

Abstract
Aquaculture production of the Pacific white shrimp is the largest in the world for crustacean species. Crucial to the sustainable global production of this important seafood species is a fundamental understanding of the shrimp gut microbiota and its relationship to the microbial ecology of shrimp pond. This is especially true, given the recently recognized role of beneficial microbes in promoting shrimp nutrient intake and in conferring resistance against pathogens. Unfortunately, aquaculture-related microbiome studies are scarce in Southeast Asia countries despite the severe impact of early mortality syndrome outbreaks on shrimp production in the region. In this study, we employed the 16S rRNA amplicon (V3–V4 region) sequencing and amplicon sequence variants (ASV) method to investigate the microbial diversity of shrimp guts and pond water samples collected from aquaculture farms located in Malaysia and Vietnam. Substantial differences in the pond microbiota were observed between countries with the presence and absence of several taxa extending to the family level. Microbial diversity of the shrimp gut was found to be generally lower than that of the pond environments with a few ubiquitous genera representing a majority of the shrimp gut microbial diversity such as Vibrio and Photobacterium, indicating host-specific selection of microbial species. Given the high sequence conservation of the 16S rRNA gene, we assessed its veracity at distinguishing Vibrio species based on nucleotide alignment against type strain reference sequences and demonstrated the utility of ASV approach in uncovering a wider diversity of Vibrio species compared to the conventional OTU clustering approach.
Keywords: Metagenomics, Aquaculture, Litopenaeus vannamei, 16S ribosomal RNA amplicons sequencing, Vibrio parahaemolyticus
Introduction
Litopenaeus vannamei (Boone, 1931), also known as the Pacific white shrimp or Whiteleg shrimp, is a major aquaculture commodity with a production of 3.69 million tonnes valued at 18 billion USD revenue ( FAO, 2016). In recent years, significant outbreaks of acute hepatopancreatic necrosis disease (AHPND), also known as early mortality syndrome (EMS) have been reported in a number of white shrimp-producing countries. EMS was first reported in China in 2009 and subsequently spread to Southeast Asian countries including Vietnam, Malaysia, and Thailand (Foo et al., 2017; Kondo et al., 2014; Tran et al., 2013). The causative agent of EMS has been reported to be Vibrio parahaemolyticus strains harbouring a plasmid containing the pirA- and pirB- like genes encoding for toxins capable of severely damaging the shrimp gut (Han et al., 2015; Lee et al., 2015). To date, EMS has caused an estimated one billion USD of losses to the shrimp industry worldwide (De Schryver, Defoirdt & Sorgeloos, 2014; Lee et al., 2015).
Monitoring and control of pond water quality play a crucial role in managing and preventing disease outbreak in aquaculture. However, the current practice of water quality monitoring usually focuses on the measurement of chemical and physical parameters such as oxygen, pH, temperature, salinity, turbidity and nitrogen compounds. The importance of microbial communities in influencing or responding to variation in aquaculture pond water quality has only been recognized in recent years (Bentzon-Tilia, Sonnenschein & Gram, 2016). This is especially relevant to managing the water quality of aquaculture ponds and their cultured biomass because microbes carry out important biological services in aquaculture environment including nutrient cycling, probiotic/pathogenic activity and nutrient acquisition in addition to potentially acting as a rapid biological indicator of critical chemical changes in the rearing water (Cardona et al., 2016; Cornejo-Granados et al., 2017; Costa, Pérez & Kreft, 2006; Emerenciano, Gaxiola & Cuzon, 2013; Grotkjær et al., 2016; Jinbo et al., 2017; Liu et al., 2015; Wright, Konwar & Hallam, 2012; Zeng et al., 2017; Zhu et al., 2016). Microbes can also be used to improve the water quality of ponds. For example, adding denitrifying bacteria to biofilters has been shown to reduce the concentration of ammonia and its immediate derivatives, which are detrimental to shrimp health (Saffran et al., 2001).
Recognizing the importance of microbial biomass and diversity on the health and production of cultured invertebrates, several microbiome studies have analysed the gut microbiome of wild-caught shrimps (Cornejo-Granados et al., 2017; Phayungsak et al., 2018; Rungrassamee et al., 2016) as well as cultured shrimps under different abiotic and biotic factors. However, most studies have been restricted to a specific country especially China, focusing only on one or very few localized ponds (Cornejo-Granados et al., 2017; Huang et al., 2016; Rungrassamee et al., 2016; Tang et al., 2014; Xiong et al., 2015; Zhang et al., 2014; Zhu et al., 2016). A study in Thailand used denaturing gradient gel electrophoresis profiling and barcoded pyrosequencing to demonstrate a greater survivability of Litopenaeus vannamei and improved the resilience of its gut microbiome upon Vibrio harveyi exposure relative to that of Penaeus monodon (Rungrassamee et al., 2016). Another more recent study in Vietnam utilised a standard Illumina 16S rRNA gene amplicon sequencing method to investigate the effect of EMS outbreak on the microbial interaction networks in shrimp guts (Chen et al., 2017b). Thus, despite the emergence of Southeast Asia (SEA) as an aquaculture hub, studies on any significant geographic scale are relatively scarce in this region. Further, to our knowledge, all recent shrimp aquaculture microbiome studies still employ the operational taxonomic units (OTU) clustering approach in marker-gene data analysis despite recent calls for the replacement of this approach with exact sequence variants which can resolve single-base differences among biological sequences in the sample thus providing a more comprehensive and accurate view of microbial communities (Callahan, McMurdie & Holmes, 2017; Eren et al., 2014; Utter, Mark Welch & Borisy, 2016).
To initiate a more broad-based investigation of shrimp microbiomes directly relevant to the aquaculture industry in SEA, we performed Illumina 16S rRNA gene amplicon sequencing of L. vannamei guts and rearing water from aquaculture farms located in two SEA countries with contrasting climates at the time of sampling i.e., Malaysia (warm and humid, 30 °C) and Vietnam (cool and dry, 20 °C). For the first time in a shrimp aquaculture microbiome study, we applied the recently advocated ASV-method (Edgar, 2016) to: (1) compare shrimp intestinal microbial diversity and their pond environments; (2) compare these microbial communities between Malaysia and Vietnam; and (3) assess the performance of the 16S rRNA V3–V4 hypervariable region and clustering approaches in capturing the genetic diversity of Vibrio.
Methods
Sample collection
Sampling in Vietnam was performed at two separate shrimp farms in the Quang Ninh province (approximately 20 km apart), while sampling in Malaysia was performed at a large shrimp farm with multiple pond systems located in Perak state (Table 1). Two mL of pond water was sampled from 2–4 distant location (corners) of each pond, pelleted via centrifugation at 7,000 rpm for 10 min and resuspended in RNA/DNA shield (ZymoResearch, Irvine, CA, USA). Shrimp intestinal samples were collected by dissecting out the shrimp guts followed by homogenization in RNA/DNA shield (ZymoResearch, Irvine, CA, USA). Sampling was performed with the permission and under the supervision of the aquaculture manager from the respective farms. No field permit was required for this study because samples were collected from private fields. As per the request of the aquaculture manager, exact sampling location of some farms was not disclosed in this study to protect the identity of the farm.
Table 1. Summary of field collection and sampling design.
| Farm | # Pond (Replicate per pond) | # Shrimp Sampled | Location | Collection date | Mean temperature (°C) | Reported age (days post hatching) |
|---|---|---|---|---|---|---|
| Aquaculture Research Station of Fisheries College | 4 (2–3) | 6 | Quang Yen, Quang Ninh, Vietnam | December 2015 | 20 | 97 |
| Private | 1 (4) | 4 | Quang Yen, Quang Ninh, Vietnam | December 2015 | 20 | 115 |
| Private | 9 (2–3) | 14 | Sitiawan, Perak, Malaysia | March 2016 | 30 | 65 |
DNA extraction, amplification, purification and sequencing
Genomic DNA was extracted from the RNA/DNA shield lysate using DNA Clean & Concentrator™-5 (ZymoResearch, Irvine, CA, USA) according to the manufacturer’s instructions. The V3–V4 region of the 16S rRNA gene was amplified using forward primer 5′–TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG –3′ and reverse primer 5′–GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC–3′ containing partial Illumina Nextera adapter. PCR reaction (∼10 ng input DNA/ reaction) and barcode incorporation were performed as previously described (Watts et al., 2017). Constructed libraries were quantified, normalized, pooled, denatured and subsequently sequenced on the Illumina MiSeq (Illumina, San Diego, CA, USA) located at Monash University Malaysia Genomics Facility using a 2 × 250 bp run configuration.
Sequence data analysis and observation table construction
Primer sequences corresponding to the 16S rRNA gene were removed from the raw paired-end reads using Cutadapt (Martin, 2011). Trimmed forward and reverse reads were overlapped with fastq_mergepairs followed by quality and length filtering with fastq_filter (maximum expected error = 0.5; min length = 250 bp) as implemented in USearch10 (Edgar, 2010). Sequence dereplication and denoising was done using uNoise3 to generate amplicon sequence variants (ASVs) (Edgar, 2016). Aside from “-minuniquesize 2″ parameter during sequence dereplication, the processes of the pipeline were done using default parameters. Taxonomic assignment and observation table construction rarefied at 8,000 reads were performed in RDP classifier 2.12 and QIIME 1.9.1 (Caporaso et al., 2010; Cole et al., 2009). ASVs that failed RDP taxonomic assignment were re-classified using SINA 1.3.1 against SILVA SSU Ref database (release 132) with default parameters (Data S3 and Data S4) (Pruesse, Peplies & Glöckner, 2012; Pruesse et al., 2007). ASVs with lower than 0.01% fraction of the total normalized observation and/or identified as chloroplast were not included in subsequent analyses. Normalization of sequencing depth per sample (8,000 reads/sample), rarefaction curves construction (10 replicates/depth) as well as alpha diversity estimation (Simpson’s evenness and Shannon diversity indices), were performed using the “core_diversity.py” python script in QIIME 1.9.1. Core genera of the shrimp gut microbiome were investigated and defined as genera with at least 0.1% relative abundance per sample in more than 50% shrimp samples. The prevalence and relative abundance of the shrimp gut core genera were visualised using the R ggplot2 package (Wickham & Wickham, 2007).
Principal coordinates analysis and relative differential abundance analysis
The proportion of sequences assigned to the lowest possible taxonomic level was calculated based on the rarefied observation table and the implemented taxonomic assignment method mentioned above. Principal coordinates analysis (PCoA) was also constructed based on weighted Unifrac and unweighted Unifrac using ordination method in R phyloseq package (Lozupone & Knight, 2005; McMurdie & Holmes, 2013). The resulting PCoA plots were then visualized using R ggplot2 package (Wickham & Wickham, 2007). Strength and significance of grouping were calculated using the compare_categories.py python script in QIIME which implements ANOSIM analysis using the default 999 permutations. Relative differential abundance test was also conducted at phylum and family levels using Tukey-Kramer post-hoc in conjunction with analysis of variance (ANOVA) statistical test in STAMP (Parks et al., 2014). Multiple hypothesis testing was done for the four generic groups namely Malaysian Farm, Malaysian Shrimp, Vietnamese Farm and Vietnamese Shrimp. A significant differential abundance of phylum distribution was defined as Benjamini–Hochberg-corrected probability p-value of ≤ 0.01 and was observed only for the top 10 most abundant phyla. A significant differential abundance of family distribution was defined as Benjamini–Hochberg corrected p-value of ≤ 0.01 and eta squared ≥ 0.3.
Comparison of Vibrio diversity using different clustering methods
Vibrio diversity was compared using different methods of 16S marker-gene data analysis, namely conventional operational taxonomic unit (OTU) clustering and amplicon sequence variants (ASV), using UParse and uNoise3 respectively (Edgar, 2013; Edgar, 2016). Except for -minuniquesize of 2 during sequence dereplication, both pipelines were conducted using default parameters. ASVs and OTUs assigned to the genus Vibrio with at least cumulative read abundance of more than 200 were retained for blastN similarity search (E-value < 1e−100) against 16S rRNA sequences of Vibrio type strain curated in EzBioCloud (as of 18th May 2018) (Yoon et al., 2017).
Results
Shrimp intestinal and pond microbial communities are distinct
A total of 2,731,818 successfully merged reads were generated in this study with 2,144,192 reads (median of 32,648 reads/sample; min = 9,948; max = 66,590) confidently mapped to the ASVs. 92.12% and 7.77% of the mapped reads correspond to RDP-classified and SINA-classified ASVs respectively, while the remaining mapped reads belong to ASVs without confident taxonomic assignment at the kingdom rank. A majority of reads recovered from shrimp intestine and ponds were assigned to members from the phyla Proteobacteria, Actinobacteria, Bacteroidetes and Fusobacteria (Fig. 1). Significant differences in the relative abundance of bacteria phyla were observed among samples isolated from ponds and shrimp guts. Reads mapping to Actinobacteria and Bacteroidetes are more abundant in ponds (p < 0.01 and p < 0.001, respectively), while shrimp guts have a significantly higher relative abundance of Proteobacteria (p < 0.001). At a finer level, Malaysian shrimp intestinal microbiome contains more reads mapping to the phylum Fusobacteria (p < 0.01). Notable, this phylum is also near absent in three out of four Malaysian shrimps noted to be unhealthy based on morphological observation by the aquaculture manager (Fig. 1).
Figure 1. Distribution of abundant phyla in all samples.

Significantly abundant phyla are annotated at the legends. Phyla abundance of pond water replicates were collapsed according to the location of sampling (see Table S1). Sample type (MF, Malaysian rearing water; VF, Vietnamese rearing water; MS, Malaysian shrimp; VS, Vietnamese shrimp) with significantly higher phylum abundance than that of another group (Superscript) were shown in bracket next to their associated phylum legend. For example, Actinobacteria (MF*; VFV S, MS) indicates that this phylum is significantly more abundant in Malaysian rearing water samples compared to all three other groups and that it is more abundant in Vietnamese rearing water samples compared to shrimp samples from both countries. Hash and asterisk signs next to y-axis labels indicate mussel-infested and suspected diseased samples, respectively.
Rarefaction curves based on alpha diversity metrics, number of observed ASVs and PD (Phylogenetic diversity) whole tree, indicated that 8,000 sequences per sample are sufficient for capturing the alpha diversity of microbial communities in both shrimp guts and ponds. Inverse Simpson’s and Shannon’s indices showed higher species richness and evenness in the pond microbiome compared to that of shrimp gut (Figs. 2A and 2B). Beta diversity analyses based on both weighted and unweighted UniFrac indicated that shrimp gut and pond microbial communities from the same sampling site are significantly different (Vietnam: R > 0.67, p-value < 0.001; Malaysia: R > 0.89, p-value < 0.001) (Table S2). An even stronger separation was also observed among pond samples from different sampling sites/ countries (Fig. 3, Table S2). Furthermore, samples from one of the Malaysian ponds (MF_Pond11) noted by the shrimp farmer to be infested by mussels (Table S1) was distinct from other Malaysian pond sample samples (Fig. 3). It is worth noting that rearing water samples collected from different parts of the same pond have minimal spatial variation in microbial composition as evidenced by the general tight clustering of pond replicates, suggesting homogenous microbial community in the rearing water and indicating that the sampling protocols are efficient for capturing pond diversity. Although the separation between Malaysian and Vietnamese shrimp gut samples was less obvious in both PCoA plots with occasional overlap, their microbial community structure appears to differ moderately (R < 0.67) with good statistical support (p-value < 0.001) based on ANOSIM analysis (Table S2).
Figure 2. Rarefaction curves and alpha diversity plots of each sample group.

(A) Rarefaction curve constructed based on observed ASVs. (B) Rarefaction curve constructed based on phylogenetic distance (PD_whole_tree). (C) Shannon’s evenness index box plot. (D) Shannon index box plot.
Figure 3. Principal component analysis of (A) weighted Unifrac and (B) unweighted Unifrac distances.

Red circular outline shows ‘Malaysian pond’ cluster, yellow circular outline shows ‘Vietnamese pond’ cluster, blue circular outline marks ‘Malaysian shrimp’ cluster, and green circular outline represents ‘Vietnamese shrimp sample. The sample points were coloured according to “Country_Source_Location” information (Table S1). Country and source information of each points were abbreviated; for example, Malaysian farm sample from Pond 1 was labelled as MF_Pond1.
Comparison of relative abundance at the microbial family level reveals fine-level microbiota dynamics
Given that more than 70% and 85% of the reads derived from pond and shrimp intestinal samples, respectively, could be assigned to the family level (Data S3 and S4), we defined the core and unique microbiomes among sample groups at this taxonomic level and used STAMP to identify statistically significant differences in the relative abundance of microbial families. A total of six microbial families (Alcaligenaceae, Flavobacteriaceae, Microbacteriaceae, Acidimicrobiaceae and Rhodobacteraceae) are shared across shrimp and rearing water samples (Fig. 4A and Data S5) with seven and 10 microbial families uniquely present in Malaysian and Vietnamese rearing water, respectively, corroborating with their higher alpha diversity (Figs. 2C and 2D). In addition, four microbial families are uniquely shared by the Malaysian and Vietnamese rearing water samples, suggesting their common affiliation with shrimp rearing water. Certain microbial families are significantly more abundant in samples of the same isolation source, regardless of the country of origin. For example, Microbacteriaceae and Flavobacteriaceae are more abundant in pond water, while Vibrionaceae is highly enriched in the shrimp gut (Fig. 4B). On the other hand, Rhodobacteraceae is more abundant in Vietnamese samples (shrimp gut and rearing water) while Cyanobacteria-Family II is more abundant in Malaysian samples, suggesting possible affiliation to regional climate and/or pond environment. In addition, four microbial families (Acidomicrobiaceae, Actinomarinaceae, Comamonadaceae, and Fusobacteriaceae) showed significantly higher abundance only in a specific country and isolation source (Fig. 4B).
Figure 4. Microbial community dynamics at the family level.
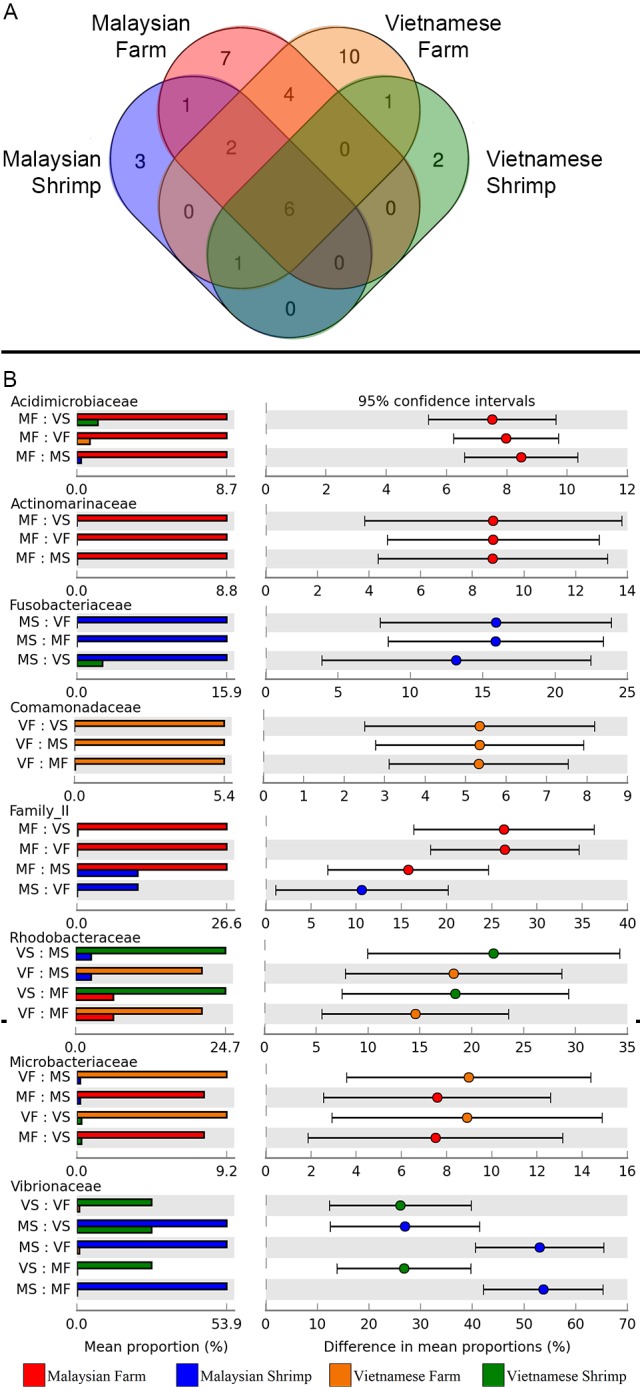
(A) Venn diagram illustrating the number of unique and overlapping microbial families among shrimp guts and rearing water. To be considered as present, a microbial family must be detected in at least 90% of the samples from the same group. (B) Tukey post-hoc pairwise comparison in conjunction with analysis of variance (ANOVA) between “Country_Source” groups of nine significant microbial families. Mean proportion values of families that are significantly different are shown in the bar plots on the left section, while the differences in the pairwise comparison mean proportion with 95% confidence interval are shown on the right section.
High prevalence and relative abundance of ASVs belonging to the genera Vibrio in shrimp gut
16S rRNA reads corresponding to 11 bacterial genera belonging to five phyla (Actinobacteria, Chloroflexi, Cyanobacteria, Planctomycetes and Proteobacteria) were detected in more than 50% of the shrimp gut samples (Fig. 5) with Vibrio being the only genus present in all shrimp gut samples with relatively similar relative abundance across samples(mean/median relative abundance per sample = 33.7%/ 27.7%). On the contrary, Photobacterium, a genus related to Vibrio at the family level (Vibrionaceae) was detected in all but two Vietnamese shrimps (mean/median relative abundance per sample = 11.6%/ 6.5%). The prevalence of the core genera was independently correlated with relative abundance e.g., the higher the prevalence of a core genus, the more likely it is to have a higher relative abundance. Rhodopirellua and Gimesia all belonging to the phylum Planctomycetes are more prevalent and abundant in Vietnamese shrimps than in the Malaysian shrimps, consistent with statistical analysis showing a significantly higher abundance of Planctomycetes in Vietnamese shrimps compared to Malaysian shrimps (Fig. 1).
Figure 5. Abundance and prevalence of core genera in shrimp guts.

(A) Normalized read counts of the selected genera among Malaysian and Vietnamese shrimp gut samples (B) Percentage of shrimp gut samples harbouring the core genera
Substantial underestimation of Vibrio diversity using the OTU clustering approach
The high cumulative abundance of reads assigned to the genus Vibrio in shrimp guts indicates that some members of this genus are endogenous to the shrimp gut microbiota. UPARSE using the default 97% sequence similarity cut-off setting identified two abundant OTUs assigned to Vibrio. This contrasts greatly with the ASV approach which identified substantially more biological sequences classified as Vibrio (Fig. 6, Data S3 and S4). A majority of the constructed ASVs do not have an exact sequence match to the constructed OTUs and more importantly, some could be assigned to a single Vibrio species (ASV22, Vibrio jasicida TCFB 0772T; ASV19, Vibrio neocaledonicus NC470T). OTU2 and OTU10 are identical in both sequence length and identity to ASV2 and ASV14, respectively. Similarity search of OTU2/ASV2 revealed an exact sequence match to V. rotiferianus LMG21460T and V. campbellii CAIM519T, indicating a limitation to the use of V3–V4 hypervariable region in delimiting some Vibrio species (Fig. 6). The high ratios of ASVs-to-OTUs observed for OTU2 and OTU10 strongly suggests that imposing a fixed dissimilarity threshold using conventional OTU clustering underestimates the true microbial diversity for Vibrio species in this study and that resolving amplicon sequence variants (ASV) from amplicons data which is sensitive down to single-nucleotide differences, through read de-replication and error correction, substantially increased the number of observed Vibrio species from the identical dataset. Unlike the ASVs associated with OTU2, nearly all ASVs associated with OTU10 have at least two mismatches to known Vibrio species, indicating the presence of unculturable or yet-to-be-cultured Vibrio strains in the shrimp gut. Of even more interest, the ASV approach revealed the presence of V. parahaemolyticus (ASV235 in Fig. 6) that was missed by the OTU clustering approach presumably due to its overall low relative abundance across samples (Table S5).
Figure 6. Cumulative normalized read counts of OTUs/ASVs classified as Vibrio and similarity table against selected type-strain Vibrio sequences.

Empty and filled bars indicate shrimp gut and rearing water samples, respectively. Different Vibrio groups consisting of one OTU and its associated ASVs were separated by grey horizontal lines.
Discussion
Despite the immense scale of shrimp aquaculture in South East Asia and the major impacts of aquaculture disease outbreaks in tropical regions, we are only starting to understand the microbial composition of the shrimp guts and their relationship to rearing water in the region (Leung & Bates, 2013). Most shrimp-related microbiome studies have been limited to a few farms in a particular country; most of which have been conducted in countries outside of SEA such as China and Mexico. Litopenaeus vannamei microbiome studies have so far investigated the microbial composition of wild-type shrimps serving as an important baseline for future comparative studies (Cornejo-Granados et al., 2017) as well as the impacts of disease exposure (Chen et al., 2017b; Cornejo-Granados et al., 2017; Jinbo et al., 2017; Rungrassamee et al., 2016; Xiong et al., 2015; Zhu et al., 2016), developmental stages (Huang et al., 2016), nutrition (Zhang et al., 2014) and temperature (Tang et al., 2014) on shrimp intestinal microbiome. We have contributed new findings to the growing literature by providing the first data on gut and pond water microbiome of Malaysian cultured shrimps. Furthermore, we compared bacterial communities of shrimp guts and pond water from multiple aquaculture farms in two distinct climatic regions (Malaysia and north Vietnam) while standardizing DNA extraction and sequencing protocols. This provides a new perspective on our current understanding on the range of normal microbial composition of a healthy shrimp gut microbiome community. Our principal findings provide evidence supporting microbiota plasticity in shrimp ponds, but in contradistinction, find a much more limited diversity of the adult shrimp intestinal microbiota.
Comparison of the pond water microbiome between the Malaysian and Vietnamese samples reveals significant dissimilarities at the phylum level as shown in Fig. 1. Although Planctomycetes was identified as a significant phylum in this study, it was not commonly found in other metagenomic studies of similar environments (Li et al., 2016; Xiong et al., 2015; Zeng et al., 2017) (Table S4). The only sample site with near-zero relative abundance of Plantomycetes was a mussel-infested Malaysian aquaculture pond (Pond11 in Fig. 1). Mussels are known ecosystem engineers that can substantially modify their habitats through biological processes such as sediment filtration and biodeposition of fecal matters (Bril et al., 2014). The presence of mussels has been reported to cause to a 4-fold decrease in the relative abundance of Plantomycetes in a freshwater system in Mississippi, USA (Black, Chimenti & Just, 2017).
Although Flavobacteriaceae and Microbacteriaceae are prevalent in the rearing water and shrimp gut samples, they exhibited significantly higher relative abundance in the rearing water samples. Albeit initially associated with a fairly general ecological function e.g., simple mineralization-based commensalism (Kirchman, 2002), emerging evidence suggests that some members within the marine Flavobactericeae clade are algal-associated species that exhibit growth promoting and inhibiting effects to its host and other algal species, respectively (Bowman, 2006). Genera such as Cellulophaga, Psychroserpens and Formoasa have been previously reported to produce toxic secondary metabolites against dinoflagellates, commonly associated with algal bloom (Adachi et al., 2002; Egan et al., 2000). Thus, the significant abundance of Flavobacteriacea in both rearing water could be linked to the natural occurrence of algal and diatom species in the rearing water some of which were co-amplified by the V3–V4 primers in this study (Data S3 and Table S3). On the contrary, most described members from the family Microbacteriaceae were not associated with marine environmental and were typically isolated from the terrestrial environment (Evtushenko & Takeuchi, 2017). High abundance of Microbacteriaceae (unclassified at the genus level) in shrimp rearing water particularly during the post-larvae stage has been previously observed in a commercial marine shrimp hatchery in Hainan, China (Zeng et al., 2017) which was suggested to be a temporal-specific bacterial family caused by changes in the shrimp diet during different growth stages. On the contrary, four microbial families namely, Cryomorphaceae, Rhodospirillaceae, Bacteriovoracaceae and Saprospiraceae, are exclusively found across both Malaysian and Vietnamese shrimp rearing water samples, indicating their specific adaptation to shrimp rearing water or more generally the marine aquatic environment. For example, members of the family Rhodospirillaceae are purple non-sulfur and mostly nitrogen-fixing photosynthetic bacteria. Their absence in the shrimp gut is consistent with their strict requirement for light to grow and proliferate which is not sufficiently present in the shrimp gut environment.
Despite the conspicuous difference in the shrimp pond microbiota between two countries, the shrimp intestinal microbiota are more similar to each other which is presumably due to host selection for microbial strains that adapt to or exploit the shrimp gut environment as corroborated by their lower alpha diversity indices compared to that of rearing water samples (Xiong et al., 2017). However, the presence of several microbial families in both shrimp and rearing water samples indicates that the shrimp gut microbiota maybe significantly affected by the microbial communities present in their aquatic environment e.g., rearing water and pond sediments (Chen et al., 2017a; Cornejo-Granados et al., 2017) as opposed to being maternally influenced as observed in some mammals and other animals with forms of parental care (Jakobsson et al., 2014; Kohl & Dearing, 2012; Zhang et al., 2014). Moulting e.g., shedding of shrimp ectodermal gut tissue also provides a new opportunity for shrimp stomach and guts to be colonised by the bacterial community of the pond (Moss, LeaMaster & Sweeney, 2000). Furthermore, crustaceans, including L. vannamei, also consume their exuvia, which provides another opportunity for microbial recolonization of the shrimp guts and also the transmission of intestinal microbiome across shrimps throughout their developmental stages (Martínez-Córdova & Peña Messina, 2005).
Although the micro-clustering of shrimp gut samples based on country and/or farm of origin may be associated with the difference in their respective growth environment, variation in developmental stage may also contribute to the observed clustering as the shrimps in this study were collected at different adult growth stages (Table 1). The abundance of members from the family Fusobacteriaceae is highly dependent on the shrimp developmental stage with a near-zero abundance in young shrimp larvae gut and subsequently making up a substantial portion of the microbiome in the adult stage (75-day post-hatching) (Chen et al., 2017b; Zeng et al., 2017). Intriguingly, although most of the Vietnamaese shrimps were 97-day post-hatching during sampling, the low abundance and prevalence of Fusobactericeae in the their guts indicates microbiome resemblance to that of younger shrimps (Zeng et al., 2017). In contrast, Fusobacteriaceae is prevalent in Malaysian shrimps that are relatively young e.g., 65-day post- hatching. However, it is worth noting that Sitiawan, the closest city to where the Malaysian farm is located, is warm (30 oC) throughout the year. Such a climate may support faster shrimp growth thus enabling them to reach adulthood earlier (Kumlu, Türkmen & Kumlu, 2010; Wyban, Walsh & Godin, 1995). Members from the family Fusobacteriaceae are microaerotolerant to obligate anaerobic Gram-negative rods bacteria that derive energy through the fermentation of a variety of carbohydrates, amino acids and peptides. Such a metabolic profile is consistent with the higher prevalence and abundance of Fusobacteriaceae in mature shrimp intestinal systems that typically exhibit a better digestive ability (Parte et al., 2011; Schock et al., 2013). Unfortunately, despite exhibiting a wide ecological diversity as evidenced by their diverse isolation source, the symbiotic relationship of Fusobacteriaceae towards its host has yet been properly demonstrated (Nelson, Rogers & Brown, 2013). Future work consisting of metatranscriptome and metagenome sequencing of the shrimp gut microbiota will be necessary to shed light on the role of Fusobactericeae in the shrimp gut.
Vibrio and Photobacterium belonging to the Vibrionaceae family are both abundant and prevalent in nearly all of the shrimp samples, an observation that is consistent with previous reports (Cornejo-Granados et al., 2017; Rungrassamee et al., 2016; Xiong et al., 2017). In contrast, Zeng et al. (2017) did not identify any Vibrio-specific OTUs in their sampling. Such anomalies are unlikely to be biological but rather due to technical and analytical differences, such as the choice of the 16S rRNA gene region sequenced (V4- vs. V3–V4-hypervariable region) and bioinformatic analysis settings. High abundance and prevalence of Vibrio and Photobacterium genera in shrimp guts suggest that they are more likely to be endogenous rather than pathogenic strains (Kriem et al., 2015). However, this also reflects the persistence and adaptation of members from these genera to the shrimp gut environments and may potentially explain the susceptibility of shrimps to non-native pathogenic Vibrio and Photobacterium strains (Kondo et al., 2014; Wang & Chen, 2006).
The use of ASVs reveals a wider diversity of Vibrio species, suggesting that previous shrimp microbiome analyses that employed the common 97% similarity cut-off for clustering will risk masking the true Vibrio diversity in the shrimp gut (Callahan, McMurdie & Holmes, 2017; Chen et al., 2017b). Fortuitously, despite the observed low resolution of the V3–V4 hypervariable region for Vibrio species, this region appears to be distinct in V. parahaemolyticus, which exhibits at least two diagnostic nucleotides that are absent from all known type strains of Vibrio species (Fig. 6). The lack of an OTU with exact match to ASV235 suggests that analysis using the OTU clustering approach will fail to report the presence of V. parahaemolyticus and/or undescribed Vibrio strains sharing the same 16S rRNA gene sequence with V. parahaemolyticus if they are present at low abundance in the dataset. This can have critical implications for aquaculture microbial management especially in the early detection of V. parahaemolyticus infection. Since shrimp gut can harbour both pathogenic and native Vibrio species, complementing 16S rRNA-based amplicon sequencing with an alternative genetic marker such as pyrH may enable a more accurate quantification of Vibrio diversity and abundance in aquaculture environment (Tall et al., 2013; Thompson et al., 2005). Given the high diversity of shrimp gut-associated Vibrio as revealed for the first time by the ASV approach, shallow shotgun metagenome sequencing will also be instructive to obtain species/strain-level taxonomic resolution of the abundant endogenous microbes in shrimp guts particularly those belonging to the genera Vibrio and Photobacterium.
Shrimp gut microbiomes vary due to biological differences (shrimp strains), differences in environmental or farming practice (temperature, diet, probiotic, wild capture) or even biases from different laboratory procedures such as the sequencing platform and the different partial 16S sequence target (Cornejo-Granados et al., 2017; Tremblay et al., 2015). Considering the crucial functions undertaken by microbial communities and the potential use of the pond microbiome for pond health surveillance, investment in measuring a wide variety of chemical and physical parameters would allow us to better correlate the relationship between microbiomes and rearing water quality and therefore improving our understanding of aquaculture microbiomes.
Conclusions
Using a standardized Illumina 16S rRNA amplicons sequencing protocol, we report for the first time, amplicon sequence variants (ASV)-based analysis of aquaculture rearing water and shrimp gut microbiota from two South East Asia countries with different climates. Despite substantial difference in the microbial composition of shrimp rearing water between farms in Malaysia and Vietnam, adult shrimp guts are more similar and exhibit a genus level core microbiome with the genus Vibrio being the most prevalent and abundant group. In addition, compared to OTU clustering approach, the ASV method improved the identification of closely related and/or rare Vibrio species, which is of relevance to the shrimp aquaculture industry. The high abundance of Vibrio in shrimp gut also suggests that some Vibrio species are endogenous and non-virulent to shrimps with functional and ecological roles that remain to be elucidated in the future.
Supplemental Information
ASVs assigned by SINA are annotated with an asterisk, while the remaining were annotated with the default RDP classifier.
OTUs assigned by SINA are annotated with an asterisk, while the remaining were annotated with the default RDP classifier.
Acknowledgments
We thank the Monash University Malaysia Genomics Facility for the provision of computational resources. We are also extremely grateful to the aquaculture managers for providing access to their farms and sharing information.
Funding Statement
This work was supported by the Tropical and Medicine Biology Platform, Monash University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests.
Author Contributions
Muhammad Zarul Hanifah Md Zoqratt performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Wilhelm Wei Han Eng performed the experiments, approved the final draft.
Binh Thanh Thai conceived and designed the experiments, contributed reagents/materials/analysis tools, approved the final draft.
Christopher M. Austin conceived and designed the experiments, contributed reagents/materials/analysis tools, authored or reviewed drafts of the paper, approved the final draft.
Han Ming Gan conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
DNA Deposition
The following information was supplied regarding the deposition of DNA sequences:
All FastQ raw data may be accessed through SRA accession number SRP126985 or NCBI BioProject PRJNA422950.
Data Availability
The following information was supplied regarding data availability:
FASTA files for OTU and ASV in addition to their taxonomic assignments are available in the Supplemental File.
References
- Adachi et al. (2002).Adachi M, Fukami K, Kondo R, Nishijima T. Identification of marine algicidal Flavobacterium sp. 5 N-3 using multiple probes and whole-cell hybridization. Fisheries Science. 2002;68:713–720. doi: 10.1046/j.1444-2906.2002.00484.x. [DOI] [Google Scholar]
- Bentzon-Tilia, Sonnenschein & Gram (2016).Bentzon-Tilia M, Sonnenschein EC, Gram L. Monitoring and managing microbes in aquaculture—towards a sustainable industry. Microbial Biotechnology. 2016;9:576–584. doi: 10.1111/1751-7915.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, Chimenti & Just (2017).Black EM, Chimenti MS, Just CL. Effect of freshwater mussels on the vertical distribution of anaerobic ammonia oxidizers and other nitrogen-transforming microorganisms in upper Mississippi river sediment. PeerJ. 2017;5:e3536. doi: 10.7717/peerj.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman (2006).Bowman JP. The marine clade of the family Flavobacteriaceae: the genera Aequorivita, Arenibacter, Cellulophaga, Croceibacter, Formosa, Gelidibacter, Gillisia, Maribacter, Mesonia, Muricauda, Polaribacter, Psychroflexus, Psychroserpens, Robiginitalea, Salegentibacter, Tenacibaculum, Ulvibacter, Vitellibacter and Zobellia. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors. The prokaryotes: volume 7: Proteobacteria: delta, epsilon subclass. Springer New York; New York: 2006. pp. 677–694. [Google Scholar]
- Bril et al. (2014).Bril JS, Durst JJ, Hurley BM, Just CL, Newton TJ. Sensor data as a measure of native freshwater mussel impact on nitrate formation and food digestion in continuous-flow mesocosms. Freshwater Science. 2014;33:417–424. doi: 10.1086/675448. [DOI] [Google Scholar]
- Callahan, McMurdie & Holmes (2017).Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. The ISME Journal. 2017;11:2639–2643. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso et al. (2010).Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona et al. (2016).Cardona E, Gueguen Y, Magré K, Lorgeoux B, Piquemal D, Pierrat F, Noguier F, Saulnier D. Bacterial community characterization of water and intestine of the shrimp Litopenaeus stylirostris in a biofloc system. BMC Microbiology. 2016;16:157. doi: 10.1186/s12866-016-0770-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al. (2017a).Chen C-Y, Chen P-C, Weng FC-H, Shaw GT-W, Wang D. Habitat and indigenous gut microbes contribute to the plasticity of gut microbiome in oriental river prawn during rapid environmental change. PLOS ONE. 2017a;12:e0181427. doi: 10.1371/journal.pone.0181427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al. (2017b).Chen W-Y, Ng TH, Wu J-H, Chen J-W, Wang H-C. Microbiome dynamics in a shrimp grow-out pond with possible outbreak of acute hepatopancreatic necrosis disease. Scientific Reports. 2017b;7:9395. doi: 10.1038/s41598-017-09923-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole et al. (2009).Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Research. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornejo-Granados et al. (2017).Cornejo-Granados F, Lopez-Zavala AA, Gallardo-Becerra L, Mendoza-Vargas A, Sánchez F, Vichido R, Brieba LG, Viana MT, Sotelo-Mundo RR, Ochoa-Leyva A. Microbiome of pacific whiteleg shrimp reveals differential bacterial community composition between wild, aquacultured and AHPND/EMS outbreak conditions. Scientific Reports. 2017;7:11783. doi: 10.1038/s41598-017-11805-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, Pérez & Kreft (2006).Costa E, Pérez J, Kreft J-U. Why is metabolic labour divided in nitrification? Trends in Microbiology. 2006;14:213–219. doi: 10.1016/j.tim.2006.03.006. [DOI] [PubMed] [Google Scholar]
- De Schryver, Defoirdt & Sorgeloos (2014).De Schryver P, Defoirdt T, Sorgeloos P. Early mortality syndrome outbreaks: a microbial management issue in shrimp farming? PLOS Pathogens. 2014;10:e1003919. doi: 10.1371/journal.ppat.1003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar (2010).Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Edgar (2013).Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- Edgar (2016).Edgar RC. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv. 2016081257
- Egan et al. (2000).Egan S, Thomas T, Holmström C, Kjelleberg S. Phylogenetic relationship and antifouling activity of bacterial epiphytes from the marine alga Ulva lactuca. Environmental Microbiology. 2000;2:343–347. doi: 10.1046/j.1462-2920.2000.00107.x. [DOI] [PubMed] [Google Scholar]
- Emerenciano, Gaxiola & Cuzon (2013).Emerenciano M, Gaxiola G, Cuzon G. Biomass now-cultivation and utilization. InTech; London: 2013. Biofloc technology (BFT): a review for aquaculture application and animal food industry. [Google Scholar]
- Eren et al. (2014).Eren AM, Morrison HG, Lescault PJ, Reveillaud J, Vineis JH, Sogin ML. Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. The Isme Journal. 2014;9:968–979. doi: 10.1038/ismej.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evtushenko & Takeuchi (2017).Evtushenko LI, Takeuchi M. The family microbacteriaceae. Growth. 2017;18:2–35. [Google Scholar]
- Foo et al. (2017).Foo SM, Eng WWH, Lee YP, Gui K, Gan HM. New sequence types of Vibrio parahaemolyticus isolated from a Malaysian aquaculture pond, as revealed by whole-genome sequencing. Genome Announcements. 2017;5:e00302–17. doi: 10.1128/genomeA.00302-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO (2016).Food and Agriculture Organization (FAO) The state of world fisheries and aquaculture 2016. FAO, Romehttp://www.fao.org/fi/oldsite/eims_search/1_dett.asp?pub_id=316888 Contributing to food security and nutrition for all. 2016
- Grotkjær et al. (2016).Grotkjær T, Bentzon-Tilia M, D’Alvise P, Dourala N, Nielsen KF, Gram L. Isolation of TDA-producing Phaeobacter strains from sea bass larval rearing units and their probiotic effect against pathogenic Vibrio spp. in Artemia cultures. Systematic and Applied Microbiology. 2016;39:180–188. doi: 10.1016/j.syapm.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Han et al. (2015).Han JE, Tang KFJ, Tran LH, Lightner DV. Photorhabdus insect-related (Pir) toxin-like genes in a plasmid of Vibrio parahaemolyticus, the causative agent of acute hepatopancreatic necrosis disease (AHPND) of shrimp. Diseases of Aquatic Organisms. 2015;113:33–40. doi: 10.3354/dao02830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang et al. (2016).Huang Z, Li X, Wang L, Shao Z. Changes in the intestinal bacterial community during the growth of white shrimp, Litopenaeus vannamei. Aquaculture Research. 2016;47:1737–1746. doi: 10.1111/are.12628. [DOI] [Google Scholar]
- Jakobsson et al. (2014).Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C, Björkstén B, Engstrand L, Andersson AF. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut. 2014;63:559–566. doi: 10.1136/gutjnl-2012-303249. [DOI] [PubMed] [Google Scholar]
- Jinbo et al. (2017).Jinbo X, Jinyong Z, Wenfang D, Chunming D, Qiongfen Q, Chenghua L. Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environmental Microbiology. 2017;19:1490–1501. doi: 10.1111/1462-2920.13701. [DOI] [PubMed] [Google Scholar]
- Kirchman (2002).Kirchman DL. The ecology of Cytophaga—Flavobacteria in aquatic environments. FEMS Microbiology Ecology. 2002;39:91–100. doi: 10.1111/j.1574-6941.2002.tb00910.x. [DOI] [PubMed] [Google Scholar]
- Kohl & Dearing (2012).Kohl KD, Dearing M-D. Experience matters: prior exposure to plant toxins enhances diversity of gut microbes in herbivores. Ecology Letters. 2012;15:1008–1015. doi: 10.1111/j.1461-0248.2012.01822.x. [DOI] [PubMed] [Google Scholar]
- Kondo et al. (2014).Kondo H, Tinwongger S, Proespraiwong P, Mavichak R, Unajak S, Nozaki R, Hirono I. Draft genome sequences of six strains of Vibrio parahaemolyticus isolated from early mortality syndrome/acute hepatopancreatic necrosis disease shrimp in Thailand. Genome Announcements. 2014;2:e00221–00214. doi: 10.1128/genomeA.00221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriem et al. (2015).Kriem MR, Banni B, El Bouchtaoui H, Hamama A, El Marrakchi A, Chaouqy N, Robert-Pillot A, Quilici ML. Prevalence of Vibrio spp. in raw shrimps (Parapenaeus longirostris) and performance of a chromogenic medium for the isolation of Vibrio strains. Letters in Applied Microbiology. 2015;61:224–230. doi: 10.1111/lam.12455. [DOI] [PubMed] [Google Scholar]
- Kumlu, Türkmen & Kumlu (2010).Kumlu M, Türkmen S, Kumlu M. Thermal tolerance of Litopenaeus vannamei (Crustacea: Penaeidae) acclimated to four temperatures. Journal of Thermal Biology. 2010;35:305–308. doi: 10.1016/j.jtherbio.2010.06.009. [DOI] [Google Scholar]
- Lee et al. (2015).Lee C-T, Chen I-T, Yang Y-T, Ko T-P, Huang Y-T, Huang J-Y, Huang M-F, Lin S-J, Chen C-Y, Lin S-S, Lightner DV, Wang H-C, Wang AH-J, Wang H-C, Hor L-I, Lo C-F. The opportunistic marine pathogen Vibrio parahaemolyticus becomes virulent by acquiring a plasmid that expresses a deadly toxin. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:10798–10803. doi: 10.1073/pnas.1503129112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung & Bates (2013).Leung TL, Bates AE. More rapid and severe disease outbreaks for aquaculture at the tropics: implications for food security. Journal of Applied Ecology. 2013;50:215–222. doi: 10.1111/1365-2644.12017. [DOI] [Google Scholar]
- Li et al. (2016).Li L, Yan B, Li S, Xu J, An X. A comparison of bacterial community structure in seawater pond with shrimp, crab, and shellfish cultures and in non-cultured pond in Ganyu, Eastern China. Annals of Microbiology. 2016;66:317–328. doi: 10.1007/s13213-015-1111-4. [DOI] [Google Scholar]
- Liu et al. (2015).Liu S, Ren H, Shen L, Lou L, Tian G, Zheng P, Hu B. pH levels drive bacterial community structure in sediments of the Qiantang River as determined by 454 pyrosequencing. Frontiers in Microbiology. 2015;6:285. doi: 10.3389/fmicb.2015.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone & Knight (2005).Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin (2011).Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Martínez-Córdova & Peña Messina (2005).Martínez-Córdova LR, Peña Messina E. Biotic communities and feeding habits of Litopenaeus vannamei (Boone 1931) and Litopenaeus stylirostris (Stimpson 1974) in monoculture and polyculture semi-intensive ponds. Aquaculture Research. 2005;36:1075–1084. doi: 10.1111/j.1365-2109.2005.01323.x. [DOI] [Google Scholar]
- McMurdie & Holmes (2013).McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLOS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, LeaMaster & Sweeney (2000).Moss SM, LeaMaster BR, Sweeney JN. Relative abundance and species composition of gram-negative, aerobic bacteria associated with the gut of juvenile white shrimp Litopenaeus vannamei reared in oligotrophic well water and eutrophic pond water. Journal of the World Aquaculture Society. 2000;31:255–263. doi: 10.1111/j.1749-7345.2000.tb00361.x. [DOI] [Google Scholar]
- Nelson, Rogers & Brown (2013).Nelson TM, Rogers TL, Brown MV. The gut bacterial community of mammals from marine and terrestrial habitats. PLOS ONE. 2013;8:e83655. doi: 10.1371/journal.pone.0083655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks et al. (2014).Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parte et al. (2011).Parte A, Krieg NR, Ludwig W, Whitman WB, Hedlund BP, Paster BJ, Staley JT, Ward N, Brown D. Bergey’s manual of systematic bacteriology: volume 4: the Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. Springer; New York: 2011. [Google Scholar]
- Phayungsak et al. (2018).Phayungsak M, Phimsucha B, Wanilada R, Sopacha A, Sirawut K, Piamsak M, Sage C. Bacterial community composition and distribution in different segments of the gastrointestinal tract of wild-caught adult Penaeus monodon. Aquaculture Research. 2018;49:378–392. doi: 10.1111/are.13468. [DOI] [Google Scholar]
- Pruesse, Peplies & Glöckner (2012).Pruesse E, Peplies J, Glöckner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse et al. (2007).Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungrassamee et al. (2016).Rungrassamee W, Klanchui A, Maibunkaew S, Karoonuthaisiri N. Bacterial dynamics in intestines of the black tiger shrimp and the Pacific white shrimp during Vibrio harveyi exposure. Journal of Invertebrate Pathology. 2016;133:12–19. doi: 10.1016/j.jip.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Saffran et al. (2001).Saffran K, Cash K, Hallard K, Neary B, Wright R. Canadian water quality guidelines for the protection of aquatic life. CCME Water Quality Index. 2001;1:34–31. [Google Scholar]
- Schock et al. (2013).Schock TB, Duke J, Goodson A, Weldon D, Brunson J, Leffler JW, Bearden DW. Evaluation of Pacific White Shrimp (Litopenaeus vannamei) health during a superintensive aquaculture growout using NMR-based metabolomics. PLOS ONE. 2013;8:e59521. doi: 10.1371/journal.pone.0059521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall et al. (2013).Tall A, Hervio-Heath D, Teillon A, Boisset-Helbert C, Delesmont R, Bodilis J, Touron-Bodilis A. Diversity of Vibrio spp. isolated at ambient environmental temperature in the Eastern English Channel as determined by pyrH sequencing. Journal of Applied Microbiology. 2013;114:1713–1724. doi: 10.1111/jam.12181. [DOI] [PubMed] [Google Scholar]
- Tang et al. (2014).Tang Y, Tao P, Tan J, Mu H, Peng L, Yang D, Tong S, Chen L. Identification of bacterial community composition in freshwater aquaculture system farming of Litopenaeus vannamei reveals distinct temperature-driven patterns. International Journal of Molecular Sciences. 2014;15:13663–13680. doi: 10.3390/ijms150813663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson et al. (2005).Thompson F, Gevers D, Thompson C, Dawyndt P, Naser S, Hoste B, Munn C, Swings J. Phylogeny and molecular identification of vibrios on the basis of multilocus sequence analysis. Applied and Environmental Microbiology. 2005;71:5107–5115. doi: 10.1128/AEM.71.9.5107-5115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran et al. (2013).Tran L, Nunan L, Redman RM, Mohney LL, Pantoja CR, Fitzsimmons K, Lightner DV. Determination of the infectious nature of the agent of acute hepatopancreatic necrosis syndrome affecting penaeid shrimp. Diseases of Aquatic Organisms. 2013;105:45–55. doi: 10.3354/dao02621. [DOI] [PubMed] [Google Scholar]
- Tremblay et al. (2015).Tremblay J, Singh K, Fern A, Kirton E, He S, Woyke T, Lee J, Chen F, Dangl J, Tringe S. Primer and platform effects on 16S rRNA tag sequencing. Frontiers in Microbiology. 2015;6:771. doi: 10.3389/fmicb.2015.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utter, Mark Welch & Borisy (2016).Utter DR, Mark Welch JL, Borisy GG. Individuality, stability, and variability of the plaque microbiome. Frontiers in Microbiology. 2016;7:564. doi: 10.3389/fmicb.2016.00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang & Chen (2006).Wang F-I, Chen J-C. Effect of salinity on the immune response of tiger shrimp Penaeus monodon and its susceptibility to Photobacterium damselae subsp. damselae. Fish & Shellfish Immunology. 2006;20:671–681. doi: 10.1016/j.fsi.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Watts et al. (2017).Watts MP, Spurr LP, Gan HM, Moreau JW. Characterization of an autotrophic bioreactor microbial consortium degrading thiocyanate. Applied Microbiology and Biotechnology. 2017;101:5889–5901. doi: 10.1007/s00253-017-8313-6. [DOI] [PubMed] [Google Scholar]
- Wickham & Wickham (2007).Wickham H, Wickham MH. The ggplot package. 2007. https://cran.r-project.org/web/packages/ggplot2/index.html https://cran.r-project.org/web/packages/ggplot2/index.html
- Wright, Konwar & Hallam (2012).Wright JJ, Konwar KM, Hallam SJ. Microbial ecology of expanding oxygen minimum zones. Nature Reviews Microbiology. 2012;10:381–394. doi: 10.1038/nrmicro2778. [DOI] [PubMed] [Google Scholar]
- Wyban, Walsh & Godin (1995).Wyban J, Walsh WA, Godin DM. Temperature effects on growth, feeding rate and feed conversion of the Pacific white shrimp (Penaeus vannamei) Aquaculture. 1995;138:267–279. doi: 10.1016/0044-8486(95)00032-1. [DOI] [Google Scholar]
- Xiong et al. (2015).Xiong J, Wang K, Wu J, Qiuqian L, Yang K, Qian Y, Zhang D. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Applied Microbiology and Biotechnology. 2015;99:6911–6919. doi: 10.1007/s00253-015-6632-z. [DOI] [PubMed] [Google Scholar]
- Xiong et al. (2017).Xiong J, Zhu J, Dai W, Dong C, Qiu Q, Li C. Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environmental Microbiology. 2017;19:1490–1501. doi: 10.1111/1462-2920.13701. [DOI] [PubMed] [Google Scholar]
- Yoon et al. (2017).Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. International Journal of Systematic and Evolutionary Microbiology. 2017;67:1613–1617. doi: 10.1099/ijsem.0.001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng et al. (2017).Zeng S, Huang Z, Hou D, Liu J, Weng S, He J. Composition, diversity and function of intestinal microbiota in pacific white shrimp (Litopenaeus vannamei) at different culture stages. PeerJ. 2017;5:e3986. doi: 10.7717/peerj.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al. (2014).Zhang M, Sun Y, Chen K, Yu N, Zhou Z, Chen L, Du Z, Li E. Characterization of the intestinal microbiota in Pacific white shrimp, Litopenaeus vannamei, fed diets with different lipid sources. Aquaculture. 2014;434:449–455. doi: 10.1016/j.aquaculture.2014.09.008. [DOI] [Google Scholar]
- Zhu et al. (2016).Zhu J, Dai W, Qiu Q, Dong C, Zhang J, Xiong J. Contrasting ecological processes and functional compositions between intestinal bacterial community in healthy and diseased shrimp. Microbial Ecology. 2016;72:975–985. doi: 10.1007/s00248-016-0831-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ASVs assigned by SINA are annotated with an asterisk, while the remaining were annotated with the default RDP classifier.
OTUs assigned by SINA are annotated with an asterisk, while the remaining were annotated with the default RDP classifier.
Data Availability Statement
The following information was supplied regarding data availability:
FASTA files for OTU and ASV in addition to their taxonomic assignments are available in the Supplemental File.