

Abstract
Background
Dendritic cell (DC)-derived exosomes (Dexs) have been proved to induce and enhance antigen-specific T cell responses in vivo, and previous clinical trials have shown the feasibility and safety of Dexs in multiple human cancers. However, there is little knowledge on the efficacy of Dexs against hepatocellular carcinoma (HCC) until now.
Methods
In this study, human peripheral blood-derived DCs were loaded with recombinant adeno-associated viral vector (rAAV)-carrying alpha-fetoprotein (AFP) gene (rAAV/AFP), and high-purity Dexs were generated. Then naive T cells were stimulated with Dexs to investigate the specific T cell-mediated immune responses against HCC.
Results
Our findings showed that Dexs were effective to stimulate naive T cell proliferation and induce T cell activation to become antigen-specific cytotoxic T lymphocytes (CTLs), thereby exhibiting antitumor immune responses against HCC. In addition, Dex-sensitized DC precursors seemed more effective to trigger major histocompatibility complex class I (MHC I)-restricted CTL response and allow DCs to make full use of the minor antigen peptides, thereby maximally activating specific immune responses against HCC.
Conclusion
It is concluded that Dexs, which combine the advantages of DCs and cell-free vectors, are promising to completely, or at least in part, replace mature DCs (mDCs) to function as cancer vaccines or natural antitumor adjuvant.
Keywords: hepatocellular carcinoma, dendritic cell, exosome, immune response, cytotoxic T lymphocyte
Liver cancer is the sixth most common cancer and the second most common causes of cancer-related death worldwide,1 with 788,000 deaths reported in 2015.2 Hepatocellular carcinoma (HCC), the most common type of liver cancer, is characterized by rapid progression, high mortality, and poor prognosis, which causes a huge burden of disease.3 Currently, the treatment of HCC mainly includes surgical resection, liver transplantation, transarterial chemoembolization (TACE), radiofrequency ablation, and molecular targeted therapy.4–10 These treatments have shown survival benefits; however, the overall survival and clinical outcomes of HCC remain unsatisfactory.11
The development, progression, and recurrence of HCC have been strongly linked with cancer cell immune escape and human immune functions.12 Since it has been proved to be effective to both directly remove the remaining tumor cells and enhance the overall immune functions,13–15 immunotherapy, as a critical part of multimodality therapy for HCC,16 is therefore considered as the most promising strategy for curing HCC.17
As antigen-presenting cells (APCs) in the mammalian immune system, dendritic cells (DCs) may trigger adaptive immune response and induce effective antitumor immune response.18–20 DC-based immunotherapy is reported to induce tumor antigen-specific cytotoxic T lymphocyte (CTL) responses against multiple cancers.21 In addition, a variety of DC-based vaccines have been developed and tested for the efficacy against cancers,22,23 and both laboratory and clinical studies have been effective to mediate specific antitumor immune responses against and mediate the regression of lung cancer,24,25 intrahepatic cholangiocarcinoma,26,27 gastrointestinal cancer,28–30 liver cancer,31–33 breast cancer,34,35 melanoma,36,37 glioma,38,39 renal cancer,40,41 prostate cancer,42,43 ovarian cancer,44,45 and others.46,47
Exosomes are a recently discovered subtype of membrane vesicle either released from the cell when multivesicular bodies fuse with the plasma membrane or released directly from the plasma membrane, which are reported to have a diameter of 50–100 nm.48 Previous studies have demonstrated that exosomes may affect the cancer biological behaviors and cancer drug resistance and have potential for diagnosis, therapy, and prognosis.49,50 DC-derived exosomes (Dexs) are found to express major histocompatibility complex class I (MHC I) and class II (MHC II), as well as co-stimulatory molecules, and have been proved to be able to induce and enhance antigen-specific T cell responses in vivo.51 Previous clinical trials have shown the feasibility and safety of Dexs in advanced non-small cell lung cancer (NSCLC), advanced colorectal cancer, and metastatic melanoma.52,53 However, there is little knowledge on the efficacy of Dexs against HCC until now.
In this study, human peripheral blood-derived DCs were loaded with recombinant adeno-associated viral vector (rAAV)-carrying alpha-fetoprotein (AFP) gene (rAAV/AFP), and high-purity Dexs were generated. Then naive T cells were stimulated with Dexs to investigate the specific T cell-mediated immune responses against HCC.
Materials and methods
Ethical consideration
This study was approved by the ethics review committee of Fujian Cancer Hospital (permission no, SQ2016-021-01). Signed informed consent was obtained from all volunteers included in this study.
Cell culture
Human HCC HepG2 (human leukocyte antigen [HLA]-A2 positive, AFP secretion positive) and SMMC-7721 (HLA-A2 positive, AFP secretion negative) cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Thermo Fisher Scientific) at 5% CO2 saturated humidity at 37°C. Following growth to 70%–80% confluence, HepG2 and SMMC-7721 cells were digested with 0.25% pancreatin and passaged every 2–3 days at a ratio of 1:3. Log-phase cells were harvested from the subsequent experiments.
DC isolation and rAAV/AFP transfection
Approximately 30 mL of peripheral blood was sampled from each healthy volunteer (HLA-A2 positive) without hematopoietic disorders, lymphatic diseases, or infectious diseases and with normal AFP levels. Peripheral blood mononuclear cells (PBMCs) were isolated, seeded onto six-well plates (Corning Life Sciences, Tewksbury, MA, USA) at a density of 4×106 cells/mL, and cultured at 37°C containing 5% CO2 for 3 hours. After PBMCs were adherent fully to the plate wall, the plate was gently shaken to suspend the non-adherent cells, and the medium was gently transferred to 50-mL sterile centrifuge tubes. Then cells were washed three times in PBS to remove the suspension cells, and serum-free RPMI 1640 medium (Thermo Fisher Scientific) containing 100 ng/mL recombinant human granulocyte macrophage colony-stimulating factor (rhGM-CSF; PeproTech, Rocky Hill, NJ, USA) and 50 ng/mL rhIL-4 (PeproTech) was added to the culture plate for further culture and incubated at 37°C with 5% CO2. Cells were then loaded with rAAV/AFP (Virovek, Inc., Nanjing, China) at a titer of 1×1010 vg/mL and incubated at 37°C with 5% CO2 for 2 hours 1 day post-stimulation with rhGM-CSF and rhIL-4. The virus supernatant was removed, added with serum-free RPMI 1640 medium containing 100 ng/mL rhGM-CSF and 50 ng/mL rhIL-4, and incubated at 37°C with 5% CO2. Cells were stimulated with 50 ng/mL lipopolysaccharide (LPS; Sigma-Aldrich, St. Louis, MO, USA) 5 days after the first stimulation. The efficiency of rAAV/AFP transfection was observed under a fluorescence microscope 7 days after the first stimulation. Mature DCs (mDCs) were harvested post-transfection with rAAV/AFP, added with PBS, centrifuged at 300× g for 5 minutes at room temperature, resuspended, and adjusted to a density of 1×106 cells/mL. The mDCs and the supernatant were harvested for the subsequent experiments.
Characterization of mDCs morphology and phenotype
The morphology of mDCs was examined by microscopy on day 7, and mDC isolation was validated by surface staining with CD11C-APC, CD209-PerCP-Cy5.5, CD54-phycoerythrin (PE), CD86-APC, CD80-PE, HLA-DR-PE, and corresponding isotype-matched antibodies (BD Biosciences, San Jose, CA, USA) and analysis on a FACSCanto II flow cytometer (BD Biosciences) using the software FlowJo version 7.6.2 (TreeStar, Inc., Ashland, OR, USA).
Dex isolation and characterization
Dexs were isolated using the protocol described previously.54 Briefly, the culture supernatant of rAAV-empty-infected and rAAV/AFP-transfected mDCs was collected and centrifuged at 37°C, 300× g for 10 minutes. The supernatant was harvested and centrifuged at 4°C, 2,000× g for 20 minutes. The supernatant was collected and centrifuged at 10,000× g for 30 minutes at low temperature. The supernatant was transferred to 100-kDa MWCO Amicon Ultra-15 Centriplus centrifugal ultrafiltration (EMD Millipore, Billerica, MA, USA) and centrifuged at 4°C, 1,500× g for 15 minutes. The floating exosome solution, together with sucrose–deuteroxide mixture containing 30% sucrose/D2O (W/V; Sigma-Aldrich) and PBS (3:3:4 ratio), was transferred to Polyallomer Bell-top Centrifuge Tubes (Beckman Coulter, Inc., Brea, CA, USA) and centrifuged on an L-100XP ultracentrifuge (Beckman Coulter, Inc.) at 4°C, 100,000× g for 1 hour. The cushion containing exosomes were washed twice with PBS at 100,000× g for 70 minutes at 4°C, and the obtained Dex pellets were finally resuspended in 100 µL PBS, filtered, and degermed by 0.22 µm filter (Nordic Biosite, Ta¨by, Sweden). The protein content of Dex was quantified with a bicinchoninic acid assay (Thermo Fisher Scientific), and then Dexs were stored at −80°C for the subsequent experiments.
For transmission electron microscopy (TEM) analysis of Dex, approximately 20 µL Dex was transferred onto a pioloform-coated copper grid and allowed to stand at room temperature for 5 minutes. Then, excess fluid was sucked into filter paper. The sample was stained by a drop of 5 µL 2% methyl cellulose (Sigma-Aldrich) containing 2% uranyl acetate (Sigma-Aldrich) under an incandescent light bulb to dry for 1–2 minutes before viewing by TEM (HT7650; Hitachi Ltd., Tokyo, Japan) at 80 kV. The Dex size was measured using a Malvern NanoSight NS300 system (Malvern Instruments, Malvern, UK) following the manufacturer’s instructions.
In addition, the Dex target protein expression was determined using Western blotting. Briefly, pre-enriched Dex samples were lysed in RIPA buffer supplemented with complete Protease Inhibitor Cocktail Tablets (Roche Applied Science, Mannheim, Germany). Lysates (30 µg/lane) were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). The exosome-negative protein was probed with specific rabbit antihuman calnexin antibody (1:1000; Abcam, Cambridge, UK). Antibodies used for probing exosome target proteins included specific mouse antihuman Alix (1:1,000; Abcam), CD81 (1:3,000; Abcam), CD9 (1:1,000; Abcam), and CD63 (1:1,000; Abcam) primary monoclonal antibodies. For quantifying Dex target protein expression, mouse antihuman MHC-I (1:500; Abcam), MHC-II (1:500; Abcam), CD86 (1:500; Abcam), and AFP (1:1,000; R&D Systems, Inc., Minneapolis, MN, USA) monoclonal antibodies were used as primary antibodies, and horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG antibody (1:1,000; Sigma-Aldrich) was used as a secondary antibody, while GAPDH (Cell Signaling Technologies, Danvers, MA, USA) served as a loading control. The corresponding bands were then visualized via chemiluminescence.
Induction of CTL
PBMCs were routinely isolated, and DCs were induced from PBMCs and cultured. DCs were infected with rAAV/AFP 1 day after culture (DC-rAAV/AFP), and DC precursors were sensitized with 100 µg Dex (DC-Dex) 5 days after culture to prepare DC vaccines. DC-rAAV/AFP, DC-Dex, and non-transfected DCs after 7 days of induction were adjusted to a density of 1×105 cells/mL and incubated with 25 µg/mL mitomycin C at 37°C for 45 minutes. After being washed three times in PBS, cells were resuspended in RPMI 1640 medium. DC-rAAV/AFP (Group A), Dex (Group B), DC-Dex (Group C), and non-transfected DCs (Group D) were mixed with naive T cells, which were isolated by negative selection using Naive T cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s instructions, at a ratio of 1:10, respectively. Cells in Group B (containing 1×106 naive T cells per well) were co-incubated with 100 µg/well Dex at 37°C with 5% CO2 for 10 days.
Detection of DC-Induced naive T cell proliferation
Naive T cells were harvested, transferred to pre-warmed medium, and adjusted to a density of 1×106 cells/mL. Cells were co-incubated with 2 µL/mL 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) stock solution (Thermo Fisher Scientific) at 37°C for 30 minutes. Then the cooled medium with 5-fold volumes was added, and cells were incubated on ice for 5 minutes, harvested, and centrifuged. The sediment was collected and washed three times with fresh medium. Cells in the four groups (Group A, B, C, and D) were co-incubated with CFSE-stained naive T cells for 96 hours. The naive T cell proliferation was determined using flow cytometry, and the proliferating cell colony formation was observed under a microscope.
Detection of expression of immune effector molecules
Cells in the four groups (Group A, B, C, and D) were co-incubated with naive T cells for 10 days. The effector cells were incubated with mouse antihuman CD45RA-fluorescein isothiocyanate (FITC)/CD45RO-PE antibody (BD Biosciences), mouse antihuman lymphocyte function-associated antigen 1alpha (LFA-1a) (CD11a)-PE/CD244-FITC antibody (BD Biosciences), mouse antihuman CD8-APC/CD28-FITC antibody (BD Biosciences), mouse antihuman CD8-APC/OX40 (CD134)-PE antibody (BD Biosciences), mouse antihuman HLA-DR-PE antibody (BD Biosciences), mouse antihuman CD69-PE antibody (BD Biosciences), mouse antihuman CD71-APC antibody (BD Biosciences), mouse antihuman CD95-PE antibody (BD Biosciences), mouse antihuman CD8-PE/PD1-APC antibody (BD Biosciences), mouse antihuman CD8-APC/CTLA-4-PE antibody (BD Biosciences), and isotype antibody at room temperature in darkness. Following incubation for 30 minutes, cells were washed in PBS and centrifuged at 1,200 r/min for 5 minutes. The supernatant was discarded, and the sediment was added with 500 µL PBS and the flow cytometry data were analyzed using the software FlowJo version 7.6.2 (TreeStar, Inc.).
To detect granzyme B (GrB)/perforin expression, cells were incubated with mouse antihuman CD8-APC antibody (BD Biosciences) at room temperature in darkness for 20 minutes, incubated in fixation buffer (BD Biosciences) at room temperature in darkness for 20 minutes, mixed evenly with permeabilization buffer (BD Biosciences), and centrifuged at 1,200 r/min for 5 minutes. The supernatant was discarded, and 200 µL permeabilization buffer, 5 µL mouse antihuman granzyme-B-FITC antibody (BD Biosciences), and 5 µL mouse antihuman perforin-PE antibody (BD Biosciences) were added to the sediment and mixed evenly using vortex. Following incubation at room temperature in darkness for 20 minutes, cells were washed with permeabilization buffer and centrifuged. The supernatant was discarded, and 200 µL PBS containing 0.5% paraformaldehyde was added to the sediment and subjected to flow cytometry.
Enzyme-linked immune absorbent spot (ELISPOT) assay
The ELISPOT assay was performed to determine the frequency of interferon (IFN)-γ-producing CTLs stimulated by target tumor cells using 96-well culture plates pre-coated with antihuman IFN-γ antibody (Dakewe Biotech Co., Ltd., Shenzhen, China) following the manufacturer’s instructions. CTLs (2×105 cells per well) induced by DC-rAAV/AFP (Group A), Dex (Group B), DC-Dex (Group C), and non-transfected DCs (Group D) were co-cultured with HepG2 cells (1×104 cells per well) at an E/T ratio of 20:1, while the phytohemagglutinin group served as a positive control. After incubation at 37°C for 24 hours without moving the plate, cells were removed and plates were washed. Biotinylated IFN-γ monoclonal antibody (1 hour at 37°C) was added, followed by the addition of streptavidin-HRP (1 hour at 37°C) and then pre-mixed 3-amino-9-ethylcarbazole solutions with washes between each step. The reaction was stopped with distilled water on development of the spots, which were subsequently quantified on an ImmunoSpot® Series 6 Analyzer (Cellular Technology Limited, Cleveland, OH, USA).
Detection of cytokines in effector and HepG2 cell Co-culture supernatant
The effector cells in the four groups (Group A, B, C, and D) were harvested and centrifuged at 400× g for 5 minutes. The effector cell density was adjusted to 5×106 cells/mL, and the target HepG2 cell density was adjusted to 1×106 cells/mL. The effector cells and target cells were co-cultured at a ratio of 20:1 at 37°C with 5% CO2 for at least 5 hours and centrifuged, and the cell co-culture supernatant was collected. The interleukin (IL)-2, IL-4, IL-6, IL-10, tumor necrosis factor (TNF), IFN-γ, and IL-17A levels were detected in the effector and HepG2 cell co-culture supernatant using the Cytometric Bead Array (CBA) Human Th1/Th2/Th17 Cytokine Kit (BD Biosciences) on an FACSCanto II flow cytometer (BD Biosciences) following the manufacturer’s instructions. All flow cytometric data were processed with the software CBA FCAP Array (BD Biosciences).
Detection of the killing action of Dex-Induced CTL against HCC cells
HepG2 and SMMC7721 cells served as target cells and were adjusted to a density of 1×106 cells/mL. The effector cells in the four groups (Group A, B, C, and D) were harvested, centrifuged at 400×g for 5 minutes, and adjusted to a density of 5×106 cells/mL. The effector and target cells were co-cultured at a ratio of 25:1 at 37°C with 5% CO2 for 5 hours and centrifuged at 400× g for 5 minutes. The supernatant was discarded, washed in pre-cooled 1× PBS, and resuspended in 500 µL binding buffer (BD Biosciences). Cells were then incubated in mouse antihuman CD3-APC antibody and Annexin-V-FITC (BD Biosciences) at 4°C for 30 minutes, washed in pre-cooled binding buffer, stained with 7-aminoactinomycin D (7-AAD) for 10 minutes, and subjected to flow cytometry. The killing rate of CTL was calculated using the following formula. The killing rate of CTL (%)=(APC− cell count–APC−FITC−7-AAD− cell count)/APC− cell count×100%.
The CD107a expression was determined in CTL using flow cytometry. Briefly, the CTL density was adjusted to 1×106 cells/mL, and CTL was co-cultured with HepG2 cells at a ratio of 20:1 at 37°C with CO2 for 2 hours. The cell co-culture was incubated with 2 µM monensin for 3.5 hours, incubated with mouse antihuman CD107a-PE antibody and mouse antihuman CD8-PerCP antibody for 30 minutes, washed three times in PBS, and subjected to flow cytometry.
Statistical analyses
All measurement data were described as mean±SD, and all statistical analyses were performed using the statistical software SPSS version 17.0 (SPSS Inc., Chicago, IL, USA). Differences of mean among the four groups were compared with one-way ANOVA, and stepwise multiple comparisons were done with Fisher’s least significant difference (LSD) method and Dunnett’s test. Differences of mean between groups were tested for statistical significance with Student’s t-test, and differences of proportions were compared with chi-squared test. A P value <0.05 was considered statistically significant.
Results
mDCs morphology and phenotypes
PBMC-derived DCs were cultured in RPMI 1640 medium supplemented with the appropriate cytokine cocktail. On day 7 post-culture, the suspension cell clusters were found to increase and typical morphology gradually appeared, such as branch-like protrusions and aggregation of branching structure among cells (Figure 1A). In addition, the mDCs post-transfection with rAAV/AFP showed positive staining for CD11C, CD209, CD54, MHC-II, and T cell co-stimulatory molecules (CD80, CD86), and the expressions of CD11C, CD209, CD54, CD86, CD80, and HLA-DR were 99.25%, 92.96%, 95.63%, 90.06%, 86.48%, and 99.54% in mDCs, respectively (Figure 1B).
Figure 1.

The morphologic and phenotypic features of mDCs.
Note: (A) mDCs post-transfection with rAAV/AFP were obtained by LPS stimulation on day 7, and the typical mDC morphology was observed under a light microscope (×40); (B) mDCs were stained with CD11C, CD209, CD54, CD86, CD80, and HLA-DR or isotype-matched control antibodies and analyzed by flow cytometry.
Abbreviations: AFP, alpha-fetoprotein; HLA, human leukocyte antigen; LPS, lipopolysaccharide; mDC, mature dendritic cell; rAAV, recombinant adeno-associated viral vector.
Dex morphology, size, and target protein expression
TEM showed that Dexs appeared as oval–biconcave-shaped membranous vesicles, which measured 50–100 nm in diameter and showed scattered or clustered distribution (Figure 2A). The Dexs were deeply stained bilayer lipid membrane, and the interior contained low-intensity electronic components. To further investigate the size distribution profile of Dex, the size distribution analysis was performed using the Malvern NanoSight NS300 system, which showed a size peak of 91 nm in mature Dex (Figure 2B). To test the purity of exosomes, the expression of calnexin, a negative marker of exosomes, was determined, and the expression of exosomal protein markers Alix, CD81, CD9, and CD63, was determined using Western blotting. Higher MHC-I, MHC-II, and CD86 expressions were detected in Dex lysates, and Dex was found to carry AFP antigen in mDC-derived exosomes (Figure 2C), which allowed the immunological activity in Dex.
Figure 2.

Validation of successful Dex isolation.
Notes: (A) Dex ultrastructure displayed by transmission electron microscopy (red arrows). Scale bar =200 nm; (B) the size distribution profile of Dex using the Malvern NanoSight NS300 system, showing a size peak of 91 nm in mature Dex; (C) the expression of the exosome-negative marker calnexin and positive markers, Alix, CD81, CD9, and CD63. In addition, the functional protein expressions of MHC-I, MHC-II, CD86, and AFP were detected in mDCs and Dex lysates by Western blotting. A total of 30 µg protein from the lysates of mDCs and Dex was loaded into each lane. Data were normalized to the GAPDH levels. The bands for targeted proteins were analyzed using the software Gel-Pro analyzer version 4.1.
Abbreviations: AFP, alpha-fetoprotein; Dex, dendritic cell-derived exosome; mDC, mature dendritic cell; MHC, major histocompatibility complex.
DC- and Dex-induced Naive T cell proliferation
Naive T cells were sorted with the magnetic-activated cell sorting (MACS) system. Following co-incubation of the cells in the four groups (Group A, B, C, and D) with CFSE-stained naive T cells for 96 hours, we observed the proliferating naive T cell colony formation under a microscope, and flow cytometry detected naive T cell proliferation in all four groups. Flow cytometry detected a higher rate of CFSE-negative naive T cells in DC-rAAV/AFP (39.73%±4.59%), Dex (45.50%±2.32%), and DC-Dex (51.46%±3.38%) than in non-transfected DCs (28.60%±2.48%; P<0.05), and a higher proliferative ability was seen in Group C than in other three groups (P<0.05; Figure 3).
Figure 3.

DC- and Dex-induced naive T cell proliferation.
Note: (A and E) DC-rAAV/AFP; (B and F) Dex; (C and G) DC-Dex; (D and H) non-transfected DCs; (A–D) CFSE staining; (E–H) flow cytometry.
Abbreviations: AFP, alpha-fetoprotein; CFSE, 5,6-carboxyfluorescein diacetate succinimidyl ester; DC, dendritic cell; Dex, dendritic cell-derived exosome; rAAV, recombinant adeno-associated viral vector.
Expression of immune effector molecules
After the co-incubation of the DC-rAAV/AFP, Dex, DC-Dex, and non-transfected DCs with naive T cells for 10 days, respectively, flow cytometry was performed to detect the expression of surface markers of immune effector cells, including naive/memory cell surface markers CD45RA/CD45RO; co-stimulatory activation; and adhesion molecules LFA-1a/CD244, CD28, OX40, HLA-DR, CD69, and CD71; apoptotic molecule CD95; immunosuppressive molecules PD-1 and CTLA-4; and cell killing effector molecules GrB/perforin. Flow cytometry detected significantly higher expressions of CD45RO, LFA-1a/CD244, GrB, and perforin in DC-rAAV/AFP, Dex, and DC-Dex than in non-transfected DCs (P<0.05). DC-Dex was found to induce higher expressions of CD28, OX40, and HLA-DR relative to non-transfected DCs (P<0.05), while the CD28, OX40, and HLA-DR expressions in non-transfected DCs were not significantly different from that in DC-rAAV/AFP and Dex (P>0.05). In addition, significantly lower CD45RA expression was detected in Dex and DC-Dex than in non-transfected DCs (P<0.05), and the PD-1 expression was significantly higher in DC-Dex relative to DC-rAAV/AFP and non-transfected DCs (P<0.05). However, there were no significant differences in the CD69, CD71, CD95, or CTLA-4 expression among the four groups (P>0.05; Table 1 and Figure 4).
Table 1.
Expression of immune effector molecules detected by flow cytometry (mean±SD,%)
| Effector cell molecule | DC-rAAV/AFP | Dex | DC-Dex | Non-transfected DCs |
|---|---|---|---|---|
| CD45RO | 35.46±1.56a,b | 32.24±4.58a,b | 53.83±3.05a | 24.96±0.53 |
| CD45RA | 40.46±7.94a,b | 32.63±0.95a | 27.8±3.3a | 54.33±2.9 |
| CD11a/CD244 | 56±2.49a,b | 50.93±3.41a,b | 70.06±3.49a | 42.86±6.01 |
| CD28 | 49.3±1.15 | 49.83±3.22 | 56.1±2.87a | 43.96±0.98 |
| OX40 | 15.79±3.86 | 21.47±7.66 | 28.06±2.67a | 10.35±3.25 |
| HLA-DR | 53.83±3.53 | 55.16±3.78 | 59.36±5.78a | 52.56±4.71 |
| CD69 | 4.08±0.61 | 5.3±1.31 | 7.06±0.86 | 4.53±1.55 |
| CD71 | 63.9±3.98 | 61.26±4.99 | 64.53±6.84 | 57.26±4.84 |
| CD95 | 52.3±3.28 | 54.61±2.74 | 55.95±4.62 | 48.86±3.12 |
| PD-1 | 8.69±1.21b | 13.94±2.11 | 17.67±2.4a | 8.63±1.48 |
| CTLA-4 | 1.26±0.06 | 1.27±0.17 | 1.4±0.28 | 1.38±0.05 |
| CD8/GrB | 53.56±1.67a,b | 57.36±1.35a | 61.5±1.17a | 43.36±2.48 |
| CD8/perforin | 31.96±1.79a,b | 32.2±0.46a,b | 37.6±1.53a | 19.46±1.47 |
Notes:
P<0.05 vs non-transfected DCs;
P<0.05 vs DC-Dex.
Abbreviations: AFP, alpha-fetoprotein; DC, dendritic cell; Dex, dendritic cell-derived xosome; rAAV, recombinant adeno-associated viral vector.
Figure 4.

Flow cytometry detects surface markers on immune effector cells.
Notes: Cells in the four groups of DC-rAAV/AFP, Dex, DC-Dex and non-transfected DCs were co-incubated with naive T cells for 10 days. Expression of surface proteins was tested by flow cytometric analysis. Data from one experiment representative of three performed. *P<0.05; #P<0.05.
Abbreviations: AFP, alpha-fetoprotein; DC, dendritic cell; Dex, dendritic cell-derived exosome; HLA, human leukocyte antigen; rAAV, recombinant adeno-associated viral vector.
IFN-γ secretion
ELISPOT assay was used to detect IFN-γ secretion, representing specific activation, by CTLs induced by DC-rAAV/AFP, Dex, DC-Dex, and non-transfected DCs in the presence of IL–2 together with tumor target cells. Since the antigen-specific cytotoxic effect may only be elicited by the tumor cells, detection of the frequency of IFN-γ-secreting CTLs was performed in DC-rAAV/AFP, Dex, DC-Dex, and non-transfected DCs with HepG2 cells (target cells) at an effector/target ratio of 20:1. As shown in Figure 5, CTLs induced by DC-rAAV/AFP, Dex, and DC-Dex showed a significantly higher level of IFN-γ (>5-fold) than non-transfected DCs (P<0.01).
Figure 5.
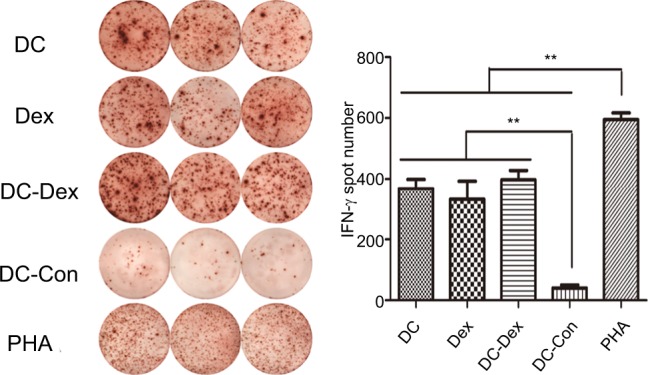
IFN-γ-secreting CTLs are quantified by the ELISPOT assay.
Notes: CTLs induced by DC-rAAV/AFP, Dex, DC-Dex, and non-transfected DCs were co-cultured with 1×104 HepG2 cells at an E/T ratio of 20:1. Compared with those in the non-transfected DCs group, the CTLs in the DC-rAAV/AFP, Dex, and DC-Dex groups showed a significantly greater frequency of IFN-γ secretion (**P<0.01), while the PHA group serves as a positive control.
Abbreviations: AFP, alpha-fetoprotein; CTL, cytotoxic T lymphocyte; DC, dendritic cell; Dex, dendritic cell-derived exosome; ELISPOT, enzyme-linked immune absorbent spot; IFN, interferon; rAAV, recombinant adeno-associated viral vector.
Cytokine production in effector cell and HepG2 cell Co-Culture supernatant
The effector cells in the four groups and HepG2 cells were co-cultured at a ratio of 20:1 at 37°C with 5% CO2 for at least 5 hours and centrifuged, respectively, and the cell co-culture supernatant was collected. The IL-2, IL-4, IL-6, IL-10, TNF, IFN-γ, and IL-17A levels were detected in the cell co-culture supernatant. Flow cytometry detected a significant reduction in the IL-4 and IL-17A levels in Dex relative to the other three groups (P<0.05), and a higher IFN-γ level was observed in DC-rAAV/AFP, Dex, and DC-Dex than in non-transfected DCs (P<0.05). The IL-2, IL-6, IL-10, and TNF levels in non-transfected DCs were not significantly different from those in the other three groups (P>0.05; Figure 6).
Figure 6.
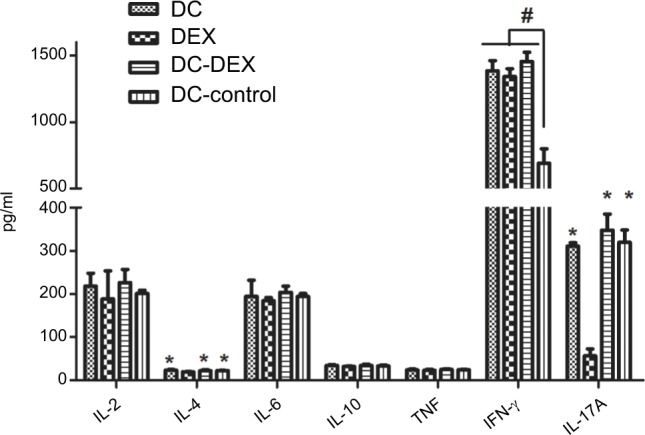
Flow cytometry detects the IL-2, IL-4, IL-6, IL-10, TNF, IFN-γ, and IL-17A expressions in the effector cells and HepG2 cell co-culture supernatant.
Note: *P<0.05 vs Dex group; #P<0.05 vs non-transfected DC group.
Abbreviations: DC, dendritic cell; Dex, dendritic cell-derived exosome; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
In vitro Dex-induced cytotoxicity against HCC cells
Flow cytometry revealed that DC-rAAV/AFP, Dex, and DC-Dex induced greater CTL cytotoxicity to HCC cells than non-transfected DC (P<0.05), and DC-rAAV/AFP-, Dex-, and DC-Dex-induced CTLs showed 39.33%±0.75%, 40.53%±1.23%, and 51.43%±1.45% killing rates of AFP-positive HepG2 cells and 21.10%±0.70%, 26.10%±1.15%, and 29.63%±0.66% killing rates of AFP-negative SMMC-7221 cells, respectively (P<0.05). There was no significant difference in the killing rate of HCC cells between Dex- and DC-rAAV/AFP-induced CTLs (P>0.05), and a higher killing rate of HCC cells was detected in DC-Dex than in DC-rAAV/AFP and Dex (P>0.05; Figure 7).
Figure 7.

The killing action of Dex-induced CTL against HCC HepG2 and SMMC7721 cell lines.
Notes: (A) Flow cytometry; (B) bar chart. A, DC-rAAV/AFP; B, Dex; C, DC-Dex; D, non-transfected DCs; E, HepG2/SMMC-7721 cells. *P<0.05. (C) flow cytometry detects the cell surface marker CD107a expression on the surface of DC- and Dex-induced CTLs. A, DC-rAAV/AFP; B, Dex; C, DC-Dex; D, non-transfected DC.
Abbreviations: AFP, alpha-fetoprotein; DC, dendritic cell; Dex, dendritic cell-derived exosome; HCC, hepatocellular carcinoma; rAAV, recombinant adeno-associated viral vector; CTL, cytotoxic T lymphocyte.
Flow cytometry detected 29.30%±2.45%, 30.30%±1.53%, and 31.90%±2.23% CD107a expression on the surface of DC-rAAV/AFP-, Dex-, and DC-Dex-induced CTLs, which was significantly higher than that (19.23%±0.30%) on the surface of non-transfected DC-induced CTL (P<0.05). However, there was no significant difference in the CD107a expression on the surface of Dex- and DC-rAAV/AFP-induced CTLs (P>0.05; Figure 7).
Discussion
It has been proved that DC-based immunotherapy may trigger tumor antigen-specific CTL responses against multiple human cancers 21. However, DC-based vaccines suffer from the following problems that may limit their wide clinical application. 1) Limited DCs proliferation, which cannot meet the requirements of clinical therapy; 2) live cell vaccine is complicated in quality control, weak in production and storage, and high in cost; and 3) the DC function and antigen-presenting ability vary in tumor antigen loading strategies of DCs.55 Previous studies showed that DC-rAAV-AFP may sustainably and effectively present tumor antigens to T cells, thereby mediating DCs to expression functional molecules and stimulating the production of immune response-related cytokines.56 This strategy seems to solve the problem of antigen sources; however, the safety of gene delivery for human diseases remains controversial.57 A search for more effective biological cancer vaccines is urgently needed and has been given a high priority.
In addition to the participation in immune regulation through the direct cell–cell interaction and secretion of cyto-kines, DCs may elicit T cell immune responses through the release of biologically active exosomes.58 In tumor-bearing mice, tumor peptide-pulsed Dexs were found to prime specific CTLs and eradicate or suppress the growth of the established murine tumors in a T cell-dependent manner.59 Since then, Dex, as a novel subcellular vaccine, has shown potential in immunotherapy for cancers.60 Murine DCs were found to have a reduction in antigen uptake ability and an increase in antigen-presenting ability with the development from immaturation to maturation, which was characterized by a remarkable rise in MHC molecules, adhesion molecule intercellular cell adhesion molecule-1 (ICAM-1), and co-stimulatory molecule B7, and a slight decrease in Dex secretion.61 Exosomes secreted by mDCs (mDexs) are therefore to exhibit a stronger ability to stimulate T cell proliferation than immature DC (imDC)-derived exosomes (imDexs).62 In the current study, PBMCs-derived DCs were loaded with recombinant AAV-carrying AFP gene (DC-rAAV/AFP) and then induced with recombinant human IL-4 (rhIL-4), rhGM-CSF, and LPS. Following ultrafiltration and sucrose density gradient ultracentrifugation, high-purity exosomes secreted from mDCs (DC-Dex) were obtained.
Results from Phase I clinical trials have shown that Dex therapy is feasible and well tolerated in patients with advanced NSCLC,63 metastatic melanoma,64 and advanced colorectal cancer.65 However, there is little knowledge on the clinical efficacy of Dex for HCC, and whether Dex directly elicits T cell-mediated immune responses is still in dispute to date. Antigen-loaded Dex was reported to highly express MHC–peptide complex, ICAM-1, and B7-2 (CD86) and may directly induce the production of antigen-specific CTLs and promote their secretion of IFN-γ in the absence of DCs.66 There is also evidence showing that Dex may cross-present functional MHC–peptide complexes to DCs, and following uptake of MHC–peptide complexes, DCs may exhibit strong antitumor immune responses.67 In addition, immunologic adjuvants have shown effective to enhance the function of Dex, including TLR3/9 ligand, CpG ODN, and Ampligen.68 In this study, we detected naive T cell proliferation in all four groups, and higher naive T cell proliferation was detected in the DC-rAAV/AFP, Dex, and DC-Dex than in non-transfected DCs, while greater naive T cell proliferation was induced in DC-Dex than in the other three groups. Flow cytometry showed significantly higher expressions of CD45RO, CD11a (LFA-1a)/CD244, GrB, and perforin in DC-rAAV/AFP, Dex, and DC-Dex than in non-transfected DCs (P<0.05), and significantly higher CD28, OX40 (CD134), and HLA-DR expressions were induced in DC-Dex than in non-transfected DCs (P<0.05). However, no significant differences were observed in the CD69, CD71 (TfR1), CD95, or CTLA-4 expression among the four groups (P>0.05). Moreover, flow cytometry detected significantly lower IL-4 and IL-17A expressions in Group B than in other three groups (P<0.05), and higher IFN-γ expression was seen in DC-rAAV/AFP, Dex, and DC-Dex than in non-transfected DCs (P<0.05). ELISPOT assay revealed an increase of IFN-γ secretion in antigen-sensitized T cells activated by DCs and Dex, which contributed to the presentation of tumor antigen, activation of naive T cells, and priming of AFP-specific antitumor immune responses.
T-cell receptor (TCR)/MHC–peptide complex, LFA-1/ICAM-1, and CD28/B7 are required for naive T cell proliferation, and the binding of MHC–peptide complex on mDex to TCR and ICAM-1 binding to LFA-1 promote T cell activation and proliferation in the presence of CD28/B7 co-stimulation.69 CD71, a 95-kD transferrin receptor, is a cell surface proliferation marker that is involved in the cellular uptake of iron.70 Since cell proliferation requires more irons, the rise of transferring on T cell surface may increase the iron transport, thereby promoting cell proliferation.71 Following exposure to DC stimulation for 20–72 hours, persistent high expression of transferrin receptor is detected in T cells, with expression to more than 75%.70 In addition, CD71 has been identified as a marker for middle- and late-stage cell activation and is expressed in a large number of proliferative cells, including activated lymphocytes, monocytes, and primitive red blood cells; however, it is not expressed in resting lymphocytes.71 Our data preliminarily indicated that Dex stimulated T cell proliferation and promoted T cells to express functional molecules that exhibit immune effects. Dex and DC-rAAV/AFP were found to upregulate the expression of granzyme and perforin, the two major effectors for CTL cytotoxicity, and activate co-stimulatory molecules CD28, OX40 (CD134), HLA-DR, and CD71. The early activation molecule CD69 was lowly expressed, while stable HLA-DR and CD71 expressions were detected 10 days post-stimulation. Following T cell activation, the CD95 (apoptosis antigen ligand 1/Fas) expression rapidly rises, and the apoptosis of T cells may occur via CD95/CD95L signaling. In addition, we detected low PD-1 and CTLA-4 expressions. Our findings showed that DC-Dex enhanced immune activities; exhibited a greater ability to stimulate T cell proliferation and to induce T cell maturation, activation, and cytotoxicity; and induced higher IFN-γ production than non-transfected DCs. It is indicated that the functional molecules on exosome surface are involved in DCs recruitment and exosome endocytosis, and Dexs, as vectors to transfer specific MHC I– or MHC II–peptide complexes between APC and T cells and between APCs, may initiate T cell immune responses. In addition, the immunogenicity of Dex may be transferred to other APCs, and antigen cross-presentation may occur among APCs through exosome secretion and uptake, resulting in the expansion of the breadth of the immune responses using weak antigens.72,73
In this study, DC-rAAV/AFP, Dex, and DC-Dex were found to induce greater CTL cytotoxicity to HCC cells than non-transfected DCs, and higher killing actions were detected against AFP-positive HepG2 cells than AFP-negative SMMC-7721 cells. In addition, DC-Dex induced a higher killing rate of HCC cells than DC-rAAV/AFP and Dex, and no significant difference was observed in the killing rate of HCC cells between the DC-rAAV/AFP and Dex. Flow cytometry detected higher CD107a expression in DC-rAAV/AFP, Dex, and DC-Dex than in non-transfected DCs, while no significant difference was observed in the CD107a expression between DC-rAAV/AFP and Dex. CD107a is rarely expressed on normal CD8 cell surface; however, CD107a may be highly expressed on CD8 cell membrane surface following stimulation with target cells, which is characterized by the release of intracellular granzyme and perforin and transfer of CD107a to cell membrane. Therefore, it is considered that the CD107a expression by CTLs may indicate CTL cytotoxicity, which indirectly reflects the killing action of CTLs.
Conclusion
The results of this study demonstrate that like DCs, Dex, as a cell-free vaccine that carries tumor antigens and exhibit antigen-presenting activity, is effective to stimulate naive T cell proliferation in vitro and induce T cell activation to become antigen-specific CTLs to exhibit in vitro antitumor immune responses against HCC. In addition, DC-Dex seems more effective to trigger MHC I-restricted CTL response and allow DCs to make full use of the minor antigen peptides, thereby maximally activating specific immune responses against HCC. It is concluded that Dex, which combines the advantages of DCs and cell-free vectors, is promising to completely, or at least in part, replace mDCs to function as cancer vaccines or natural antitumor adjuvant. In addition, antigen-modified Dex have shown beneficial for tumor suppression in diverse HCC mouse models,74,75 which is in agreement with the findings from this study. Taken together, the efficacy of Dex against HCC merits further preclinical investigations. Hence, the need for further studies to investigate the most effective doses of Dex, the precise active status of DC, and the preclinical feasibility and safety of Dex for HCC seems justified.
Acknowledgments
The authors would like to thank the staff from the Fujian University of Traditional Chinese Medicine for their help during the transmission electron microscopy. The present study was supported by the grants from the Natural Science Foundation of Fujian Province (grant no. 2017J01178), the Research Talent Training Program of Fujian Provincial Health Commission (grant no. 2017-ZQN-14), and the Medical Innovation Project of Fujian Provincial Health Commission (grant no. 2016-CX-11).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.GBD 2015 Mortality and Causes of Death Collaborators Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1459–1544. doi: 10.1016/S0140-6736(16)31012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 4.Mendizabal M, Reddy KR. Current management of hepatocellular carcinoma. Med Clin North Am. 2009;93(4):885–900. doi: 10.1016/j.mcna.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Akoad ME, Pomfret EA. Surgical resection and liver transplantation for hepatocellular carcinoma. Clin Liver Dis. 2015;19(2):381–399. doi: 10.1016/j.cld.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Bruix J, Sherman M, Practice Guidelines Committee, American Association for the Study of Liver Diseases Management of hepatocellular carcinoma. Hepatology. 2005;42(5):1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 7.Bruix J, Llovet JM, Castells A, et al. Transarterial embolization versus symptomatic treatment in patients with advanced hepatocellular carcinoma: results of a randomized, controlled trial in a single institution. Hepatology. 1998;27(6):1578–1583. doi: 10.1002/hep.510270617. [DOI] [PubMed] [Google Scholar]
- 8.Morimoto M, Sugimori K, Shirato K, et al. Treatment of hepatocellular carcinoma with radiofrequency ablation: radiologic-histologic correlation during follow-up periods. Hepatology. 2002;35(6):1467–1475. doi: 10.1053/jhep.2002.33635. [DOI] [PubMed] [Google Scholar]
- 9.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48(4):1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stotz M, Gerger A, Haybaeck J, Kiesslich T, Bullock MD, Pichler M. Molecular targeted therapies in hepatocellular carcinoma: past, present and future. Anticancer Res. 2015;35(11):5737–5744. [PubMed] [Google Scholar]
- 11.Sangiovanni A, Colombo M. Treatment of hepatocellular carcinoma: beyond international guidelines. Liver Int. 2016;36(Suppl 1):124–129. doi: 10.1111/liv.13028. [DOI] [PubMed] [Google Scholar]
- 12.Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12(12):681–700. doi: 10.1038/nrgastro.2015.173. [DOI] [PubMed] [Google Scholar]
- 13.Finn OJ. Cancer immunology. N Engl J Med. 2008;358(25):2704–2715. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 14.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29(36):4828–4836. doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sturm JW, Keese M. Multimodal treatment of hepatocellular carcinoma (HCC) Onkologie. 2004;27(3):294–303. doi: 10.1159/000077982. [DOI] [PubMed] [Google Scholar]
- 17.Breous E, Thimme R. Potential of immunotherapy for hepatocellular carcinoma. J Hepatol. 2011;54(4):830–834. doi: 10.1016/j.jhep.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. 2017;45:43–51. doi: 10.1016/j.coi.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabrilovich DI, Ciernik IF, Carbone DP. Dendritic cells in antitumor immune responses. I. Defective antigen presentation in tumor-bearing hosts. Cell Immunol. 1996;170(1):101–110. doi: 10.1006/cimm.1996.0139. [DOI] [PubMed] [Google Scholar]
- 20.Apetoh L, Locher C, Ghiringhelli F, Kroemer G, Zitvogel L. Harnessing dendritic cells in cancer. Semin Immunol. 2011;23(1):42–49. doi: 10.1016/j.smim.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Colaco CA. DC-based cancer immunotherapy: the sequel. Immunol Today. 1999;20(4):197–198. doi: 10.1016/s0167-5699(98)01407-8. [DOI] [PubMed] [Google Scholar]
- 22.Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117(5):1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilboa E, Nair SK, Lyerly HK. Immunotherapy of cancer with dendritic-cell-based vaccines. Cancer Immunol Immunother. 1998;46(2):82–87. doi: 10.1007/s002620050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hradilova N, Sadilkova L, Palata O, et al. Generation of dendritic cell-based vaccine using high hydrostatic pressure for non-small cell lung cancer immunotherapy. PLoS One. 2017;12(2):e0171539. doi: 10.1371/journal.pone.0171539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Um SJ, Choi YJ, Shin HJ, et al. Phase I study of autologous dendritic cell tumor vaccine in patients with non-small cell lung cancer. Lung Cancer. 2010;70(2):188–194. doi: 10.1016/j.lungcan.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu K, Kotera Y, Aruga A, Takeshita N, Takasaki K, Yamamoto M. Clinical utilization of postoperative dendritic cell vaccine plus activated T-cell transfer in patients with intrahepatic cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2012;19(2):171–178. doi: 10.1007/s00534-011-0437-y. [DOI] [PubMed] [Google Scholar]
- 27.Marks EI, Yee NS. Immunotherapeutic approaches in biliary tract carcinoma: Current status and emerging strategies. World J Gastrointest Oncol. 2015;7(11):338–346. doi: 10.4251/wjgo.v7.i11.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narita M, Kanda T, Abe T, et al. Immune responses in patients with esophageal cancer treated with SART1 peptide-pulsed dendritic cell vaccine. Int J Oncol. 2015;46(4):1699–1709. doi: 10.3892/ijo.2015.2846. [DOI] [PubMed] [Google Scholar]
- 29.Zhu H, Yang X, Li J, et al. Immune response, safety, and survival and quality of life outcomes for advanced colorectal cancer patients treated with dendritic cell vaccine and cytokine-induced killer cell therapy. Biomed Res Int. 2014;2014:603871–5. doi: 10.1155/2014/603871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burgdorf SK, Fischer A, Myschetzky PS, et al. Clinical responses in patients with advanced colorectal cancer to a dendritic cell based vaccine. Oncol Rep. 2008;20(6):1305–1311. [PubMed] [Google Scholar]
- 31.Sun TY, Yan W, Yang CM, et al. Clinical research on dendritic cell vaccines to prevent postoperative recurrence and metastasis of liver cancer. Genet Mol Res. 2015;14(4):16222–16232. doi: 10.4238/2015.December.8.12. [DOI] [PubMed] [Google Scholar]
- 32.Shimizu K, Kotera Y, Aruga A, et al. Postoperative dendritic cell vaccine plus activated T-cell transfer improves the survival of patients with invasive hepatocellular carcinoma. Hum Vaccin Immunother. 2014;10(4):970–976. doi: 10.4161/hv.27678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Ansary M, Mogawer S, Elhamid SA, et al. Immunotherapy by autologous dendritic cell vaccine in patients with advanced HCC. J Cancer Res Clin Oncol. 2013;139(1):39–48. doi: 10.1007/s00432-012-1298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi CJ, Ning YL, Han YS, et al. Autologous dendritic cell vaccine for estrogen receptor (ER)/progestin receptor (PR) double-negative breast cancer. Cancer Immunol Immunother. 2012;61(9):1415–1424. doi: 10.1007/s00262-011-1192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gelao L, Criscitiello C, Esposito A, et al. Dendritic cell-based vaccines: clinical applications in breast cancer. Immunotherapy. 2014;6(3):349–360. doi: 10.2217/imt.13.169. [DOI] [PubMed] [Google Scholar]
- 36.López MN, Pereda C, Segal G, et al. Prolonged survival of dendritic cell-vaccinated melanoma patients correlates with tumor-specific delayed type IV hypersensitivity response and reduction of tumor growth factor beta-expressing T cells. J Clin Oncol. 2009;27(6):945–952. doi: 10.1200/JCO.2008.18.0794. [DOI] [PubMed] [Google Scholar]
- 37.Nakai N, Hartmann G, Kishimoto S, Katoh N. Dendritic cell vaccination in human melanoma: relationships between clinical effects and vaccine parameters. Pigment Cell Melanoma Res. 2010;23(5):607–619. doi: 10.1111/j.1755-148X.2010.00736.x. [DOI] [PubMed] [Google Scholar]
- 38.Garg AD, Vandenberk L, Koks C, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016;8:328:ra27. doi: 10.1126/scitranslmed.aae0105. [DOI] [PubMed] [Google Scholar]
- 39.Li M, Han S, Shi X. In situ dendritic cell vaccination for the treatment of glioma and literature review. Tumour Biol. 2016;37(2):1797–1801. doi: 10.1007/s13277-015-3958-1. [DOI] [PubMed] [Google Scholar]
- 40.Pal SK, Hu A, Figlin RA. A new age for vaccine therapy in renal cell carcinoma. Cancer J. 2013;19(4):365–370. doi: 10.1097/PPO.0b013e31829d74b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Liao L, Tan J. Dendritic cell-based vaccination for renal cell carcinoma: challenges in clinical trials. Immunotherapy. 2012;4(10):1031–1042. doi: 10.2217/imt.12.107. [DOI] [PubMed] [Google Scholar]
- 42.Kongsted P, Borch TH, Ellebaek E, et al. Dendritic cell vaccination in combination with docetaxel for patients with metastatic castration-resistant prostate cancer: A randomized phase II study. Cytotherapy. 2017;19(4):500–513. doi: 10.1016/j.jcyt.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Reyes D, Salazar L, Espinoza E, et al. Tumour cell lysate-loaded dendritic cell vaccine induces biochemical and memory immune response in castration-resistant prostate cancer patients. Br J Cancer. 2013;109(6):1488–1497. doi: 10.1038/bjc.2013.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanyi JL, Chu CS. Dendritic cell-based tumor vaccinations in epithelial ovarian cancer: a systematic review. Immunotherapy. 2012;4(10):995–1009. doi: 10.2217/imt.12.100. [DOI] [PubMed] [Google Scholar]
- 45.Cannon MJ, Goyne H, Stone PJ, Chiriva-Internati M. Dendritic cell vaccination against ovarian cancer–tipping the Treg/TH17 balance to therapeutic advantage? Expert Opin Biol Ther. 2011;11(4):441–445. doi: 10.1517/14712598.2011.554812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui Y, Yang X, Zhu W, Li J, Wu X, Pang Y. Immune response, clinical outcome and safety of dendritic cell vaccine in combination with cytokine-induced killer cell therapy in cancer patients. Oncol Lett. 2013;6(2):537–541. doi: 10.3892/ol.2013.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15(7):e257–e267. doi: 10.1016/S1470-2045(13)70585-0. [DOI] [PubMed] [Google Scholar]
- 48.Butler JS. The yin and yang of the exosome. Trends Cell Biol. 2002;12(2):90–96. doi: 10.1016/s0962-8924(01)02225-5. [DOI] [PubMed] [Google Scholar]
- 49.Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013;32(3-4):623–642. doi: 10.1007/s10555-013-9441-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Srivastava A, Filant J, Moxley KM, Sood A, McMeekin S, Ramesh R. Exosomes: a role for naturally occurring nanovesicles in cancer growth, diagnosis and treatment. Curr Gene Ther. 2015;15(2):182–192. doi: 10.2174/1566523214666141224100612. [DOI] [PubMed] [Google Scholar]
- 51.Taïeb J, Chaput N, Zitvogel L. Dendritic cell-derived exosomes as cell-free peptide-based vaccines. Crit Rev Immunol. 2005;25(3):215–223. doi: 10.1615/critrevimmunol.v25.i3.30. [DOI] [PubMed] [Google Scholar]
- 52.Pitt JM, André F, Amigorena S, et al. Dendritic cell-derived exosomes for cancer therapy. J Clin Invest. 2016;126(4):1224–1232. doi: 10.1172/JCI81137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pitt JM, Charrier M, Viaud S, et al. Dendritic cell-derived exosomes as immunotherapies in the fight against cancer. J Immunol. 2014;193(3):1006–1011. doi: 10.4049/jimmunol.1400703. [DOI] [PubMed] [Google Scholar]
- 54.Viaud S, Terme M, Flament C, et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One. 2009;4(3):e4942. doi: 10.1371/journal.pone.0004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brody JD, Engleman EG. DC-based cancer vaccines: lessons from clinical trials. Cytotherapy. 2004;6(2):122–127. doi: 10.1080/14653240410005267. [DOI] [PubMed] [Google Scholar]
- 56.Zhou J, Ma P, Li J, Song W. Comparative analysis of cytotoxic T lymphocyte response induced by dendritic cells pulsed with recombinant adeno-associated virus carrying α-fetoprotein gene or cancer cell lysate. Mol Med Rep. 2015;11(4):3174–3180. doi: 10.3892/mmr.2014.3059. [DOI] [PubMed] [Google Scholar]
- 57.Giacca M, Zacchigna S. Virus-mediated gene delivery for human gene therapy. J Control Release. 2012;161(2):377–388. doi: 10.1016/j.jconrel.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 58.Bianco NR, Kim SH, Morelli AE, Robbins PD. Modulation of the immune response using dendritic cell-derived exosomes. Methods Mol Biol. 2007;380:443–455. doi: 10.1007/978-1-59745-395-0_28. [DOI] [PubMed] [Google Scholar]
- 59.Zitvogel L, Regnault A, Lozier A, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4(5):594–600. doi: 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]
- 60.Yin W, Ouyang S, Li Y, Xiao B, Yang H. Immature dendritic cell-derived exosomes: a promise subcellular vaccine for autoimmunity. Inflammation. 2013;36(1):232–240. doi: 10.1007/s10753-012-9539-1. [DOI] [PubMed] [Google Scholar]
- 61.Segura E, Amigorena S, Théry C. Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cells Mol Dis. 2005;35(2):89–93. doi: 10.1016/j.bcmd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 62.Utsugi-Kobukai S, Fujimaki H, Hotta C, Nakazawa M, Minami M. MHC class I-mediated exogenous antigen presentation by exosomes secreted from immature and mature bone marrow derived dendritic cells. Immunol Lett. 2003;89(2–3):125–131. doi: 10.1016/s0165-2478(03)00128-7. [DOI] [PubMed] [Google Scholar]
- 63.Morse MA, Garst J, Osada T, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3(1):9. doi: 10.1186/1479-5876-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Escudier B, Dorval T, Chaput N, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. 2005;3(1):10. doi: 10.1186/1479-5876-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dai S, Wei D, Wu Z, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther. 2008;16(4):782–790. doi: 10.1038/mt.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.André F, Chaput N, Schartz NE, et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol. 2004;172(4):2126–2136. doi: 10.4049/jimmunol.172.4.2126. [DOI] [PubMed] [Google Scholar]
- 67.Segura E, Guérin C, Hogg N, Amigorena S, Théry C. CD8+ dendritic cells use LFA-1 to capture MHC-peptide complexes from exosomes in vivo. J Immunol. 2007;179(3):1489–1496. doi: 10.4049/jimmunol.179.3.1489. [DOI] [PubMed] [Google Scholar]
- 68.Chaput N, Schartz NE, André F, et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol. 2004;172(4):2137–2146. doi: 10.4049/jimmunol.172.4.2137. [DOI] [PubMed] [Google Scholar]
- 69.Gilliet MF, Nestle FO. Generation of blood-derived human dendritic cells for antitumor immunotherapy. Methods Mol Med. 2001;64:297–303. doi: 10.1385/1-59259-150-7:297. [DOI] [PubMed] [Google Scholar]
- 70.Marsee DK, Pinkus GS, Yu H. CD71 (transferrin receptor): an effective marker for erythroid precursors in bone marrow biopsy specimens. Am J Clin Pathol. 2010;134(3):429–435. doi: 10.1309/AJCPCRK3MOAOJ6AT. [DOI] [PubMed] [Google Scholar]
- 71.Speeckaert MM, Speeckaert R, Delanghe JR. Biological and clinical aspects of soluble transferrin receptor. Crit Rev Clin Lab Sci. 2010;47(5–6):213–228. doi: 10.3109/10408363.2010.550461. [DOI] [PubMed] [Google Scholar]
- 72.Théry C, Duban L, Segura E, Véron P, Lantz O, Amigorena S. Indirect activation of naïve CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol. 2002;3(12):1156–1162. doi: 10.1038/ni854. [DOI] [PubMed] [Google Scholar]
- 73.Sobo-Vujanovic A, Munich S, Vujanovic NL. Dendritic-cell exosomes cross-present Toll-like receptor-ligands and activate bystander dendritic cells. Cell Immunol. 2014;289(1–2):119–127. doi: 10.1016/j.cellimm.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rao Q, Zuo B, Lu Z, et al. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology. 2016;64(2):456–472. doi: 10.1002/hep.28549. [DOI] [PubMed] [Google Scholar]
- 75.Lu Z, Zuo B, Jing R, et al. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J Hepatol. 2017;67(4):739–748. doi: 10.1016/j.jhep.2017.05.019. [DOI] [PubMed] [Google Scholar]