as the carrier gas with a constant flow setting of 1 mL/min. The injector temperature was set at 250 oC. The initial oven temperature was set at 40 oC, followed by a linear ramp of 8 oC/min to a final temperature of 300 oC. The test substances were injected and the retention times of the compounds were then correlated to the log(VP) values.
2.4. Determination of Water Solubility and Henry’s Law Constant
Measurements of WS were generated using high-performance liquid chromatography-ultraviolet (HPLC-UV) analysis. The test substances were dissolved in HPLC grade acetonitrile to approximately 1 mg/mL. The stock solutions were further diluted to 100 μg/mL, 10 μg/mL and 1 μg/mL for use as calibration curves to quantitate the amount of material soluble in water. For the test item substance, 1 mL of water was added to approximately 5 mg of material74. The aqueous solutions were equilibrated at room temperature for 48 hours. Prior to analysis, the aqueous solution was carefully transferred to a separate vial avoiding any particulates present in the vial. VP results obtained from the GC experiment were used in conjunction with WS results to generate HLC results. HLC measurements were based on the relationship of vapor pressure and water solubility describe in Equation (3):
| Eq. 3 |
where MW is the molecular weight of the substance.
2.5. Determination of Acid Dissociation Constant
For high-throughput determination of pKa values, spectrophotometric titration was conducted75. An ultraviolet (UV) spectrum from 210–400 nm of a target analyte was acquired at each unit pH datum point of the titration. The change in UV with pH absorbance was then plotted. This method is suitable for compounds that contain a chromophore close to the ionization center such that the spectrum absorbance changes as a function of ionization. Although a rapid measurement method for pKa was attempted (SI Methods), it was considered to be unsuccessful due to a failed experimental procedure.
2.6. Previous Experimental Data versus New Experimental measurements
Previously measured properties for log(Kow), VP, WS, and HLC were compiled from the curated version of the publicly available PHYSPROP56,66 database. In this curated version structures are best matched to their respective experimental properties based on a series of data accuracy scoring mechanisms developed at the USEPA National Center for Computational Toxicology (NCCT). This database does not contain any pKa values. A linear regression (in log-space) was employed to illustrate the correlation between the new and previous experimental values along with the Pearson76 coefficient (R2) (using the ‘ggplot 2’77 R package), root mean squared error (RMSE), and mean absolute error (MAE) were reported for each of these. RMSEs are reported in addition to the R2 in order to emphasize the average deviation of new and previous data versus the use of a model to explain the percentage of response variation.
2.7. Identification of Features in Chemicals Lacking New Experimental Data
In order to identify key features of the chemicals for which some or all experimental measurements were unsuccessful, odds ratios were calculated for each fingerprint bit (i.e., substructure) between chemicals that had new experimental results and those that did not. The odds ratio is defined as:
| Eq. 4 |
where n0,N is the number of chemicals where the bit of the fingerprint was not present and the chemical could not be measured, n0,Y is the number of chemicals where the bit was not present and the chemical could be measured, n1,N is the number of chemicals where the bit was present and the chemical could not be measured, and n1,Y is the number of chemicals where the bit was present and the chemical could be measured. Using this definition, if the OR for a substructure is greater than 1, then that bit’s presence in a chemical’s structure is associated with an inability to measure that property. Conversely, if the OR is less than 1, then that bit’s absence from a chemical’s structure is associated with the inability to measure a property. The closer an OR value is to 1 the less significant that bit’s association with either the ability or inability to measure a chemical’s property.
2.8. Predicted Physicochemical Properties
Model predictions for the properties were obtained from sources that were publicly available and/or using commercially licensed products available in the authors laboratories: ACD/Labs59,61 (log(Kow), WS, and pKa), ChemProp (v6.5)62 (log(Kow), WS, and VP), EPI Suite (v4.11)53,61 (log(Kow), VP, WS, and HLC), NICEATM61,63 (log(Kow), and WS), and OPERA (v1.5)55,56,61 (log(Kow), VP, WS, and HLC). ChemProp (v6.5)62 predictions were obtained via multiple algorithms each for log(Kow), VP, and WS. Sources for the EPI Suite (v4.11) predictions for log(Kow), VP, and HLC are the from KOWWIN (v1.68), MPBPWIN (v1.43), and HENRYWIN (v3.20) modules, respectively. Predictions from ACD/Labs59,61, EPI Suite53,61 and NICEATM61,63 were collected from the USEPA’s CompTox Chemistry Dashboard61,78 (https://comptox.epa.gov/dashboard/) using its batch mode functionality. OPERA’s predictions, which are also available on the CompTox Chemistry Dashboard61,78, can be obtained via a batch mode prediction using the command line application recently developed at USEPA’s NCCT and available on GitHub (https://github.com/kmansouri/OPERA).55,56,61 Values and links to OPERA predictions are provided in Supplemental Table S2. Unit conversions were applied (where necessary) such that all data sources for a given property were reported in consistent units. Supplemental Table S3 summarizes the scale and units for reported values from all seven sources of physicochemical property data.
2.9. Previous Experimental Data versus QSPR Model Predictions
Analysis was performed in order to compare QSPR model predictions (if applicable) to experimental measurements for all five physicochemical properties. Final model predictions of chemicals with both new measurement and PHYSPROP values for log(Kow), VP, WS, HLC, and pKa are provided in Tables S4-S8, respectively. Results from ACD/Labs models used to determine if a compound could be ionized and absorbance measurements used for determining pKa values are provided in Tables S9 and S10, respectively. For the compounds that had available experimental data, in the literature or in accessible databases, for log(Kow), VP, WS, and HLC properties, new measurement data were aggregated with previously measured (PHYSPROP) data and model predictions from ACD/Labs59,61, ChemProp62, EPI Suite53,61, NICEATM61,63, and OPERA.55,56,61 Linear regression plots were used to illustrate the correlation between new experimental data and predicted values. RMSEs and MAEs were reported for each property in logarithmic-space. In order to achieve a one-to-one comparison between previous measurements and model predictions, only the subset of compounds that had previous PHYSPROP values along with newly obtained data, were included in this analysis. As ChemProp provided multiple predictive models for log(Kow), VP, and WS, the models with the fewest missing predictions and the lowest RMSE value were selected. Thus, via ChemProp, for log(Kow) and VP, the unpublished read-across methods yielded the lowest RMSE value while the model that yielded the lowest RMSE for WS was that of Huuskonen (2001)79.
3. RESULTS
3.1. Comparison of Experimental Measurements with Previous Measurements
200 chemicals reflecting a mix of chemicals with traditionally measured physicochemical properties, unmeasured chemicals with structures expected to be similar to those with measured properties, and more diverse (i.e., challenging) chemicals were characterized using high-throughput property measurement methods. As summarized in Table 1, the log(Kow) method was successful for 176 of the 200 because measurements were obtained. Only 32 of these have PHYSPROP56,66 values. The rapid VP method was successful for 168 of the 200, of which only 32 of these had PHYSPROP values. For WS, 129 of 200 were successfully measured and 36 had new experimental measurements. For HLC, 23 of the 110 compounds that could be measured also had experimental values reported in PHYSPROP. The structural similarity of the chemicals which had both new experimental measurements and PHYSPROP values for a given property is shown in Supplemental Figures S1 – S4.
Table 1.
For the total number of new experimental measurements (Ntotal), coefficient of determination (R2), root mean square error (RMSE) and mean absolute error (MAE) values between the number of new experimental measurements and previously measured values (Nprevious) for octanol-water partition coefficient (log(Kow)), vapor pressure (VP), water solubility (WS), and Henry’s Law Constant (HLC).
| Ntotal | Nprevious | R2 | RMSE | MAE | |
|---|---|---|---|---|---|
| log(Kow) | 176 | 32 | 0.77 | 7.71E-01 | 6.21E-01 |
| log(VP) | 168 | 33 | 0.66 | 2.10E+00 | 1.49E+00 |
| log(WS) | 129 | 36 | 0.55 | 9.32E-01 | 8.12E-01 |
| Iog(HLC) | 110 | 23 | 0.69 | 2.07E+00 | 1.82E+00 |
The correspondence between the previous measurements and the rapid experimental measurements is illustrated in Figure 2 and Tables 1 and 2. RMSE and MAE values were computed along with linear regressions for each property that was measured. For log(Kow) and WS the RMSE in Table 1 indicates concordance within a factor of 10 between the rapid measurements and the traditionally measured values. The VP of the selected chemicals are somewhat difficult to measure because they are all non- or semi-volatile chemicals amenable to high-throughput in vitro testing. Experimentally measured compounds that show the most consistency are closest to their PHYSPROP counterparts as illustrated in Figure 3. All experimentally measured data and PHYSPROP values were compared with OPERA model predictions.
Figure 2.
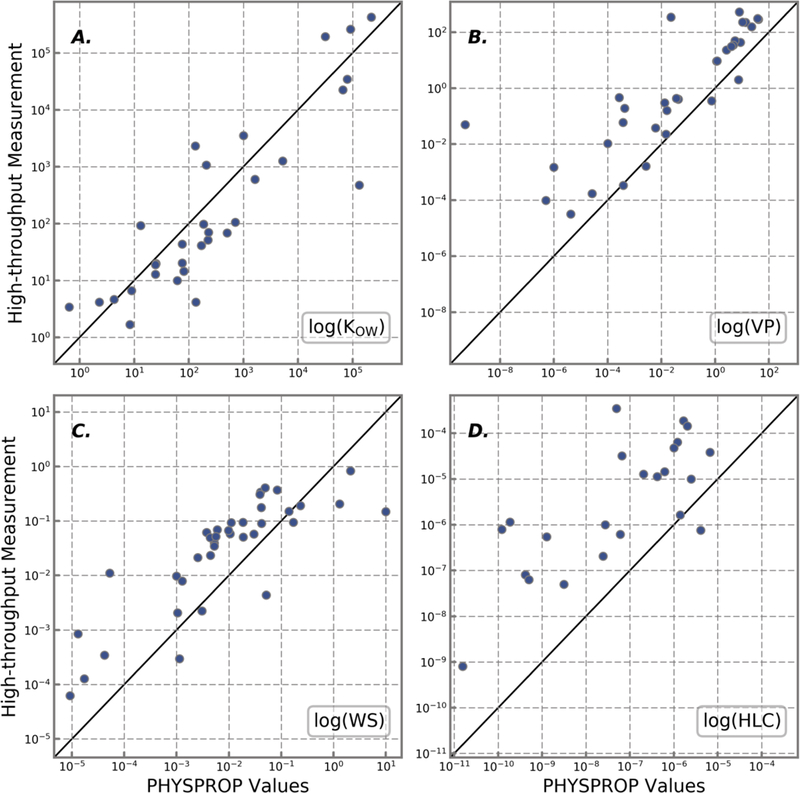
Plot of high-throughput measurements versus PHYSPROP values for A) octanol:water partition coefficient (log(Kow)), B) vapor pressure (VP), C) water solubility (WS), and D) Henry’s Law constant (HLC). All axes are shown in a log-scale value of the physicochemical property. The identity line (black) represents a perfect predictor. All axes are shown in a log-scale value of the physicochemical property.
Table 2.
R2, RMSE and MAE values (rectilinear-space) between new experimental measurements and predicted or previously measured values for octanol-water partition coefficient (log(Kow)), vapor pressure (VP), water solubility (WS), Henry’s Law Constant (HLC), and acid dissociation constant (pKa).
| N | R2 |
RMSE |
MAE |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACD/Labs | ChemProp* | EPI Suite | NICEATM | OPERA | PHYSPROP | ACD/Labs | ChemProp* | EPI Suite | NICEATM | OPERA | PHYSPROP | ACD/Labs | ChemProp* | EPI Suite | NICEATM | OPERA | PHYSPROP | |
| 32 | 0.71 | 0.76 | 0.75 | 0.74 | 0.78 | 0.77 | 8.30E-01 | 7.91E-01 | 8.02E-01 | 7.75E-01 | 7.36E-01 | 7.71E-01 | 6.42E-01 | 6.41E-01 | 6.78E-01 | 5.73E-01 | 5.78E-01 | 6.21E-01 |
| 33 | - | 0.61 | 0.74 | - | 0.69 | 0.66 | - | 1.44E+02 | 1.38E+02 | - | 1.36E+02 | 1.38E+02 | - | 6.89E+01 | 6.44E+01 | - | 6.37E+01 | 6.40E+01 |
| 23 | - | - | 0.30 | - | 0.67 | 0.55 | - | - | 5.99E-04 | - | 8.77E-05 | 8.96E-05 | - | - | 1.63E-04 | - | 3.89E-05 | 3.92E-05 |
| 36 | 0.54 | 0.76 | 0.60 | 0.59 | 0.71 | 0.69 | 1.20E+00 | 2.33E-01 | 3.07E-01 | 2.28E-01 | 5.07E-01 | 1.67E+00 | 4.26E-01 | 1.11E-01 | 1.32E-01 | 1.07E-01 | 1.85E-01 | 4.00E-01 |
| 76 | - | - | - | - | - | - | 6.15E+00 | - | - | - | - | - | 4.20E+00 | - | - | - | - | - |
For ChemProp, two predictions for VP were removed because the prediction produced a null value.
Previous pKa values were predicted and not measured. RMSE and MAE are based on removing one measurement outlier.
Figure 3.
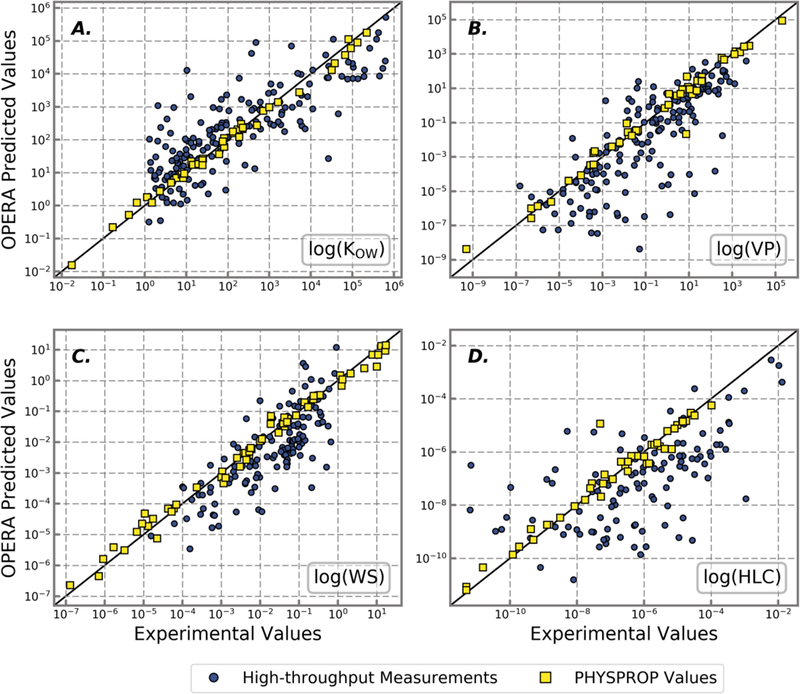
Experimental values of A) octanol:water partition coefficient (log(Kow)), B) vapor pressure (VP), C) water solubility (WS), and D) Henry’s Law constant (HLC) compared to the predicted values of those same properties from the OPERA models. High-throughput measurement values are shown as blue circles while traditionally measured values retrieved from PHYSPROP are shown as yellow squares. The identity line (black) represents a perfect predictor. All axes are shown in a log-scale value of the physicochemical property.
3.2. Comparison of Experimental Measurements with Model Predictions
Linear regressions were performed, comparing the new measurements to each of the 5 (or fewer) QSPR sources. The chemicals used in this comparison are the same as those used in the previous section (i.e., those chemicals having both traditional measurements and new experimental measurements). For log(Kow), the order of increasing RMSE values are as follows: OPERA < NICEATM < ChemProp < EPI Suite < ACD/Labs. For VP, there were data available from three predictive models whereby the RMSEs were ranked: OPERA < EPI Suite < ChemProp. For WS, there were predictions from five models. RMSE rankings for WS models were as follows: ACD/Labs < NICEATM < ChemProp < EPI Suite < OPERA. For HLC there were only two available models and the RMSE rankings were OPERA < EPI Suite. RMSE and MAE values are summarized in Table 2. In order to illustrate correlations between new rapid experimental values, previous traditional experimental values, and all predicted values, statistics of linear regressions (i.e., R2, RMSE and MAE) for log(Kow), VP, WS, HLC, and pKa (all in logarithmic-space), are provided in Table 2 (see also Supplemental Figures S5 – S8 for with linear regression equations and 95th percent confidence intervals). Using ACD/Labs to predict pKa values for the 200 pilot chemicals and to determine whether a compound was acidic or basic (Supplemental Table S9), we observed a balanced accuracy of 0.38 for acids (a “true positive” prediction occurs when experimental data show any hydrogen donor ionization equilibrium and this pKa was also determined), 0.61 for bases, and 0.49 for any ionization (acid or base). Because of the low balanced accuracies, we did not consider this assay to have performed successfully and thus did not further analyze the data.
3.3. Identification of Features in Chemicals Where Experimental Measurements Failed
The impact of chemical structure on the success or failure of experiments was then evaluated by calculating odds ratios (OR) for substructural features. Here OR describes the odds that a specific substructural feature in the fingerprint is common to chemicals that were unsuccessfully measured for one or more of the physicochemical properties. There were 47 features that were positively associated with experimental failures with respect to log(Kow). Seven of these features had 10-fold or higher odds of being present in chemicals where the log(Kow) experiment of a property was unsuccessful. The fingerprint feature with the highest odds of contributing to whether the log(Kow) experiment was unsuccessful was a linear chain where the second and third carbons are bound by an alkene bond (OR=25). Here, this feature was present in the fingerprints of 21 of the 23 chemicals that did not have a new experimental measurement of log(Kow). Conversely, the aromatic benzene ring feature had the lowest OR (0.0182) for log(Kow), indicating that the odds of this feature being in a compound that has a successful experiment was higher than that of a compound that was unsuccessful. For the group of 8 structurally similar chemicals where all five property measurements were unsuccessful, both the presence of 21 features and the absence of two alternative features may play a role in their experimental success (Figure 4). The features whose presence contributed to the failure of all property experiments, had much higher odds than those whose absence contributed to the failure to produce new experimental data. Structural similarity between chemicals that had common unsuccessful experimental measurements is shown in Figure S9; odds ratio results are provided in Supplemental Table S11.
Figure 4.
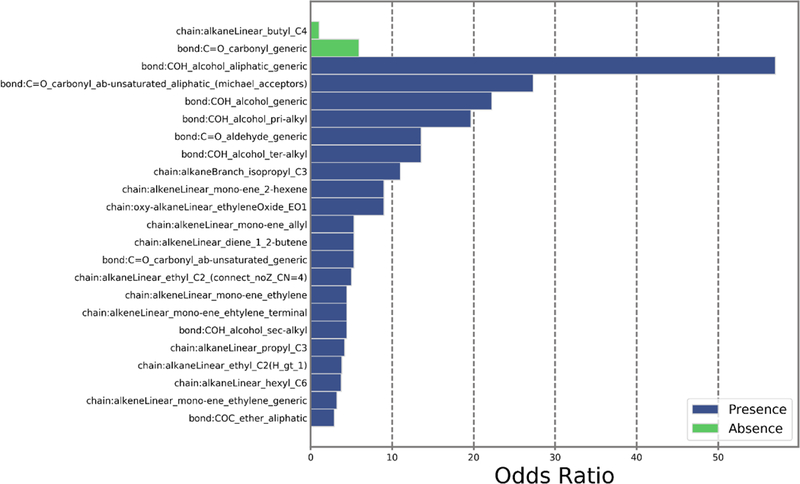
Odds ratio values for all ToxPrint bits either positively (presence; blue bars) or negatively contributing (absence; green bars) to failure of all five physicochemical property measurements.
4. DISCUSSION
While physicochemical properties impact the total environment from how chemicals emit from their sources80–82 to how they ultimately affect their environmental targets1,3,83, resources are not available to determine physicochemical properties for all of the thousands of chemicals of interest to the USEPA using traditional measurement methods. QSPRs provide a means to address this problem but rely upon training sets that may not necessarily reflect all chemicals of interest14,45,55,63,78. As an intermediary, high-throughput measurements can evaluate the performance of QSPRs for chemicals that may be different from training sets. If high-throughput methods reasonably reproduce the more resource-intensive traditional measurement methods, then the data provided may replace QSPR estimates and augment QSPR training sets to expand their ADs. Here, a diverse set of two hundred chemicals was used as part of a pilot study to assess methods of high-throughput experimental measurements of five physicochemical properties: octanol:water partition coefficient, vapor pressure, Henry’s law constant, water solubility, and acid dissociation constant. These properties are of highest interest because these data are needed to prioritize the risk of thousands of chemicals6.
We recognize that a high-throughput method for property measurement will trade some degree of precision for speed. We neither expect nor require that these new methods work for all classes of chemicals. For these reasons, we have attempted to characterize bias and examine a range of chemicals to establish the strengths and weaknesses of the available high-throughput approaches. We summarized structural features whose presence or absence may lend themselves to experimental success, which may provide insight into new methods development for failed compounds. Also, experimental measurements were compared against previous low-throughput measurements (as represented by the data in the PHYSPROP56,66 database), which illustrated experimental inconsistencies. This highlights a need for insight as to which experimental measurements must be used in cases where multiple experimental measurements for the same compound disagree. Upon comparing new experimental measurements to predicted values from a selection of both publicly, and commercially available QSPR models, we found that some models are relatively similar in predictive ability for compounds whose experimental measurements were successful. Although AD information is not available in all models, we did not observe marked differences in prediction accuracies between chemicals inside and outside the domain of the models.
The physicochemical properties studied here are relevant to many areas of risk assessment. Any refinements to both the QSPR models that predict these properties as well as QSAR models for hazard, exposure, and toxicokinetics that use these properties as inputs, may impact human and environmental health chemical risk prioritization. These data can help prioritize chemicals for more traditional testing by identifying the regions of chemical space most in need of further study84.
4.1. High-Throughput Experimental Measurements of Physicochemical Properties:
The high-throughput property measurement methods described here rely on calibrations of an experiment developed with a few reference chemicals and well-established measured values (e.g., two phthalates for water solubility). If a database of high-throughput experimental values for a large number of reference chemicals could be established, then structural similarity could potentially be used to identify a handful of reference chemicals to build a chemical-specific calibration for each new test chemical. The high-throughput method for characterizing ionization (pKa) attempted here (high-throughput UV–visible spectrophotometry) has been demonstrated for pharmaceuticals and pesticide-like compounds75,85,86. However, this method appears to require structural features that are not as common within the broader chemical space explored here. Until new high-throughput methods can be developed, it may be that traditional, lower-throughput methods will be needed to characterize pKa for arbitrary chemical structures.
4.2. Use of New Experimental Data to Expand QSPR Model Training Sets:
This new pilot dataset showed good agreement with the curated version of PHYSPROP used in this analysis. PHYSPROP data were the basis for the training sets of some of the QSPR models picked for this study; PHYSPROP was used to train EPISUITE models, while the curated PYHYSPROP database was used to train NICEATM and OPERA models. In the case of log(Kow), experimental measurements that were not contained in the PHYSPROP database were mostly within expected ranges of the models. Thus, these new data can be merged with the PHYSPROP data and used to recalibrate and refine the QSPR models. Although both OPERA and EPI Suite models predicted log(Kow), VP, WS, and HLC, only OPERA provided insight into the reliability of a prediction via its AD and confidence level metrics for each prediction (which EPI Suite does not provide). After further analysis, OPERA and other models may incorporate this pilot study data in order to improve AD coverage and reduce the prediction uncertainty for environmentally relevant chemical classes.
4.3. Impact of Expanding the Training Sets of the Models:
As the ExpoCast project depends on physicochemical properties (either measured or QSPR-predicted) to parameterize a number of exposure models6,54,87, 23 of the 200 pilot chemicals were found in reported consumer product material safety and data sheets or ingredient disclosures that have been collected by the USEPA from publicly available sources4,5,88,89. Also, 9 of the 200 pilot chemicals were identified across 32 consumer products in a recent suspect screening analysis of 100 consumer products90. Many more of these pilot chemicals were found within ~4000 products within a larger database consisting mostly of personal care products and products used to clean indoor residential areas. The ability to have accurate measurements or predictions of these properties will facilitate better exposure predictions in two ways: 1) direct input of these experimental measurements into exposure models; and 2) construction of QSAR/QSPR models with a wider AD, as their predicted physicochemical values can be used to parameterize exposure models in the absence of experimental data.
5. CONCLUSION
Following a rigorous pilot chemical selection process, 200 chemicals now have new physicochemical data for up to 5 parameters (log(Kow), VP, WS, HLC, and pKa). Novel rapid experimental methods were implemented to 1) collect physicochemical property values for data poor chemicals, 2) compare traditional versus novel experimental methods, and 3) inform in silico QSPR models that predict these parameters. Applicability domains of the various experimental methods and QSPR models were explored in each of these cases and can further be explored on a larger scale for prioritizing chemicals for the next wave of data collection. Furthermore, QSPR models that are trained on broader environmental chemical spaces, may serve to increase the utility of computational toxicology tools that depend on these parameters to help inform environmental health risk-based decision making.
Supplementary Material
HIGHLIGHTS.
High-throughput measurements of five physicochemical properties for 200 compounds were attempted
New data are now available for optimizing physicochemical property QSPR models.
Data gathered from rapid physicochemical property measurement methods will help reduce uncertainty in QSAR models that are relevant for informing chemical risk assessment.
6. ACKNOWLEDGEMENTS
We acknowledge exceptional efforts from Elaine Cohen-Hubal, Peter Egeghy, David Murphy, Nicole Hairston, Matthew T. Martin, and Stephen Little in developing and awarding the ExpoCast contract between SWRI and the U.S. EPA. The authors would like to thank Drs. Grace Patlewicz and John Nichols for their helpful review of the manuscript. Data were analyzed and plotted with Pandas91, Matplotlib92, and the R93 statistical computing language via the RStudio94 graphic user interface (including ggplot295).
Footnotes
7. DISCLAIMER
The United States Environmental Protection Agency, through its Office of Research and Development, funded and managed the research described here. However, it may not necessarily reflect official Agency policy, and reference to commercial products or services does not constitute endorsement.
This work was performed under a Memorandum of Understanding between ScitoVation and the EPA. The participation of the ScitoVation authors in this research were in part funded by the American Chemistry Council Long-range Research Initiative and in part by the Research Participation Program at the Office of Research and Development, US Environmental Protection Agency, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between US Department of Energy and US Environmental Protection Agency.
Disclaimer: The views expressed in this publication are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Reference to commercial products or services does not constitute endorsement.
REFERENCES
- 1.Lipinski CA, et al. , Advanced drug delivery reviews, 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- 2.Martin TM, et al. , Prediction of Solvent Physical Properties using the Hierarchical Clusering Method, American Chemical; Society Fall Meeting, Boston, MA, 2010. [Google Scholar]
- 3.Arnott JA, et al. , Expert opinion on drug discovery, 2012, 7, 863–875. [DOI] [PubMed] [Google Scholar]
- 4.Isaacs KK, et al. , Toxicology Reports, 2016, 3, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillips KA, et al. , Green Chemistry, 2017, 19, 1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnot JA, et al. , Environmental health perspectives, 2012, 120, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnot JA, et al. , Environmental science & technology, 2006, 40, 2316–2323. [DOI] [PubMed] [Google Scholar]
- 8.MacLeod M, et al. , Environmental science & technology, 2010, 44, 8360–8364. [DOI] [PubMed] [Google Scholar]
- 9.Nichols JW, et al. , Environmental toxicology and chemistry, 2013, 32, 1611–1622. [DOI] [PubMed] [Google Scholar]
- 10.Peyret T, et al. , Toxicology and applied pharmacology, 2010, 249, 197–207. [DOI] [PubMed] [Google Scholar]
- 11.Rosenbaum RK, et al. , The International Journal of Life Cycle Assessment, 2008, 13, 532–546. [Google Scholar]
- 12.Schmitt W, Toxicology in Vitro, 2008, 22, 457–467. [DOI] [PubMed] [Google Scholar]
- 13.MacLeod M, et al. , Environmental science & technology, 2014, 48, 11057–11063. [DOI] [PubMed] [Google Scholar]
- 14.Cronin MT, et al. , Environmental health perspectives, 2003, 111, 1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanderson H, et al. , Toxicology letters, 2003, 144, 383–395. [DOI] [PubMed] [Google Scholar]
- 16.Price DA, et al. , Expert opinion on drug metabolism & toxicology, 2009, 5, 921–931. [DOI] [PubMed] [Google Scholar]
- 17.Mackay D, et al. , SAR and QSAR in Environmental Research, 2009, 20, 393–414. [DOI] [PubMed] [Google Scholar]
- 18.USGAO, Chemical Regulation: Options for Enhancing the Effectiveness of the Toxic Substances Control Act, 2009.
- 19.USGAO, Toxic Substances: EPA Has Increased Efforts to Assess and Control Chemicals but Could Strengthen Its Approach, 2013.
- 20.Schymanski EL, et al. , Analytical and bioanalytical chemistry, 2015, 407, 6237–6255. [DOI] [PubMed] [Google Scholar]
- 21.Rager JE, et al. , Environment International, 2016, 88, 269–280. [DOI] [PubMed] [Google Scholar]
- 22.Park YH, et al. , Toxicology, 2012, 295, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wambaugh JF, et al. , Toxicological Sciences, 2015, 147, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnot JA, et al. , Environ Sci Technol, 2008, 42, 4648–4654. [DOI] [PubMed] [Google Scholar]
- 25.Nabholz JV, Sci Total Environ, 1991, 109-110, 649–665. [DOI] [PubMed] [Google Scholar]
- 26.Walker JD, et al. , SAR QSAR Environ Res, 2002, 13, 713–725. [DOI] [PubMed] [Google Scholar]
- 27.Swanson MB, et al. , Environmental toxicology and chemistry, 1997, 16, 372–383. [Google Scholar]
- 28.Reuschenbach P, et al. , Chemosphere, 2008, 71, 1986–1995. [DOI] [PubMed] [Google Scholar]
- 29.European Chemicals Agency, Guidance on information requirements and chemical safety assessment, European Chemicals Agency, Helsinki, Finland, 2012. [Google Scholar]
- 30.Benfenati E, et al. , Chem Cent J, 2011, 5, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Netzeva TI, et al. , Altern Lab Anim, 2005, 33, 155–173. [DOI] [PubMed] [Google Scholar]
- 32.Cohen Hubal EA, et al. , Journal of Toxicology and Environmental Health, Part B, 2010, 13, 299–313. [DOI] [PubMed] [Google Scholar]
- 33.Kerns EH, Journal of pharmaceutical sciences, 2001, 90, 1838–1858. [DOI] [PubMed] [Google Scholar]
- 34.Camenisch GP, Pharmaceutical research, 2016, 33, 2583–2593. [DOI] [PubMed] [Google Scholar]
- 35.Potts RO, et al. , Pharmaceutical research, 1992, 9, 663–669. [DOI] [PubMed] [Google Scholar]
- 36.Krämer SD, Pharmaceutical science & technology today, 1999, 2, 373–380. [DOI] [PubMed] [Google Scholar]
- 37.Yun Y, et al. , Xenobiotica, 2013, 43, 839–852. [DOI] [PubMed] [Google Scholar]
- 38.Pearce R, et al. , J Pharmacokinet Pharmacodyn, 2017, 44, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu X-W, et al. , Pharmaceutical research, 2013, 30, 1790–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ingle BL, et al. , Journal of chemical information and modeling, 2016, 56, 2243–2252. [DOI] [PubMed] [Google Scholar]
- 41.Armitage JM, et al. , Environmental science & technology, 2014, 48, 9770–9779. [DOI] [PubMed] [Google Scholar]
- 42.Fischer FC, et al. , Chemical Research in Toxicology, 2017, 30, 1197–1208. [DOI] [PubMed] [Google Scholar]
- 43.Tong W, et al. , Environmental health perspectives, 2004, 112, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaworska J, et al. , ATLA-NOTTINGHAM-, 2005, 33, 445. [Google Scholar]
- 45.Tropsha A, et al. , Current pharmaceutical design, 2007, 13, 3494–3504. [DOI] [PubMed] [Google Scholar]
- 46.Golbraikh A, et al. , Journal of chemical information and modeling, 2014, 54, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dearden JC, et al. , SAR QSAR Environ Res, 2009, 20, 241–266. [DOI] [PubMed] [Google Scholar]
- 48.Mansouri K, Estimating degradation and fate of organic pollutants by QSAR modeling, Saarbrucken, Germany: LAP LAMBERT Academic Publishing, 2013. [Google Scholar]
- 49.Todeschini R, and Consonni V, Handbook of Molecular Descriptors, Wiley- VCH Verlag GmbH, Weinheim, Germany, 2008. [Google Scholar]
- 50.Sahigara F, et al. , Molecules, 2012, 17, 4791–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Box GE, Robustness in statistics, 1979, 1, 201–236. [Google Scholar]
- 52.Walker JD, et al. , Molecular Informatics, 2003, 22, 346–350. [Google Scholar]
- 53.USEPA, United States Environmental Protection Agency, Washington, DC, USA., Editon edn, 2017. [Google Scholar]
- 54.Wambaugh JF, et al. , Environmental Science and Technology, 2013, 47, 8479–8488. [DOI] [PubMed] [Google Scholar]
- 55.Mansouri K, et al. , Journal of Cheminformatics, 2018, 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mansouri K, et al. , SAR QSAR Environ Res, 2016, 27, 939–965. [DOI] [PubMed] [Google Scholar]
- 57.OCHEM, Online chemical database with modeling environment, https://ochem.eu/home/show.do.
- 58.Simulations Plus, ADMET Predictor, www.simulations-plus.com.
- 59.Advanced Chemistry Development Inc. (ACD/Labs), www.acdlabs.com, Accessed August 11, 2017.
- 60.ChemAxon Ltd., Chemicalize, https://chemicalize.com/welcome.
- 61.USEPA, Chemistry Dashboard, https://comptox.epa.gov/dashboard/, Accessed August 10, 2017.
- 62.UFZ Department of Ecological Chemistry, ChemProp 6.5, http://www.ufz.de/ecochem/chemprop.
- 63.Zang Q, et al. , Journal of chemical information and modeling, 2017, 57, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richard AM, et al. , Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 2002, 499, 27–52.11804603 [Google Scholar]
- 65.Berthold MR, et al. , KNIME: The Konstanz Information Miner, Springer, 2007. [Google Scholar]
- 66.SRC Inc., PHYSPROP database., https://www.srcinc.com/what-we-do/environmental/scientific-databases.html, Accessed August 11, 2017.
- 67.OECD, Test No. 117: Partition Coefficient (n-octanol/water), HPLC Method, OECD Publishing, 2004.
- 68.Donovan SF, Journal of Chromatography A, 1996, 749, 123–129. [Google Scholar]
- 69.Martínez CHR, et al. , ACS medicinal chemistry letters, 2013, 4, 142–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanimoto TT, Elementary mathematical theory of classification and prediction, International Business Machines Corporation, New York, 1958. [Google Scholar]
- 71.Steinbeck C, et al. , Journal of chemical information and computer sciences, 2003, 43, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.USEPA, OPPTS 830.7570 Partition Coefficient (n-Octanol/Water), Estimation By Liquid Chromatography, 1996.
- 73.ASTM International, Annual Book of ASTM Standards, https://www.astm.org/.
- 74.ASTM International, Standard test method for measurements of aqueous solubility, ASTM International, West Conshohocken, PA, 2008. [Google Scholar]
- 75.USEPA, OPPTS 830.7370 Dissociation Constants in Water, 1996.
- 76.Pearson K, Proceedings of the Royal Society of London, 1895, 58, 240–242. [Google Scholar]
- 77.Wickham H, ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag, New York, 2009. [Google Scholar]
- 78.Williams AJ, et al. , J Cheminform, 2017, 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huuskonen J, Comb Chem High Throughput Screen, 2001, 4, 311–316. [DOI] [PubMed] [Google Scholar]
- 80.Phillips KA, et al. , Environ Sci Technol, 2018, 52, 3125–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Isaacs KK, et al. , J Expo Sci Environ Epidemiol, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Biryol D, et al. , Environ Int, 2017, 108, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lipinski CA, Drug Discovery Today: Technologies, 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- 84.Mansouri K, et al. , Environ Health Perspect, 2016, 124, 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bharate SS, et al. , Comb Chem High Throughput Screen, 2016, 19, 461–469. [DOI] [PubMed] [Google Scholar]
- 86.Settimo L, et al. , Pharm Res, 2014, 31, 1082–1095. [DOI] [PubMed] [Google Scholar]
- 87.Isaacs KK, et al. , Environmental science & technology, 2014, 48, 12750–12759. [DOI] [PubMed] [Google Scholar]
- 88.Goldsmith MR, et al. , Food Chem Toxicol, 2014, 65, 269–279. [DOI] [PubMed] [Google Scholar]
- 89.Dionisio KL, et al. , Toxicology Reports, 2015, 2, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Phillips KA, et al. , Editon edn, 2017. [Google Scholar]
- 91.McKinney W, Data structures for statistical computin in python, Proceedings of the 9th Python in Science Conference, 2010. [Google Scholar]
- 92.Hunter JD, Computing in Science & Engineering, 2007, 9, 90–95. [Google Scholar]
- 93.R Core Team R: A Language and Environment for Statistical Computing, Vienna, Austria, 2015. [Google Scholar]
- 94.Team RStudio, RStudio: Integrated Development for R, RStudio, Inc, Boston, MA, 2016 [Google Scholar]
- 95.Wickham H, ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag, New York, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.