

Diffuse intrinsic pontine glioma (DIPG) and other histone H3 K27M (H3K27M) mutated diffuse midline gliomas (DMGs)1–5 are aggressive and universally fatal pediatric brain cancers6. Chimeric antigen receptor (CAR)-expressing T-cells have mediated impressive clinical activity in B-cell malignancies7–10, and recent results suggest benefit in CNS malignancies11–13. Here, we report that patient-derived H3K27M-mutant glioma cell cultures exhibit uniform, high expression of the disialoganglioside GD2. Anti-GD2 CAR T-cells incorporating a 4-1BBz costimulatory domain14 demonstrated robust antigen-dependent cytokine generation and DMG cell killing in vitro. In five independent patient-derived H3K27M+ DMG orthotopic xenograft models, systemic administration of GD2-CAR T-cells cleared engrafted tumors, save a small number of residual GD2-low glioma cells. To date, GD2-targeting CAR T-cells have been well-tolerated in clinical trials15–17. While GD2-CAR T-cell administration was tolerated in the majority of animals, peri-tumoral neuroinflammation during the acute phase of antitumor activity resulted in hydrocephalus that was lethal in a fraction of animals. Given the precarious neuroanatomical location of midline gliomas, careful monitoring and aggressive neurointensive care management will be required for human translation. With a cautious multidisciplinary clinical approach, GD2-CAR T-cell therapy for H3K27M+ diffuse gliomas of the pons, thalamus and spinal cord could prove transformative for these lethal childhood cancers.
Immune checkpoint inhibitors mediate substantial benefit in adult cancers refractory to traditional therapies, but have not yet demonstrated widespread benefit in sporadic childhood cancers, possibly due to the paucity of neoantigens in these diseases18. In contrast, CAR T cell therapies have mediated highly potent effects in childhood B-cell acute lymphoblastic leukemia (B-ALL). To identify potential targets for CAR T-cell immunotherapy in DIPG, we screened cell surface antigens using an antibody array in patient-derived DIPG cultures (Fig. 1a, Supplementary Table 1). Significant overlap between independent patient-derived cultures (Fig. 1b), suggests conservation of a core group of surface markers across DIPG patients. From these common targets, we observed that the disialoganglioside GD2 was expressed at high levels on each of the four patient-derived DIPG cultures screened (Fig. 1a). Hit validation by flow cytometry in six H3K27M+ DIPG cultures confirmed uniform, high GD2 expression in all H3K27M+ DIPG cultures examined, including those with the H3F3A K27M mutation (SU-DIPG-6, 13, 17, 25, 29) and the less-common HIST1H3B K27M mutation (SU-DIPG-21)2,19,20 (Fig. 1c). GD2 expression was far lower in two histone-3 WT pediatric high-grade gliomas (pHGG), including a case of H3WT DIPG (Fig. 1c). To assess whether transcriptional perturbations resulting from the H3K27M mutation might be linked to GD2 overexpression, we profiled gene expression of ganglioside synthesis enzymes in patient-derived DIPG and pHGG cultures and found higher expression of upstream ganglioside synthesis enzymes in H3K27M+ cultures (Supplementary Figure 1). Double immunostaining of primary human DIPG tissue for H3K27M to identify infiltrating malignant cells and GD2 confirmed local expression of GD2 in the native tumor context (Fig. 1d).
Figure 1. GD2 is an immunotherapy target in DIPG.
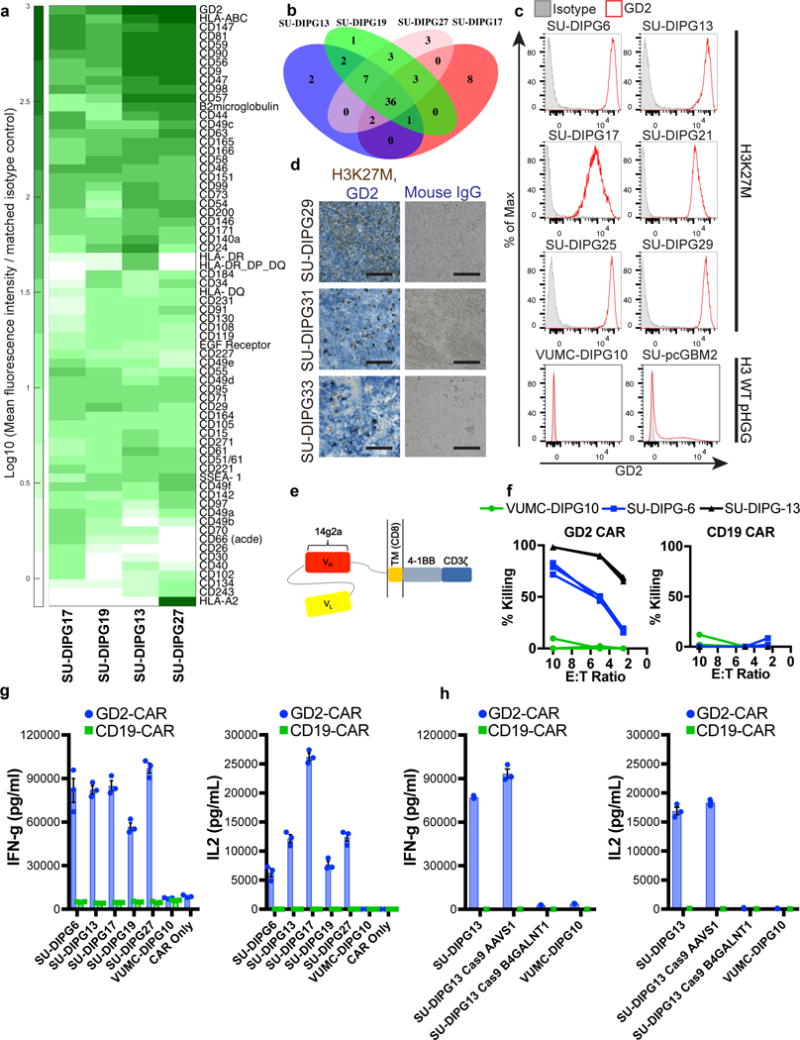
(a) Top 68 cell surface antigens expressed on DIPG as determined using flow cytometry screening of a monoclonal antibody panel in patient-derived DIPG cell cultures (complete data available in Supplementary Table 1). (b) Assessment of hit overlap between screened cultures identified a total of 36 hits present at a mean fluorescence intensity (MFI) of at least 10 times isotype control in all screened cultures. (c) Flow cytometry staining of histone 3 K27M DIPGs reveals high, generally homogeneous GD2 expression in contrast to histone 3 WT pediatric high-grade glioma cultures VUMC-DIPG10, diagnosed as a DIPG, and SU-pcGBM2, which arose in cortex. (d) Double immunohistochemistry of primary DIPG tumor specimens utilizing an antibody against mutant H3K27M (brown) to identify tumor cells and the anti-GD2 mAb 14g2a (blue) reveals extensive local GD2 expression in primary DIPG (scale bar = 100 microns). (e) Schematic of the GD2.4-1BB.z-CAR utilized in functional experiments. (f/g) GD2-CAR, but not CD19-CAR T-cells, mediate potent lysis (f) and produce high levels of IFN-gamma and IL-2 (g) following co-culture with GD2hi H3K27M DIPG cells, but not GD2lo/neg H3WT tumor cells. (h) GD2-CAR T-cells do not produce substantial levels of IFN-gamma or IL-2 following co-culture with H3K27M GD2neg line generated using CRISPR/Cas9 to knockout GD2 synthase compared with unmodified control cells or Cas9 targeting the control AAVS1 locus. Data as shown are mean±SEM, n=3 for in vitro cytokine and cell lysis experiments. In (f–h), n=3 independent samples; experiments in (c–d) were repeated twice.
GD2-targeting immunotherapies are currently under clinical and preclinical investigation in several diseases, including neuroblastoma, osteosarcoma, and melanoma14–17,21–24. Unlike monoclonal antibodies which do not efficiently cross the blood-brain barrier, activated T-cells can infiltrate the CNS following adoptive transfer7,25. We generated human GD2-targeting CAR T-cells incorporating a 4-1BBz costimulatory domain (GD2-CAR)14 (Fig. 1e) and observed significant GD2-dependent killing (Fig. 1f) and cytokine generation (Fig. 1g) upon exposure to patient-derived DIPG cultures relative to control CD19-CAR T-cells incorporating 4-1BBz (CD19-CAR). Notably, GD2-CAR T-cells do not produce significant cytokines or induce cell killing when exposed to the H3WT, GD2-negative VUMC-DIPG10 patient-derived DIPG culture, providing evidence of therapeutic specificity of GD2-CAR T-cells toward H3K27M DIPG. To further confirm the targeting specificity of GD2-CAR T-cells, we used CRISPR-Cas9-mediated deletion of GD2 synthase (B4GALNT1) in patient-derived DIPG cells to generate GD2 knockout DIPG cells (Supplementary Figure 2). Loss of GD2 antigen expression eliminated cytokine production by the GD2-CAR T-cells in comparison to untreated or DIPG cells electroporated with a control guide sequence targeting the AAVS1 locus (Fig. 1h). While GD2 expression in normal brain has been previously reported26, GD2-CAR T-cell therapy in human trials in which the targeting domain is derived from the same monoclonal antibody (14g2a) have reported no cases of significant neurotoxicity15–17. Moreover, intrathecal and intraventricular administration of anti-GD2 antibody radioconjugates in patients has been well-tolerated in clinical trials27,28. Taken together, these findings indicate specific reactivity of GD2-CAR T-cells to H3K27M+ glioma cells.
To evaluate in vivo efficacy of GD2-CAR T-cells against DIPG, we prepared orthotopic mouse xenografts of DIPG cultures derived from post-mortem patient tissue. DIPG cultures were transduced with a luciferase-expressing construct to enable longitudinal monitoring of tumor burden. These xenograft models faithfully recapitulate the diffusely infiltrating histology of DIPG29,30. Mice were distributed by tumor burden into equivalent treatment and control groups before receiving 1×107 GD2-CAR or CD19-CAR T-cells by a single intravenous injection 7-8 weeks after establishment of pontine xenografts. Within 40 days post-treatment (DPT), marked reductions in tumor burden were observed across two independent GD2-CAR T-cell treated cohorts of mice bearing SU-DIPG6 xenografts31 (Fig. 2a). Similar results were observed in a second patient-derived xenograft model, SU-DIPG13FL30 (Fig. 2e). All GD2-CAR treated animals demonstrated complete tumor clearance by bioluminescence imaging (Supplementary Figure 3). By contrast, no mice in the CD19-CAR T-cell control groups exhibited significant tumor regression. At 50 DPT brains were harvested, and immunostaining for the mutant histone H3K27M – present in all engrafted tumor cells – revealed that GD2-CAR treated tumors had been largely eradicated (Fig. 2c,d,g,h,i). The small number of H3K27M+ tumor cells that remain after treatment are negative for GD2 by immunostaining (Supplementary Figure 4). We hypothesize that the potency of the GD2-CAR in this model is driven by very high expression of the target antigen in H3K27M mutant DIPG, which was consistently higher than that present on GD2+ neuroblastoma and sarcoma cell lines (Supplementary Figure 5). This study adds to the growing evidence suggesting that CAR T-cells are more efficacious at high levels of target antigen expression32,33.
Figure 2. GD2-CAR T-cells mediate potent and lasting antitumor response in DIPG orthotopic xenografts.
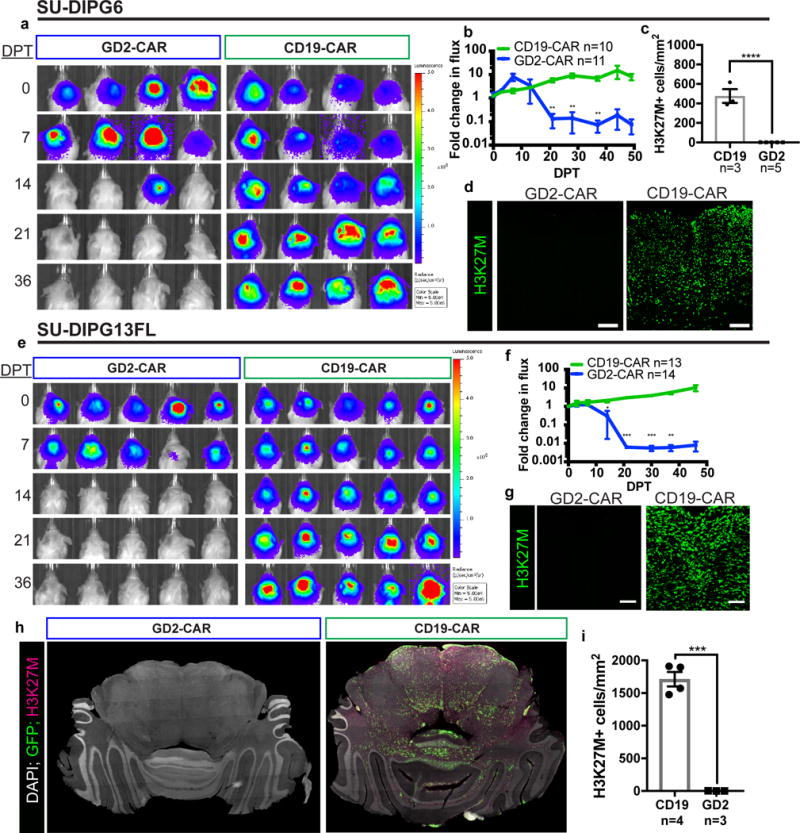
(a) Bioluminescence imaging of NSG mice xenografted with luciferase-expressing SU-DIPG6 into the pons (color map for all images: radiance, min = 5E4, max = 5E6) and infused intravenously with 1×107 GD2-CAR or CD19-CAR T-cells as designated. Each column represents one mouse; each row represents time point of imaging. Antitumor response was observed between 14 and 28 days post treatment (DPT) in GD2-CAR T-cell treated mice. (b) Tumor burden over time expressed as fold change in flux. (c) Quantification of H3K27M+ tumor cell density within infiltrated brainstem regions of SU-DIPG6 GD2-CAR (n=5) vs. CD19-CAR (n=3) T-cell treated mice. Within GD2-CAR T-cell-treated SU-DIPG6 xenografts, we identified approximately 36 H3K27M+ cells remaining per mouse in the sampled volume, compared with approximately 18,596 cells per mouse in the sampled volume of CD19-CAR T-cell treated controls. (d) Representative immunofluorescence confocal microscopy of CD19-CAR and GD2-CAR treated SU-DIPG6 tumors staining for the mutant histone H3K27M (green). (e–i) GD2-CAR activity in a second patient-derived orthotopic xenograft model of DIPG, SU-DIPG13FL. (e–f) Bioluminescent imaging over time as above. (g) Representative immunofluorescent confocal microscopy of SU-DIPG13FL xenografts treated with CD19- or GD2-CAR T-cells reveals clearance of H3K27M+ tumor cells. (h) Tiled immunofluorescence images across engrafted regions. (i) Quantification of H3K27M+ tumor cell density within infiltrated brainstem regions of SU-DIPG13FL. In SU-DIPG13FL xenografts, approximately 32 total H3K27M+ cells remained in the sampled volume of each GD2-CAR T-cell treated mouse, compared to approximately 31,953 cells per mouse in the sampled volume of CD19-CAR T-cell treated controls. Data as shown are mean±SEM. ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05 by unpaired 2-tailed Student’s t-test with Holm-Sidak correction for multiple comparisons applied for bioluminescence imaging data. Scale bars = 100 microns. Experiments were replicated in two independent cohorts of mice. In all panels, n values indicate independent animals.
Most patient-derived orthotopic DIPG xenograft models require many months for lethality, limiting the ability to monitor survival benefit due to development of xenogeneic graft versus host disease (GVHD) after treatment with human T-cells34. We therefore used SU-DIPG-13P*, a model that exhibits a dense pattern of growth histologically35, and is consistently lethal within one month. Substantial improvement in survival was seen in GD2-CAR treated animals compared with CD19-CAR treated controls (Fig. 3a). However, in one out of three independent cohorts, lethal toxicity occurred in several GD2-CAR T-cell treated animals, while all GD2-CAR T-cell treated animals in the other cohorts survived to endpoint (Supplementary Figure 6). GD2-CAR treated animals that survived the initial phase of glioma clearance returned to a visibly healthy state indistinguishable from untreated immunodeficient mice until the onset of GVHD symptoms 4+ weeks after CAR administration that invariably triggered endpoint criteria (Supplementary Figure 7). Histologic analysis of the brains of endpoint GD2-CAR treated animals reveals clearance of this high-burden tumor while surrounding neural tissues appear grossly normal (Fig. 3b).
Figure 3. GD2-CAR T-cell therapy improves survival in DIPG orthotopic xenografts.
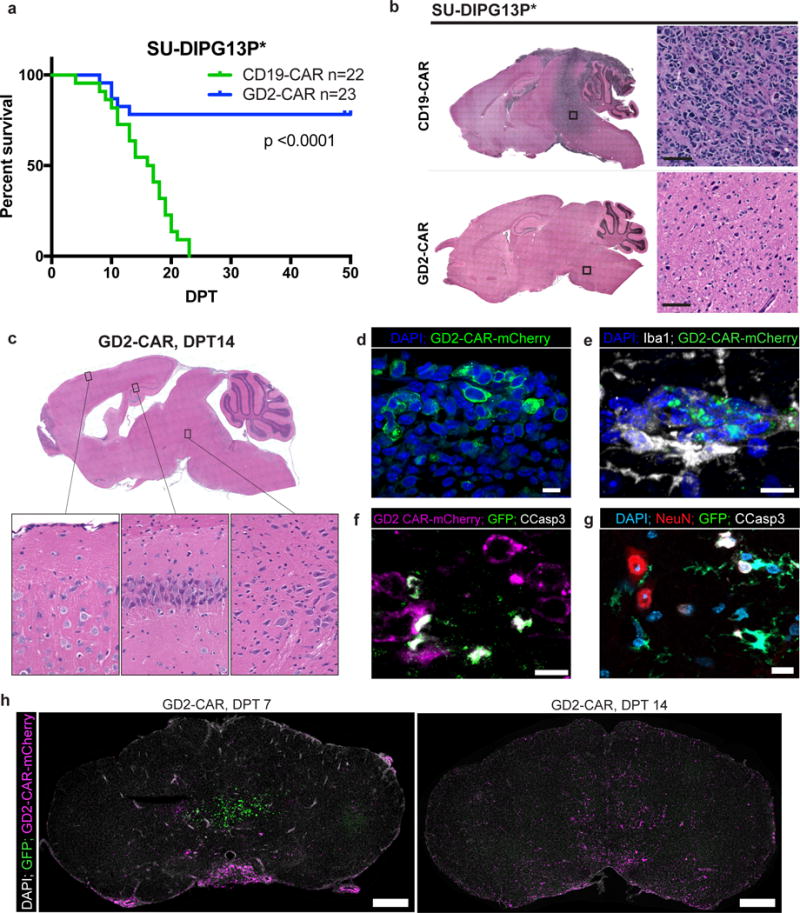
(a) Survival analysis of GD2-CAR T-cell treated orthotopic xenografts in SU-DIPG-13P*, a particularly aggressive patient-derived xenograft model of DIPG that is lethal within one month of engraftment, reveals a robust survival improvement in GD2-CAR T-cell treated animals (p<0.0001 Log-rank (Mantel-Cox) test, n=22 CD19-CAR and 23 GD2 CAR across 3 independent cohorts (Supplementary Figure 6)). While CD19-CAR T-cell treated xenografts were universally lethal by study endpoint, all GD2-CAR T-cell-treated animals that survived the acute toxicity of therapy survived to study endpoint at which time they manifested GVHD-like symptoms (Supplementary Figure 7). (b) Hematoxylin-eosin staining of SU-DIPG13P* xenografts at DPT50 demonstrate clearance by GD2-CAR T-cells of highly-infiltrative parenchymal tumor observed throughout the brain in CD19-CAR T-cell-treated controls and normal gross tissue architecture. (c) Hematoxylin-eosin staining of SU-DIPG6 GD2-CAR T-cell-treated xenograft analyzed at DPT14 demonstrates ventriculomegaly but histologically normal-appearing neurons in cortex, hippocampus, and brainstem (inset images). (d) Fluorescence microscopy of DPT7 SU-DIPG13FL xenografts reveals intravenously-administered GD2-CAR-mCherry T-cells infiltrating the engrafted tumor. (e) Representative image of infiltrating GD2-CAR-mCherry T-cells at DPT14 in a SU-DIPG13FL xenografted medulla demonstrates spatial association with Iba1+ macrophages. (f) Representative image of GD2-CAR-mCherry T-cell-mediated tumor cell killing with apoptosis of GFP+ tumor cells as evidenced by co-localization with cleaved caspase 3+. (g) Tumoricidal activity occurs in proximity to non-apoptotic NeuN+ neurons in the xenografted pons, shown here at DPT7 (Supplementary Figure 9). (h) Representative images of GD2-CAR-mCherry T-cells infiltrating the parenchyma of SU-DIPG13FL xenografts during the period of acute antitumor activity (Supplementary Figure 8). Experiments in (b–h) were repeated twice.
To better understand the etiology of treatment-related toxicity in these DIPG xenograft models, we examined the brains of treated SU-DIPG6 xenograft-bearing mice acutely at DPT14 (Fig. 3c). GD2-CAR treatment was accompanied by a widespread inflammatory infiltrate involving brain parenchyma, meninges and ventricles that was most prominent in the brainstem. Ventriculomegaly was observed, consistent with hydrocephalus. We observed histologically normal-appearing neurons present throughout the pons, hippocampus, and cortex of GD2-CAR T-cell-treated animals with no evidence of neuronal cell killing nor other tissue destruction in this model (Fig. 3c). Thus, neuropathological evaluation indicates that the toxicity described above results from brainstem inflammation and hydrocephalus due to fourth ventricular compression during the tumor-clearing interval and not on-target, off-tumor toxicity of GD2-CAR T-cells.
To visualize CAR T-cell infiltration into the parenchyma and tumor, we generated GD2-4-1BBz-mCherry and CD19-4-1BBz-mCherry fusion constructs (Fig. 3d). By DPT7, GD2-CAR T-cells are extensively distributed throughout the leptomeninges of treated animals, leptomeningeal tumor has been largely eradicated, and few mCherry+ cells are present within the brain parenchyma (Fig. 3h, Supplemental Figure 8). By DPT14, mCherry+ GD2-CAR T-cells had widely infiltrated throughout the parenchyma and numerous foci of Iba1+ macrophages (Fig. 3e) are present in the xenografted site, along with extensive apoptotic cleaved caspase 3+ cells (Fig. 3f). Notably, very few cleaved caspase 3+ apoptotic cells are neurons as identified by NeuN double immunostaining (10 total apoptotic neurons identified across 4 mice; Fig. 3g, Supplementary Figure 9). By DPT21, mCherry+ GD2-CAR T-cells remain present throughout the CNS; whereas few CD19-CAR T-cells infiltrate the parenchyma (Supplementary Figure 8). This supports a model in which intravenously administered GD2-CAR T-cells enter through the meningeal lymphatic system36, then subsequently infiltrate brain parenchyma, although the mechanism of CAR T-cell trafficking to the tumor remains to be defined. Given that resolution of tumor clearance and ventriculomegaly temporally coincide in treated animals, it is likely that antigen-specific antitumor activity, rather than on-target, off-tumor cell killing, precipitates neuroinflammation and edema during active tumoricidal activity that results in hydrocephalus. Understanding systemic and microenvironmental mechanisms that contribute to the resolution of acute CAR T-induced neuroinflammation is a critical area of future study that may be best addressed in immunocompetent models.
Recent WHO criteria place DIPG within a larger classification of diffuse midline gliomas (DMG) expressing the H3K27M mutation (Fig. 4a)37. In patient-derived cultures of pediatric H3K27M thalamic (QCTB-R059, derived from resection)4, and spinal cord (SU-pSCG1, derived post-mortem) DMGs, GD2 is also highly and uniformly expressed (Fig. 4b) and triggers IFNγ and IL-2 production by GD2-CAR T-cells (Fig. 4c). While these data might reasonably predict similar efficacy of GD2-CAR T-cells in these midline H3K27M gliomas, we reasoned that the neuroanatomical site of disease could impact outcomes of CAR T-cell therapy. To explore in vivo GD2-CAR T-cell efficacy in these H3K27M DMGs, we generated patient-derived orthotopic xenograft models of spinal cord (SU-pSCG1) and thalamic (QCTB-R059) glioma. When engrafted in the medulla to avoid the paralysis induced by injection into the spinal cord, widespread SU-pSCG1 growth is observed throughout the CNS (Fig. 4d). Systemic administration of GD2-CAR T-cells achieves potent and lasting tumor-clearing in this spinal cord glioma xenograft model, assessed both by longitudinal bioluminescence imaging (Fig. 4d,e) and endpoint histology, where approximately 16 residual H3K27M+ cells per animal remained across the sampled volume of 3 GD2-CAR T-cell treated animals (Fig. 4f,g). No mice from this cohort died during the tumor-clearing phase.
Figure 4. GD2 CAR T-cell therapy effectively clears other midline H3K27M mutant pediatric diffuse midline gliomas but is associated with toxicity in thalamic xenografts.
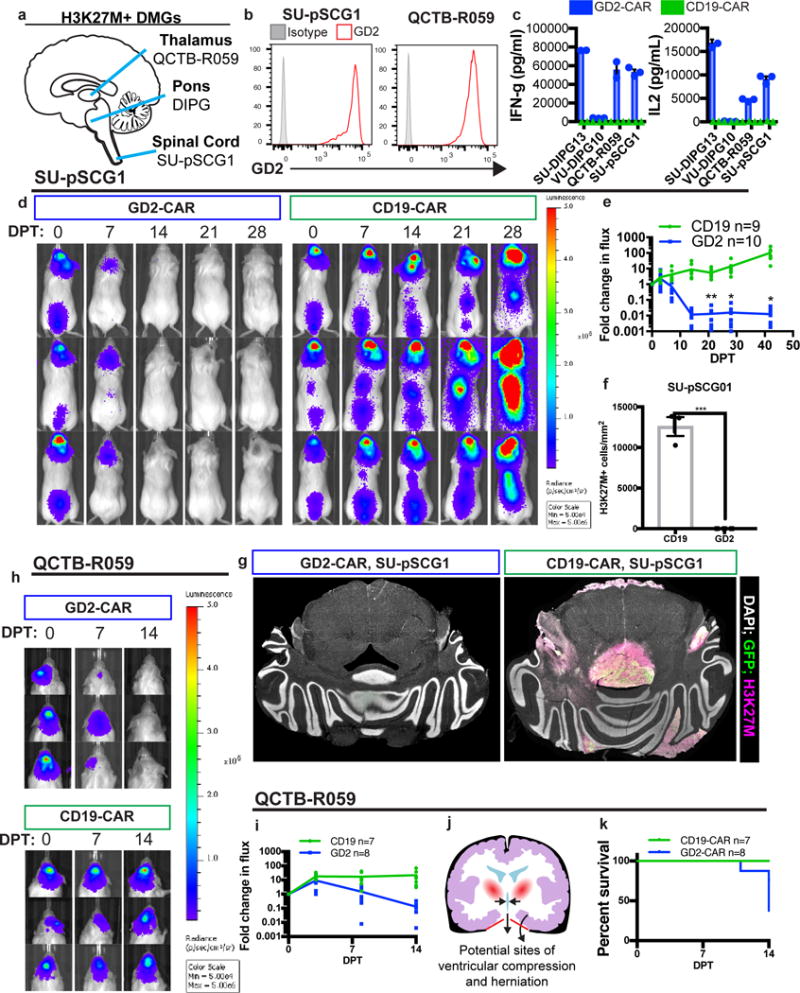
(a) Anatomic sites of origin of H3K27M+ DMGs. (b,c)Patient-derived culture models of H3K27M mutant tumors that arose in the thalamus (QCTB-R059) and spinal cord (SU-pSCG1) highly and uniformly express GD2 as assessed by flow cytometry (b) and induce antigen-dependent secretion of IFNγ and IL-2 when incubated with GD2 or CD19-CAR T-cells in vitro (c). (d,e) SU-pSCG1 cells stably transduced to express GFP and luciferase were engrafted into the medulla of NSG mice and treated with intravenous infusion of 1×107 GD2-CAR T-cells (n=10) or CD19-CAR T-cells (n=9), and substantial clearance of engrafted tumor was observed by DPT14. Each row represents one mouse over time. (f) Quantification of H3K27M+ cells remaining in SU-pSCG1 xenografts at study endpoint revealed near complete clearance of engrafted tumor in GD2-CAR T-cell treated (n=3) animals compared to CD19-CAR T-cell controls (n=3). (g) Tiled immunofluorescence images across affected regions (GFP = green; H3K27M = red, DAPI = white). (h–i) The H3K27M mutant patient-derived cell culture QCTB-R059 was orthotopically engrafted into the thalamus of NSG mice and treated by systemic administration of GD2 or CD19-CAR T-cells as described for SU-pSCG1. Tumor burden over time as determined by bioluminescence imaging illustrates marked reduction by DPT 14, and histological clearance in surviving animals at DPT 30 (Supplementary Figure 10). (j) Diagram showing the risk for 3rd ventricular compression and herniation through the tentorium cerebelli (red) accompanying inflammation in the thalamus. (k) GD2-CAR T-cell therapy-associated deaths in mice with thalamic xenografts observed by DPT14 highlight the hazards of immunotherapy for midline tumors. Data as shown are mean±SEM. ***p<0.001, *p<0.05 by unpaired 2-tailed Student’s t-test with Holm-Sidak correction for multiple comparisons, n=3 independent samples for in vitro cytokine experiments. Experiments in (b,c,e,f,g) were performed twice.
To evaluate efficacy in H3K27M thalamic glioma, we engrafted QCTB-R059 cells orthotopically in the thalamus (Fig. 4h). Tumor clearance was observed in this model (Fig. 4h,i, Supplementary Figure 10) on a similar time scale as observed for DIPG and spinal cord tumors discussed above. However substantial toxicity occurred in GD2-CAR T-cell treated animals during the period of maximal therapeutic effect (Fig. 4k). The results are reminiscent of “pseudoprogression”, well described following immunotherapy with checkpoint inhibitors38, and highlight the danger of a robust immunotherapeutic response and subsequent neuroinflammation in neuroanatomical locations intolerant of swelling. The thalamus – located just above the cerebellum tentorial notch – is a precarious location for edema, particularly when already expanded by tumor, and swelling in this location can precipitate hydrocephalus from third ventricular compression, increased intracranial pressure and lethal transtentorial herniation (Fig. 4j). Meticulous clinical monitoring and neurointensive management of edema will be required for successful clinical translation of this powerful immunotherapy, particularly for thalamic tumors, but may be insufficient to mitigate the peril of neuroinflammation at midline brain locations susceptible to herniation.
Although stark tumor clearance occurs in these xenograft models, the persistence of small numbers of tumor cells that are negative for GD2 expression by immunofluorescence staining suggests that multimodal therapy could be required to circumvent potential antigen escape (Supplementary Figure 4). While GD2 antibody-associated neuropathy has been observed in clinical trials, it is critical to note that in GD2-CAR T-cell human trials to date in which the targeting domain is derived from the same antibody as used here, treatment-associated neuropathy has not been observed15–17. The effects of robust T-cell infiltration and potent killing with associated inflammation within the tumor site represents a CNS tumor-specific element of the larger category of possible neurotoxic complications of CAR T-cell therapy, distinct from the observed CAR T-cell therapy-associated encephalopathy syndrome (CRES) that occurs in the context of cytokine release and appears to result from endothelial dysfunction39. The importance of careful monitoring for hydrocephalus and signs of increased intracranial pressure cannot be overemphasized and will require careful inpatient monitoring with frequent neurological and fundoscopic exams, and neuroimaging as indicated. Neurosurgical interventions, such as intraventricular shunt placement for relief of hydrocephalus or even craniectomy for decompression, may be required to support children through the phase of tumoricidal neuroinflammation.
In summary, we have identified GD2 as a novel immunotherapy target in H3K27M mutant DMGs, and demonstrated potent antitumor efficacy of GD2-CAR T-cells delivered systemically in five independent patient-derived orthotopic xenograft models representing three distinct H3K27M mutant glioma subtypes of the pons, thalamus and spinal cord. Upregulation of ganglioside synthesis pathway component genes in H3K27M mutant pediatric high-grade gliomas relative to H3 WT pediatric high-grade gliomas suggests this mutation drives GD2 overexpression. Tumor associated inflammation generated by CAR T-cell activity is tolerated by the majority of mice in this setting, suggesting the possibility for a therapeutic window with neurointensive management despite the precarious anatomic sites of these midline tumors, but putative tolerability of this approach remains to be demonstrated in children with H3K27M+ DMG. If the results in mouse models are predictive of results in humans, this immunotherapeutic strategy could be transformative for the outcomes of children with H3K27M-mutant diffuse midline gliomas.
Methods
DMG cultures
Patient-derived glioma cell cultures were generated as previously described40. Briefly, tumor tissue was dissociated mechanically and enzymatically (Liberase DH, Roche) prior to separation of myelin and debris by sucrose centrifugation. Neurosphere-generating cultures were maintained in serum-free media supplemented with B27 (ThermoFisher), EGF, FGF, PDGF-AA, PDGF-BB (Shenandoah Biotechnology), and Heparin (StemCell Technologies). All cultures were validated and monitored by STR-fingerprinting (Supplementary Table 2) and verified to be mycoplasma-free within the previous 6 months (MycoAlert Plus, Lonza). SU-DIPG6 and SU-DIPG13 have been previously referred to as and are identical to SU-DIPG-VI and SU-DIPG-XIII, respectively. Clinical characteristics and STR fingerprints of all DIPG and pSCG cultures30 along with QCTB-R0594 used here have been previously reported. For all studies using human tissue, informed consent was obtained per guidelines of the approved Stanford Institutional Review Board protocol.
Cell surface screening
Cell surface markers present on DIPG cell cultures were screened using a panel of monoclonal antibodies against human cell surface markers (Lyoplate, BD Biosciences). Low passage (<12) DIPG cultures expanded from tumor tissue collected at autopsy in serum-free, neurosphere forming conditions40 were allotted to 96 well plates and blocked with 1ug of goat IgG per million cells to reduce nonspecific binding of secondary antibodies subsequently used in the assay. Cells were then incubated sequentially with primary and secondary antibodies with intermediate wash steps according to the manufacturer’s instructions. Dead cells were then labeled with a Live/Dead violet stain (ThermoFisher), and following washes cells were fixed in 1% PFA for 10 minutes at room temperature. The following day, stained cells were analyzed by flow cytometry. Doublets and dead cells were excluded by gating, and the mean fluorescence intensity of antibody labeling for each target on the panel was normalized to the mean fluorescence intensity for the matched isotype control per the manufacturer’s recommendations.
Immunohistochemistry and light microscopy
Primary DIPG patient tumor samples were fixed overnight in 4% paraformaldehyde/PBS upon arrival, and then transferred to 30% sucrose until the tissue samples sank (2-3 days). Tissues were then transferred to cryomolds and embedded in OCT (TissueTek). 10 micron cryosections were generated on a cryostat (Leica), and endogenous peroxidase activity was neutralized (Bloxall, Vector Laboratories) prior to permeabilization (0.3% Triton X-100, TBS) and blocking (5% horse serum, Vector Laboratories). Sequential double-immunohistochemistry was conducted for H3K27M (Abcam ab190631, 1:1000, 1hr RT) and GD2 (14g2a, BD, 1:500, 1hr at RT). H3K27M was developed with a peroxidase secondary (ImmPRESS VR anti-rabbit IgG, Vector Laboratories, 30 minutes at RT) and DAB substrate (BD). Under these conditions, H3K27M+ cells could be routinely identified in multiple tissues confirmed to bear both H3F3A and HISTH1B3 mutations by Sanger sequencing, but staining was absent in tissues from non-DIPG tumor samples confirmed to be H3 WT by Sanger sequencing. After quenching the DAB substrate development in TBS and staining with the 14g2a primary antibody, GD2 signal was developed using a polymer-based alkaline phosphatase secondary (ImmPRESS AP anti-mouse IgG, Vector Laboratories, 30 minutes at RT) and blue alkaline phosphatase substrate (Vector Blue AP substrate kit, Vector Laboratories, 150 seconds at RT). AP development was quenched in TBS, and samples were mounted and imaged (Zeiss AxioObserver). For hematoxylin-eosin staining, mice were deeply anesthetized by ip injection of tribromoethanol and perfused transcardially with cold PBS, brains were removed and fixed overnight in 4% paraformaldehyde/PBS. Brains were then transferred to 70% ethanol and subsequently embedded in paraffin, sectioned, and stained with hematoxylin/eosin. H&E histology was analyzed by an expert neuropathologist (H.V.)
Immunofluorescence and confocal microscopy
Mice were deeply anesthetized with tribromoethanol (Avertin) before being perfused transcardially with cold PBS. Brains were then removed and fixed overnight in 4% PFA/PBS before being transferred to 30% sucrose and allowed to sink (2-3 days). Serial 40 micron coronal sections were then cut on a freezing microtome and floated in a tissue cryoprotectant solution (glycerol, ethylene glycol, phosphate buffer) before storage at -20C. Serial sections were then stained overnight at 4C. Primary antibodies used were: rabbit anti-H3K27M (Abcam, 1:1000), rabbit anti-cleaved caspase-3 (Cell Signaling Technology, 9661, 1:250), mouse anti-NeuN (Millipore, MAB377, 1:500), and rabbit Iba1 (Wako, 019-19741, 1:500). Secondary antibodies raised in donkey and conjugated with AlexaFluor 594 or 647 were used at 4C overnight to detect primary labeling (Jackson ImmunoResearch, 1:500). Mounted samples were imaged by confocal microscopy (Zeiss LSM710), and acquired Z stacks through the tumor region were flattened by maximum intensity projection (ImageJ). To quantify tumor cell density, cells within the borders of infiltrating tumor in acquired micrographs were counted and normalized to the tumor area (ImageJ), and the sum of all cells was normalized to the total area investigated across 3-4 sections for each animal in a 1:12 series.
RT-qPCR
Cultures were plated in triplicate under standard growth conditions and harvested in Trizol 24 hours later. After DNAse treatment, extracted RNA was reverse transcribed (Maxima first strand, ThermoFisher) and utilized as template for qPCR reactions (Maxima SYBR green, ThermoFisher). Primers utilized are listed in Supplementary Table 3. No-template and RT- controls did not significantly amplify. For each of the triplicate cultures, technical duplicates were assayed and averaged. Relative expression was then calculated by the deltaCt method.
CAR construction, retroviral vector production and T-cell transduction
The GD2-CAR incorporates the scFv derived from the 14g2a monoclonal antibody, a CD8 transmembrane domain, and 4-1BB and TCR zeta signaling endodomains. The CD19-CAR was similarly configured but incorporated the FMC63 scFv. The GD2-4-1BBz and CD19-4-1BBz CAR retroviral vectors were constructed as previously described41. GD2-4-1BBz and CD19-4-1BBz CAR-encoding retroviral supernatants were produced via transient transfection of the 293GP cell line, as described42. Briefly, 293GP cells were transfected on poly-d-lysine coated plates via Lipofectamine 2000 (Life Technologies) with the plasmids encoding the CARs and the RD114 envelope protein. Supernatants were collected 48 and 72 h after transfection. Isolated human T-cells were activated with anti-CD3/CD28 beads (Life Technologies) in a 3:1 bead:cell ratio with 40 IU/ml IL-2 for 3 days. Activated T-cells were then retrovirally transduced on days 3 and 4 as described14 using Retronectin (Takara) coated plates, and cultured in 300 IU/ml IL-2. Anti-CD3/CD28 beads were removed on day 5, and media and IL-2 were changed every 2-3 d. Flow cytometry using the 1A7 anti-idiotype antibody43 for the GD2-4-1BBz CAR and the FMC63 anti-idiotype antibody 136.20.144 or Protein L (Pierce) for the CD19-4-1BBz CAR was used to assess transduction efficiency for each batch of CAR T cells.
In vitro cytokine generation and cell killing
In vitro cell killing activity of GD2 and CD19-CAR T-cells was assessed with a luciferase-based assay as previously described41. Tumor cells lentivirally transduced to stably express firefly luciferase (10,000 tumor cells per well) were co-incubated with GD2 or CD19-CAR T-cells for 24 hours at effector-to-target (E:T) ratios ranging from 10:1 to 2.5:1. The Steady-Glo® Luciferase Assay System (Promega) was used to measure residual luciferase activity from remaining tumor target cells, and lysis was calculated as follows: percent lysis = 100 – [[(average signal from T-cell–treated wells)/(average signal from untreated target wells)] × 100].
Cytokine production by CAR T-cells in vitro was evaluated by co-incubation of CAR+ T-cells with target tumor cells at a 1:1 ratio (100,000 cells each), with CAR+ T-cell counts incorporating the transduction efficiency as assessed by anti-idiotype staining and flow cytometry. The total number of T-cells used for the control CD19-4-1BBz CAR T-cells matched the number used for the GD2-4-1BBz CAR T-cells to ensure that the total number of T-cells remained consistent across groups. After 24 hours, supernatants were harvested and cytokine levels measured by ELISA for IL-2 and IFN-gamma (BioLegend).
T-cell proliferation
GD2-CAR T-cells on Day 15 of culture were labeled with Cell Trace Violet (ThermoFisher) according to manufacturer protocol. Labeled GD2-CAR T-cells were either incubated with no tumor, VUMC-DIPG10 (GD2 negative, H3K27M negative) or SU-DIPG13 (GD2 positive, H3K27M positive). After five days of incubation, cells were collected and analyzed by flow cytometry for proliferation. Analysis was performed on CAR+ T-cells only as identified by anti-idiotype staining.
Orthotopic xenograft generation and treatment
Orthotopic DIPG xenografts were generated as previously described31. All in vivo experiments were approved by the Stanford University Institutional Care and Use Committee and performed in accordance with institutional guidelines. Animals were housed according to institutional guidelines with free access to food and water on a 12-hour light/dark cycle. Briefly, patient-derived DIPG cell cultures (SU-DIPG6 and SU-DIPG13-FL) previously transduced with a lentivirus expressing eGFP and firefly luciferase driven by the CMV promoter were infused by stereotaxic injector (Stoelting) into the pons (coordinates lambda AP-3mm, DV -3mm, 100k cells) of cold-anesthetized newborn (P0-2) NOD-scidIL2rg−/− (NSG) mice (Jax). Orthotopic pediatric spinal cord glioma xenografts were generated by stereotaxic injection of SU-pSCG1 transduced with a lentivirus expressing eGFP and firefly luciferase driven by the CAG promoter into the medulla of isoflurane-anesthetized P35 NSG mice (coordinates lambda ML+0.7mm, AP-3.5mm, DV-4.5mm, 600k cells). Orthotopic thalamic glioma xenografts were generated by stereotaxic injection of QCTB-R059 transduced with a lentivirus expressing eGFP and firefly luciferase driven by the CMV promoter into the thalamus of isoflurane-anesthetized P35 NSG mice (coordinates bregma ML+0.8mm, AP-1mm, DV -3.5mm, 600k cells). Tumors were then allowed to develop for 60 days. Prior to treatment, tumor burden was assessed by in vivo luminescence imaging (IVIS Spectrum, PerkinElmer), and total flux was calculated by included software (Living Image, Perkin Elmer) as the radiance through standard circular ROIs centered on the animal’s head. Paired background regions were quantified using circular ROIs over the animal’s flank where no significant luminescence was detected above background. Animals were rank-ordered by tumor burden and distributed sequentially into GD2 or CD19-CAR treatment groups, such that populations of equivalent initial tumor burden underwent each arm of therapy. Initial burden assessed in this manner was equivalent across treatment groups and engrafted cell lines (Supplementary Figure 3). SU-DIPG13P* cells were injected into the pons of isoflurane-anesthetized P35 NSG mice (coordinates lambda ML+1mm, AP-0.8mm, DV-5mm, 600k cells) and allowed to develop for 14 days before T-cell administration. CAR T-cells with concentrations adjusted to deliver 1×107 transduced cells in 200 microliters of PBS (assessed by idiotype staining using flow cytometry, routinely >60%) were then administered by intravenous injection into the tail vein of animals. Where transduction efficiencies varied between GD2-CARs and CD19-CARs, the concentration of CD19-CAR cells was adjusted to match the total dose of human T-cells present in the GD2-CAR infusion. Tumor burden was monitored longitudinally by in vivo luminescence imaging. Due to the obvious nature of GD2-CAR response, blinding in initial cohorts was deemed ineffective and subsequently not performed. All images were scaled to display minimum flux intensity as 5E4 and maximum as 5E6, then images of individual animals were arranged with like-treated animals in the cohort for display in figures. Trial endpoint at 50 days post treatment was determined in initial cohorts where substantial hair loss, reduced activity, and weight loss in both GD2-CAR and CD19-CAR groups that triggered morbidity criteria for euthanasia.
CRISPR/Cas9-mediated deletion of GD2 synthase
Deletion of GD2 synthase (B4GALNT1) in SU-DIPG13 cells was accomplished by electroporation of DIPG13 with Cas9:sgRNA ribonucleoprotein complexes as previously described45. Briefly, guide RNAs targeting exon1 of B4GALNT1 (CGUCCCGGGUGCUCGCGUAC and CCGGCUACCUCUUGCGCCGU, Synthego) were incubated with Cas9 nuclease to form ribonucleoprotein complexes and electroporated with an Amaxa 4-D nucleofector (SE Buffer, program DS-112). In parallel, a control gRNA targeting the AAVS1 locus46 (GGGGCCACUAGGGACAGGAU) was electroporated with Cas9 nuclease as a ribonucleoprotein complex using identical parameters. GD2-negative cells electroporated with B4GALNT1-targeting gRNAs were isolated by FACS sorting, and deletion was confirmed by Sanger sequencing and TIDE analysis47.
Statistics and Reproducibility
Statistical tests were conducted using Prism (GraphPad) software unless otherwise indicated. Gaussian distribution was confirmed by the Shapiro–Wilk normality test. For parametric data, unpaired, two-tailed Student’s t-tests and one-way ANOVA with Tukey’s post hoc tests to further examine pairwise differences were used. For survival analysis, a log-rank (Mantel-Cox) test was used. A level of P < 0.05 was used to designate significant differences. On the basis of the variance of xenograft growth in control mice, we used at least 3 mice per treatment group to give 80% power to detect an effect size of 20% with a significance level of 0.05. For all animal experiments, the number of independent mice used is listed in figure legend. For each of the five patient-derived xenograft models used, at least two independent cohorts were tested (i.e. independent litters of mice on different days with independent batches of cells.) For cytokine and in vitro cell killing experiments, n=3 and experiments were repeated twice.
Data Availability Statement
The datasets generated during the current study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank the following for generously providing cell cultures: Andrew Moore and Chris Jones for QCTB R059, Robert Seeger for CHLA136, 255, Chand Khanna for MG63-3, and Lee Helman for EW8 and TC32.
This work was supported by a Stand Up To Cancer—St Baldrick's—National Cancer Institute Pediatric Dream Team Translational Cancer Research Grant (C.L.M.). Stand Up To Cancer is a programme of the Entertainment Industry Foundation administered by the American Association for Cancer Research. C.L.M is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program. The authors gratefully acknowledge support from the National Institute of Neurological Disorders and Stroke (F31NS098554 to C.W.M. and R01NS092597 to M.M.), Abbie’s Army Foundation (M.M.), Unravel Pediatric Cancer (M.M.), Maiy’s Miracle Foundation (E.P.A.), Stella S. Jones Foundation (M.M.), McKenna Claire Foundation (M.M.), Alex’s Lemonade Stand Foundation (M.M.), The Cure Starts Now Foundation and DIPG Collaborative (M.M.), Lyla Nsouli Foundation (M.M.), Declan Gloster Memorial Fund (M.M.), N8 Foundation (M.M.), Fly a Kite Foundation (M.M.), Liwei Wang Research Fund (M.M.), Virginia and D.K. Ludwig Fund for Cancer Research (M.M.), Sam Jeffers Foundation (M.M.), Michael Mosier DEFEAT DIPG Foundation (M.M.), ChadTough Foundation (M.M.), Child Health Research Institute at Stanford and SPARK program (M.M.), and the Anne T. and Robert M. Bass Endowed Faculty Scholarship in Pediatric Cancer and Blood Diseases (M.M.).
Footnotes
Author Contributions
C.W.M and E.P.A performed the antibody array screening. C.W.M. and M.M. identified GD2 as a target in H3K27M+ diffuse midline gliomas. C.W.M. and S.S. performed IHC and immunofluorescence microscopy on primary and xenograft tissue. L.L. and R.G.M. designed CAR constructs. R.G.M. and M.K. prepared CAR T cells for in vivo experiments. C.W.M. and P.J.W. conducted in vivo experiments. R.G.M., M.K. and S.P.R. performed in vitro T cell experiments and flow cytometry. E.H. contributed VUMC-DIPG10 and data on ganglioside synthesis pathway expression. H.V. performed neuropathological review of brain tissue. S.H. performed CRISPR/Cas9-mediated gene editing. C.W.M., R.G.M., H.V., M.M. and C.L.M contributed to data analysis and interpretation. C.W.M., M.M., R.G.M., and C.L.M wrote the manuscript. C.W.M. and R.G.M. made the figures. M.M. and C.L.M. supervised all aspects of the work.
Competing Financial Interests Statement
C.L.M, M.M., R.G.M., and C.W.M. are inventors on a patent application for GD2-directed CAR use for H3K27M DMG.
References
- 1.Paugh BS, et al. Novel Oncogenic PDGFRA Mutations in Pediatric High-Grade Gliomas. Cancer Research. 2013;73:6219–6229. doi: 10.1158/0008-5472.CAN-13-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buczkowicz P, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46:451–456. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor KR, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46:457–461. doi: 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu G, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones C, et al. Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro-oncology. 2016 doi: 10.1093/neuonc/now101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee DW, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davila ML, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maude SL, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. New Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardner RA, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown CE, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. New Engl J Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed N, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017 doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Rourke DM, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long AH, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature Medicine. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pule MA, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louis CU, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heczey A, et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther. 2017;25:2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majzner RG, Heitzeneder S, Mackall CL. Harnessing the Immunotherapy Revolution for the Treatment of Childhood Cancers. Cancer Cell. 2017;31:476–485. doi: 10.1016/j.ccell.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Khuong-Quang DA, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 21.Thomas S, Straathof K, Himoudi N, Anderson J, Pule M. An Optimized GD2-Targeting Retroviral Cassette for More Potent and Safer Cellular Therapy of Neuroblastoma and Other Cancers. PLoS One. 2016;11:e0152196. doi: 10.1371/journal.pone.0152196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long AH, et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol Res. 2016;4:869–880. doi: 10.1158/2326-6066.CIR-15-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu AL, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perez Horta Z, Goldberg JL, Sondel PM. Anti-GD2 mAbs and next-generation mAb-based agents for cancer therapy. Immunotherapy. 2016;8:1097–1117. doi: 10.2217/imt-2016-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong JJ, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16:4892–4898. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki K. The pattern of mammalian brain gangliosides. II. Evaluation of the extraction procedures, postmortem changes and the effect of formalin preservation. J Neurochem. 1965;12:629–638. doi: 10.1111/j.1471-4159.1965.tb04256.x. [DOI] [PubMed] [Google Scholar]
- 27.Kramer K, et al. Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neurooncol. 2010;97:409–418. doi: 10.1007/s11060-009-0038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kramer K, et al. A phase II study of radioimmunotherapy with intraventricular 131 I-3F8 for medulloblastoma. Pediatr Blood Cancer. 2017 doi: 10.1002/pbc.26754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monje M, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A. 2011;108:4453–4458. doi: 10.1073/pnas.1101657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin EY, et al. Neural Precursor-Derived Pleiotrophin Mediates Subventricular Zone Invasion by Glioma. Cell. 2017;170:845–859 e819. doi: 10.1016/j.cell.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grasso CS, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21:555–559. doi: 10.1038/nm.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fry TJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2017 doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker AJ, et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol Ther. 2017;25:2189–2201. doi: 10.1016/j.ymthe.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ali N, et al. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rgammanull mice display a T-effector memory phenotype. PLoS One. 2012;7:e44219. doi: 10.1371/journal.pone.0044219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagaraja S, et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell. 2017;31:635–652 e636. doi: 10.1016/j.ccell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Louveau A, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louis DN, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 38.Wolchok JD, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 39.Gust J, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017 doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 40.Lin GL, Monje M. A Protocol for Rapid Post-mortem Cell Culture of Diffuse Intrinsic Pontine Glioma (DIPG) J Vis Exp. 2017 doi: 10.3791/55360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lynn RC, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. 2015;125:3466–3476. doi: 10.1182/blood-2014-11-612721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haso W, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sen G, Chakraborty M, Foon KA, Reisfeld RA, Bhattacharya-Chatterjee MB. Induction of IgG antibodies by an anti-idiotype antibody mimicking disialoganglioside GD2. J Immunother. 1998;21:75–83. doi: 10.1097/00002371-199801000-00010. [DOI] [PubMed] [Google Scholar]
- 44.Jena B, et al. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS One. 2013;8:e57838. doi: 10.1371/journal.pone.0057838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hendel A, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu VT, et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 47.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the corresponding author upon reasonable request.