

Abstract
Recent efforts by both academic and pharmaceutical researchers have focused on the HIV-1 capsid (CA) protein as a new therapeutic target. An interprotomer pocket within the hexamer configuration of the CA, which is also a binding site for key host dependency factors, is the target of the most widely studied CA inhibitor compound PF-3450074 (PF-74). Despite its popularity, PF-74 suffers from properties that limit its usefulness as a lead, most notably it’s extremely poor metabolic stability. To minimize unfavorable qualities, we investigated bioisosteric modification of the PF-74 scaffold as a first step in redeveloping this compound. Using a field-based bioisostere identification method, coupled with biochemical and biological assessment, we have created four new compounds that inhibit HIV-1 infection and that bind to the assembled CA hexamer. Detailed mechanism of action studies indicates that the modifications alter the manner in which these new compounds affect HIV-1 capsid core stability, as compared to the parental compound. Further investigations are underway to redevelop these compounds to optimize potency and drug-like characteristics and to deeply define the mechanism of action.
Keywords: Bioisosteres, HIV-1 capsid protein, Antiviral, Surface plasmon resonance, Computer-aided drug design
INTRODUCTION
Drug-resistance and toxicity to antiretroviral drugs is a significant problem that drives the search for new inhibitors of HIV. Therefore, identifying new targets and developing therapeutic compounds remains a continuing research priority. Recent efforts by both academic and pharmaceutical researchers have focused on the HIV-1 capsid (CA) protein [1] as a new therapeutic target. An inter-protomer pocket within the hexamer configuration of the CA, which is also a binding site for key host dependency factors [2–4], is the target of the very high potency compound GS-CA1 [5] and the most widely studied CA inhibitor compound PF-3450074 (PF-74) [2–4,6–8]. Even though these compounds demonstrate that high-affinity compounds can be designed to this binding site, they, unfortunately, suffer from problems. GS-CA1 suffers from low solubility/bioavailability [9], and PF-74 is incredibly metabolically labile (Supplemental Data, Table 1 and Figure 4).
Our group has previously targeted the inter-protomer pocket, attempting to find new compounds that would bind and disrupt HIV-1 replication [10]. This effort provided a compound that targeted the hexameric configuration of the HIV-1 CA but lacked the requisite potency to be a viable lead, without significant modifications. Ideally, we are eventually aiming for a compound that exhibits the following attributes: (i) Biological activity: potency with median IC50 values ≤ 10 nM against HIV-1 subtypes A, B, and C; (ii) Toxicity: CC50 ≥ 500 μM against uninfected PBMCs; (iii) Drug-like properties: solubility ≥ 100 mg/ml and cLogP between 1 and 3.5; and an in vitro half-life (T½) in human liver microsomes of greater than 90 minutes. Therefore, to continue in our goal towards a viable candidate compound that targets this inter-protomer pocket on HIV-1 CA, and inhibits viral replication, we chose to explore modification of the PF-74 scaffold, testing how tolerant to modification it is. We chose PF-74 due to the availability of multiple crystal structures of it complexed with HIV-1 CA [2–4] and its established mode of action. Moreover, as we have demonstrated its utility in the modification of HIV1 entry inhibitors, we chose to utilize field-based bioisosteric replacement to guide the redesign [11].
MATERIALS AND METHODS
Chemical compounds
PF-3450074 (PF-74) was purchased from MilliporeSigma (Burlington, MA). The CX compounds were synthesized de novo according to the schemes and details that follow.
Synthetic scheme of CX03:
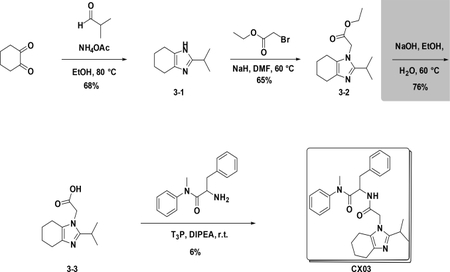
Synthetic scheme of CX04:
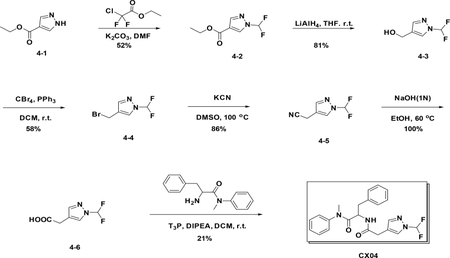
Synthetic scheme of CX05:
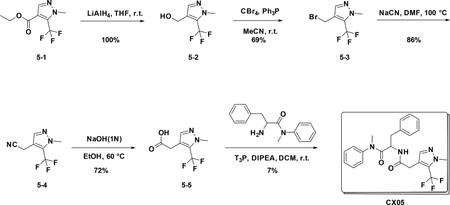
Synthetic scheme of CX06:
Preparation of CX03
Preparation of 2-isopropyl-4,5,6,7-tetrahydro-1H-benzo[d]imidazole (3–2):
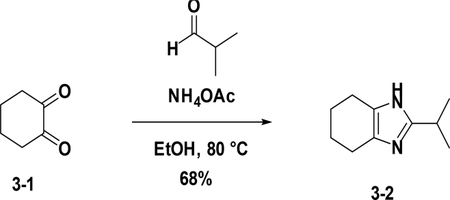
To a solution of 3–1 (1.0 g, 8.9 mmol) in EtOH (20 mL) were added NH4OAc (4.1 g, 53.4 mmol) and aldehyde (634 mg, 8.9 mmol), the resulting mixture was heated to 80°C for 4 hours. After cooled down to room temperature, the reaction was poured into water (30 mL) and then extracted with EA (25 mL×3). The combined organic phases were washed with brine (25 mL×3), dried over NaSO4, filtered and concentrated. The residue was purified by flash chromatography (silica gel, ethyl acetate (10%) in petroleum ether) to provide 3–2 (992 mg, 68%).
Preparation of ethyl 2-(2-isopropyl-4,5,6,7-tetrahydro-1H-benzo[d]imidaz -ol-1-yl)acetate (3–3):
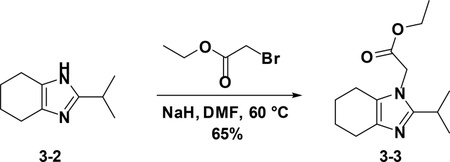
To a solution of 3–2 (1 g, 6.0 mmol) in DMF (10 ml) was added NaH (264 mg, 6.6 mmol), the resulting mixture was stirred at 60°C for 4 hours. After cooled down to room temperature, the reaction was poured into water (30 mL) and then extracted with EA (25 mL×3). The combined organic phases were washed with brine (25 mL×3), dried over NaSO4, filtered and concentrated. The residue was used in the next step without further purification (65%).
Preparation of 2-(2-isopropyl-4,5,6,7-tetrahydro-1H-benzo[d]imidazol-1-yl)acetic acid (3–4):
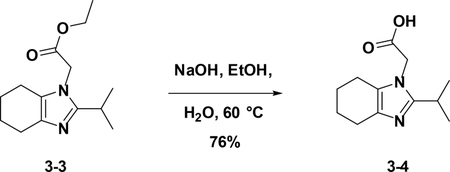
To a solution of crude 3–3 in EtOH (10 mL) and H2O (5 ml) was added NaOH, the resulting mixture was stirred at stirred at 60°C for 8 hours. After cooled down to room temperature, the reaction was poured into water (30 mL) and then extracted with EA (25 mL×3). The combined organic phases were washed with brine (25 mL×3), dried over NaSO4, filtered and concentrated. The residue was used in the next step without further purification (76%).
Preparation of 2-(2-(2-isopropyl-4,5,6,7-tetrahydro-1H-benzo[d]imidazol-1-yl)acetamido)-N-methyl-N,3-diphenylpropanamide (CX03):
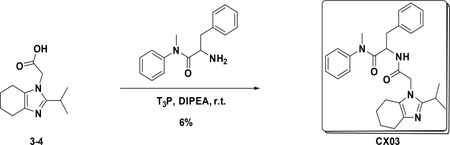
At room temperature, to a solution of 3–4 (223 mg, 1 mmol) in DCM (5 mL) were added amine (280 mg, 1.1 mmol), T3P (1.2373 g, 2.0 mmol) and DIPEA (516 mg, 4.0 mmol), the resulting mixture was stirred at room temperature overnight. Then it was washed with water and 5% HCl aqueous solution and concentrated to give crude product which was purified by Pre-HPLC to afford CX03 (27 mg, 6%) as white solid. LC-MS (ESI): m/z 459.55 [M+1]1H NMR (400 MHz, CDCl3) δ 7.44 – 7.25 (m, 3H), 7.16 – 7.05 (m, 3H), 7.02 – 6.90 (m, 2H), 6.77 – 6.59 (m, 3H), 4.74 (td, J = 8.4, 6.1 Hz, 1H), 4.37 (s, 2H), 3.74 – 3.54 (m, 4H), 3.30 – 3.11 (m, 3H), 2.90 – 2.74 (m, 2H), 2.64 – 2.43 (m, 3H), 2.33 – 2.04 (m, 2H), 1.31 – 1.10 (m, 6H).
Preparation of CX04
Preparation of ethyl 1-(difluoromethyl)-1H-pyrazole-4-carboxylate (4–2):
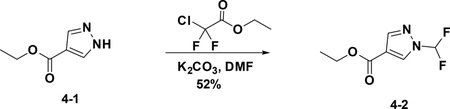
To a solution of 4–1 (100 mg, 0.351 mmol) in DMF (1.2 mL) were added 2-chloro-2,2-difluoroacetate (136 mg, 0.428 ml) and K2CO3 (197 mg, 0.714 mmol), the resulting mixture was stirred at 60°C for 5 hours. The reaction was quenched with water, and extracted with EA. The combined organic phases were washed with brine (15 mL), dried over NaSO4 and filtered. The filtrate was concentrated,and the residue was purified by flash chromatography (silica gel, MeOH (6%) in DCM) to provide 4–2 (69 mg, 52%).
Preparation of (1-(difluoromethyl)-1H-pyrazol-4-yl) methanol (4–3):

To a solution of 4–2 (40 mg, 0.21 mmol) in THF (1 mL) was added LAH (40 mg, 1.05 mmol), the resulting mixture was stirred at room temperature for 1 hour. The reaction was quenched with water, and extracted with EA. The combined organic phases were washed with brine (15 mL), dried over NaSO4 and filtered. The filtrate was concentrated, and the residue was used in next step without purification (81%).
Preparation of 4-(bromomethyl)-1-(difluoromethyl)-1H-pyrazole (4–4):
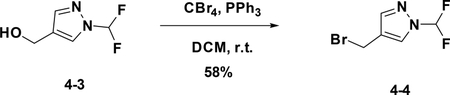
A solution of the 4–3 (600 mg, 4.02 mmol) in DCM was added CBr4 (2.0 g, 6 mmol) and PPh3 (1.6 g, 6 mmol). After stirred at r.t. for 12h, the reaction mixture was filtered and concentrated to give the crude product which was purified by flash chromatography (silica gel, 10%~50% EA in PE) to afford the product 4–4 (500 mg). Yield: 58%. LC-MS (ESI): m/z M / M+2=211.10 / 213.01
Preparation of 2-(1-(difluoromethyl)-1H-pyrazol-4-yl) acetonitrile (4–5):
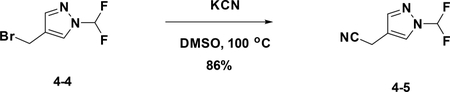
A solution of 4–4 (500 mg, 2.3 mmol) in DMSO was added KCN (184 mg, 2.8 mmol). After stirred at r.t. for 15 min, the reaction mixture was concentrated to give the crude product which was purified by flash chromatography (silica gel, 10%~50% EA in PE) to afford the product 4–5 (320 mg). Yield: 86%. LC-MS (ESI): m/z M+1=159.32
Preparation of 2-(1-(difluoromethyl)-1H-pyrazol-4-yl) acetic acid (4–6):
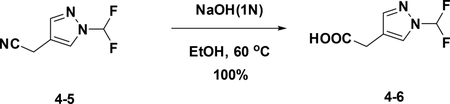
A solution of 4–5 (320 mg, 2.03 mmol) in EtOH was added 1N NaOH (2.03 mL). After stirred at 60°C for 24h, the reaction mixture concentrated to give the crude product (200 mg) without further purification. LC-MS (ESI): m/z M+1=177.21
Preparation of 2-(2-(1-(difluoromethyl)-1H-pyrazol-4-yl) acetamido)-N-methyl-N,3-diphenylpropanamide (CX04):
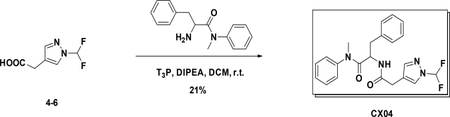
A solution of 4–6 (100 mg, 0.57 mmol) in DCM was added amine (160 mg, 0.62 mmol), T3P (361 mg, 1.14 mmol) and DIPEA (300 mg, 2.28 mmol). After stirred at r.t. for 1h, the reaction mixture was concentrated to give the crude product which was purified by Gilson (C18, 20%~100% MeCN in H2O with 0.1% HCOOH) to afford the product (40 mg). Yield: 21 %. LC-MS (ESI): m/z M+1=413.53, 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 7.47 – 7.36 (m, 1H), 7.34 – 7.25 (m, 3H), 7.17 – 7.08 (m, 3H), 6.94 – 6.83 (m, 2H), 6.77 (dd, J = 7.2, 2.0 Hz, 2H), 6.43 (d, J = 8.3 Hz, 1H), 4.77 (dd, J = 15.3, 7.4 Hz, 1H), 3.30 (s, 2H), 3.16 (s, 3H), 2.83 (dd, J = 13.4, 6.8 Hz, 1H), 2.62 (dd, J = 13.4, 7.5 Hz, 1H).
Preparation of CX05
Preparation of (1-methyl-5-(trifluoromethyl)-1H-pyrazol-4-yl) methanol (5–2):
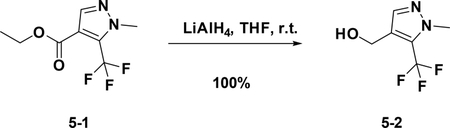
At 0°C, to a solution of 5–1 (200 mg, 0.9 mmol) in THF (4 mL) was added LiAlH4 (171 mg, 4.5 mmol), the resulting mixture was stirred at room temperature for 3 hours. The reaction was quenched with water, and extracted with Et2O. The combined organic phases were washed with brine (15 mL), dried over NaSO4 and filtered. The filtrate was concentrated,and the residue was used for the next step directly (190 mg, 100%).
Preparation of 4-(bromomethyl)-1-methyl-5-(trifluoromethyl)-1H-pyrazole (5–3):
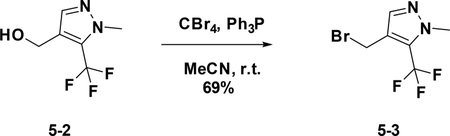
To a solution of 5–2 (500 mg, 2.8 mmol) in anhydrous MeCN (15 mL) were added PPh3 (1.5 g, 5.72 mmol) and CBr4 (1.88 g, 5.67 mmol) at 0°C. The resulting mixture was stirred at room temperature overnight. The reaction was concentrated and purified by flash chromatography (silica gel, 30% ethyl acetate in petroleum ether) to provide 5–3 ( 469 mg, 69%).
Preparation of 2-(1-methyl-5-(trifluoromethyl)-1H-pyrazol-4-yl) acetonitrile (5–4):
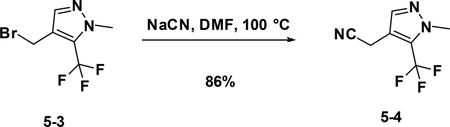
A solution of 5–3 (420 mg, 2.2mmol) in DMF was added KCN (286 mg, 4.4mmol). After stirred at r.t. for 4 hours, the reaction mixture was concentrated to give the crude product which was purified by flash chromatography (silica gel, 10%~50% EA in PE) to afford the product 5–4 ( 357 mg, 86%).
Preparation of 2-(1-methyl-5-(trifluoromethyl)-1H-pyrazol-4-yl) acetic acid (5–5):
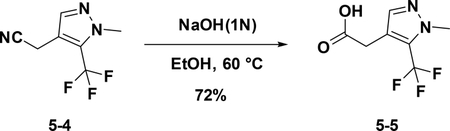
To a solution of 5–4 (150 mg, 0.7 mmol) in EtOH (8 mL), KOH (18 mg, 4mmol) and water (2 ml) were added. The resulting mixture was stirred at 60°C for 8 hours. Then the solution was concentrated to give the crude product which was used for the next step without further purification (104 mg, 72 %).
Preparation of N-methyl-2-(2-(1-methyl-5-(trifluoromethyl)-1H-pyrazol-4-yl) acetamido)-N,3-diphenylpropanamide (CX05)
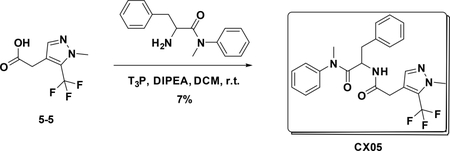
To a solution of 5–5 (140 mg, 0.673 mmol) in DCM (3 mL) were added amine (188 mg, 0.74 mmol), T3P (860 mg, 2.7 mmol) and DIPEA (350 mg, 2.69 mmol), the resulting mixture was stirred at room temperature for 12 hours. Then it was washed with water and 5% HCl aqueous solution and concentrated to give crude product which was purified by Pre-HPLC to afford CX05 (25 mg, 7%) as white solid. LC-MS (ESI): m/z 445.42 [M+1], 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.22 (m, 4H), 7.16 – 7.06 (m, 3H), 6.84 – 6.66 (m, 4H), 6.03 (d, J = 8.2 Hz, 1H), 4.76 (dd, J = 15.1, 7.6 Hz, 1H), 3.91 (d, J = 0.8 Hz, 3H), 3.38 (d, J = 1.0 Hz, 2H), 3.13 (s, 3H), 2.80 (dd, J = 13.3, 7.6 Hz, 1H), 2.62 (dd, J = 13.2, 6.7 Hz, 1H)..
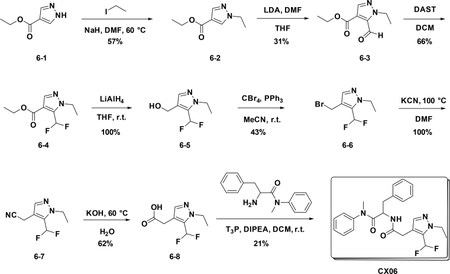
Preparation of CX06
Preparation of ethyl 1-ethyl-1H-pyrazole-4-carboxylate (6–2):
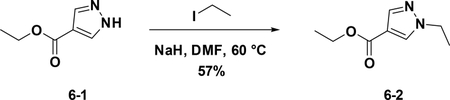
A solution of 6–1 (500 mg, 3.57 mmol) in DMF (5 mL) was added NaH (314 mg, 3.93 mmol) at 0°C. After 20 mins, iodoethane was added,and the mixture was stirred at room temperature for 16 hours. The reaction was quenched with water (30 mL), and extracted with EA (30 mL×3). The combined organic phases were washed with brine (15 mL), dried over NaSO4 and filtered. The filtrate was concentrated, and the residue was used for the next step directly (57%).
Preparation of ethyl 1-ethyl-5-formyl-1H-pyrazole-4-carboxylate (6–3):
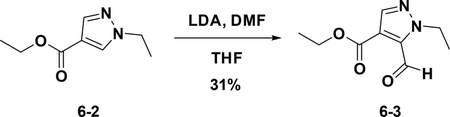
At −78°C, to a solution of LDA (127 mg, 1.2 mmol) in THF (10 mL) was added 6–2 (100 mg, 0.6 mmol). The resulting mixture was stirred at −78°C for 5 mins. Then DMF (346 mg, 0.8 mmol) was added, and the mixture was warmed to room temperature for another 1 hour. The resulting solution was diluted with water (10 mL) and extracted with EA (15 mL×3). The combined organic phases were washed with brine (25 mL×3), dried over NaSO4, filtered and concentrated. The residue was purified by flash chromatography (silica gel, 30% ethyl acetate in petroleum ether) to provide 6–3 (36 mg, 31%).
Preparation of ethyl 5-(difluoromethyl)-1-ethyl-1H-pyrazole-4-carboxylate (6–4):
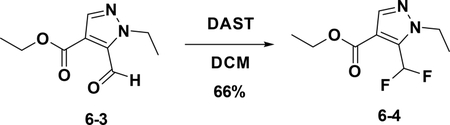
To a solution of 6–3 (50 mg, 0.255 mmol) in DCM (1 mL) was added DAST (102.8 mg, 0.638 mmol) was added at 0°C. The resulting mixture was stirred at room temperature for 16 hours. Then the solution was diluted with water (10 mL) and extracted with EA (15 mL×3). The combined organic phases were washed with brine (25 mL×3), dried over NaSO4, filtered and concentrated. The residue was used in the next step without purification (66%).
Preparation of (5-(difluoromethyl)-1-ethyl-1H-pyrazol-4-yl) methanol(6–5):
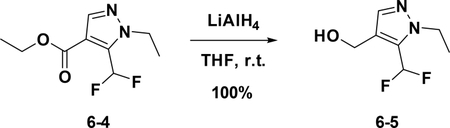
To a solution of 6–4 (370 mg, 1.697 mmol) in anhydrous THF (5 mL) was added LiAlH4 (322 mg, 8.486 mmol) at 0°C. The resulting mixture was stirred at room temperature for 30 mins. Then the reaction was quenched with water and extracted with Et2O; the combined organic phases were dried over NaSO4, filtered and concentrated to give the crude product which was used for the next step without further purification (100%).
Preparation of 4-(bromomethyl)-5-(difluoromethyl)-1-ethyl-1H-pyrazole (6–6):
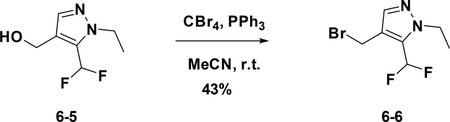
To a solution of 6–5 (150 mg, 0.852 mmol) in anhydrous MeCN (15 mL) were added PPh3 (565 mg, 1.704 mmol) and CBr4 (447 mg, 1.704 mmol) at 0°C. The resulting mixture was stirred at room temperature overnight. The reaction was concentrated and purified by flash chromatography (silica gel, 20% ethyl acetate in petroleum ether) to provide 6–6 ( 87 mg, 43%).
Preparation of 2-(5-(difluoromethyl)-1-ethyl-1H-pyrazol-4-yl)acetonitrile (6–7):
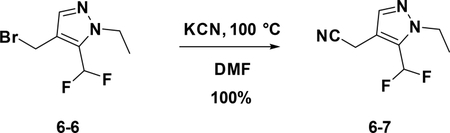
A solution of 6–6 (100 mg, 0.418 mmol) in DMF was added KCN (30 mg, 0.418 mmol). After stirred at r.t. for 15 min, the reaction mixture was concentrated to give the crude product which was purified by flash chromatography (silica gel, 10%~50% EA in PE) to afford the product 6–7 ( 77 mg, 100%).
Preparation of 2-(5-(difluoromethyl)-1-ethyl-1H-pyrazol-4-yl) acetic acid (6–8):
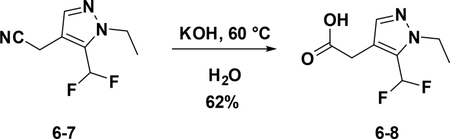
To a solution of 6–7 (80 mg, 0.4 mmol) in EtOH (4 mL), NaOH (120 mg) and water (2 ml) were added. The resulting mixture was stirred at 60°C overnight. Then the solution was concentrated to give the crude product which was used for the next step without further purification (62%).
Preparation of 2-(2-(5-(difluoromethyl)-1-ethyl-1H-pyrazol-4-yl) acetamido)-N-methyl-N,3-diphenylpropanamide (CX06)
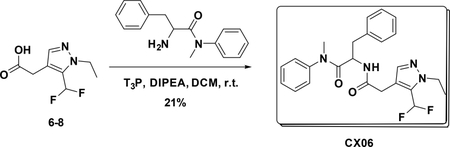
To a solution of 6–8 (55 mg, 0.127 mmol) DCM (5 mL) were added amine (70 mg, 0.27 mmol), T3P (350 mg, 0.54 mmol) and DIPEA (140 mg, 1.08 mmol), the resulting mixture was stirred at room temperature for 12 hours. Then it was washed with water and 5% HCl aqueous solution and concentrated to give crude product which was purified by Pre-HPLC to afford CX06 (25 mg, 21%) as white solid. LC-MS (ESI): m/z 441.42 [M+1], 1H NMR (400 MHz, CDCl3) δ 7.38 – 7.23 (m, 4H), 7.18 – 7.03 (m, 3H), 6.90 – 6.73 (m, 4H), 6.68 (s, 1H), 6.15 (d, J = 8.2 Hz, 1H), 4.74 (dd, J = 15.4, 7.1 Hz, 1H), 4.21 (q, J = 7.2 Hz, 2H), 3.32 (s, 2H), 3.18 – 3.03 (m, 3H), 2.79 (dd, J = 13.3, 7.3 Hz, 1H), 2.60 (dd, J = 13.3, 6.9 Hz, 1H), 1.45 – 1.35 (m, 3H).
Cells
Human embryonic kidney 293T cells (a gift from Dr. Irwin Chaiken, Drexel University, Philadelphia, PA) and TZM-bl reporter cell lines (obtained through the NIH AIDS reagent program from Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc.) [12] were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), 10% Fetal Bovine Serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine. Human astroglioma U87 cells stably expressing CD4/CCR5 or CD4/CXCR4 were obtained from Professor Hongkui Deng, Peking University, and Prof. Dan Littman, New York University, USA, through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NI [13,14]. U87 cells were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 2mM L-glutamine, 300 μg/ml G418 (Thermo Scientific, Waltham, MA) and 1 μg/ml Puromycin (Thermo Scientific). All cells were incubated continuously, unless otherwise stated, at 37°C in a humidified chamber containing 5% CO2 air environment.
Antibodies and peptides
Antibodies were obtained from the following sources: Mouse anti-p24 monoclonal antibody and rabbit anti-p24 polyclonal antibody from Abcam, Cambridge, United Kingdom (Cat numbers: ab9071 and ab63913, respectively); HRP-conjugated goat antirabbit antibody from Life Technologies, Carlsbad, CA (Cat numbers: 31460). Antibodies were used at a concentration of 0.5 μg/ml unless otherwise indicated. N-terminal biotinylated CPSF6(308–327) peptide (DRPPPPVLFPGQPFGQPPLG) and non-biotinylated CPSF6(313327) peptide (PVLFPGQPFGQPPLG) were synthesized by GenScript Corp. (Piscataway, NJ).
Elisa-based quantification of capsid (p24) content
An ELISA plate was coated with 50 ng of mouse anti-p24 (Abcam, ab9071) per well for 2 hours at room temperature, blocked with 3% BSA for 2 hours at room temperature and washed with PBST buffer (0.1% Tween-20 in PBS). Pseudoviral stocks were lysed with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) at 37°C for 1 hour and added to the plate overnight at 4°C. Simultaneously, p24 protein (produced and purified as described in below) was added for the generation of a standard curve. Following the overnight incubation, the plate was washed with PBST buffer and 1:5000 dilution of rabbit anti-p24 (Abcam, ab63913) was added for 2 hours at room temperature. After washing the unbound rabbit anti-p24 off the plate with PBST buffer, goat anti-rabbit-HRP at a 1:5000 dilution was added for 1 hour at room temperature. The plate was then extensively washed with PBST buffer. Subsequently, a solution of 0.4 mg/ml o-phenylenediamine in a phosphate-citrate buffer with sodium perborate (Sigma-Aldrich) was added and incubated in the dark for 30 minutes. Optical densities were then obtained at 450 nm in a Multiskan™ GO Microplate Spectrophotometer (Thermo Scientific).
Recombinant capsid overproduction, purification, and mutagenesis
The overproduction and purification of p24 (HIV-1 CA) were performed as previously described [15]. Briefly, the procedure is also described here: plasmid containing C-terminally His-tagged HIV-1NL4–3 CA (a gift from Dr. Eric Barklis, Oregon Health and Science University, Portland, OR) was transformed into BL21-Codon Plus (DE3)-RIL Competent Cells (Agilent Technologies, Wilmington, DE) and grown up in auto-induction medium ZYP-5052 overnight with shaking (225 rpm) at 30°C [16]. Bacterial cultures were spun down at 5000 rpm (J2-HC centrifuge, Beckman), and the supernatant was discarded. Cell pellets were resuspended in PBS and lysed via sonication. The resultant supernatant was ultracentrifuged at 40,000 rpm (Optima LE-80K ultracentrifuge, Beckman) for 1 hour and immediately applied to a Talon cobalt resin affinity column (Clonetech Laboratories, Mountain View, CA) and eluted with 0.2 M imidazolein PBS buffer. The eluted protein was dialyzed overnight into 20 mM Tris-HCl pH 8.0 at 4°C, concentrated and applied to a Q-sepharose column (HiTrap Q HP, GE 71–7149-00 AP, GE), then washed with 20mM Tris-HCl pH 8.0 and eluted with 0.3M NaCl in 20 mM Tris-HCl pH 8.0. Purified capsid protein was dialyzed into 20 mM Tris-HCl pH 8.0 at 4°C, concentrated to 120 μM, flash frozen in liquid nitrogen, aliquoted and stored at −80°C. Capsid hexamer was generated by introducing mutations at following sites: A14C, E45C, W184A, and M185A. Capsid mutagenesis was done by sitedirected mutagenesis (Stratagene).
Production of pseudotyped viruses
Single-round infectious specific envelope-pseudotyped luciferase-reporter viruses were produced by dual transfection of two plasmids (3 μg of viral backbone plasmid and 4 μg of viral envelope plasmid) in 6-well plated 293T cells (0.8×106 cells/ well) [13]. Viral backbone plasmid is an envelope-deficient HIV1 pNL4–3-LucR+E- plasmid that carries the luciferase-reporter gene [17]. Viral envelope plasmid is a plasmid expressing either the HIV-1 JR-FL envelope [18,19] or the heterologous envelope from amphotropic murine leukemia virus (AMLV) or the vesicular stomatitis virus glycoprotein (VSV-G). Transfections of these plasmids was carried out via calcium phosphate transfection (ProFection Mammalian Transfection System, Promega, Madison, WI) or using the JetPrime transfection reagent (Polyplus #71260) for 5–6 hours. Following the incubation, transfection reagent and the DNA-containing medium was removed, cells were washed with DMEM and replenished with fresh culture media. Supernatants containing produced pseudovirus were collected 48 hours post-transfection, clarified, filtered, aliquoted and stored at −80°C. Mutations in the HIV-1 pNL4–3-LucR+E- plasmid were performed by Genscript (Piscataway, NJ).
Single-round infection assay
The details of the single-round HIV-1 infection assay for detecting viral infectivity have been published previously [17,20,21]. Briefly, U87.CD4.CCR5 (1.2 × 104 cells/well) target cells were seeded in 96-well luminometer-compatible tissue culture plates (Greiner bio-one). After 24 hours incubation at 37°C, a compound or DMSO (vehicle control for the compound, Sigma) were mixed with pseudotyped viruses (normalized to p24 content) and the mixture was added to the target cells and incubated for 48 hours at 37°C. Subsequently, the media was removed from each well, and the cells were lysed by the addition of 50 μl of luciferase lysis buffer (Promega) and one freeze-thaw cycle. A GloMax 96 microplate luminometer (Promega) was used to measure the luciferase activity of each well after the addition of 50 μl of luciferase assay substrate (Promega). For the timeof-addition assay, DMSO or compounds were added at indicated time points post-infection and the resulting luciferase signal was measured after 48 hours.
Viral late-stage infection assay
Single-round infectious specific envelope-pseudotyped luciferase-reporter viruses were produced from 293T cells [13] in the presence of a compound (from 0.1 mM to 0.1 μM with same DMSO concentration) or DMSO (a vehicle control for compounds, Sigma). After 48 hours incubation at 37°C, the result culture supernatants containing pseudotype virus stocks were diluted ten-fold and then used to infect U87.CD4.CCR5 target cells. Target cells with or without pseudotyped viruses were incubated for 48 hours at 37°C. Subsequently, the media was removed from each well, and the cells were lysed by the addition of 50 μl of luciferase lysis buffer (Promega) and one freeze-thaw cycle. A GloMax 96 microplate luminometer (Promega) was used to measure the luciferase activity of each well after the addition of 50 μl of luciferase assay substrate (Promega). Compoundinduced effects are manifested as a decrease in infectivity in the target cells (measured as luciferase activity), normalized against the infectivity of virus produced from DMSO (vehicle control) treated cells. The effects of a compound on viral late stage were also ascertained by effect on viral production as monitored by the p24 assay as described above.
Competition ELISA assay
A 96-well ELISA plate was coated with 200 ng of neutravidin per well for overnight at 4°C. The next day the plate was blocked with 3% BSA for overnight at 4°C. The plates were washed five times with PBST buffer (0.1% Tween in PBS). Biotinylated CPSF6 peptide (final concentration 0.1 μg/ml) in blocking buffer (0.5% BSA in PBS) was added in and incubated for 4 hours. The plate was then washed five times with PBST buffer. Capsid hexamer (final concentration 0.4 μg/ml) with or without a compound were mixed and added to the plate in blocking buffer (0.05% BSA in PBS). The mixture was incubatedfor overnight at 4°C. Following the overnight incubation, the plate was washed with PBST buffer and 1:5000 dilution of rabbit anti-p24 (Abcam, ab63913) was added for 2 hours at room temperature. After washing the unbound rabbit anti-p24 off the plate with PBST buffer, goat anti-rabbit-HRP at a 1:5000 dilution was added for 1 hour at room temperature. The plate was then extensively washed with PBST buffer. Subsequently, a solution of 0.4 mg/ml o-phenylenediamine in a phosphate-citrate buffer with sodium perborate (Sigma-Aldrich) was added and incubated in the dark for 30 minutes. Optical densities were then obtained at 450 nm in a Multiskan™ GO Microplate Spectrophotometer (Thermo Scientific). The binding of capsid hexamer to the CPSF6 peptide in the presence of a compound was normalized to the binding without a compound (only in the presence of DMSO, as vehicle control for all compounds). A non-biotinylated CPSF6 peptide and compound PF-74 were used as positive binding controls for this assay.
Surface plasmon resonance assay
All binding assays were performed on a ProteOn XPR36 SPR Protein Interaction Array System (Bio-Rad Laboratories, Hercules, CA). The instrument temperature was set at 25°C for all kinetic analyses. ProteOn GLH sensor chips were preconditioned with two short pulses each (10 seconds) of 50 mM NaOH, 100 mM HCl, and 0.5% sodium dodecyl sulfide. Then the system was equilibrated with PBS-T buffer (20 mM sodium phosphate, 150 mM NaCl, and 0.005% polysorbate 20, pH 7.4). The surface of a GLH sensor chip was activated with a 1:100 dilution of a 1:1 mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (0.2 M) and sulfo-N-hydroxysuccinimide (0.05 M). Immediately after chip activation, the HIV-1 disulfide stabilized NL4–3 capsid protein, purified as outlined in Pornillos et al. [22], was prepared at a concentration of 100 μg/ml in 10 mM sodium acetate, pH 5.0 and injected across ligand flow channels for 5 min at a flow rate of 30 μl/min. Then, after unreacted protein had been washed out, excess active ester groups on the sensor surface were capped by a 5 minutes injection of 1 M ethanolamine HCl (pH 8.0) at a flow rate of 5 μl/min. A reference surface was similarly created by immobilizing a non-specific protein (IgG b12 anti HIV1 gp120; was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti-HIV-1 gp120 Monoclonal (IgG1 b12) from Dr. Dennis Burton and Carlos Barbas) and was used as a background to correct non-specific binding.
To prepare a compound for direct binding analysis, compound stock solutions, along with 100% DMS, and totaling 30 μl was made to a final volume of 1 ml by addition of sample preparation buffer (PBS, pH 7.4). Preparation of analyte in this manner ensured that the concentration of DMSO was matched with that of running buffer with 3% DMSO. Serial dilutions were then prepared in the running buffer (PBS, 3% DMSO, 0.005% polysorbate 20, pH 7.4) and injected at a flow rate of 100 μl/min, for a 1 minute association phase, followed by up to a 5 minutes dissociation phase using the “one shot kinetics” capability of the Proteon instrument [23]. Data were analyzed using the ProteOn Manager Software version 3.0 (Bio-Rad). The responses of a buffer injection and responses from the reference flow cell were subtracted to account for the nonspecific binding and injection artifacts. The equilibrium dissociation constant (KD) for the interactions, and derived from a minimum of four experiments, were calculated in ProteOn Manager Version 3.1.0.6 (Bio-Rad, Hercules, CA), using the equilibrium analysis function.
For the mutant analysis, responses at a single concentration of compound, either PF-74 or CX06, were expressed as a percentage of Rmax, to take into account differences in molecular weights between the compound and differences in immobilization densities.
In vitro capsid assembly assay
The effect of compounds on the assembly of HIV-1 CA was measured by monitoring turbidity at 350 nm using a modification of the method of our lab and others [15,24,25]. Briefly, 1.0 μl of concentrated compound (5 mM) in 100% DMSO was added to a 74-μl aqueous solution (solution was made by mixing 2 ml of 5 M NaCl with 1 ml of 200 mM NaH2PO4, pH 8.0). To initiate the assembly reaction, 25 μl of purified capsid protein (120 μM) was added. An identical reaction mixture was prepared, omitting the compound (i.e., just DMSO as a vehicle control). Samples were allowed to equilibrate for 2 minutes before reading. Readings were taken at 350 nm every one minute for 39 minutes. Capsid was used at a final concentration of 30 μM.
Nuclear import assay
Fluorescently labeled HIV-1 particles were produced in 293T/17 cells (from ATCC, Manassas, VA) by co-transfecting plasmids expressing the viral genome (pR9ΔEnv), VSV-G glycoprotein (pMD2.G, Add gene, Cat#1259) and the Vpr-INsfGFP plasmid (Francis et al., 2014). Fluorescent viruses were collected at 36 hours post-transfection, filtered through a 0.45 μm filter and quantified for RT activity. TZM-bl cells were infected at MOI 2 for 4 hours. Cells were fixed with 2% PFA and immune-labeled with a mouse antibody against the nuclear membrane marker Lamin A/C (Pierce# MA3–1000) and secondary anti-mouse Cy5 fluorescent antibodies (Southern Biotech). Cells were imaged on a Zeiss LSM780 laser scanning confocal microscope using a C-Apo 63x/1.4NA oil-immersion objective. Images of the nuclear volume (~21 Z-stacks spaced by 0.5 μm) were acquired with multiple line averaging. INsfGFP and Lamin/Cy5 fluorescence was excited using 488 and 633 nm laser lines, respectively. 3D-image series were processed off-line using ICY image analysis software (http://icy.bioimageanalysis.org/) (de Chaumont et al., 2012), and the number of INsfGFP complexes per nucleus was determined manually for 30 nuclei (from 4 randomly selected fields of view) for each independent experiment.
HIV-1 capsid core-stability measurements in vitro
VSV-G pseudotyped HIV-1 particles produced in 293T/17 cells were labeled with INsfGFP and CypA-DsRed, as described previously [26]. Single virus particles were bound to a poly-Llysine treated coverglass and mildly permeabilized with saponin (100 μg/ml) for 1 min. Saponin was washed away, permeabilized virions transferred into in a phosphate buffer containing 10 μM of the CX compounds or 5 μM of PF-74 and imaged on a DeltaVision (GE Healthcare) wide-field microscope at room temperature. Images were acquired from 4 neighboring fields of view at 1 min intervals. CyclosporinA (CsA, 5 μM) was added at 21 min after permeabilization/onset of imaging to displace CypA-DsRed from permeabilized viral cores that have not disassembled during the imaging time window. The number of INsfGFP and CypA-DsRed puncta was detected using the ICY image analysis software (http://icy.bioimageanalysis.org/). The total number of INsfGFP puncta in each field of view remained constant over the 30 min imaging period. The kinetics of CA disassembly was determined by normalizing the number of CypA-DsRed puncta to that at t=0 min subtracting the background non-permeabilized or immature virus detected through CsA addition.
Q-PCR analysis of viral DNA
Detection of early (RU5) and late (U5Ψ) viral reverse transcription products, 2-LTR circles, and integrated pro-virus was performed as previously described [27]. Briefly, whole-cell DNA was isolated after 48 hours (RU5 and U5Ψ) or 24 hours (2-LTR) pseudotyped virus infection in cells (treated with a compound or only with vehicle control, DMSO) via DNeasy Blood and Tissue Kit (Qiagen). Real-time PCR analysis was performed in an Applied Biosystems 7300 Real-Time PCR System (Thermo Scientific) with TaqMan Universal Master Mix II, no UNG (Thermo Scientific), 200ng of whole cell DNA and the indicated viral DNA primer/probe sets along with the simultaneous detection of an endogenous housekeeping gene porphobilinogen deaminase (PBGD). Primers and probes (sequences listed in Appendix 2) used in the following concentrations: 500 nM hRU5-F2 and hRU5-R, 100 nM hRU5-P, 900 nM MH531 and MH532, 360 nM LRT-P, 900 nM nPBGD- For and nPBGD-Rev, 360 nM nPBGD-Pr, 50 nM MH535 and MH536, 100 nM MH603, 250 nM LTR-Rev and LTR-rev and 200 nM LTRp. The first initial amplification step in the Alu-LTR integrated assay used 100 ng of total DNA, 100 mM Alu-F’ and 600 mM Gag-rev with Tth DNA polymerase (Promega), 5x RT buffer (Promega), 25 mM MgCl2 and 10 mM dNTPs (Thermo Scientific) [28]. Reaction mixtures were subjected to 3 min incubation at 94°C, 35 cycles of 94°C for 30 s, 50°C for 30 sec, 72°C for 1:40 min with a final extension at 72°C for 3 min in a T100 Thermo Cycler (BioRad). As controls for reverse transcription and integration inhibition, cells were treated with TMC278 (also referred to as Rilpivirine, obtained via AIDS Research and Reference Reagent Program [29]).
Computational docking analyses
Docking of CX06 into the capsid dimer:
Docking calculations were carried out on a CA dimer extracted from PDB 4XFZ. The CA protein was prepared by the Protein Preparation Wizard implemented with Maestro (Schrödinger Maestro Version 11.5.011, MM share Version 4.1.011, Release 2018–1, Platform Darwin-x86_64). The Grid box was centered on the co-crystalized PF-74. For validating the glide docking protocol, the original ligand PF-74 was built using the LigPrep tool (Schrödinger Maestro Version 11.5.011) and docked using glide-XP mode. The predicted docked pose matched the original co-ordinates of the co-crystalized PF-74 with an RMS value of 0.3. CX06 was docked using the same glide-XP mode and the top ranked pose was selected as a representation of the predicted binding mode.
Docking of PF-74 and CX06 into the capsid dimer Q63A mutant:
Residues Gln63, in both protomers, were mutated to Ala in silico (Schrödinger Maestro Version 11.5.011). The mutated protein was then energy minimized using MacroModel (water as a solvent and OPLS3 force field). The same grid preparation steps and docking protocol were used to dock PF-74 and CX06 into the Q63A CA. The top ranked pose for each was selected as a representation of the predicted binding mode.
RESULTS AND DISCUSSION
Computational design of head group variants of PF-74
To explore modifications of PF-74, as a lead into our overarching goal, we chose to start fairly small and choose a limited number of compounds to design and focused upon the changes to the methylindole head group of PF-74. We first imported the structure of PF-74 bound to the native HIV-1 capsid hexamer structure (PDB ID 4XFZ) [2] into Spark Version 10.4 (Cresset®, Litlington, Cambridgeshire, UK; http://www.cresset-group.com/spark/) to derive a field-based high-content pharmacophore [30,31]. Two protomers, extracted from the native hexameric HIV-1 capsid protein structure, were added as excluded volume to discourage the selection of bioisosteric fragments that would clash with the protein within the interprotomer pocket during the search (Figure 1). Spark searches a database of including fragments derived from multiple databases to find non-classical bioisosteres that exhibit similar shape and electronic properties as the region of interest when placed in the context of the final molecule [11]. The results of this search were analyzed,and structures that displayed low 2D similarity, while retaining a sufficiently high BIF% value (a factor that indicates how good the replacement is in the context of the conformation of the entire molecule) were favored. Finally, the suggested molecules were analyzed for their predicted ADME properties (absorption, distribution, metabolism, and excretion) and compared to PF-74. The in silico prediction of drug-like metrics was achieved using StarDrop 6.4 (Optibrium, Ltd., Cambridge, UK) [32], implementing the oral non–central nervous system (CNS) drug profile [33], supplemented with an additional parameter for logD. Details for the specific models are provided in the StarDrop Reference Guide from Optibrium and are online at the StarDrop FAQs (http://www.optibrium.com/community/faq/adme-qsar-models). A probabilistic scoring algorithm [34] is then used to combine the model predictions in the oral nonCNS drug profile into an overall score. For reference, scores range from 0 to 1, with 0 suggesting extremely non–drug-like, and 1 suggesting the perfect drug. Following these analyses, four compounds containing 2-(propan-2-yl)-4,5,6,7-tetrahydro-1H1,3-benzodiazole, 1-(difluoromethyl)-1H-pyrazole, 1-methyl5-(trifluoromethyl)-1H-pyrazole, and 5-(difluoromethyl)-1ethyl-1H-pyrazole head groups were chosen for synthesis and evaluation. All of these compounds had improved predicted ADME properties over that of PF-74 (Figure 2). The syntheses of these compounds, termed CX03, CX04, CX05, and CX06, are outlined in supplemental data. PF-74 MilliporeSigma (Burlington, MA) served as a control compound for these studies. All CX compounds were synthesized as racemic mixtures. The metabolic stability of these new compounds were tested and compared to PF-74. Although they did not display a huge increase in stability, all of the CX compounds did have greater half-lives as compared to PF-74, with the greatest increase being 4-fold for CX04 (Supplemental Data, Table 2).
Figure 1.

PF-74 bound in the inter-protomer pocket. Positive field surfaces of the compounds are depicted in red; negative field surfaces of the compounds are depicted in blue, and hydrophobic field surfaces of the compounds are depicted in gold. Visualized in Flare Version 2.0 (Cresset®, Litlington, Cambridgeshire, UK; http://www.cresset-group.com/flare/) [30]. The green cross represents a chloride ion present in the structure.
Figure 2.

(A) The individual models that comprise the modified oral non-CNS (central nervous system) drug profile and their respective importance to the profile. HIA = human intestinal absorption; hERG (human ether-à-go-go-related gene); IC50 = half-maximal inhibitory concentration; BBB = blood–brain barrier.(B) Plot showing the StarDrop (Optibrium, Ltd., Cambridge, UK)– derived logS versus the score from a multimetric, modified oral non- CNS profile for PF-74 and the CX compounds.
The CX compounds inhibit HIV-1 from distinct genetic subtypes replicating in human primary peripheral blood mononuclear cells
Following successful syntheses, we next sought to quantify the potency of CX compounds against the fully infectious virus. Moreover, this assay was performed using healthy, primary cells to gain further insight into the potential toxicities of the compound towards natural target cells. Additionally, as a key issue in the development of novel HIV drugs is their ability to inhibit the replication of genetically diverse isolates (especially isolates from the most globally prevalent subtypes A, B, and C), we chose to assess the potency of the CX compounds to inhibit the replication of the primary isolates (i.e., not previously passaged in cell lines prior to cloning) from subtypes A (HIV-193RW025), B (HIV-1JR-CSF), and C (HIV-193MW965), in primary human peripheral blood mononuclear cells (PBMCs) [11,13,15,35]. The toxicity of the CX compounds to the PBMCs using an MTS assay was evaluated in parallel [33]. PF-74 was tested at the same time for comparison, and as to our knowledge, it hasn’t been tested in this particular format, against these particular isolates. The results of these analyses are summarized in Table 1. As can be observed from the IC50 values, the CX compounds displayed antiviral activity against all of the isolates tested, despite their varying degrees of 2D structure dissimilarity (based on Tanimoto similarity scores based on Merck Atom Pairs) [36]. Moreover, the CX compounds demonstrated no toxicity over the concentration range under investigation, in contrast to PF-74 (Supplemental data, Figure 5). Compounds CX03 and CX06 displayed the best potency, with a median IC50 value over the three isolates tested of 5.9 μM, as compared to the median IC50 of PF-74 of 0.9 μM (Table 1). CX03 demonstrates a Spark-derived shape score of 0.948, which is indicative of it being 3-dimensionally the closest to PF74. Interestingly, CX06 displays the lowest 3D similarity to PF74, despite retaining almost identical potency as the most similar compound, CX03.
Table 1:
Potency of the CX compounds and PF-74 against isolates from subtypes A, B, and C. The therapeutic index for each compound was calculated using the median IC50 across all three isolates tested. Chemical structures were drawn using ChemAxon software (Budapest, Hungary).
| Compound | 2D similarity to PF-74 | IC50 HIV-1 92RW025 (μM) Clade A | IC50 HIV-1 JR-CSF (μM) Clade B | IC50 HIV-1 93MW965 (μM) Clade C | Median IC50 (μM) | CC50 (μM) | Therapeutic Index (CC50/IC50) |
|---|---|---|---|---|---|---|---|
 |
1 | 1.5 ± 0.9 | 0.6 ± 0.20 | 0.6 ± 0.10 | 0.9 ± 0.5 | 90.5 ± 5.9 | 93.9 |
 |
0.489 | 6.5 ± 2.2 | 4.9 ± 1.4 | 6.4± 2.1 | 5.9 ± 0.9 | >100 | >16.9 |
 |
0.482 | 18.7 ± 3.9 | 18.7 ± 2.9 | 14.0 ± 5.7 | 17.1 ± 2.7 | >100 | >5.84 |
 |
0.498 | 23.1 ± 11.1 | 16.2 ± 6.5 | 14.5 ± 3.7 | 18.0 ± 4.6 | >100 | >5.58 |
 |
0.494 | 6.4 ± 0.4 | 6.5 ± 0.7 | 4.7 ± 2.1 | 5.9 ± 1.0 | >100 | >17.0 |
The CX compounds retain target specificity of the parental molecule, PF-74
Having demonstrated antiviral activity of the CX compounds, we next sought to verify that the compounds still bind to HIV-1 CA. We, therefore, overproduced and purified the HIV-1 NL4–3 disulfide stabilized CA hexamer, as outlined previously [22], for use in surface plasmon resonance (SPR) interaction analysis using the ProteOn™ XPR36 Protein Interaction Array System (Bio-Rad, Hercules, CA). Detailed methods are provided in the material and methods section. Briefly, the pure CA hexamer was immobilized to the surface of a GLH to a density of 16,048 RU using standard amine coupling procedures. A reference surface was similarly created using IgG b12 [37] and was matched with the experimental CA hexamer surface density to within 3%. First, we tested the activity of the CA hexamer surface by performing binding analyses using a peptide derived from nucleoporin 153 (NUP153) [38]. The NUP1531407–1423 peptide [3] was synthesized by Genscript (Piscataway, NJ) and dissolved in pure DMSO to a concentration of 50 mM. This stock was then used to make a concentration series, diluted to match that of the running buffer for the experiment (PBS, pH 7.4, 0.005% polysorbate 20, 3% DMSO). Figure 1, Panel A shows representative sensorgrams from this analysis. The equilibrium dissociation constant (KD) for this interaction, and derived from a minimum of four experiments, was calculated in ProteOn Manager Version 3.1.0.6 (Bio-Rad, Hercules, CA), using the equilibrium analysis function and was found to be 202 ± 22 μM. This is in excellent agreement with the published values obtained using isothermal titration calorimetry (ITC) [3,4] and fluorescence polarization [4] and demonstrates the activity of the CA hexamer surface. Next, we characterized the interaction of PF-74 with the CA hexamer under the same buffer conditions as for the NUP153 peptide. Representative sensorgrams for this interaction are shown in Figure 3, panel B. The KD for the interaction between PF-74 and the CA hexamer, derived in the same manner as for NUP153, was determined to be 176 ± 78nM. This value, again, is in excellent agreement with previously published values using ITC [3,4]. Having demonstrated the activity of our CA hexamer surface and our ability to correctly derive KD values for the control molecules NUP153 and PF-74, we set about to characterize the interaction of our CX compounds with the CA hexamer. Representative sensorgrams and the KD values derived from a minimum of four data sets as described for NUP153 are shown in Figure 1, panels C-F. The KD values obtained are in very good agreement (within 2-fold) with the IC50 values obtained in our multiple round infection assays (Table 1). Moreover, the kinetic profile of the CX compounds versus PF-74 indicates that the increased potency of PF-74 is likely correlated with the slower off rate of PF-74.
Figure 3.

Sensorgrams depicting the interaction of (A) NUP1531407– 1423 peptide, (B) PF-74, (C) CX03, (D) CX04, (E) CX05, and (F) CX06, with immobilized HIV-1NL4–3 CA hexamer. Concentration shown: NUP1531407–1423 peptide (450, 225, 113, 56.3, 28.1 and 0μM); PF- 74 (20, 5, 1.25, 0.313, 0.0.78 and 0μM); CX03 (225, 56.3, 14.1, 3.52, 0.9 and 0μM); CX04 and CX05 (300, 150, 75, 37.5, 18.8 and 0μM); CX06 (100, 33.3, 11.1, 3.7, 1.2 and 0μM).
CX06 inhibits the replication of HIV-1 in both the early and late stages
Having demonstrated target engagement and antiviral activity, we next sought to gain some understanding regarding the mechanism of antiviral action of the CX compounds. Due to its low Tanimoto similarity and low Spark-derived shape score, we chose to focus our mechanisms of action studies solely on compound CX06.
We first sought to determine which stage or stages of the viral life-cycle. CX06 exerts its antiviral action, as HIV-1 CA functions in both pre- and post-integration events. To achieve this, we used the modular nature of the single round infection assay, in which singly-round infective HIV-1 particles are generated recombinantly in one cell type, and then used to infect another cell type: effectively separating early and late stages of the replication cycle. We have successfully used this system to determine the stage-specific inhibition of other compounds generated by our group [13].
Effects on assembly were identified by incubating the viral producer cells in the absence or presence of various concentrations of CX06. Supernatants containing HIV-1JR-FL envelope pseudotyped virus (which encodes for firefly luciferase as a reporter gene) were then diluted tenfold and used to infect U87–CD4–CXCR4 target cells. Compound-induced effects are manifested as a decrease in infectivity in the target cells, compared with those infected with the virus from untreated cells. Effects on early-stage events were determined by using virus produced in the absence of compound for infection of target cells in the absence or presence of various concentrations of CX06. As can be seen in Figure 4, CX06 displays effects in both the late, assembly and early, infective stages of the HIV-1 replication cycle. This dual-stage inhibition profile is shared with the parental molecule PF-74 (Supplemental Data, Figure 7).
Figure 4.

CX06 functions in the late and early stages of HIV-1 replication. (A) To assess the effect of CX06 on late-stage events, recombinant virus was produced from 293T cells either in the absence or presence of CX06, and the culture supernatants containing pseudotype stocks were diluted tenfold and then used to infect target cells in the absence of test compound. Compound-induced effects are manifested as a decrease in infectivity in the target cells (measured as luciferase activity), normalized against the infectivity of virus from untreated cells. (B) Effects of CX06 on early-stage events were determined by producing virus via transfection of 293T cells (as described in the materials and methods) for infection of U87.CD4. CCR5 target cells in the absence or presence of various concentrations of CX06. The antiviral effects of CX06were determined to be due to a specific effect on the virus as the compound was not cytotoxic to 293T or U87.CD4. cells over the concentration range tested.
Elucidating the late stage mechanism
Having demonstrated that CX06 exerts an antiviral effect upon HIV-1 replication at the late stage, we next sought to determine the nature of this block. PF-74 has been demonstrated to also have an antiviral effect in the late, assembly stages of HIV1 replication, producing misformed virions, rather than reducing the amount of virus produced [6]. Therefore, we chose to first look at the amount of virus produced in the presence of CX06 before looking more closely. Quantification of the virus produced in the presence of 20 μM of the compound indicates that it functions by simply decreasing the amount of virus released from the cells, as judged by p24 content (Figure 5).
Figure 5.

The effect of compound CX06 on viral production. The effects of compound CX06 (20 μM) on the production of HIV-1JR-FL Envpseudotyped viruses relative to vehicle DMSO control as shown by p24 quantitation. Data shown are mean values of three individual experiments done in triplicate, with error bars depicting the standard error of the mean (SEM). Statistical significance was assessed by student t-test (p<0.005).
Elucidating the early stage mechanism
The results shown in Figure 4B, demonstrate that CX06 also exerts an antiviral effect at the early stage of replication. In the assay used, this early stage encompasses every process from the entry of the virus, to the final integration of the proviral DNA into the host genome. Therefore, we next sought to hone in on the exact process or processes that are disrupted in the phase by CX06. To rule out an effect on the entry process as a point of intervention for this compound. To do this, we performed the single round infection assay, but pseudotyped the HIV-1 virions withthe amphotropic murine leukemia virus (AMLV) Env. As can be seen in Figure 6A, CX06 inhibited this AMLV Envpseudotyped HIV-1 with almost identical potency as when the HIV-1JR-FLEnv was used (Figure 4), suggesting that CX06 targets a post-entry process or processes. This finding is corroborated by a time-ofaddition study, in which CX06 is only found to be effective within the first 6 hours post-infection (Figure 6B).
Figure 6.

(A) Effect of CX06 on AMLV Envpseudotyped HIV-1 infection. (B) Time of addition study. TZM-bl cells were infected with VSV-pseudotyped HIV-1 virus. Drugs Nevirapine (10 μM), Raltegravir (10 μM), PF-74 (2 μM) and CX06 (20 μM) were added at indicated time points. Infectivity was measured by the luciferase assay (Promega) at 48 hpi and normalized to DMSO control. Data shown are mean values of three individual experiments done in triplicate. Error bars = STD. dev.
The CA-mediated process of uncoating is an essential but poorly understood process but is tightly associated with the process of reverse transcription [39]. As such, a hallmark of the disruption of uncoating in HIV-1 replication is the inability of the virus to initiate and or complete reverse transcription. Unintegrated viral DNA synthesized during HIV-1 infection includes linear and circular forms, and each of these distinct viral cDNAs can be used as a surrogate marker for events surrounding the completion of reverse transcription and for nuclear import of viral DNA during replication [15]. We, therefore, used polymerase chain reaction (PCR)-based detection and quantitation of HIV-1 early, late and 2-long-terminal repeat (LTR) circle reverse transcriptase products as a convenient and sensitive means to assess the effect of CX06 upon completion of reverse transcription and nuclear import of viral DNA. Additionally, we utilized Alu-PCR to quantify the amount of integrated provirus. As can be seen in Figure 7, treatment with CX06 slightly diminished early RT products, but late RT products, 2-LTR circles, and integrated pro-virus significantly affected. This, coupled with our direct interaction data, is consistent with disruption of uncoating. However, we cannot rule out a direct effect on RT itself. Therefore, to rule this possibility out, we tested the effect of the compounds TMC278, PF-74, and CX06 directly on purified recombinant RT using an RT assay kit (Roche Diagnostics, IN, USA). From Figure 8, it is evident that while the control reverse transcriptase inhibitor TMC278 inhibits RT, neither PF-74 nor CX06 does so to any appreciable level. We can, therefore, be fairly confident that CX06’s effect on RT in the context of infection is indirect via its effect upon uncoating.
Figure 7.

CX06 treatment drastically reduces late reverse transcriptase products, 2-LTR circles, and integrated provirus. Total DNA at 24 h (2- LTR) or 48 h (RU5, U5Ψ, integrated provirus) isolated from HIV-1JR-FLEnv infected U87.CD4.CCR5 cells. Real-time PCR was performed as described in Materials and Methods. Results were normalized to the endogenous control PBGD and reflect the relative fold change compared to DMSO, TMC278, PF-74, and CX06 treatment. Data shown are mean values of three individual experiments done in triplicate, with error bars depicting the standard error of the mean (SEM). Statistical significance was assessed by student t-test (p<0.005). TMC278 was used at a concentration of 1.5 nM, PF-74 was used at 1.5 μM, and CX06 was used at 15 μM. Nuclear import of HIV-1 INsfGFP complexes was visualized in TZM-bl cells at 4 hpi. Infections (MOI2) were treated with vehicle control (DMSO), RT inhibitor Nevirapine (Nevi 10μM) or CA binding drugs PF-74 (5μM) and CX06 (20μM). (E) Representative images, scale bar = 5 μm. (F) Quantification of nuclear INsfGFP complexes. Error bars represent SEM from 4fields of view.
Figure 8.

The effect of compound CX06 on RT activity in vitro. CompoundsCX06 (15 μM) and PF-74 (1.5 μM) have no effect on RT activity relative to vehicle DMSO control group. TMC278 (1 nM) has significant inhibitory effect on RT activity. Assay was performed using a colorimetric reverse transcriptase assay kit (Roche Diagnostics, IN, USA). Data shown are mean values of three individual experiments done in triplicate, with error bars depicting the standard error of the mean (SEM). Statistical significance was assessed by student t-test (p<0.005).
The nuclear import of HIV-1 complexes, which is independent of reverse transcription [40–42], was visualized by confocal microscopy, as described in (Francis and Melikyan 2018). In this experiment, TZM-bl cells were infected with a VSV-G pseudotyped HIV-1 fluorescently labeled with a marker for viral ribonucleoprotein complexes the integrase-superfolder GFP (INsfGFP). The number of INsfGFP complex that entered the cell nuclei was determined in the presence of drugs or DMSO (control). Analysis of HIV-1 nuclear import revealed that PF-74 (2 μM) and CX06 (20 μM) efficiently inhibited the appearance of INsfGFP spots in the nucleus, while the RT inhibitor Nevirapine was without effect. This result suggests that both PF-74 and CX06 abrogate CA-dependent steps of virus nuclear import.
Mutation of residues in the inter-protomer pocket reduces the sensitivity of HIV-1 to CX06 by decreasing its interaction with HIV-1 CA
Having demonstrated thus far that CX06 interacts with HIV1 CA and inhibits HIV-1 replication in a manner consistent with disruption of CA function, we next sought to establish a direct link between its interaction with the CA protein and its antiviral effect. As a first step towards this, we chose to establish whether or not CX06 truly shares the same binding pocket as PF-74. To achieve this, we designed and implemented a competition ELISA using a peptide from CPSF6 [43]. It has been determined structurally and through competitive binding assays, that PF-74 and CPSF6 share a common binding site and that PF-74 interaction inhibits the binding of CPSF6 to CA [2–4]. Therefore, we capitalized on this to determine whether or not CX06 would inhibit CPSF6 interaction with CA (Figure 9). The non-biotinylated CPSF6 peptide was used as a control to demonstrate that we could see inhibition in this assay. As can be seen, CX06 inhibits the binding of CA to CPSF6 to the same degree as PF-74 in this competition ELISA. As such, we can be reasonably confident that CX06 shares the same interprotomer binding pocket as does CPSF6 and PF-74.
Figure 9.

CX06 competes with a peptide from CPSF6 for binding to the CA hexamer. Using competition ELISA, non-biotinylated CPSF6 peptide CPSF6313–327 (25 μM) competes with biotinylated CPSF6 peptide CPSF6308–327 in binding with capsid hexamer as does CX06 (50 μM) and PF-74 (5 μM). Data shown are mean values of three individual experiments done in triplicate, with error bars depicting the standard error of the mean (SEM). Statistical significance was assessed by student t-test (p<0.05).
As CX06 and PF-74 share a common binding site, we next sought to establish if they share common interactions with residues within this site and demonstrate that the antiviral effect of CX06 is mediated through its interaction with HIV-1 CA. Therefore, we investigated mutation of residues shown to be contact residues for PF-74, and tested the sensitivity of these mutant viruses to inhibition by CX06. Also, we included two other mutations, P90A and N74D, which have shown to block the interaction of CA with cyclophilin A and to alter nuclear entry pathways, respectively [44,45]. HIV-1 pseudovirus containing CA wild-type (WT), CA N57A, CA Q63A, CA K70A, CA N74D, and CA P90A were produced and equal amounts of p24 were utilizedto infect the cell for a single-round infection assay. Figure 10 shows the relative infectivity of these mutants, establishing that they are infectious enough to utilize in this assessment. As shown in Table 2, PF-74 is relatively resistant to these changes, as demonstrated by the minor changes in the IC50 values between wild-type and the mutant viruses. This is consistent with resistant virus selection studies that demonstrated to have a large effect on PF74 potency, a mutant must have more than a single mutation [6]. CX06 showed a similar tolerance to mutations within the interprotomer pockets, with the exception of Q63A, whose mutation resulted in an almost 7-fold increase in the IC50 value relative to wild-type. To investigate whether this is simply by reducing the interaction of CX06 with CA, or something more convoluted, we introduced the Q63A mutation into the disulfide stabilized hexameric CA construct and performed SPR. Figure 11 shows that the mechanism by which the Q63A mutation desensitizes the virus harboring that mutation to CX06 is by reducing the CA protein’s interaction with the compound. Taken together, the discovery that mutation of CA glutamine 63 to alanine increases the half maximal inhibitory concentration of CX06 relative to wild-type, most likely by reducing interaction with the compound, demonstrates that the antiviral effect of CX06 is mediated through the CA.
Figure 10.

Infectivity of HIV-1 with mutations in the CA region of the genome as compared to wild-type. Data shown are mean values of three individual experiments done in triplicate, with error bars depicting the standard error of the mean (SEM). Statistical significance was assessed by student t-test (p<0.05).
Table 2:
Potency of PF-74 and CX06 against wild-type and CA mutant viruses.
| Virus | PF-74 IC50 (μM) | Fold increase | CX06 IC50 (μM) | Fold increase |
|---|---|---|---|---|
| Wild-type | 0.22 ± 0.05 | 1 | 3.1 ± 0.5 | 1 |
| CA N57A | 0.25 ± 0.04 | 1.14 | 4.3 ± 0.5 | 1.4 |
| CA Q63A | 0.40 ± 0.05 | 1.82 | 21.4 ± 1.4 | 6.9 |
| CA K70A | 0.25 ± 0.08 | 1.14 | 3.2 ± 0.3 | 1.03 |
| CA N74D | 0.50 ± 0.10 | 2.3 | 2.4 ± 0.7 | 0.8 |
| CA P90A | 0.21 ± 0.04 | 0.95 | 3.1 ± 0.5 | 1 |
Figure 11.

Reduced interaction of compound CX06 with a capsid hexamer mutant Q63A. The interaction of compound CX06 (25 μM) and control compound PF-74 (10 μM) with wild-type and mutant Q63A capsid hexamer in SPR. Both responses in wild-type and mutant Q63A were normalized to the theoretical Rmax as percent maximal response. Data shown are mean values of three individual experiments done in triplicate, with error bars depicting the standard error of the mean (SEM) (p<0.005).
CX06 reduces CA assembly and destabilizes the assembled core
A previous study by Lemke et al. [46], demonstrated that inhibitors that bind to the same pocket on the HIV-1 capsid can have distinct binding modes and mechanism of action. This, coupled with the difference in mutant sensitivity, prompted us to look at the effects of CX06 compounds on both the in vitro assembly of the HIV-1 CA [15] and the in vitro stability of intact viral cores using a previously published methodology [26].
First, we chose to look at whether or not CX06 accelerated CA assembly in vitro. Using the same turbidity assay utilized by Blair et al. [6], we assessed the effect of CX06 in this assay. Figure 12A shows the results of this assay. In line with its previously shown effect of accelerating assembly of HIV-1 CA, PF-74 (red) increased the rate of assembly of HIV-1 CA under the in vitro conditions of the assay as compared to DMSO alone (black). In stark contrast, CX06 in the same assay reduced the multimerization of HIV-1 CA in the assay compared to both PF-74 and DMSO. This reduction in assembly could easily explain the reduced amount of virions produced in the presence of the CX06.
Figure 12.

Compound CX06 reduces the rate of capsid assembly and destabilizes the assembled capsid core in vitro. (A) Capsid assembly was monitored by an increase in turbidity using a spectrophotometer at 350 nm. Capsid was used at a final concentration of 30 μM, and compounds CX06 and PF74 at a final concentration of 50 μM. The presence of compound CX06 prevents the assembly of the capsid. (B) IN-GFP and CypA-DsRed double labeled HIV-1 viruses were bound to a coverglass, permeabilized with saponin (100ug/ml, 1 min) and washed. Viral Cores were imaged in the presence of a compound (5μM PF74 or 10μM CX06) or vehicle DMSO control at the indicated time. Average number of CypA-DsRedspots from four random fields was calculated at initial time point (0 min) as 100% value and calculated every minute for over 25 minutes. The CypA-DsRedspot numbers were divided by the number at initial time point and were presented as percent intact cores.
Next, we looked at the effect of CX06 on the assembled mature HIV-1 capsid. Briefly, HIV-1 pseudoviruses produced in 293T cells were labeled with INsfGFP and the CA marker cyclophilin A-DsRed (CypA-DsRed) (Francis et al 2016). Single viral particles were immobilized onto coverslips and mildly permeabilized by saponin to allow core disassembly (loss of CypA-DsRed/CA) in the presence or absence of indicated compounds. Viruses were continuously imaged to measure the loss of CypA-DsRed (red) puncta, while the number of INsfGFP puncta remained stable over time (Supplemental data, Figure 6). Figure 12B shows that, unlike PF-74, compound CX06 accelerated the loss of CypADsRed/CA and thus destabilized the HIV-1 core. Destabilization of the conical capsid shell in the cytoplasm would likely inhibit the completion of reverse transcription and block nuclear import [42].
Rotational freedom of the pyrazole ring may explain the different affinity and mechanism of action CX06
To help to rationalize the observed differences between the affinity and mechanism of action of CX06 as compared to PF74, we performed molecular docking analyses. Compound CX06 was docked into the inter-protomer pocket using Glide-XP mode and its binding pose compared to that of the co-crystal structure of PF-74 [2]. Figure 13 shows the overlay of the CX06 docked model on the co-crystalized PF-74. The two compounds appear to share a number of the same protein contacts (mainly Asn57 and Lys70) but differ significantly in the orientation of the indole ring of PF-74 and the pyrazole ring of CX06. The side chain carbonyl of Gln63 can accept a hydrogen bond (~ 1.7 Å) from the PF-74 indole ring, whereas this bond is not possible with the pyrazole ring of CX06. Moreover, the hydrogen bond between Gln63 and the PF-74 indole ring appears to restrict the rotation of the indole ring, therefore stabilizing the compound between the two CA protomers. This stabilizing hydrogen bonding interaction is not maintained in the CX06 docking models, and slightly lower energy poses of CX06 show the possible rotation of the CX06 pyrazole ring by 180 degrees. This rotation of CX06 pyrazole moiety causes steric clashes and may be the reason for the destabilization of the CA hexamer by this compound.
Figure 13.

Overlay of the dockedCX06 (pink) on the co-crystalized PF-74 (white).
Next, we sought to see whether or not molecular modeling could help us understand why the Q63A mutation causes a 7-fold decrease in antiviral potency, despite CX06 not directly interacting with Gln63. After in silico mutation, protein relaxation, and docking, we found that replacing the Gln63 side chain with the smaller Ala methyl side chain generated a hydrophobic sub-pocket at the main inter-protomer pocket. Not having the Gln63 hydrogen bond with the PF-74 allows the rotation of the indole ring, with the benzo-moiety now facing the new hydrophobic sub-pocket (Figure 14A,B), while other protein contacts are maintained (Asn57 and Lys70). However, in case of CX06, interestingly the new sub-pocket appears to force the compound to flip and adopt a different binding mode altogether in order to fill the pocket with a phenyl group (Figure 14C,D). Contacts with Asn57 and Lys70 are maintained with this pose (Figure 14D), however, in a vastly different way to that of the wild-type interaction. It is therefore conceivable that either this different binding mode, or the possibility of dynamically having two binding modes to the Q63A mutated CA, are the reasons behind the reduced interaction affinity and consequent reduction in antiviral potency.
Figure 14.

Molecular modeling of PF-74 and CX06 with wild-type and CA Q63A mutant CA protein. Panels A and C show a comparison between the co-crystalized PF-74 (A) and the CX06 model (C) with the wild-type CA. Panels B and D show a comparison of the docked models of PF-74(B) and CX06(D) with the Q63A mutant CA protein.
CONCLUSIONS
In summary, we have designed and tested a number of bioisosteric variants of PF-74 and demonstrated antiviral activity and maintenance of target specificity. This demonstrates that the methylindole region of PF-74 is more tolerant to variation than previously appreciated. Mechanism of action studies with a representative compound, CX06, also demonstrate that the head group region of the compounds is a primary determinant of both affinity and mechanism of action. Taken together, this work further highlights the potential of the inter-protomer pocket in the CA hexameric configuration as an attractive target for the design of further inhibitors. The continued exploration of modification to the PF-74 inhibitor scaffold to increase potency and drug-like metrics is ongoing within our research group.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH/NIAID grant 1 R56 AI118415–01A1 (Cocklin, PI) and R01 GM054787 (Melikian, PI). We’d like to thank Cresset (UK; http://www.cresset-group.com/) for access to their software via an academic license.
REFERENCES
- 1.Campbell EM, Hope TJ. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol. 2015; 13: 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, Sarafianos SG. STRUCTURAL VIROLOGY. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science. 2015; 349: 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, et al. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014; 10: e1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A. 2014; 111: 18625–18630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrier M, Bertine M, Le Hingrat Q, Joly V, Visseaux B, Collin G, et al. Prevalence of gag mutations associated with in vitro resistance to capsid inhibitor GS-CA1 in HIV-1 antiretroviral-naive patients. J Antimicrob Chemother. 2017; 72: 2954–2955. [DOI] [PubMed] [Google Scholar]
- 6.Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010; 6: e1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saito A, Ferhadian D, Sowd GA, Serraob E, Shic J, Halambage UD, et al. Roles of Capsid-Interacting Host Factors in Multimodal Inhibition of HIV-1 by PF74. J Virol. 2016; 90: 5808–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Price AJ, Halambage UD, James LC, Aiken C. HIV-1 Resistance to the Capsid-Targeting Inhibitor PF74 Results in Altered Dependence on Host Factors Required for Virus Nuclear Entry. J Virol. 2015; 89: 9068–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarvis LM., Conquering HIV’s capsid, in Chemical & Engineering News. Americal Chemistry Society. 2017; 95: 23–25. [Google Scholar]
- 10.Xu JP, Branson JD, Lawrence R, Cocklin S. Identification of a small molecule HIV-1 inhibitor that targets the capsid hexamer. Bioorg Med Chem Lett. 2016; 26: 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuyishime M, Lawrence R, Cocklin S. Core chemotype diversification in the HIV-1 entry inhibitor class using field-based bioisosteric replacement. Bioorg Med Chem Lett. 2016. 26: 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998; 72: 2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zentner I, Sierra LJ, Fraser AK, Maciunas L, Mankowski MK, Vinnik A, et al. Identification of a small-molecule inhibitor of HIV-1 assembly that targets the phosphatidylinositol (4,5)-bisphosphate binding site of the HIV-1 matrix protein. Chem MedChem. 2013; 8: 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Björndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, et al. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol. 1997; 71: 7478–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kortagere S, Madani N, Mankowski MK, Schön A, Zentner I, Swaminathan G, et al. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J Virol. 2012; 86: 8472–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005; 41: 207–234. [DOI] [PubMed] [Google Scholar]
- 17.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995; 206: 935–944. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann W, Schubert D, LaBonte J, Munson L, Gibson S, Scammell J, et al. Species-specific, postentry barriers to primate immunodeficiency virus infection. J Virol. 1999; 73: 10020–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcon L, Choe H, Martin KA, Farzan M, Ponath PD, Wu L, et al. Utilization of C-C chemokine receptor 5 by the envelope glycoproteins of a pathogenic simian immunodeficiency virus, SIVmac239. J Virol. 1997; 71: 2522–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cocklin S, Gopi H, Querido B, Nimmagadda M, Kuriakose S, Cicala C, et al. Broad-spectrum anti-human immunodeficiency virus (HIV) potential of a peptide HIV type 1 entry inhibitor. J Virol. 2007; 81: 3645–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Billich A, Hammerschmid F, Peichl P, Wenger R, Zenke G, Quesniaux V, et al. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: interference with HIV protein-cyclophilin A interactions. J Virol. 1995; 69: 2451–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pornillos O, Ganser-Pornillos BK, Banumathi S, Hua Y, Yeager M. Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J Mol Biol. 2010; 401: 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG. Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal Biochem. 2006; 358: 281–288. [DOI] [PubMed] [Google Scholar]
- 24.Abdurahman S, Végvári A, Levi M, Höglund S, Högberg M, Tong W, et al. Isolation and characterization of a small antiretroviral molecule affecting HIV-1 capsid morphology. Retrovirology. 2009; 6: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boutwell CL, Rowley CF, Essex M. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxicT-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J Virol. 2009; 83: 2460–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Francis AC, Marin M, Shi J, Aiken C, Melikyan GB. Time-Resolved Imaging of Single HIV-1 Uncoating in vitro and in Living Cells. PLoS Pathog. 2016; 12: e1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mbisa JL, Delviks-Frankenberry KA, Thomas JA, Gorelick RJ, Pathak VK. Real-time PCR analysis of HIV-1 replication post-entry events. Methods Mol Biol. 2009; 485: 55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swaminathan G, Rossi F, Sierra LJ, Gupta A, Navas-Martín S, MartínGarcía J. A role for microRNA-155 modulation in the anti-HIV-1 effects of Toll-like receptor 3 stimulation in macrophages. PLoS Pathog. 2012; 8: e1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh K, Marchand B, Rai DK, Sharma B, Michailidis E, Ryan EM, et al. , Biochemical mechanism of HIV-1 resistance to rilpivirine. J Biol Chem. 2012; 287: 38110–38123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheeseright T, Mackey M, Rose S, Vinter A. Molecular field extrema as descriptors of biological activity: definition and validation. J Chem Inf Model. 2006; 46: 665–676. [DOI] [PubMed] [Google Scholar]
- 31.Cheeseright TJ, Mackey MD, Scoffin RA. High content pharmacophores from molecular fields: a biologically relevant method for comparing and understanding ligands. Curr Comput Aided Drug Des. 2011; 7: 190–205. [DOI] [PubMed] [Google Scholar]
- 32.Segall M, Champness E, Obrezanova O, Leeding C. Beyond Profiling: Using ADMET Models to Guide Decisions. Chemistry & Biodiversity. 2009; 6: 2144–2151. [DOI] [PubMed] [Google Scholar]
- 33.Tuyishime M, Danish M, Princiotto A, Mankowski MK, Lawrence R, Lombart HG, et al. Discovery and optimization of novel smallmolecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorg Med Chem Lett. 2014; 24: 5439–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Segall MD. Multi-Parameter Optimization: Identifying High Quality Compounds with a Balance of Properties. Curr Pharm Des. 2012; 18: 1292–1310. [DOI] [PubMed] [Google Scholar]
- 35.Zentner I, Sierra LJ, Maciunas L, Vinnik A, Fedichev P, Mankowski MK, et al. Discovery of a small-molecule antiviral targeting the HIV-1 matrix protein. Bioorg Med Chem Lett. 2013; 23: 1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheridan RP, Hunt P, Culberson JC. Molecular transformations as a way of finding and exploiting consistent local QSAR. J Chem Inf Model. 2006; 46: 180–192. [DOI] [PubMed] [Google Scholar]
- 37.Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW, et al. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science. 1994; 266: 1024–1027. [DOI] [PubMed] [Google Scholar]
- 38.McMorrow I, Bastos R, Horton H, Burke B. Sequence analysis of a cDNA encoding a human nuclear pore complex protein, hnup153. Biochim Biophys Acta. 1994; 1217: 219–223. [DOI] [PubMed] [Google Scholar]
- 39.Ambrose Z, Aiken C. HIV-1 uncoating: connection to nuclear entry and regulation by host proteins. Virology. 2014; 454-455: 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burdick RC, Hu WS, Pathak VK. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proc Natl Acad Sci U S A. 2013; 110: E4780–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Francis AC, Melikyan GB. Live-Cell Imaging of Early Steps of Single HIV-1 Infection. Viruses. 2018; 10: E275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Francis AC, Melikyan GB. Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe. 2018; 23: 536–548 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rüegsegger U, Beyer K, Keller W. Purification and characterization of human cleavage factor Im involved in the 3’ end processing of messenger RNA precursors. J Biol Chem. 1996; 271: 6107–6113. [DOI] [PubMed] [Google Scholar]
- 44.Malikov V, da Silva ES, Jovasevic V, Bennett G, de Souza Aranha Vieira DA, Schulte B, et al. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat Commun. 2015; 6: 6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011; 7: e1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemke CT, Titolo S, von Schwedler U, Goudreau N, Mercier JF, Wardrop E, et al. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J Virol. 2012; 86: 6643–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.