

Summary
Androgen receptor splice variant 7 (AR-V7) is crucial for prostate cancer progression and therapeutic resistance. We show that, independent of ligand, AR-V7 binds both androgen-responsive elements (ARE) and non-canonical sites distinct from full-length AR (AR-FL) targets. Consequently, AR-V7 not only recapitulates AR-FL’s partial functions but regulates an additional gene-expression program uniquely via binding to gene promoters rather than ARE enhancers. AR-V7 binding and AR-V7-mediated activation at these unique targets do not require FOXA1 but rely on ZFX and BRD4. Knockdown of ZFX or select unique targets of AR-V7/ZFX, or BRD4 inhibition, suppresses growth of castration-resistant prostate cancer cells. We also define an AR- V7 direct target gene signature that correlates with AR-V7 expression in primary tumors, differentiates metastatic prostate cancer from normal, and predicts poor prognosis. Thus, AR-V7 has both ARE/FOXA1 canonical and ZFX-directed non-canonical regulatory functions in the evolution of anti-androgen therapeutic resistance, providing information to guide effective therapeutic strategies.
eTOC Blurb
By cistrome profiling of endogenous androgen receptor (AR) versus an AR splice variant, AR-V7, Cai et al uncovered non-canonical pathways uniquely targeted by ARV7 and ZFX, a previously unknown AR-V7 partner. Targeting cofactors (ZFX or BRD4) or non-canonical downstream pathways of AR-V7 provides potential therapeutic ways for treating prostate cancer.
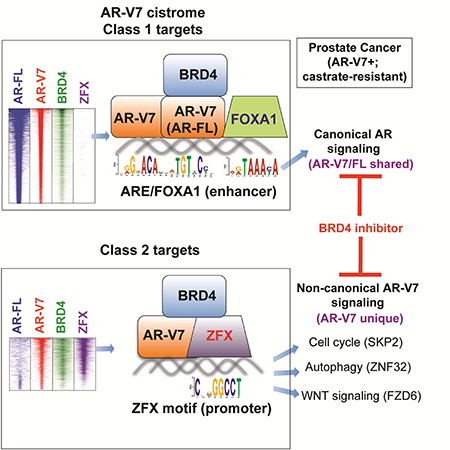
Graphical Abstract
Introduction
Androgen and androgen receptor (AR)-mediated signaling and gene transcription programs are pivotal for prostate tumorigenesis (Watson et al., 2015). Androgen deprivation based therapy of prostate cancer continues to improve with the FDA- approved inhibitors abiraterone (de Bono et al., 2011) and enzalutamide (MDV3100) (Scher et al., 2012; Tran et al., 2009). However, therapy resistance develops ultimately owing to various mechanisms, including AR gene amplification or mutation (Chen et al., 2004; Gottlieb et al., 2012; Taylor et al., 2010; Visakorpi et al., 1995), intratumoral androgen production (Montgomery et al., 2008), expression of AR splice variants (AR- Vs) such as AR-V7 (Antonarakis et al., 2014; Dehm et al., 2008; Guo et al., 2009; Hornberg et al., 2011; Hu et al., 2009; Li et al., 2013; Sun et al., 2010; Watson et al., 2010), and cell lineage switch (Ku et al., 2017; Mu et al., 2017). AR-Vs are detected by sensitive methods in early-stage prostate cancer and their expression appears to increase substantially in castration-resistant prostate cancer (CRPC) patients, indicating a tumor evolution process involving AR-V7 (Antonarakis et al., 2014; Dehm et al., 2008; Guo et al., 2009; Hu et al., 2009; Hu et al., 2011; Miyamoto et al., 2015; Sun et al., 2010; Watson et al., 2010). AR-V7 (also known as AR3) contains the N-terminal transactivation and DNA-binding domains but lacks the ligand-binding domain that exists in full-length AR (AR-FL) (Figures 1A and S1A). AR-V7 exhibits a ligandindependent, constitutive activation function and its expression is correlated with resistance to abiraterone or enzalutamide treatment in the clinic (Antonarakis et al., 2014). Profiling of AR-Vs revealed a ligand-independent recruitment to androgen- responsive elements (AREs), providing a mechanism by which AR-Vs sustain tumor growth without ligand (Chan et al., 2015; Lu et al., 2015); however, antibodies used in these studies cannot differentiate AR-Vs from AR-FL, raising a question of whether AR- Vs have regulatory functions distinctive from AR-FL. Indeed, the broader alterations in the phenotype of CRPC occur seemingly beyond the simple maintenance of persistent ARE-dependent transcriptional control. Thus, the full oncogenic mechanisms by which AR-V7 mediates development of advanced prostate cancer remain elusive. Elucidating mechanisms underpinning drug resistance may provide therapeutic approaches for targeting AR-V7 in advanced disease.
Figure 1. ChIP-Seq of endogenous AR-FL, AR-V7 and BRD4 in CRPC cells under different compound treatment conditions.

(A) Exons encoding the AR-FL or AR-V7 isoform. CE, cryptic exon; DBD, DNA-binding domain; LBD, ligand-binding domain. Epitopes recognized by isoform-specific antibodies are labeled in red.
(B) Antibody specificity shown by immunoblotting of 293 cells transfected with AR-FL or AR-V7.
(C) Antibody specificity confirmed by ChIP-qPCR of canonical ARE enhancers (e) in three prostate cancer cell lines that express AR-FL only (LNCaP), or both (22Rv1) or neither (PC3) of AR-FL and AR-V7.
(D) Heatmap of AR-FL (blue), AR-V7 (red) and BRD4 (green) ChIP-Seq signals at enhancers (top) and promoters (bottom) in ligand-starved 22Rv1 cells after a 6-hour treatment with vehicle, 10 nM of DHT, DHT plus 10 pM of MDV3100 (+D/M), or DHT plus 500 nM of JQ1 (+D/J).
(E) Venn diagram shows common and solo binding for AR-V7 identified in ligand- starved 22Rv1 cells (vehicle), relative to AR-FL peaks identified in DHT-treated 22Rv1 cells.
(F) Heatmap showing ChIP-Seq peaks that are common to AR-V7 in ligand-starved 22Rv1 cells (vehicle) and AR-FL in DHT-stimulated cells, with the corresponding BRD4 binding shown on the right.
(G) Averaged AR-FL (left) and AR-V7 (right) ChIP-Seq read density for the AR-FL/V7 common peaks (black) or AR-V7 solo peaks (red) as defined above in E.
(H) Pie chart showing distribution of the defined common and solo sites of AR-FL and AR-V7 among promoters and distal or intergenic enhancers in 22Rv1 cells.
(I) The most enriched motif at AR-FL or AR-V7 ChIP-Seq peaks.
(J) ChIP-Seq profile of AR-FL, AR-V7 and BRD4 at AR canonical targets, PSA/KLK3 and KLK2, in 22Rv1 cells that were ligand-starved followed by treatment of the indicated compound.
(K) Heatmap of AR-FL, AR-V7 and BRD4 ChIP-Seq signals at the AR-V7-solo binding sites defined in E.
(L) ChIP-Seq profile of AR-FL, AR-V7 and BRD4 at SKP2 in 22Rv1 cells under the indicated treatment condition.
(M) Venn diagram illustrates common and solo binding for AR-V7 identified in ligand- starved VCaP cells, compared to AR-FL peaks in DHT-treated VCaP cells.
(N) Venn diagram shows overlap between the AR-V7 unique sites identified in 22Rv1 and VCaP cells.
See also Figures S1–3 and Table S1.
To this end, we performed genomic profiling of AR-V7 and AR-FL targets in same CRPC cells, which includes chromatin immunoprecipitation followed by sequencing (ChIP-Seq) with AR isoform-specific antibodies and RNA-Seq after isoform-specific knockdown (KD). We found that AR-V7 has previously unappreciated oncogenic activities, in addition to its established mechanism stimulating ligandindependent gene programs via binding to canonical AREs. Specifically, our study revealed a family of AR-V7 binding sites that are not targeted by AR-FL; these previously unknown AR-V7 binding sites are referred to as the unique AR-V7 targets. Moreover, we identified a zinc finger protein, ZFX, as a crucial AR-V7 partner co-occupying a vast majority of AR-V7 unique binding sites. Integration of datasets from prostate cancer cell lines and patients further defined clinical relevance of our finding as we derived an AR-V7-associated direct target signature, which correlates with AR-V7 expression levels in primary tumors, separates metastatic prostate cancer from normal, and predicts poorer clinic outcomes. Lastly, we show that inhibition of AR-V7 co-factors (ZFX or BRD4), or KD of downstream targets uniquely co-activated by AR-V7 and ZFX, suppressed the AR-V7-dependent CRPC cell growth. Thus, besides canonical ARE- FOXA1 signaling, this study unveils a crucial, yet unexplored pathway by which AR-V7 enforces the phenotypic alterations seen in men failing potent androgen deprivation.
Results
AR-V7 exhibits ligand-independent binding in the genome of CRPC cells co-expressing AR-V7 and AR-FL.
In order to dissect redundant and distinctive functions of AR-V7 and AR-FL in CRPC, we used two antibodies against a unique epitope of either AR-FL or AR-V7 (Figure 1A) and validated their isoform-specificity first by immunoblot (Figure 1B and S1A-B). By ChIP-qPCR of canonical AREs, we further confirmed antibody specificity in three prostate cancer lines with differential AR expression— LNCaP expressing high AR-FL and almost none of AR-V7, 22Rv1 co-expressing AR-FL and AR-V7, and PC3 lacking AR expression (Guo et al., 2009; Hu et al., 2009) (Figure 1C). We next used these antibodies to map genomic binding of endogenous AR-V7 and AR-FL by ChIP-Seq in 22Rv1 cells. We also did ChIP-Seq for BRD4, an AR cofactor mediating gene activation (Asangani et al., 2014). Cells were ligand-starved, followed by a 6-hour treatment with vehicle, dihydrotestosterone (DHT), DHT plus MDV3100, or DHT plus a BRD4 inhibitor JQ1. ChIP-Seq peaks and overall binding are summarized in Figures 1D and S1C-F. As expected and without ligand, AR-FL showed weak but detectable binding to ~1,600 sites; DHT treatment dramatically enhanced genomic binding of AR-FL, an effect almost completely abolished by MDV3100 co-treatment (Fig. 1D, blue; and Fig. S1D-E). Without ligand, AR-V7 displayed significant chromatin occupancy across the genome (Fig. 1D, red and Fig. S1D-E). DHT further enhanced AR-V7 binding, likely due to AR- V7 heterodimerization with ligand-activated AR-FL (Xu et al., 2015b), whereas MDV3100 had little effect on ligand-independent binding of AR-V7 (Fig. 1D and S1D-E). Genomic binding of BRD4 was also induced by DHT and reduced by JQ1 treatment (Fig. 1D, green and Fig. S1F).
Compared to AR-FL, AR-V7 exhibits both redundant and distinctive binding in two independent CRPC cell models.
ChIP-Seq profiling of endogenous AR-FL and AR-V7 in the same CRPC cells allowed direct comparison of their binding. First, we found the AR-V7 binding in ligand-starved cells largely overlap that of DHT-stimulated AR-FL at 15,162 out of a total of 17,409 sites (Fig. 1E-G and S2A). These AR-FL/V7 common sites were mainly at enhancers enriched with motifs of ARE and FOXA1, an AR-interacting pioneer factor (Lupien et al., 2008) (Fig. 1H-I and S2B-C), such as those of classic AR targets KLK3/PSA, KLK2 and FKBP5 (Fig. 1J and S2D-E). This is consistent to reports that AR-V7 recapitulates AR- FL functions and that the two also form heterodimers (Chan et al., 2015; Lu et al., 2015; Xu et al., 2015b).
Unexpectedly, we identified a significant portion of AR-V7 peaks (12.8%; 2,221 out of 17,409; Table S1) lacking AR-FL binding (Fig. 1E, 1G, 1K and S2F), as exemplified by those at SKP2 and ZFY (Fig. 1L and S2G-I). The overlapped binding with AR-FL and the distinct solo binding of AR-V7 were also seen in DHT-treated 22Rv1 cells, with the latter accounting for 19.3% of peaks (7,537 out of 39074; Fig. S2J-K). In contrast to AR-FL binding at enhancers, the AR-V7-solo binding was mainly found at promoters (Fig. 1H and S2L), indicating a distinct recruitment mechanism. Also, we did AR-FL and AR-V7 ChIP-Seq in VCaP cells (Fig S1C, right), another CRPC model with AR amplification and AR-V7 co-expression (Hu et al., 2009), and identified similar AR-V7-solo binding, relative to AR-FL (Fig. 1M and S3A). Importantly, there is significant overlap between AR-V7-solo sites identified in 22Rv1 and VCaP cells (Figure 1N), suggesting a common feature for AR-V7-solo binding in CRPC.
We found AR-V7 solo-peaks enriched with genes showing MYC binding or frequent aberration in cancer including metastatic prostate tumor (Fig. S3B), indicating that these previously unappreciated, non-canonical AR-V7 sites may be biologically crucial. Additionally, we found both AR-V7 and AR-FL peaks significantly overlapped with BRD4 peaks, supporting their role in gene activation (Fig. 1D and S3C). By ChIP- qPCR, we verified AR-FL/V7 common and AR-V7 solo binding in five different cell lines, with either the isoform-specific antibodies used in endogenous ChIP-Seq (Fig. S3D-E) or additional antibodies (e.g. HA ChIP with cells expressing an HA-tagged AR-V7) as independent verification approaches (Fig. S3F-H). Together, these results show that AR-V7 had non-canonical targets in CRPCs that are not targeted by AR-FL upon ligand stimulation.
Integrated RNA-Seq and ChIP-Seq analyses reveal the AR-V7 associated gene signature in CRPC cell models and primary patients.
To dissect the role of AR-V7 in gene regulation, we specifically knocked down AR-V7 and not AR-FL in ligand-starved 22Rv1 cells (Fig. 2A-B). Transcriptome analysis by RNA-Seq identified 1,178 genes up and 648 down regulated by AR-V7 (Fig. 2C). Consistent to AR-V7 and BRD4 co-occupancy (Fig. S3C), significantly more of AR-V7- activated genes showed AR-V7 binding, relative to randomized control or AR-V7- repressed genes (Fig. S4A). GSEA analysis support involvement of AR-V7 in activation of androgen-responsive, oncogenic (MYC, MYB), cell cycle progression (E2F), and cancer progression-associated genes (Fig. 2D-G and S4B-G). Integration of RNA-Seq and ChIP-Seq data identified 475 of AR-V7-activated genes as direct AR-V7 targets in 22Rv1 cells (Fig. 2H and Table S2). To further define clinically relevant signatures for AR-V7, we turned to the public patient datasets and found 41 of the AR-V7 directly activated genes in 22Rv1 cells significantly correlating positively to the relative AR-V7 expression level in the TCGA prostate cancer cohort (Cancer Genome Atlas Research, 2015) (Fig. 2I and S4H; Table S3). This 41-gene AR-V7 direct target signature also positively correlates to the AR-V7 level in an independent CRPC patient cohort (Beltran et al., 2016) (Fig. 2J), differentiates tumor from normal (Fig. 2K and S4I), and predicts worse prognosis in a clinical prostate cancer cohort with long-term followup (Taylor et al., 2010) (Fig. 2L).
Fig 2. Integration of genomic datasets from 22Rv1 CRPC cell model and clinical prostate cancer samples reveals the AR-V7 direct or unique target signature predicting prognosis.

(A-B) RT-qPCR (A) and immunblot (B) show selective KD of AR-FL or AR-V7 in 22Rv1 cells. Used in B are AR N-terminus (top; pan-AR) and AR-V7-specific (middle) antibodies. NS, not significant, **, p<0.01; ***, p<0.001.
(C) Heatmap showing expression of genes down (left) and up regulated (right) after AR- V7 KD (sh_V7) relative to vector in ligand-starved 22Rv1 cells (2 biological replicates per group). Threshold of differential expression is adjusted DESeq p value (adj. p) of <0.01 and fold-change (FC) of >1.5. Color bar, log2(FC).
(D-G) GSEA shows negative correlation of the indicated gene set with selective KD of AR-V7, relative to mock (sh_Vec).
(H) Averaged AR-V7 and BRD4 ChIP-Seq signals at the 475 genes that are upregulated by AR-V7 and also have direct AR-V7 binding in 22Rv1 cells.
(I) Heatmap showing that the 41 genes directly upregulated by AR-V7 in 22Rv1 cells also positively correlates (spearman rank r >0.2 and BH FDR <0.01) with AR-V7 expression level in the TCGA prostate cancer cohort. Top of panel I shows the ratio of RNA-Seq read counts specific to AR-V7 (i.e. those of CE in Fig 1A) to those common to all AR isoforms (i.e. AR N-terminal domain).
(J-K) Box plots shows mean expression values of the 41-gene AR-V7 direct targets (41- gene signature defined in I) in patient cohorts reported in (Beltran et al., 2016) (J) or (Yu et al., 2004) (K). V7-low/high, bottom/top one-third of patients ranked by AR-V7 expression in the cohort. The p-value and test are denoted on top.
(L) Kaplan-Meier curve for the above defined 41-gene AR-V7 direct target signature in a patient cohort reported in (Taylor et al., 2010).
Genomic profiling also identifies downstream genes uniquely regulated by AR- V7, compared to AR-FL, promoting CRPC cell growth.
Next, to further characterize unique regulatory functions of AR-V7, we compared transcriptome perturbations caused by specific KD of either AR-V7 or AR-FL in 22Rv1 cells (Fig. 2A-B). Despite significant overlap between genes regulated by the two isoforms (Fig. 3A-D), supporting their cooperativity in AR signaling (Guo et al., 2009; Watson et al., 2010), AR-V7 and AR-FL also had differential gene-regulatory roles. For instance, the AR and GR signature genes (Arora et al., 2013) were preferentially regulated by AR-FL, relative to AR-V7 (Fig. S4J-K). Importantly, we also found 329 transcripts uniquely or preferentially up-regulated by AR-V7, compared to AR-FL (Fig. 3E and Table S4). By RT-qPCR, we validated cooperative (Fig. 3F) and isoform- preferential effect (Fig. 3G) by AR-FL and AR-V7 on downstream gene activation. Besides SKP2, an E3 ligase complex subunit recently shown to be crucial for tumorigenesis including CRPC (Chan et al., 2013; Ruan et al., 2017), the downstream targets uniquely activated by AR-V7 and not by AR-FL (Fig 3G) included ZNF32, a Kruppel-like transcription factor associated with autophagy (Li et al., 2015), and FZD6, a non-canonical WNT receptor. Non-canonical WNT signaling and autophagy were implicated in prostate tumorigenesis and castration resistance (Miyamoto et al., 2015; Nguyen et al., 2014). ZNF32 expression positively correlates to AR-V7 levels in TCGA prostate tumors (Fig. S4L). KD of either ZNF32 or FZD6 significantly suppressed androgen-independent proliferation of 22Rv1 cells (Fig. 3H-K), demonstrating a role for non-canonical targets of AR-V7 in sustaining CRPC growth. Collectively, we have defined AR-V7-associated gene signatures and demonstrated a growth-related requirement of transcripts uniquely upregulated by AR-V7.
Fig. 3. RNA-Seq profiling reveals common and distinctive pathways regulated by AR-V7 and AR-FL isoforms.

(A-D) Venn diagram and heatmap show overlap and relative expression of transcripts co-activated (A-B) or co-repressed (C-D) by AR-FL and AR-V7 in ligand-starved 22Rv1 cells as revealed by RNA-Seq (2 replicates per group). The thresholds for differential expression are adjusted p value (adj. p) of <0.01 and FC of >1.5. Color bar, log2FC.
(E) Heatmap shows relative expression of 329 genes identified by RNA-Seq to be uniquely or preferentially upregulated by AR-V7, compared to AR-FL, in 22Rv1 cells, with the thresholds set to be down-regulated (adj-p<0.01 and log2FC<−0.58) in samples with sh_V7, compared to mock and to AR-FL-specific KD (sh_FL). Color bar, log2FC. (F-G) RT-qPCR of the indicated genes co-activated by AR-V7/FL (F) or uniquely activated by AR-V7 (G) in ligand-starved 22Rv1 cells. Data of three independent experiments are plotted as mean ± SD after normalization to those of GAPDH and to mock treated. NS, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001. (H-K) KD of ZNF32 (H-I) or FZD6 (J-K) by either of two independent shRNAs interferes with androgen-independent growth of 22Rv1 cells, relative to mock.
ZFX, a conserved zinc finger transcription factor, interacts with AR-V7 and co-occupies the AR-V7 unique binding sites at target gene promoters.
AR-V7 recruitment to non-canonical solo peaks was previously unappreciated and the unique gene upregulation by AR-V7 (such as ZNF32, FZD6 and SKP2) promotes 22Rv1 cell malignant growth. We thus performed a motif search to identify common cis- regulatory elements at AR-V7-solo sites. The motif of ZFX, a zinc finger factor mediating transcriptional activation (Schneider-Gadicke et al., 1989), was most significantly enriched at AR-V7-solo peaks identified in both ligand starved and stimulated conditions (Fig. 4A, red and Fig. S5A-C). This contrasted with the ARE and FOXA motifs being most enriched in AR-FL sites (Lupien et al., 2008) (Fig. 4A and S2B-C,S5A). Such motif enrichment distinction was also seen when only those AR-FL/V7 common sites mapped to promoters were analyzed (Fig. S5D). Co-immunoprecipitation (CoIP) showed ZFX physically associates with AR-V7 (Fig. 4B and S5E-F), supporting a potential co-regulatory action. Next, we carried out ZFX ChIP-Seq with two separate validated antibodies in ligand-starved 22Rv1 cells, which generated consistent data (Fig. S5G). Indeed, we found co-occupancy of ZFX at AR-V7-solo peaks, and not at peaks shared by AR-V7 and AR-FL (Fig. 4C-E and S5H-I). Again, AR-V7 solo-peaks co-bound by ZFX overlapped with BRD4 peaks (Fig. 4F and S5H) and were mainly at promoters (Fig. 4G) such as those of ZNF32, FZD6 and ZFY (Fig. 4H-I and S5J-K), suggesting a role for these associated factors in gene activation. By ChIP-qPCR, we further confirmed ZFX binding specificity at the tested loci of AR-V7 unique targets, and not at canonical AREs (Fig. 4J). Together, we identified ZFX as a AR-V7 partner co-occupying the AR-V7-solo sites at gene promoters in CRPC cells.
Fig 4. ZFX interacts with AR-V7, co-occupying the solo AR-V7 sites in 22Rv1 cells.

(A) Percentage of the AR-V7 unique, AR-V7/FL common or AR-FL unique ChIP-Seq peaks (defined in Fig 1E) that contain the indicated motif as revealed by the motif analysis.
(B) CoIP of AR-V7 and ZFX in 22Rv1 cells.
(C) Averaged ZFX ChIP-Seq signals at common or unique peaks of AR-V7 and AR-FL as shown in A.
(D) Heatmap illustrates overlap of ZFX ChIP-Seq peaks with AR-V7 solo peaks (bottom panel) in ligand-starved 22Rv1 cells.
(E-F) Venn diagram shows overlap of ZFX peaks with AR-V7 solo binding in DHT- treated 22Rv1 cells (E) and BRD4 (F) in ligand-starved 22Rv1 cells.
(G) Averaged ZFX ChIP-Seq read density at the indicated peaks, located at either promoter or non-promoter and showing either AR-FL/V7 common binding or AR-V7 solo binding (as defined in A).
(H-I) ChIP-Seq profile of AR-FL, AR-V7, BRD4 and ZFX at the indicated gene in 22Rv1 cells.
(J) ChIP-qPCR of ZFX at the indicated site in ligand-starved 22Rv1 cells. tss, transcription start site; e, ARE enhancer.
See also Figures S5.
Frequently amplified in prostate cancer, ZFX is required for i) AR-V7 binding to its solo sites, ii) expression of AR-V7-regulated gene programs, and iii) AR-V7- dependent growth of CRPC cells.
ZFX is significantly amplified in multiple clinical cohorts of metastatic prostate cancers (Cerami et al., 2012; Gao et al., 2013), indicating its role in advance diseases (Fig. 5A and S6A). We next sought to study whether ZFX regulates AR-V7-mediated gene regulation. First, we found that stable KD of ZFX in 22Rv1 cells (Fig. 5B) caused significant reduction in overall AR-V7 binding to its solo sites (Fig. 5C, top) as exemplified by the ZFY, ZNF32 and FZD6 promoters (Fig. 5D). This is contrasted with the almost lack of effect of ZFX KD on AR-V7 binding to enhancers co-bound by AR-FL (Fig. 5C, bottom), such as AREs of KLK3, KLK2 and FKBP5 (Fig. 5E). By ChIP-qPCR in 22Rv1 cells with stable (Fig. 5B) and transient ZFX KD (Fig. 5F), we verified negligible effect of ZFX on AR-V7 binding to KLK2/3 AREs (Fig. 5G, left) but the significantly decreased AR-V7 binding to the ZNF32 and FZD6 promoters upon ZFX KD (Fig. 5G-H). Meanwhile, KD of FOXA1 did not affect AR-V7 binding at the tested solo sites (Fig. 5F and5H). These results support ZFX as a crucial cofactor that mediates AR-V7 recruitment and/or stabilization at its solo peaks.
Fig 5. ZFX shows gene amplification in prostate cancer and potentiates AR-V7 binding to its unique targets.

(A) AR, ZFX and MYC amplification in a prostate cancer cohort reported in (Kumar et al., 2016).
(B) ZFX immunoblot in 22Rv1 cells with stable shRNA transduction.
(C) Averaged AR-V7 ChIP-Seq signals at AR-V7 unique peaks (top) or AR-FL/V7 common peaks (bottom) in 22Rv1 cells with mock (blue) or ZFX KD (purple; sh2 used). (D-E) AR-V7 ChIP-Seq profile at the indicated AR-V7-solo (D) or AR-FL/V7 common target (E) in 22Rv1 cells with vector mock (top) or ZFX KD (bottom).
(F) Immublots after siRNA-mediated KD of ZFX or FOXA1 in 22Rv1 cells.
(G-H) ChIP-qPCR of AR-V7 (red) at the indicated AR-FL/V7 common ARE (e) targets or AR-V7 unique promoter sites (tss) in 22Rv1 cells with stable ZFX KD (G) or transient knockdown of ZFX or FOXA1 (H), relative to mock. Plotted are data of 3 independent experiments normalized to input and presented as mean ± SD. Non-specific IgG (black) and control (CTL) siRNA serve as control. NS, not significant; *, p<0.05; **, p<0.01; ***, p<0.001.
See also Figures S6 and Table S5-6.
To further characterize the role of ZFX in CRPC, we performed RNA-Seq profiling following ZFX KD in 22Rv1 cells. Genes activated and repressed by ZFX significantly overlapped those by AR-V7, supporting cooperation of the two in gene regulation (Fig. 6A). Similar to AR-V7-activated genes, ZFX-activated genes were found to be enriched with androgen responsive, cell proliferative and oncogenic gene sets (Fig. 6B-C and S6B-D). In addition, using AR-V7 and ZFX KD RNA-Seq data, we observed positive correlation between the expression of AR-V7 or ZFX and a higher overall expression level of genes associated with the AR-V7-solo sites, with p value of 0.0158 and 1.18e-21, respectively (Fig S6E and Table S5). We further confirmed an essential requirement of ZFX for activation of AR-V7 uniquely activated targets including ZNF32 and FZD6 (Fig. 6D), which we have validated as involved in CRPC cell growth (Fig. 3G-K). Importantly, ZFX KD significantly impaired androgen-independent 22Rv1 cell growth in colony forming (Fig. 6E), proliferation (Fig. 6F) and in vivo xenograft growth assays (Fig. 6G). These phenotypes are reminiscent of those seen after AR-V7 KD (Guo et al., 2009), illustrating the critical role for ZFX in AR-V7-mediated gene regulation and CRPC progression.
Fig 6. ZFX is required for AR-V7-mediated gene expression and AR-V7-dependent CRPC growth.

(A) Heatmap shows expression changes for genes co-regulated by AR-V7 and ZFX in ligand-starved 22Rv1 cells as revealed by RNA-seq of the indicated KD samples (2 replicates per group). The thresholds for differential expression are p-adj of < 0.01 and FC of >1.5. Color bar, log2FC relative to mock.
(B-C) GSEA reveals negative correlation of the indicated gene set to ZFX KD.
(D) RT-qPCR of the indicated genes uniquely upregulated by AR-V7 in 22Rv1 cells after mock or ZFX KD. **, p<0.01; ***, p<0.001; ****, p<0.0001.
(E-F) Colony formation (E) and proliferation assays (F) of 22Rv1 cells after stable KD of ZFX.
(G) Growth of xenografted 22Rv1 cells with stable mock or ZFX KD in castrated NSG mice.
Compared to anti-androgen, BRD4 inhibitors are more effective in suppressing target genes activated by AR-V7 and/or ZFX and in suppressing the AR-V7- dependent CRPC growth.
BRD4 inhibition was recently shown to suppress AR-dependent prostate tumor growth (Asangani et al., 2014). We observed that AR-V7 solo peaks overlapped with BRD4 binding (Fig. 1K and S2F) and that a 6-hour JQ1 treatment significantly decreased AR- V7 binding at solo peaks whereas MDV3100 had little inhibitory effect (Fig. 7A and S7A, panels of AR-V7 unique). RNA-Seq of 22Rv1 cells post-treatment with DHT alone or in combination of inhibitors further showed that MDV3100 largely reversed the DHT- induced changes resetting cell transcriptome back to its basal state of androgen- independence (i.e., “mock”; Fig. 7B, green versus blue); however, JQ1 treatment had more dramatic effect altering the basal transcriptome profile of 22Rv1 cells (Fig. 7B, red versus blue). Indeed, JQ1 and not MDV3100 efficiently suppressed expression of gene sets up-regulated by AR-V7 and/or AR-FL, as well as those co-activated by AR-V7 and ZFX (Fig. 7C). By RT-qPCR, we confirmed a greater suppressive effect of JQ1, relative to MDV3100, on expression of canonical AR targets co-activated by AR-FL and AR-V7 such as PSA/KLK3, KLK2 and PMEPA1 (Fig. 7D) and the unique effect of JQ1 at genes uniquely activated by AR-V7 and ZFX such as ZNF32, FZD6 and SKP2 (Fig. 7E). Consistently, BRD4 inhibitors, and not MDV3100, significantly suppressed growth of 22Rv1 cells in vitro (Fig. S7B-C) or post-xenograft in castrated NSG mice (Fig. 7F). Similar in vivo effect of BRD4 inhibitors was also observed in 22Rv1 xenografted models using non-castrated mice (Fig. S7D-E). These findings thus expand oncogenic actions of BRD4 to the non-canonical AR isoform binding sites, providing an additional explanation for BRD4 inhibition as an attractive CRPC therapeutic approach.
Fig 7. Compared to anti-androgen, BRD4 inhibitors have a superior effect on expression of AR-V7-associated signature genes and AR-V7-dependent CRPC cell growth.

(A) Averaged AR-V7 ChIP-Seq read density at AR-V7 solo peaks in ligand-starved 22Rv1 cells after a 6-hour treatment with DHT, DHT plus MDV3100, or DHT plus JQ1.
(B) Principal component analysis (PCA) plot with RNA-Seq data of ligand-starved 22Rv1 cells after a 24-hour treatment with vehicle (mock), DHT, DHT plus MDV3100, or DHT plus JQ1 (2 replicates per group).
(C) Heatmap shows overall expression changes for the indicated genes uniquely or commonly activated by AR-FL, AR-V7 and/or ZFX in 22Rv1 cells after drug treatment. Color bar, mean of the log2FC compared to mock. Direct, direct target; up, up- regulated; common, common target.
(D-E) Effect of compound treatment on expression of AR-V7/FL co-activated targets (D) and the AR-V7/ZFX uniquely activated targets (E) in 22Rv1 cells. NS, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
(F) Growth of xenografted 22Rv1 cells in castrated NSG mice treated with vehicle, 10mg/kg MDV3100, or 30mg/kg I-BET151 5 days per week. n, cohort size.
(G) Proliferation of 22Rv1 cells after FOXA1 KD versus mock.
(H) A model illustrates a canonical androgen-independent ARE/FOXA1 signaling and a previously unexplored ZFX-dependent oncogenic pathway enforced by AR-V7, both of which can be reversed by BRD4 blockade.
See also Figures S7.
Discussion
Profiling of AR-V7 and AR-FL cistromes in same CRPC cells identifies non-canonical gene pathways uniquely targeted by AR-V7.
Consistent with previous reports (Chan et al., 2015; Lu et al., 2015), our characterization of the AR-V7-regulated cistrome in CRPC cells showed the expected ligand-independent, canonical function of AR-V7 at ARE enhancers. Our endogenous AR-V7/FL ChIP-Seq in same cells further shows that AR-V7 binds part but not all of the AR-FL targets (~57%; Fig. S2J), indicating their functional difference. Importantly, our study unveiled additional, non-canonical AR-V7 functions at promoters of previously unappreciated, unique targets. To our knowledge, this study is among the first to determine genome-wide binding of endogenous AR-V7 versus AR-FL in the same CRPC cells utilizing two validated AR isoform-specific antibodies. This work differs from previous ones relying on pan-AR antibodies (Chan et al., 2015; Lu et al., 2015). In order to map binding of AR variants with pan-AR antibodies, previous studies had to genetically manipulate cells to knock out/down endogenous AR-FL (Chan et al., 2015; Lu et al., 2015); however, such manipulation alters CPRC cell transcriptome and AR-V7 associated phenotypes (Fig. 3), which may subsequently alter chromatin landscape of CPRC cells and occupancy of AR-V7. A recent work also used such a strategy of AR- FL KD to profile AR-V7-preferred binding in 22Rv1 cells (He et al., 2018); however, close comparison shows that almost all AR-V7-preferred and half of AR-FL-preferred sites defined by this work are co-bound by AR-FL/V7 according to our data (Fig S7F, left two panels), and that this recent work did not uncover non-canonical AR-V7 sites we detected by using two isoform-specific antibodies. This is most likely due to their low coverage of AR-FL/V7 binding (He et al., 2018), relative to that of our current and prior works (Asangani et al., 2014) (~7–17 times less; Fig S7G). It is also worth noting that AR-V7-expressing prostate cancers tend to express AR-FL at high levels in the clinic (Antonarakis et al., 2014; Miyamoto et al., 2015). Consistent with our ChIP-Seq results revealing both common and isoform-specific binding, our RNA-Seq studies following isoform-specific KD further substantiate both cooperative and differential roles for AR- FL and AR-V7 in gene regulation in CRPC (Fig 2A-B and Fig 3). Pharmacologic treatment with non-effective anti-androgen MDV-3100 versus the effective BRD4 inhibitor in 22Rv1 CRPC cells revealed differences in drug response at target sites uniquely bound by AR-V7 (Fig. 7A and S7A) and transcripts uniquely regulated by AR- V7 and not AR-FL (Fig. 7C). Importantly, we have carried out integrated analysis of cancer cell line and TCGA tumor datasets, deriving an AR-V7-associated gene signature that predicts worse prognosis of patients and differentiates tumor from normal (Fig. 2). Collectively, these findings validate both commonality and distinction of AR-V7 and AR-FL functions in prostate cancer. Detection of AR-Vs at early stages of prostate cancer reported in recent studies (Antonarakis et al., 2014; Miyamoto et al., 2015) suggests a cancer-evolving opportunity for selection of tumor cell clones towards not only drug resistance but a more aggressive phenotype.
Our study also identifies ZFX as a crucial cofactor colocalizing with AR-V7 at its unique binding sites and promoting CRPC cell growth.
Unlike canonical ARE enhancers enriched with FOXA1 binding (Lupien et al., 2008), the unique AR-V7 sites are most enriched with the ZFX motif. ZFX interacts with AR-V7 and ZFX KD interfered with AR-V7 binding to its unique targets (Fig. 4–5). Previously, ZFX was shown to promote stem cell self-renewal (Chen et al., 2008; Galan-Caridad et al., 2007) and carry cancer-promoting roles in leukemia (Weisberg et al., 2014) and medulloblastoma (Palmer et al., 2014). Intriguingly, ZFX shows gene amplification in ~8– 24% of prostate cancer cases in multiple cohorts (Cerami et al., 2012; Gao et al., 2013) (Fig. 5A and S6A), and ZFX KD significantly delayed malignant growth of 22Rv1 cells in vitro and in xenografted tumors (Fig. 6). These observations collectively show that ZFX acts as a crucial AR-V7 partner enforcing a previously unrecognized aspect of generegulatory networks during CRPC progression. Such an AR-V7/ZFX enforced program most likely acts in parallel with that controlled via FOXA1/ARE cis-elements as FOXA1 KD suppressed 22Rv1 cell growth as well (Fig. 7G). Our results support an unexplored mechanism and signaling network (see a model in Fig. 7H) that emerges as prostate cancer becomes resistant to even more powerful androgen deprivation agents. The HOX/homeodomain motif was found enriched similarly at overall AR-FL and AR-V7 sites, and not AR-V7-solo sites (Fig. S2B-C and S5B-C), consistent to reports that HOXB13 interacts with AR/FOXA1 at their corresponding ARE enhancers (Norris et al., 2009; Pomerantz et al., 2015; Whitington et al., 2016). Other motifs such as ETS and YY1 were found significantly enriched at AR-V7-solo sites (Fig. S5B-C) and their potential role for AR-V7 regulation warrants further investigation.
Transcripts uniquely co-regulated by AR-V7 and ZFX contributes to malignant growth of 22Rv1 CRPC cells.
Among the transcripts uniquely activated by AR-V7 and ZFX, and not AR-FL, in 22Rv1 cells included an E3 ligase factor SKP2 and a non-canonical WNT receptor FZD6. Non-canonical WNT signaling was recently suggested to be clinically relevant and potentially responsible for anti-androgen resistance, based on single-cell transcriptome studies of circulating tumor cells from prostate cancer patients (Miyamoto et al., 2015). Moreover, our result suggested involvement of ZNF32, a transcription factor mediating autophagy regulation (Li et al., 2015). Exploration of ZFX and downstream targets co-activated by AR-V7 shall identify more effective therapeutic targets of CPRC under androgen- deprived milieus. As a proof of principle, we and others show inhibition of SKP2 (Chan et al., 2013; Ruan et al., 2017), FZD6 or ZNF32 suppressed CRPC cell growth (Fig. 3H-K). Determination of the unique AR-V7/ZFX transcriptional signatures crucial for CRPC cell growth may also provide additional biomarkers for testing in both retrospective cohorts with outcome data and prospective clinical trials.
Targeting AR-V7 cofactor provides a more effective means for the treatment of CRPC showing therapy resistance.
Besides targeting ZFX, we have also shown blockade of BRD4, another cofactor of AR- V7, significantly suppressed androgen-independent growth of CRPC cells in vitro and in vivo, which is in contrast to effect of anti-androgen and consistent with recent studies (Asangani et al., 2014; Asangani et al., 2016). We further unveiled inhibitory effect of JQ1 on AR-V7 chromatin binding and transcriptional programs coactivated by AR-V7 and ZFX, providing a previously unappreciated explanation for effect of BRD4 inhibitors in CRPC (a model in Fig. 7H). Inhibitor of chromatin factors such as bromodomain proteins emerge as promising therapeutics for various tumors including prostate and blood cancer (Asangani et al., 2014; Lu and Wang, 2017; Zuber et al., 2011) and exploration of the underlying mechanisms should lead to development of more effective interventions in the future. In summary, this study provides a series of insights as to how AR-V7 leads to CRPC progression and therapy resistance through its non-canonical function mediated by ZFX.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |||
|---|---|---|---|---|---|
| Antibodies | |||||
| anti-full-length AR (AR-FL) C-terminus (C19) | Santa Cruz Biotechnology | catalog # sc-815X | |||
| anti-AR-V7 C-terminus specific | Precision Antibody | AG10008 | |||
| anti-pan-AR N-terminus (N20) | Santa Cruz Biotechnology | sc-816 | |||
| anti-BRD4 ChIP Grade | Bethyl | A301–985A100 | |||
| Mouse anti-Flag tag (M2) | Sigma | F1804 | |||
| Mouse Anti-HA tag antibody - ChIP Grade | Abcam | ab9110 | |||
| Anti-HA HA.11 (16B12) | Covance | MMS-101 P-200 | |||
| anti-ZFX | Thermo Fisher | PA5–34376 | |||
| anti-ZFX | Chen et al., 2008 | NA | |||
| Mouse anti-ZFX | Cell Signaling | mAb #5419 | |||
| anti-Tubulin (DM1A) Mouse mAb | Cell signaling tech. | 3873S | |||
| anti-FOXA1 ChIP Grade | Abcam | 23738 | |||
| goat anti-mouse IgG HRP | Santa Cruz Biotechnology | sc-2005 | |||
| goat anti-rabbit IgG-HRP | Santa Cruz Biotechnology | sc-2004 | |||
| Dynabeads™ M-280 Sheep Anti-rabbit IgG | Thermo Fisher Scientific | 11203D | |||
| Dynabeads™ M-280 Sheep Anti-Mouse IgG | Thermo Fisher Scientific | 11202D | |||
| Dynabeads™ Protein G for Immunoprecipitation | Thermo Fisher Scientific | 10003D | |||
| Bacterial and Virus Strains | |||||
| DH5a E.coli competent cells | Thermo Fisher Scientific | catalog # 18265017 | |||
| One Shot TOP10 Chemically competent E.coli | Thermo Fisher Scientific | catalog # sc-2004 | |||
| Chemicals, Peptides, and Recombinant Proteins | |||||
| Dihydrotestosterone (DHT) | Sigma-Aldrich | Catalog # D-073; CAS: 521–18-6 | |||
| Enzalutamide (MDV3100) | Selleck chemical | Catalog #.S1250; CAS: 915087–33-1 | |||
| (+)-JQ1 BET bromodomain inhibitor | Selleck chemical | S7110; CAS: 1268524–70-4 | |||
| I-BET151 (GSK1210151A) BET inhibitor | Selleck chemical | S2780; CAS: 1300031–49-5 | |||
| UltraPure™ 10mg/mL Ethidium Bromide | Invitrogen | 15–585-011 | |||
| Polybrene | Sigma | TR-1003-G | |||
| PMSF | Sigma-Aldrich | 78830 | |||
| Glycine | Sigma | G8898 | |||
| Retro-X Concentrator | Clontech | 631455 | |||
| 16% Paraformaldehyde | Electron Microscopy Sciences | 15710 | |||
| Lipofectamine 3000 Transfection Reagent | Thermo Fisher Scientific | L3000150 | |||
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | 11668–019 | |||
| Protease inhibitor COMPLETE EDTA-FREE | Roche | 11873580001 | |||
| T4 DNA Ligase | New England Biolabs | M0202S | |||
| Proteinase K | Thermo Fisher Scientific | BP1700–500 | |||
| Rnase A | Sigma-Aldrich | R4875 | |||
| Thiazolyl Blue Tetrazolium Blue | Sigma-Aldrich | M2128 | |||
| Critical Commercial Assays | |||||
| RNeasy Plus Mini Kit (250) | Qiagen | 74136 | |||
| iSCRIPT cDNA Synthesis KIT | Biorad | 1708891 | |||
| iTaq Universal SYBR Green Supermix | Biorad | 1725125 | |||
| SsoAdvanced™ Universal SYBR® Green Supermix | Biorad | 172–5270 | |||
| Bio-Rad Protein Assay Dye Reagent Concentrate | Biorad | 5000006 | |||
| MycoAlert™ PLUS Mycoplasma Detection Kit | Lonza | LT27–286 | |||
| MycoZap™ Plus-CL | Lonza | VZA-2012 | |||
| Beckman Coulter AMPURE XP PCR Purification | Beckman Coulter | A63881 | |||
| TruSeq RNA Library Preparation Kit v2, Set A | Illumina | RS-122–2001 | |||
| TruSeq RNA Library Preparation Kit v2, Set B | Illumina | RS-122–2002 | |||
| End-It™ DNA End-Repair Kit | Epicentre | ER81050 | |||
| NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) | New England Biolabs | E7335S | |||
| CellTiter 96 AQueous One Solution Cell Proliferation Assay | Promega | G3580 | |||
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent | 200521 | |||
| SureBeads™ immunoprecipitation Kit with protein A and G conjugated magnetic beads | Biorad | 161–4833 | |||
| Restore™ Western Blot Stripping Buffer | Thermo Scientific | 21059 | |||
| BD Matrigel™ Basement Membrane Matrix | BD Biosciences | 354234 | |||
| Deposited Data | |||||
| Raw and analyzed data | This paper | GEO: GSE94013 | |||
| Unprocessed immunoblotting image data (deposited as Mendeley DOI link) | This paper | htto://dx.doi.org/10.17632/vmmrvbnd4.2 | |||
| TCGA-PRAD dataset (RNA-seq of 543 samples) | Cancer Genome Atlas Research, 2015 | https://portal.gdc.cancer.gov/projects/TCGA-PRAD | |||
| Beltran et al study of prostate cancer | (Beltran et al., 2016) | dbGAP:pht004946.v1.p1 | |||
| Yu et al. study of prostate cancer | Yu et al., 2004 | GEO GSE68555 | |||
| Taylor et al. study of prostate cancer | (Taylor et al., 2010) | GEO GSE21034 | |||
| Varambally et al study of prostate cancer | Varambally et al., 2008 | GEO GSE3325 | |||
| Experimental Models: Cell Lines | |||||
| 22Rv1 cells | ATCC | CRL-2505 | |||
| VCaP cells | ATCC | CRL-2876 | |||
| LNCaP cells | ATCC | CRL-1740 | |||
| HEK293 cells | ATCC | CRL-1573 | |||
| HEK293T cells | ATCC | CRL-3216 | |||
| Experimental Models: Organisms/Strains | |||||
| NSG; NOD/scid/ IL2Rgamma-null | Jackson Laboratory | Strain: 005557 | |||
| Oligonucleotides | |||||
| shRNA for specific knockdown of AR-FL or AR-V7 | Guo et al., 2009 | N/A | |||
| RT q-PCR oligos | This paper; see Table S6 | N/A | |||
| ChIR-qPCR oligos | This paper; see Table S6 | N/A | |||
| ON-TARGETplus SMART-pool siRNA of ZFX | GE Dharmacon | N/A | |||
| ON-TARGETplus SMART-pool siRNA of FOXA1 | GE Dharmacon | N/A | |||
| recommended control siRNAs | GE Dharmacon | N/A | |||
| Recombinant DNA | |||||
| MSCV-HA-AR-V7 | This paper | N/A | |||
| pcDNA AR-FL and deletion construct | Zhou et al 1995; | N/A | |||
| Plasmid: MSCV neo; MSCV puro | Clonetech | 634401 | |||
| Plasmid: pCDNA3.1 | Thermo Scientific | V79520 | |||
| Software and Algorithms | |||||
| BWA (V0.7.12) alignment software | (Li and Durbin, 2010) | https://sourceforge.net/projects/bio-bwa/ | |||
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ | |||
| MACS2 & MACS1.4.2 | Zhang et al., 2008 | https://github.com/taoliu/MACS | |||
| seqMiner | Ye et al., 2011 | https://sourceforge.net/projects/seqminer/ | |||
| Java treeview | n/a | https://sourceforge.net/projects/jtreeview/ | |||
| Genomic Regions Enrichment of Annotations Tool (GREAT) | n/a | http://bejerano.stanford.edu/great/public/html/index.php | |||
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ | |||
| MEME-ChIP | Machanick and Bailey, 2011 | http://meme-suite.org/doc/meme-chip.html | |||
| MapSplice | Wang et al., 2010 | https://sourceforge.net/projects/mapsplice/ | |||
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR | |||
| RSEM | Li and Dewey, 2011 | https://www.encodeproject.org/software/rsem/ | |||
| DESeq/DESeq2 | (Anders and Huber, 2010; Love et al., 2014) | https://software.broadinstitute.org/gsea/index.jsp | |||
| IGV Browser | (Robinson et al., 2011) | https://software.broadinstitute.org/software/igv/ | |||
| GSEA 2–2.2.0 software | (Subramanian et al., 2005) | https://software.broadinstitute.org/gsea/index.jsp | |||
| Other | |||||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, G.G.W. (greg_wang@med.unc.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293 and HEK293T cells (acquired from ATCC) were cultured in DMEM supplemented with 10% FBS and 1% antibiotics. The human prostate cancer cell lines, 22Rv1, VCaP and LNCaP, were obtained from American Type Culture Collection (ATCC) and grown as recommended by the provider. Authentication of cell line identities, including those of parental and derived lines, was ensured by the Tissue Culture Facility affiliated to UNC Lineberger Comprehensive Cancer Center with the genetic signature profiling and fingerprinting analysis. Every 1–2 months, a routine examination of cell lines in culture for any possible mycoplasma contamination was performed using commercially available detection kits (Lonza).
Bacterial strains
DH5a and TOP10 competent cells were purchased from Thermo Fisher Scientific and used for plasmid transformation and propagation based on manufacturer’s instructions.
Mouse xenograft models
NOD/SCID/IL2Rgamma-null (NSG) mice (Jax Lab) were maintained by the Animal Studies Core, UNC at Chapel Hill Cancer Center. All animal experiments are approved by and performed in accord with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at UNC.
METHOD DETAILS
Chemicals
DHT is purchased from Sigma and MDV3100 from Selleck chemical LLC. BRD4 inhibitors used in the study are JQ1 (Filippakopoulos et al., 2010) and I-BET151 (GSK1210151A; Selleck chemical LLC).
Antibodies
Specific antibodies used in ChIP-Seq include those against the C-terminus of full-length AR (Santa Cruz Biotechnology C-19; catalog # sc-815X; AR-FL specific) or AR-V7 (Precision Antibody catalog # AG10008; AR-V7 specific), the pan-AR antibodies that recognize the N-terminus of AR (Santa Cruz, AR N20; sc-816), BRD4 (Bethyl catalog # A301–985A100), HA tag (Abcam; 9110), ZFX (Thermo Fisher; catalog# PA5–34376) and anti-ZFX serum (Chen et al., 2008) as a kind gift of Dr. Huck Hui Ng. Additional antibodies used for IP or immunoblotting include Flag tag (Sigma; F1804); ZFX (Cell Signaling; Mouse mAb #5419), FOXA1 (Abcam; 23738) and Tubulin (Cell signaling 3873S).
Plasmids
cDNA of AR-V7 (also known as AR3) was cloned from 22Rv1 cells by PCR, fused with a HA tag and then cloned into MSCV-neo retroviral expression vector (Clontech). Various mammalian expression plasmids for AR (full-length or serial deletion) were described and used previously (Zhou et al., 1995).
Stable and transient RNA interference
The shRNA system for specific knockdown of AR-FL or AR-V7 was previously described (Guo et al., 2009). The pLKO.1 lentiviral shRNA plasmids for knockdown of FZD6, ZNF32 and ZFX were obtained from Sigma, with the detailed target sequences for shRNAs provided in the supplemental Table S5. All plasmids used are verified by sequencing. Transient knockdown of ZFX and FOXA1 expression was performed using the ON-TARGETplus SMART-pool siRNAs against the gene that were purchased from GE Dharmacon, in comparison to vendor recommended control siRNAs.
Cell culture and compound treatment
For compound treatment experiments, cells are first cultured under ligand-starved conditions for three days using the phenol red-free RPMI-1640 base medium supplemented with charcoal-stripped serum, followed by treatment with vehicle, dihydrotestosterone (DHT), or DHT together with compounds.
Viral Production and stable cell line generation
Retro- or lenti-virus was prepared with the packaging system in 293T cells according to manufacturer’s instructions (Lu et al., 2016; Xu et al., 2015a). Cell line with stable overexpression of AR-V7 was generated by infection of MSCV-neo based retrovirus encoding a HA-tagged AR-V7, followed by neomycin selection in growth medium for over a week. Generation of stable knockdown lines using the pLKO.1 lentiviral shRNA- expressing system was carried out according to providers’ protocols as described before (Cai et al., 2013; Lu et al., 2016).
Quantitative PCR (qPCR)
Real-time qPCR following either RT or Chromatin immunoprecipitation (ChIP), i.e. RT- qPCR or ChIP-qPCR) was performed as described before (Cai et al., 2013; Lu et al., 2016; Xu et al., 2015a). Data from at least three independent experiments are presented as mean ± standard deviation (SD) after normalization. Primers used for ChIP-qPCR and RT-qPCR were listed in Supplementary Information.
ChIP and ChIP followed by sequencing (ChIP-Seq)
ChIP were performed as previously described (Wang et al., 2007; Wang et al., 2009) and ChIP-Seq carried out as before (Cai et al., 2013; Lu et al., 2016; Xu et al., 2015a). Briefly, 22Rv1 cells were first cultured under ligand-starved conditions for three days, followed by a 6-hour drug treatment with vehicle, or 10nM of DHT, or DHT plus 10uM of MDV3100, or DHT plus 500nM of JQ1. Cells were cross-linked with 1% formaldehyde at room temperature for 10 minutes, followed by addition of glycine to stop crosslinking. After washing, lysis and sonication, cell chromatin samples were incubated with antibody-conjugated Dynabeads (Invitrogen) overnight at 4 degree. Beads bound with chromatin were then subject to extensive washing and elution. Eluted chromatin was de-crosslinked overnight at 65 degree, followed by protein digestion with proteinase K and DNA purification with Qiagen PCR purification kit. The obtained ChIP DNA samples were submitted to the UNC-Chapel Hill High-Throughput Sequencing Facility (HTSF) for preparation of multiplexed libraries and deep sequencing with an Illumina High-Seq 2000/2500 platform according to the manufacturer’s instructions.
RNA-Seq
RNA was prepared as described before (Cai et al., 2013; Lu et al., 2016; Xu et al., 2015a), using 2 million of the 22Rv1 cells stably transduced with shRNAs or after a 24- hour drug treatment. Then, complementary DNA was generated, amplified and subjected for library construction using TruSeq RNA Library Preparation Kit v2 (Illumina; catalog# RS-122–2002). The multiplexed RNA-Seq libraries were subject to deep sequencing using the Illumina Hi-Seq 2000/2500 platform according to the manufacturer’s instructions.
Co-immunoprecipitation (CoIP)
ColP with the prepared nuclear extracts was carried out as described before (Cai et al., 2013; Xu et al., 2015a). Briefly, nuclear pellet was lysed by brief sonication in IP buffer (20mM Tris pH 7.5, 150mM NaCl, 1% Triton-X100) with protease inhibitor cocktails (Roche) and PMSF. 1 milligram of nuclear lysates were pre-cleared with protein-G Dynabeads, added with 4ug of antibody, and subject to incubation on a rotator overnight at 4 degree. Then, protein G Dynabeads were added for 2hrs. Beads were washed three times in IP buffer, resuspended in 40ul of 2 X protein loading buffer, and boiled at 90 degree for 5 min before loading onto gel. Western blot was performed with standard protocols using SDS-page gels and PVDF membrane, and signals were visualized with an ECL system as described by the manufacturer (GE healthcare).
Cell proliferation assays
3,000 cells per well were seeded in triplicate in 96-well plates for each time point. The change in cell number was measured using MTT assay kit based on instructions of the manufacturer (Promega).
Colony formation assays
Cells were plated in triplicate at a density of 50,000 cells per well of 6-well plates and grown for 3 weeks before staining with Thiazoyl Blue Tetrazolium Bromide (Sigma). Fresh medium was changed twice a week.
In vivo tumor growth in xenograft models
1 million of 22Rv1 cells with stable transduction of shRNA or control empty vector were suspended in 100ul of PBS with 50% Matrigel (BD Biosciences) and then subcutaneously (s.c.) injected in the dorsal flanks of NOD/SCID/gamma-null (NSG) mice bilaterally (carried out by the Animal Studies Core, UNC at Chapel Hill Cancer Center). For castration models, a cohort of four-week-old NSG mice was castrated before cell injection. For in vivo compound treatment experiment, 1 million of 22Rv1 cells suspended in 100ul of PBS with 50% Matrigel were implanted s.c. into the flanks of NSG mice bilaterally. Once the tumors reached a palpable stage (around 100 mm3), mice were randomized into separate groups and subject to treatment with either MDV3100 by oral gavage (with a dose of 10mg/kg body weight) or I-BET151 intraperitoneally (30mg/kg body weight) for five days a week. Tumor growth was monitored twice a week and the tumor volume was calculated.
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-Seq data analysis
ChIP-Seq reads were aligned to the human reference genome (hg19) by the BWA (V0.7.12; default parameters) alignment software (Li and Durbin, 2010). After duplicated reads were removed, MACS2 (v2.1.0; -q 0.1 -, 20 100) (Zhang et al., 2008) was used to call peaks with input as controls. Weak peaks with no base covered by at least 10 reads were excluded and peaks overlapping (>= 1 bp) with the “blacklist” regions identified by the ENCODE project (Consortium, 2011) were also removed. For ZFX ChIP-Seq, data from the two different antibodies (Thermo Fisher catalog# PA5–34376 and anti-ZFX serum from Dr. Huck Hui Ng) showed high correlation and therefore were merged for final peak calling. The separation of AR-V7 and AR-FL peaks into “common” or “unique/solo” was simply based on their overlap in genomic coordinates. In-house scripts were used to assign peaks to annotated (coding and non-coding) genes, defined as “promoter proximal” (±2kb of transcription start site, TSS), “promoter distal”, i.e., “enhancer” (−50kb to −2kb of TSS and +2kb of TSS to +5kb of transcription ends), or otherwise “intergenic” using the human RefSeq annotation as reference. Genes with either a promoter or enhancer AR peak were considered to be AR bound targets. The ChIP-Seq read densities were calculated using the program seqMiner (Ye et al., 2011), which yielded for each peak an array of the maximal number of overlapping ChIP-Seq reads (extended to 200 bp) in 50 bp bins from −500 bp to +500 bp of the peak summits. The read density matrices were converted to heatmap using Java treeview (https://sourceforge.net/projects/jtreeview/). When ChIP-Seq read densities were compared across samples, reads from each sample were randomly selected to match the smallest read depth of all the samples. The enrichment of motifs were identified by the software HOMER (Heinz et al., 2010) or MEME-ChIP (Machanick and Bailey, 2011), using 500-bp sequences centered on the peak summits.
RNA-Seq data analysis
RNA-seq was mapped with MapSplice (Wang et al., 2010) and quantified with RSEM (Li and Dewey, 2011). Read counts were upper-quantile normalized and log2 transformed. Raw read counts were used for differential gene expression analysis by DESeq (Anders and Huber, 2010). Genes with Benjamini-Hochberg (BH) adjusted false discovery rate (FDR) less than 0.01 and fold change greater than 1.5 between AR-V7 knockdown and vector controls were called as differentially expressed genes (DEG). The intersection of DEGs and those bound by AR-V7 from ChIP-Seq were carried forward as the AR-V7 directly activated genes. In a separate test, the AR-V7 uniquely activated genes were identified as genes with two criteria: (1) lower in AR-V7 knockdown relative to vector controls (FDR < 0.01 and log2(fold change) < −0.58) and (2) lower relative to AR-FL knockdown (FDR < 0.01 and log2(fold change) < −0.58).
Analysis of public prostate cancer datasets
From the TCGA database, we collected expression measurement for 20,531 genes of the 543 TCGA-PRAD samples (RNA-seq level 3 data) (Cancer Genome Atlas Research, 2015). Raw read counts were up-quantile normalized. We also obtained the bam files for each samples (RNA-seq level 1 data) to estimate the levels of AR-V7 expression which is defined by the ratio of RNA-Seq read counts within the cryptic exon CE3 (hg19: chrx: 66914515–66915580) to the read counts within the N-terminal domain (NTD; hg19: chrx: 66763874–66766604). The DEGs directly bound by AR-V7 was filtered to require positive correlation (Spearman’s rho > 0.2 and BH FDR < 0.01) with the AR-V7 ratio estimated from TCGA resulting in 41 AR-V7 directly activated genes. The AR-V7 directly activated set was then clustered using hierarchical clustering with average linkage and Pearson correlation. The two predominant clusters were identified as the TCGA AR-V7-high and AR-V7-low groups by their ratio of AR CE3 to NTD.
Other public gene expression dataset are from the Yu et al. study representing 139 samples (Yu et al., 2004) (NCBI GEO GSE68555), the Taylor et al. study (Taylor et al., 2010) (GEO GSE21034) the Varambally et al study(Varambally et al., 2008) (GEO GSE3325), and the Beltran et al study (Beltran et al., 2016). Gene expression data available for the gene set of interest were extracted, log2 transformed and summarized to the mean expression of the signature for each sample. These summarized values were tested for association with sample type (such as benign, primary or metastatic) by ANOVA. For the Taylor et al study datasets (Taylor et al., 2010), samples were also grouped into tertiles of expression and related to biochemical recurrence free survival. Differences in event rate across the three groups were tested using the Log Rank test.
Gene Set Enrichment Analysis (GSEA)
GSEA was carried out using the GSEA 2–2.2.0 software (Subramanian et al., 2005) as previously described (Xu et al., 2015a).
Genomic Regions Enrichment of Annotations Tool (GREAT) Analysis
GREAT analysis for the select ChIP-Seq peaks was performed at its website according to providers’ instructions (http://bejerano.stanford.edu/great/public/html/index.php).
Statistical Analysis
Data are presented as the mean ± s.d. for three independent experiments unless otherwise noted. Statistical analysis was performed with Student’s t-test, except for nonparametric analysis such as Kaplan-Meier survival curve and gene expression association analysis that employed the Log-rank (Mantel-Cox test) and Analysis of variance (ANOVA) test, respectively.
Supplementary Material
Acknowledgments
We thank the Sequencing, Bioinformatics and Animal Studies Cores of UNC for assistance, and Drs. H Ng and Y Qiu for providing reagents, and the Wang and Earp labs for discussions. This work was supported by National Institute of Health grants (P50-CA058223 to H.S.E., R01-EY014237 to D.Z., and R01-CA215284, R01-CA218600 and R01-CA211336 to G.G.W.), the Lineberger Professorship (to H.S.E.), a Kimmel Scholar Award (to G.G.W.), a V Scholar Award (to G.G.W.) and a Concern Foundation for Cancer Research grant (to G.G.W.). UNC Core is supported in part by the UNC Cancer Center Core Support Grant P30-CA016086, and L.C. and R.L. supported by a DoD Prostate Cancer Research Program (W81XWH-10–1-0701) and Lymphoma Research Foundation fellowship, respectively. G.G.W. is an American Cancer Society Research Scholar and a Leukemia & Lymphoma Society Scholar.
Footnotes
Declaration of Interests
We have no conflict of interest to declare.
DATA AVAILABILITY
The Genomics data produced by this study, including ChIP-Seq and RNA-Seq, have been deposited in Gene Expression Omnibus (GEO) under accession code GSE94013. The original imaging data were deposited to Mendeley Data and included in the URL: http://dx.doi.org/10.17632/vrpmrvbnd4.2
Supplemental information
Supplemental information includes seven figures and six tables and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. (2014). AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. The New England journal of medicine 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. (2013). Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155, 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, et al. (2014). Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 510, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani IA, Wilder-Romans K, Dommeti VL, Krishnamurthy PM, Apel IJ, Escara- Wilke J, Plymate SR, Navone NM, Wang S, Feng FY, et al. (2016). BET Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, et al. (2016). Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nature medicine 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Rothbart SB, Lu R, Xu B, Chen WY, Tripathy A, Rockowitz S, Zheng D, Patel DJ, Allis CD, et al. (2013). An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Molecular cell 49, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu Dv et al. (2013). Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 154, 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, Raj GV, Tilley WD, and Dehm SM (2015). Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic acids research 43, 5880–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, and Sawyers CL (2004). Molecular determinants of resistance to antiandrogen therapy. Nature medicine 10, 33–39. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. (2008). Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117. [DOI] [PubMed] [Google Scholar]
- Consortium EP (2011). A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS biology 9, e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr., Saad F, et al. (2011). Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine 364, 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, and Tindall DJ (2008). Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer research 68, 5469–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan-Caridad JM, Harel S, Arenzana TL, Hou ZE, Doetsch FK, Mirny LA, and Reizis, (2007). Zfx controls the self-renewal of embryonic and hematopoietic stem cells. Cell 129, 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, and Trifiro M (2012). The androgen receptor gene mutations database: 2012 update. Human mutation 33, 887–894. [DOI] [PubMed] [Google Scholar]
- Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper G, et al. (2009). A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer research 69, 2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Lu J, Ye Z, Hao S, Wang L, Kohli M, Tindall DJ, Li B, Zhu R, Wang L, et al. (2018). Androgen receptor splice variants bind to constitutively open chromatin and promote abiraterone-resistant growth of prostate cancer. Nucleic acids research 46, 1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, and Wikstrom P (2011). Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 6, e19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al. (2009). Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer research 69, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Isaacs WB, and Luo J (2011). A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 71, 1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe P, Gomez EC, Wang J, et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (New York, N.Y 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, et al. (2016). Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nature medicine 22, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, and Dehm SM (2013). Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer research 73, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang L, Li K, Li J, Xiang R, Zhang J, Li H, Xu Y, Wei Y, Gao Jv et al. (2015). ZNF32 inhibits autophagy through the mTOR pathway and protects MCF-7 cells from stimulus-induced cell death. Sci Rep 5, 92–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Lonergan PE, Nacusi LP, Wang L, Schmidt LJ, Sun Z, Van der Steen T, Boorjian SA, Kosari F, Vasmatzis Gv et al. (2015). The cistrome and gene signature of androgen receptor splice variants in castration resistant prostate cancer cells. J Urol 193, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, and Wang GG (2017). Pharmacologic Targeting of Chromatin Modulators As Therapeutics of Acute Myeloid Leukemia. Front Oncol 7, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Wang P, Parton T, Zhou Y, Chrysovergis K, Rockowitz S, Chen WY, Abdel-Wahab O, Wade PA, Zheng D, et al. (2016). Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer cell 30, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, and Brown M (2008). FoxA1 translates epigenetic signatures into enhancer-driven lineage- specific transcription. Cell 132, 958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, Desai R, Fox DB, Brannigan BW, Trautwein J, et al. (2015). RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science (New York, N.Y 349, 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, and Nelson PS (2008). Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer research 68, 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, et al. (2017). SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science (New York, N.Y 355, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki D, Lara PN Jr., DeVere White RW, Gao AC, and Evans CP (2014). Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 33, 4521–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JD, Chang CY, Wittmann BM, Kunder RS, Cui H, Fan D, Joseph JD, and McDonnell DP (2009). The homeodomain protein HOXB13 regulates the cellular response to androgens. Molecular cell 36, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CJ, Galan-Caridad JM, Weisberg SP, Lei L, Esquilin JM, Croft GF, Wainwright B, Canoll P, Owens DM, and Reizis B (2014). Zfx facilitates tumorigenesis caused by activation of the Hedgehog pathway. Cancer research 74, 5914–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, Cejas P, Vazquez F, Cook J, Shivdasani RA, et al. (2015). The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nature genetics 47, 1346–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan D, He J, Li CF, Lee HJ, Liu J, Lin HK, and Chan CH (2017). Skp2 deficiency restricts the progression and stem cell features of castration-resistant prostate cancer by destabilizing Twist. Oncogene 36, 4299–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. (2012). Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine 367, 1187–1197. [DOI] [PubMed] [Google Scholar]
- Schneider-Gadicke A, Beer-Romero P, Brown LG, Mardon G, Luoh SW, and Page DC (1989). Putative transcription activator with alternative isoforms encoded by human ZFX gene. Nature 342, 708–711. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, et al. (2010). Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. The Journal of clinical investigation 120, 2715–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al. (2009). Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science (New York, N.Y 324, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al. (2008). Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science (New York, N.Y 322, 1695–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, and Kallioniemi OP (1995). In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nature genetics 9, 401–406. [DOI] [PubMed] [Google Scholar]
- Wang GG, Cai L, Pasillas MP, and Kamps MP (2007). NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nature cell biology 9, 804–812. [DOI] [PubMed] [Google Scholar]
- Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, Luo JL, Patel DJ, and Allis CD (2009). Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459, 847–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. (2010). MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic acids research 38, e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, and Sawyers CL (2015). Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, and Sawyers CL (2010). Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the National Academy of Sciences of the United States of America 107, 16759–16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, Smith-Raska MR, Esquilin JM, Zhang J, Arenzana TL, Lau CM, Churchill M, Pan H, Klinakis A, Dixon JE, et al. (2014). ZFX controls propagation and prevents differentiation of acute T-lymphoblastic and myeloid leukemia. Cell Rep 6, 528–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitington T, Gao P, Song W, Ross-Adams H, Lamb AD, Yang Y, Svezia I, Klevebring D, Mills IG, Karlsson R, et al. (2016). Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nature genetics 48, 387–397. [DOI] [PubMed] [Google Scholar]
- Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, Allison DF, Cai L, Rockowitz S, Liu S, et al. (2015a). Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 125, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, Gambhir SS, Lee P, Sartor O, Flemington EK, et al. (2015b). Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer research 75, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, and Tora L (2011). seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic acids research 39, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, McDonald C, Thomas R, Dhir R, Finkelstein S, et al. (2004). Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol 22, 2790–2799. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZX, Lane MV, Kemppainen JA, French FS, and Wilson EM (1995). Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Molecular endocrinology (Baltimore, Md 9, 208–218. [DOI] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.