

Abstract
The hemoglobin of Synechococcus sp. PCC 7002, GlbN, is a monomeric group I truncated protein (TrHb1) that coordinates the heme iron with two histidine ligands at neutral pH. One of these is the distal histidine (His46), a residue displaceable by dioxygen and other small molecules. Here, we show with mutagenesis, electronic absorption spectroscopy, and NMR spectroscopy that at high pH and exclusively in the ferrous state, Lys42 competes with His46 for the iron coordination site. When b heme is originally present, the population of the lysine bound species remains too low for detailed characterization; however, the population can be increased significantly by using dimethylesterified heme. Electronic absorption and NMR spectroscopies showed that the reversible ligand switching process occurs with an apparent pKa of 9.3 and a Lys-ligated population of ~60 % at the basic pH limit in the modified holoprotein. The switching rate, which is slow on the chemical shift timescale, was estimated to be 20–30 s−1 by NMR exchange spectroscopy. Lys42–His46 competition and attendant conformational rearrangement appeared related to weakened bis-histidine ligation and enhanced backbone dynamics in the ferrous protein. The pH and redox dependent ligand exchange process observed in GlbN illustrates the structural plasticity allowed by the TrHb1 fold and demonstrates the importance of electrostatic interactions at the heme periphery for achieving axial ligand selection. Analogy is drawn to the alkaline transition of cytochrome c, in which Lys–Met competition is detected at alkaline pH, but, in contrast to GlbN, in the ferric state only.
Keywords: truncated hemoglobin, 2/2 hemoglobin, heme iron coordination, dimethyl esterified heme, conformational exchange, NMR
For Table of Contents Use Only
Heme proteins harness the heme cofactor to perform diverse chemistries using a network of interactions between the polypeptide and the prosthetic group. Among the many features that dictate reactivity, the axial ligands to the heme iron play a critical role.1 They are intimately involved in modulating the redox potential of the heme,2 the structural properties of the heme binding site, and the stability of the holoprotein.3 Most heme proteins use one or two residues to coordinate the central iron. Among the latter group, it is common for one of the ligands to undergo facile displacement, a property that adds thermodynamic, kinetic, and structural complexity to the behavior of the protein. A specific challenge in the design of functional heme proteins is not only to achieve the desired ligation scheme but also to control its stability, life time, and the consequences of its disruption.4 The hemoglobin superfamily offers several examples of endogenous hexacoordination and can be used to explore how a common tertiary structure evolved to control axial ligand lability as demanded by distinct reactivity requirements.
“Hexacoordinate” hemoglobins use as axial ligands the conserved “proximal” histidine, located on the F helix, and a residue on the E (or distal) helix. This latter residue is most frequently a histidine (yielding a bis-histidine complex), although recently a lysine ligand has also been found in a small number of proteins.5–8 Endogenous hexacoordination has explicable consequences for the properties of a globin. For example, the rate of decoordination by the labile distal ligand can limit the kinetics of dioxygen binding,9 and a preserved ligand set in the ferric and ferrous oxidation states can lower the reorganization energy associated with electron transfer compared to systems in which the ligand set is altered.10 In rare instances, pronounced kinetic stability of distal ligation in the ferric or ferrous state (or both) is observed.11,12 For globin enzymes that must accomplish both small molecule binding and redox cycling, however, a balance of ligand association and dissociation rates must be reached to ensure activity on the physiological time scale.
Many studies have elucidated the properties of model hemes with axial imidazole ligands (see for example13 and references therein). Microperoxidases, which are small proteolytic fragments of cytochrome c, have been particularly useful for describing complexes of the His–Fe–X type,14 as has been a proximal histidine variant of myoglobin for complexes of the X–Fe–imidazole and the X–Fe–amine type.15 Nevertheless, controlling the properties of axial ligation has been a persistent, and formidable, objective in the de novo engineering of heme proteins.16–19 Whereas de novo electron carrier proteins may be designed around a robust bis-histidine ligation scheme, the design of an asymmetric or labile coordination sphere is considerably more difficult.4,20 Furthermore, anticipating the axial ligand set in an arbitrary protein scaffold remains a challenge. Prediction from sequence is unreliable in part because homology modeling is rarely possible at the necessary level of resolution. In addition, the static structures used to construct these models do not capture all features that may lead to ligation such as access to alternative conformational states (for example open versus closed forms) or reduced local stability easily overcome by the formation of a coordination bond. In hemoglobins, expectation of proximal histidine coordination is invariably satisfied, but distal ligation, let alone its thermodynamic and kinetic stability, has so far escaped prediction. Experimental data are necessary to articulate the determinants of heme coordination strength and lability.
In order to bind an exogenous ligand, an endogenously hexacoordinate protein must have an energetically and kinetically accessible space for the displaced axial residue. Thus, hexacoordinate globins are generally capable of a ligand-related conformational rearrangement. Structural studies illustrate that bis-histidine globins have a range of responses to exogenous ligand binding. In human cytoglobin, the distal histidine changes rotameric state and turns away from the heme when the ferrous protein binds carbon monoxide21 (PDB IDs: 2DC3 and 3AG0). In carbonmonoxy murine neuroglobin, the distal histidine retains its orientation but the heme slides deeper in the protein matrix, lengthening the distance between the iron and the histidine Nε2 atom22 (PDB IDs: 1Q1F and 1W92). Drosophila melanogaster hemoglobin undergoes a comparatively larger rearrangement involving heme sliding and displacement of a distal region (CD corner, D helix, and E helix in the canonical description of the fold) to accommodate cyanide as a ligand23 (PDB IDs: 2BK9, 2G3H). Various modes of flexibility have therefore evolved for ligand binding in these systems.
For ionizable ligands such as lysine and histidine, bonding to iron occurs in their neutral states; for a lysine side chain at physiological pH, deligation is expected to be coupled with protonation and formation of hydrogen bonds and other interactions with solvent or the protein. Proton-coupled deligation is exemplified by the Chlamydomonas reinhardtii hemoglobin, THB1, which in the ferric state undergoes a reversible pH-dependent transition between a lysine-ligated state and a water-bound lysine-deligated state with an apparent pKa of ~ 6.5.5 In another example, the well-known alkaline transition of ferric yeast iso-1-cytochrome c involves the displacement of the native Met80 ligand by Lys7324 and Lys79,25 residues otherwise exposed to solvent. The cytochrome c alkaline transition occurs with a higher apparent pKa (~ 8.5 to 9.5, depending on the specific protein and sample conditions) than in C. reinhardtii THB1. Modification of cytochromes c by mutagenesis often favors lysine ligation, suggesting that the strongly conserved protein sequence has evolved to condition its pH response.26–29 Further investigation of how hexacoordinate globins respond to changes in pH adds insights complementary to the cytochrome c studies.
The monomeric hemoglobin from Synechococcus sp. PCC 7002 (GlbN) accesses different conformations in its endogenously hexacoordinated bis-histidine state and exogenous ligand-bound state (Figure 1).30 In addition, this cyanobacterial protein can undergo covalent attachment of the heme group,31 a post-translational modification (PTM) that generates a c-like heme and reduces the motional freedom of the heme relative to the protein matrix. The PTM mechanism is straightforward; it consists in the addition of a histidine (His117) to the α carbon of one of the two b heme vinyl groups. The reaction occurs spontaneously in the ferrous oxidation state but not in the ferric state.32,33 The modification has been extensively studied for its pH dependence,33 its influence on the stability of the protein,34 its perturbation of backbone dynamic properties,35 and its potential utility in protein engineering.36 The PTM yields a holoprotein that has heme attachment in common with cytochrome c, a feature that provides a bridge between two important groups of proteins. The structure of the modified heme is shown in Figure S1.
Figure 1.

Heme propionate interactions in (A) the His–Fe–His and (B) the His–Fe–CN structures of GlbN with PTM (GlbN-A). “6p” indicates the 6-propionate group. The E, EF, and F elements of structure are shown as ribbon. The rest of the structure is omitted for clarity. The asymmetric units of the crystals contain three (A, PDB ID: 4MAX) and two (B, PDB ID: 4L2M) monomers, which were superimposed to illustrate variability in side chain positions. The 6-propionate interacts with Lys42 in the His–Fe–His complex whereas the 7-propionate interacts with His46 in the His–Fe–CN complex. The conformational change of the E helix and EF loop is apparent in the right side of the figure. The heme–His117 cross-link (PTM) is shown in both structures.
Here, we sought to probe the stability of the bis-histidine coordination scheme in GlbN and explore the conformational space accessible to the polypeptide beyond the structures depicted in Figure 1. When displaced by cyanide, the distal histidine (His46) stacks on the heme 7-propionate (Figure 1B) whereas in the bis-histidine state, a lysine (Lys42) points to the 6-propionate to form a putative salt-bridge (Figure 1A). We therefore reasoned that perturbation of charge interactions at the heme periphery, as would be caused by a change in pH or heme modification, might reshape the energy surface of the protein in a consequential way. As will be shown, elevated pH promotes an alternative GlbN ligation scheme in a process that can be linked to the heme propionates. This observation opens new opportunities to compare and contrast ligand competition and switching in a large number of heme proteins.
MATERIALS AND METHODS
GlbN Forms Under Investigation.
The present study required the preparation of several GlbN forms. Before PTM, the native holoprotein contains iron-protoporphyrin IX (Fe PP or b heme). We represent the ferric state of this species with Fe(III) PP GlbN. We also used the dimethyl-esterified cofactor and represent the ferric protein containing it with Fe(III) PPDME GlbN. GlbN in the ferrous oxidation state spontaneously forms a covalent linkage between the Nε2 of His117 and the 2-vinyl Cα of the heme group (Figure S1C).31 For brevity, we refer to the GlbN forms with the PTM as PP GlbN-A and PPDME GlbN-A.
Protein Production and Purification.
Recombinant GlbN was obtained from E. coli essentially as described previously.31 Briefly, inclusion bodies of apoGlbN were solubilized in 8 M urea. The protein was refolded and purified by passage through a Sephadex G-50 Fine (SigmaAldrich) sizing column. Fe(III) PP chloride (SigmaAldrich) was added and the holoprotein was subjected to anion exchange chromatography on a diethylaminoethyl (DEAE) Sephacel (GE Healthcare) column. GlbN purity was confirmed using sodium dodecyl sulfate polyacrylamide gel electrophoresis and mass spectrometry. Alternatively, no cofactor was added prior to anion exchange, and the purified apoprotein was exchanged into 1 mM potassium phosphate buffer pH 7.0 before storage as lyophilized powder or immediate reconstitution with Fe(III) PPDME chloride as detailed below. Uniformly 15N labeled GlbN was purified in the same fashion from cells grown in M9 minimal medium containing 15NH4Cl (Cambridge Isotope Laboratories) as the sole nitrogen source. Synechocystis sp. PCC 6803 GlbN containing Fe(III) PP was prepared with a similar protocol, as described previously.37
The K42L, H46L, K48L, and H117A GlbN variants were produced using the QuikChange site-directed mutagenesis method (Qiagen, Valencia, CA) and primers purchased from IDT (Coralville, IA). The GlbN variants were purified and reconstituted with Fe(III) PP chloride31 prior to the anion exchange step and lyophilized for long-term storage. The K42L and H117A GlbN holoproteins were converted to the apoprotein using cold butanone (Alfa Aesar) according to the procedure of Teale.38 Following dialysis into 10 mM potassium phosphate buffer pH 7.0, the apoprotein was reconstituted with Fe(III) PPDME chloride.
Reconstitution with Fe(III) PPDME.
Fe(III) PPDME chloride (Frontier Scientific, Logan, UT) was used without further purification. Reconstitution was performed according to procedures established for iron-porphyrin derivatives with low aqueous solubility.39 Fe(III) PPDME chloride was dissolved in dry dimethylsulfoxide (DMSO, J.T.Baker) to a concentration of 5–10 mg/mL and gently added to an aqueous solution of apoGlbN (0.3–1 mM protein, 2–5 mL potassium phosphate butter, pH 7) in 1.1 molar excess. The mixture was allowed to incubate with stirring at 4 °C, and heme binding was assessed after several hours using optical absorbance spectroscopy. Multiple additions of Fe(III) PPDME chloride to a final ~5-fold molar excess were required for sufficient reconstitution. The final DMSO concentration was kept below 10% v/v, and the reaction was allowed to proceed for 12 h after the last addition. The mixture was then clarified by centrifugation and the resulting cherry-red supernatant was applied to a DEAE anion-exchange column, which precipitated any remaining free Fe(III) PPDME. The purified protein was eluted with high salt buffer (50 mM potassium phosphate buffer pH 7, 100 mM NaCl) before exchange into 1 mM potassium phosphate pH 7.0 buffer for lyophilization and storage. With this protocol, some apoprotein remained in the sample as apparent in NMR spectra.
Preparation of GlbN-A.
GlbN-A was prepared as reported previously.35 For example, a sample of PPDME GlbN (~1 mM, 50 mM potassium phosphate buffer pH 7.1) was reduced with ~5 mM dithionite (DT, SigmaAldrich) and incubated at room temperature for ~10 min. The sample was then oxidized with 10 mM K3[Fe(CN)6] and passed over a G-25 (SigmaAldrich) desalting column equilibrated in 1 mM potassium phosphate buffer pH 7.0, concentrated using centrifugation, and lyophilized for storage. Formation of the covalent linkage between the GlbN polypeptide and PPDME was confirmed using ultra-performance liquid chromatography/mass spectrometry. The same procedure was applied to GlbN variants and 15N-labeled GlbN.
pH Titration of PPDME GlbN-A by Electronic Absorption Spectroscopy.
A concentrated sample of Fe(III) PPDME GlbN-A was diluted 50-fold into the appropriate buffer and the electronic absorption spectrum was measured using a Cary50 UV-Vis spectrophotometer over the wavelength range 800–260 nm in 1 nm steps. Ferrous (“deoxy”) GlbN-A was generated by addition of 2 mM fresh DT, and spectra were collected from 750 to 350 nm every 45 s for at least 5 min after addition of DT. A series of buffers were used over a pH range of 6.4 to 11.3: 100 mM potassium phosphate (pH 6.4 to 7.8), 100 mM Tris (pH 7.8 to 8.8), 100 mM sodium borate (pH 8.8 to 9.8), 100 mM glycine (pH 9.8 to 10.8), and 100 mM 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) (pH 10.8 to 11.3). When buffers were switched, two spectra were collected at the same pH to control for the influence of the buffer. To account for small dilution errors, the ferrous GlbN-A spectra were scaled on the basis of the ferric absorbance spectra, which do not vary with pH over the studied range.
The apparent pKa of the alkaline transition observed during the pH titration of Fe(II) PPDME GlbN-A was determined by globally fitting the Henderson-Hasselbalch equation (1) to two traces (424 nm and 526 nm, approximate isosbestic points of a low-pH transition) using a routine implemented in Wolfram Mathematica version 10.0.2.0. In Equation (1), n is a Hill coefficient, λ is the wavelength of observation, Abase is the absorbance in the alkaline limit, Aacid is the absorbance in the acid limit and pKa(app) is the apparent pKa of the transition.
| (1) |
Heme–Protein Crosslinking Kinetics.
Concentrated samples of Fe(III) PP GlbN, Fe(III) PPDME GlbN, Fe(III) PP K42L GlbN, and Fe(III) PPDME K42L GlbN were diluted into 100 mM sodium borate buffer, pH 9.2. DT (2 mM) was added to initiate the reaction, and spectra were collected from 700 to 350 nm every 45 s (or every 45 s for 10 min then every 2 min for PPDME K42L GlbN) using a Cary50 UV-Vis spectrophotometer. The reaction of each protein was analyzed with singular value decomposition40 on a portion of the visible spectrum (580–540 nm). For each data set the first two singular values are significantly larger than the third, and the first two V-vectors were globally fit to a single exponential. In addition to PTM formation, the first V-vector revealed a slow, low amplitude process in the spectra that is tentatively assigned to H2O2-mediated heme bleaching.32 Inclusion of a second exponential improved the global fit but did not affect the rate constant determined for the main process.
NMR Data Acquisition and Analysis.
NMR data were collected on either a Bruker Avance-600 or a Bruker Avance II-600 spectrometer each equipped with a TXI Cryoprobe. Multidimensional data sets were processed with NMRPipe 3.0 and analyzed using Sparky3. 1D data were processed and analyzed using Topspin 3.1. 1H chemical shifts were referenced indirectly through the (residual) water signal corrected for temperature. 15N chemical shifts were referenced indirectly with the Ξ ratio.41 High-pressure data were collected to a maximum pressure of 1.5 kbar using a Daedalus Innovations Xtreme-60 pump and cell apparatus equipped with a zirconia NMR cell.
NMR Sample Preparation.
Fe(III) PPDME samples with and without PTM, and with and without 15N labeling, were prepared as reported previously for the wild-type protein.35 Details of sample composition and conditions are provided in the supplementary figures. Backbone, side chain, and heme assignments were obtained with a complement of homonuclear (DQF-COSY, TOCSY, and NOESY experiments) and 1H-15N data.
Fe(II) samples were prepared under a N2 atmosphere by first resuspending lyophilized protein in buffer that was degassed and then sparged with N2. The pH of the protein sample was measured prior to addition of ~5 mM DT to generate the ferrous state. The sample was then transferred to a Shigemi NMR tube, layered with argon and sealed with the Shigemi plunger and Parafilm M®. Samples prepared in this manner showed no indication of oxidation for weeks.
NMR Assignment of Alkaline Species.
In the ferrous state at alkaline pH (1.5 mM PPDME GlbN-A, 100 mM sodium borate buffer pH* 9.6, 99% 2H2O), a novel form of PPDME GlbN was observed in the 1H 1D spectrum compared to the neutral pH spectrum (1.2 mM PPDME GlbN-A, 50 mM potassium phosphate buffer pH* 7.1, 99% 2H2O). As detailed in the Results, this new form was identified as a His–Fe–Lys complex. Simulation of selected peaks yielded the relative population of the native His–Fe–His and new His–Fe–Lys forms as a function of pH. A 1H-1H 2D EXSY42 experiment was performed at 25 °C (τmix = 30 ms) to transfer chemical shift assignments from the native conformation to this new form. The suite of 1H-1H 2D experiments given above was used to confirm and extend these assignments at 17 °C. The kinetics and temperature dependence of the chemical exchange allowed the cross peaks arising from exchange to be distinguished from those arising from dipolar contact in the EXSY and NOESY (τmix = 80 ms) spectra. Partial backbone 1H-15N assignments (0.5 mM PPDME GlbN-A, 100 mM sodium borate buffer pH 9.8, 10% 2H2O) were made using 2D 1H-Nz-1H and 1H-Nz-15N ZZ-exchange spectra (25 °C, τmix = 50 ms) and a 3D 1H-15N-1H NOESY-HSQC (17 °C, τmix = 80 ms) spectrum.33,43 A water presaturation 1H 1D spectrum of the ferrous species was acquired at alkaline pH with a sodium carbonate-bicarbonate buffer44 (0.5 mM PPDME GlbN-A, 90 mM sodium (bi)carbonate buffer pH 10.5, 10% 2H2O) using a 5 s recycling delay for quantification of the two forms well above the apparent pKa of the transition (see Results).
Kinetics of Ligand Exchange.
The kinetics of chemical exchange were investigated with an alkaline ferrous sample (1.5 mM PPDME GlbN-A, 90 mM sodium borate buffer pH 9.3, 10% 2H2O, 25 °C) using a modified 1H-1H 2D EXSY experiment that incorporated a WATERGATE solvent suppression scheme. The relaxation delay was 1.1 s and the sorted mixing times (τmix) were (ms): 2.5, 5.0, 7.5, 10.0, 15.0, 20.0, 30.0, 50.0, 75.0, 100.0, and 150.0. A two-state equilibrium represented by His–Fe–His Δ His–Fe–Lys was assumed. The observed equilibrium constant, Keq, is Σ[His–Fe–Lys]/Σ[His–Fe–His] = kf/kr, where kf and kr are apparent unimolecular forward and reverse rate constants, respectively. The equations governing longitudinal relaxation in the presence of chemical exchange42 were best fit to the integrated peak intensity data to obtain the initial intensity of the resolved (His–Fe–Lys) diagonal peaks. The initial intensity of unresolved (His–Fe–His) diagonal peaks was constrained by the observed equilibrium constant, Keq. The initial slope method45 was applied to the build-up of the exchange cross peaks. Data at the four shortest mixing times were used to avoid excessive contamination by NOEs. The initial slope, divided by the initial diagonal intensity, returns the exchange rate constant. Additional details are included in the supporting information.
Structural Comparison.
Backbone chemical shift perturbations (CSP) were calculated using an 15N weighting factor of 0.146 according to
| (2) |
CSPs for Fe(III) PPDME GlbN used assignments published for Fe(III) PP GlbN.47 In the case of Fe(II) PPDME GlbN-A the CSPs were calculated between the two conformations (His–Fe–His and His–Fe–Lys) present in the same sample and identified with published assignments.33,47
Multiple sequence alignment.
A BLAST search was performed with the sequence of GlbN as the query and expect value set to 1E−6. Over 1,500 sequences were retrieved, which were filtered after Clustal alignment with the following criteria: 80% identity at most, 100 residues at least, and the presence of the proximal histidine, a pair of glycines at GlbN positions 9 and 10, and a tyrosine at position 22 (B10). The qualifying 341 sequences were then inspected for amino acid identity at GlbN positions 42 and 46.
RESULTS
General Features of GlbN.
Synechococcus hemoglobin is a Group I truncated hemoglobin (TrHb1). It can be prepared in pure form with (GlbN-A) and without (GlbN) the His117–heme PTM. Both species merit study as they appear to be physiologically relevant in mitigating reactive nitrogen species (RNS) stress,48 albeit with distinct reactivities toward a common RNS, nitric oxide.49 Whereas it is possible to study separately GlbN and GlbN-A in the ferric state, reduction of the ferric bis-histidine form results in spontaneous PTM,32 and therefore only GlbN-A is readily accessible in the ferrous (deoxy) state if His117 is present. Replacement of His117 with a residue incapable of nucleophilic attack on the heme vinyl gives access to analogues of Fe(II) PP GlbN and Fe(II) PPDME GlbN. For this purpose, the His117Ala variant was utilized.
GlbN has the typical TrHb1 fold composed of a 2-on-2 helical sandwich.50 The secondary structure has seven helices, labeled A to C and E to H. Beside His117 (H helix), positions of interest in this study are His70, the proximal ligand (F helix); His46, the distal ligand (E helix); and Lys42 (E helix). The available three-dimensional structures are the ferric bis-histidine protein (PDB IDs 2KSC 48 and 4MAX 30) and the cyanide-bound protein (PDB ID 4L2M 30), all with PTM. We use the Fischer nomenclature for the heme group. IUPAC and Fischer numbering are reconciled in Figure S1. As abbreviations for iron ligation schemes, we use (proximal histidine)–Fe–(distal ligand), e.g., His–Fe–His. Where necessary, we include the oxidation state of the iron as Fe(II) (ferrous) or Fe(III) (ferric). Regarding spectroscopic properties, His–Fe(III)–His complexes have one unpaired electron (paramagnetic, low-spin S = 1/2 state) whereas the His–Fe(II)–His complexes are diamagnetic (S = 0).
Response of PP GlbN and GlbN-A to High pH.
To investigate the stability of GlbN, samples were exposed to high pH and their coordination state was inspected with electronic absorption spectroscopy. Fe(III) PP GlbN and GlbN-A exhibited no change in the spectrum up to pH 10.5 (data not shown). This indicated that the affinity for hydroxide ion was too low for His46 to be displaced by near mM concentration of the anion. A previous study of Fe(II) PP GlbN-A did not detect electronic absorption changes from neutral pH to pH 9.2.33 Raising the pH to 10.5, however, alters the spectrum noticeably (Figure S2). To identify the origin of the perturbation and in light of the heme–protein interactions depicted in Figure 1, our first approach was to replace the native cofactor with its dimethylesterified form (PPDME).
Effects of Propionate Esterification at Neutral pH.
The purified PPDME GlbN complex was initially investigated by electronic absorption spectroscopy in the ferric oxidation state at neutral pH. Heme esterification does not affect directly the porphyrin macrocycle or vinyl groups, and major changes are not expected in the spectrum as long as the protein maintains its original low-spin bis-histidine state. Comparison to Fe(III) PP GlbN under the same experimental conditions (Figure S3) reveals an unchanged spectrum. Inspection of NMR data (Figure S4) shows resolved heme and axial histidine resonances to be minimally shifted, confirming that the His–Fe(III)–His scheme is retained.
Figure 2A illustrates a portion of the 1H-15N HSQC spectrum of Fe(III) PP and PPDME GlbN-A. Figure 2B shows the same region for the GlbN pair. Additional GlbN cross peaks are shown in Figure S5. The majority of amide backbone assignments obtained for the His–Fe(III)–His PP complexes47 could be transferred to the His–Fe(III)–His PPDME complexes by inspection and further verified through NOEs. Obvious chemical shift perturbations are detected for backbone amides in the proximity of the iron. Val36 shows the greatest change in 1H shift (0.25 ppm), and Gln43 shows the greatest change in 15N shift (0.41 ppm). Because the protein is paramagnetic, shifts of this small magnitude are difficult to interpret in structural terms. They could arise directly from the displacement of a nucleus within the structure or indirectly through a perturbation of the magnetic susceptibility tensor caused by a slight alteration of heme and axial ligand geometry. Larger shifts (~1 ppm) observed for the side chains of Phe35 and Leu73, near the heme 5-CH3, suggest local reorientation of these residues. An inspection of NOEs, however, does not indicate major changes in heme–protein or protein–protein distances. For example, one of the methylene protons of the heme 7-substituent remains in dipolar contact with Ala69 despite esterification. Overall, the data indicate a high degree of structural similarity for the Fe(III) PP and Fe(III) PPDME proteins, whether the heme–protein crosslink is present or not. Select chemical shifts for Fe(III) PPDME GlbN are listed in Table S1.
Figure 2.

Portion of the 1H-15N HSQC spectra of ferric (A) PPDME GlbN-A (red) overlaid with PP GlbN-A (black) and (B) PPDME GlbN (red) overlaid with PP GlbN (black) at neutral pH. This region of the spectrum contains hyperfine shifted resonances. Assignments are as determined for the Fe(III) PP GlbN and GlbN-A species.47
Turning to the ferrous state at neutral pH, esterification has a minor effect on the electronic absorption spectrum of Fe(II) PPDME GlbN-A compared to Fe(II) PP GlbN-A (Figure S3). A suite of 2D homonuclear experiments (NOESY, DQF-COSY and TOCSY) performed at pH 7.1 was used to assign signals belonging to the heme and nearby protein side chains. Backbone assignments are available for Fe(II) PP GlbN-A35 and a comparison of the 1H-15N HSQC spectra showed small differences caused by esterification (Figure S6). As in the ferric state, signals from the ring of Phe35 are affected and suggest a small displacement with respect to the macrocycle. We conclude that in both oxidation states and upon substitution of the heme cofactor, the protein experiences no major structural rearrangement. Nevertheless, the NMR spectral response does illustrate a propagation of the perturbation to positions remote from the propionates in the structure (e.g., Met40). Such long-range effects have also been observed in PPDME cytochrome b5.51
PPDME GlbN-A Undergoes an Alkaline Transition in the Ferrous State.
The electronic absorption spectrum of Fe(III) PPDME GlbN-A was monitored as a function of pH in the range 6.4 to 11. As for Fe(III) PP GlbN-A, no change was observed (Figure S7). In the same pH range, NMR spectra supported the maintenance of stable bis-histidine ligation. We note, however, that prolonged (> 1 day) incubation at pH 11 resulted in hydrolysis of the ferric heme methylesters as demonstrated by the build-up of a methanol signal at 3.33 ppm and signals attributed to singly-hydrolyzed and eventually doubly-hydrolyzed (PP) heme species (not shown). The chemical instability limited NMR data collection on the Fe(III) PPDME GlbN species at high pH.
Interestingly, the minor spectral perturbations detected in Fe(II) PP GlbN-A at pH 10.5 (Figure S2) were enhanced in Fe(II) PPDME GlbN-A. Titration from pH 6.4 to pH 11 revealed the tail end of a low pH process attributed to the deprotonation of the heme-linked His117 and a clear alkaline transition with onset above pH 7. The changes in the visible and Soret bands associated with this alkaline transition are line sharpening, a 1–2 nm hypsochromic shift, and a hyperchromic shift (Figure 3). Two wavelengths that did not respond to the low pH process (526 nm and 424 nm) were chosen to characterize the alkaline transition. A modified Henderson-Hasselbalch equation was globally fitted to the data and returned an apparent pKa of 9.26 (with error bounds 9.20, 9.32) and a Hill coefficient of 1.01 (0.88, 1.15). The best-fit curve of the absorbance data at 424 nm is shown in the inset of Figure 3. Because the spectrum is insensitive to most ionization events above pH 7 (according to the behavior of Fe(III) GlbN), and the high-spin signature expected from a five-coordinate “deoxy” complex52 is not observed, the alkaline transition is attributed to the replacement of His46 with another strong-field ligand.
Figure 3.

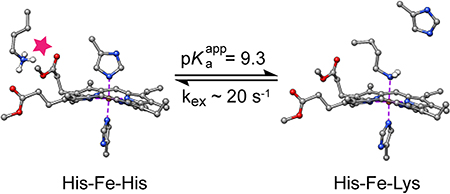
pH titration of Fe(II) PPDME GlbN-A. The electronic absorption spectrum is shown over the pH range 7.4 (red) to 11 (blue). The inset presents the absorbance at 424 nm and the fit to a modified Henderson-Hasselbalch equation (apparent pKa of ~9.3 and Hill coefficient ~1.0).
Lysine 42 Is a Ferrous Heme Distal Ligand at Alkaline pH.
NMR spectroscopy was used to determine the identity of the supplanting axial side chain in the alkaline state. NMR data collected on Fe(II) PPDME GlbN-A at pH 7.2 and pH 9.7 are shown in Figure 4A and B. At neutral pH, the most upfield peaks (Val121 methyl groups) resonate at approximately −1 ppm. As the pH is increased, a second set of signals appears for Val121 along with five new far upfield peaks. The shifted protons are J-correlated with each other, and their large negative shift must be caused by the ring current of the porphyrin macrocycle. The peak at −8.7 ppm disappears when the sample is prepared in 2H2O and exhibits J-coupling to a 15N nucleus resonating at approximately −37 ppm (Figure S8). These observations are consistent with axial lysine coordination as observed in C. reinhardtii THB15, 53 and the M100K variant of Thiobacillus versutus cytochrome c-550.54 Thus, at alkaline pH, Fe(II) PPDME GlbN-A populates two conformations in slow exchange on the chemical shift time scale: one with native His–Fe–His ligation, the other with His–Fe–Lys ligation. 1H NMR data collected on Fe(II) PP GlbN-A at pH 11 reveal a weak but analogous set of upfield peaks (Figure 4C), confirming that the changes detected by electronic absorption (Figure S2 and Figure 3) and NMR have the same origin whether or not the heme propionates are esterified.
Figure 4.

Upfield region of the 1H NMR spectrum of ferrous GlbN-A. (A) PPDME GlbN-A, pH 7.2, 25 °C; (B) PPDME GlbN-A, pH 9.7, 25 °C; (C) PP GlbN-A, pH 10.9, 25 °C; (D) Synechocystis PP GlbN-A, pH 11.0, 17 °C; (E) PP K48L GlbN-A, pH 11.0, 25 °C; (F) PP K42L GlbN-A, pH 10.9, 25 °C. Data collected in 1H2O with 5–10% 2H2O; alkaline samples were buffered with sodium borate (B) or sodium (bi)carbonate (C–F). Vertical scaling is arbitrary; however, the population of His–Fe–Lys form in (B) is higher than in (C). The Met40–Phe50 sequence is shown for each protein.
The primary structure of GlbN contains six lysines. If the heme plane is used to divide the protein in two parts, all lysines are located in the distal part (Figure S9). Three of the six lysines are found on the A and B helices, far from the heme cofactor and presumably out of contention for iron coordination barring global unfolding. The other three, Lys42, Lys44, and Lys48, are located on the E helix. In the X-ray structure of His–Fe–His GlbN-A, Lys44 and Lys48 point away from the iron whereas Lys42 stands out as the most likely candidate, with its Nζ atom near the heme 6-propionate carboxyl group (Figure 1A). To test whether Lys42 may be an axial ligand to the iron, data were acquired on Fe(II) PP K42L GlbN-A at pH 10.9. This complex showed no evidence of lysine coordination by 1H NMR spectroscopy (Figure 4F) or electronic absorption spectroscopy (not shown). Likewise, no lysine coordination is detected in the 1H NMR spectrum or the electronic absorption spectrum of Fe(II) PPDME K42L GlbN-A at pH 9.2 (Figure S10).
Additional support for the ligation of Lys42 is provided by the related globin from Synechocystis sp. PCC 6803. This protein, which displays 59% identity with GlbN, has lysines at positions 42 and 48, but not at position 44. We found that Synechocystis GlbN-A is also capable of forming a His–Fe–Lys complex in the ferrous state under alkaline conditions (Figure 4D). Finally, we prepared the K48L variant of Synechococcus GlbN in the ferrous GlbN-A state at high pH, and it too presents the signature signals of coordinated lysine (Figure 4E). Thus, Lys42 is a plausible heme ligand in Fe(II) PP GlbN-A and Fe(II) PPDME GlbN-A at high pH and is assigned as such. Propionate esterification favors the population of the His–Fe–Lys species and allows for more extensive characterization than possible with the natural cofactor.
Thermodynamics of Lysine Ligation.
From the electronic absorption data, the apparent pKa for the alkaline transition of Fe(II) PPDME GlbN-A is ~9.3 (Figure 3), but because the absorption coefficient of the His–Fe–Lys form of Fe(II) PPDME GlbN-A is not known, the limiting fractional population of the alkaline form is also unknown. An estimate can be made from integration of NMR data collected at pH 10.5: an appreciable amount of the His–Fe–His species is present, accounting for ~45% of the sample (Figure S11). This measurement was performed within 45 min after exposure of the PPDME protein to high pH and is consistent with NMR data collected over a range of pH. The titration end points are therefore the His–Fe–His state, in which Lys42 is “out” and protonated, on the acidic side, and a nearly equal mixture of His–Fe–His and His–Fe–Lys states, in which Lys42 and His46 are neutral, on the alkaline side.
A simple equilibrium model capturing the pH response of Fe(II) PPDME GlbN-A is presented in Figure 5, where the five-coordinate species is included but negligibly populated by GlbN under all conditions. The histidine–lysine competition is assigned an equilibrium constant Keq = [His–Fe–Lys]/[His–Fe–His] = K1/K2, where K1 and K2 are the decoordination constants for distal histidine and lysine, respectively. K3 is the acid dissociation constant of the lysine when not ligated to the iron. The ionization equilibrium for the axial histidine is unnecessary to consider in this pH range. With these constants, the populations of the main species can be calculated and related to the spectroscopic observables (sum of His–Fe–His species and sum of His–Fe–Lys species). The apparent equilibrium for ligand switching is then expressed as Kapp = 10−9.3 = (1 + K1/K2) K3 (derivation in Supporting information). The NMR population estimate at high pH suggests that Keq = K1/K2 ~ 1.4. Thus, pK3 is constrained to be 9.7, only ~0.4 unit above the apparent pK of 9.3 and ~ 0.7 pH unit lower than the expected pKa of 10.4 for an unperturbed lysine. Additional information will be needed to understand the origin of a shift in the pKa of the decoordinated lysine (e.g., specific interactions) or to consider a “trigger group,” the ionization of which is coupled to ligand switching.55 Regardless of the mechanistic details, the model allows for a prediction of His–Fe–His and His–Fe–Lys populations at arbitrary pH values (Figure 6).
Figure 5.

Proposed equilibria involving heme axial ligands in the ferrous state. K1 and K2 are decoordination constants. K3 is the lysine acid dissociation constant in the bis-histidine state. The five-coordinate species (bracketed) is included for generality as many hexacoordinate globins have weak distal ligand affinity.
Figure 6.

Population plot obtained with the scheme of Figure 5. The apparent pK of the alkaline transition is 9.26 and the fraction of protein with a distal lysine ligand (HK|H) is 0.55 at pH 10.5. The value of pK3 is then 9.64. K1/K2 is 1.38, corresponding to a free energy difference of 0.8 kJ mol−1 favoring the lysine bound form.
Response of the Alkaline Form to Pressure.
The structural change necessary to coordinate Lys42 is expected to result in a change in volume. In an attempt to increase the population of the His–Fe–Lys conformation for NMR characterization, a sample of Fe(II) PPDME GlbN-A at pH ~9.5 was subjected to an increase in hydrostatic pressure. The 1H NMR data presented in Figure S12 shows the disappearance of the His–Fe–Lys species and an increase of the His–Fe–His species as the pressure is raised to 1.5 kbar. The pH drift accompanying the compression of the borate buffer is significant56 but does not account fully for the shift in equilibrium. This result eliminates the use of pressure to promote lysine coordination, but shows that the system adopts a greater volume when in the lysine-ligated conformation than when in the histidine-ligated conformation.
Rate of Ligand Switching and NMR Assignments at Alkaline pH.
Further homonuclear experiments exploited the slow exchange between the His–Fe–Lys and His–Fe–His complexes. Correlations detected for the axial Lys in 1H-1H EXSY data place the CδH2 and CεH2 signals of Lys42 at 1.7 ppm and 3.1 ppm, respectively, in the His–Fe–His state (Figure 7), and, as expected from the bis-histidine crystal structure, are in agreement with exposure to solvent. The intensity of the exchange cross peaks was measured as a function of the mixing time (Figure S13) and treated with an initial slope approach.45 Combined with the equilibrium information from the pH titration, the data provide an estimate for the apparent rate constants describing the conversion of His–Fe–His to His–Fe–Lys coordination as kf ~ 8 s−1 and the reverse process as kr ~ 15 s−1 at pH 9.3 and 25 °C.
Figure 7.

A portion of the 1H-1H EXSY spectrum collected on Fe(II) PPDME GlbN-A at pH 9.3 with a mixing time of 5 ms. The upfield peaks arising from the coordinated Lys42 CδH2 and CεH2 protons have exchange cross peaks with their decoordinated counterparts at 1.7 and 3.1 ppm.
The same 1H-1H EXSY data show that the imidazole Cδ2H and Cε1H signals of His46 move from 0.5 ppm and 1.6 ppm in the His–Fe–His conformation to 7.6 ppm and 8.1 ppm, respectively, in the His–Fe–Lys conformation. Thus, when Lys42 is not coordinated, its two terminal methylenes (CδH2 and CεH2) exhibit shifts in agreement with exposure to solvent, whereas the ring protons of decoordinated His46 deviate from the expected random coil values of ~7 and ~7.7 ppm for a neutral histidine.57 This suggests that in the alkaline conformation, His46 remains in contact with the heme or protein residues.
If the neutral pH structures serve as guides, ligation of Lys42 requires considerable distortion of the E helix upstream of His46. This was investigated with additional NMR data at pH values within the alkaline transition. As mentioned above, 2D 1H-1H EXSY experiments acquired at 25 °C connect the signals of Lys42 in the two species. The same experiment complemented with the standard suite of homonuclear data allowed the transfer of heme and additional side chain assignments to the alkaline form. Data were also collected at pH 9.6 and 17 °C in an attempt to alleviate some of the overlap. To illustrate data quality at this temperature, an annotated portion of the DQF-COSY spectrum is shown in Figure S14. The heme and select side chain chemical shifts of both conformations of Fe(II) PPDME GlbN-A are listed in Table S2.
Amide assignments were obtained with 2D 1H-Nz-15N ZZ exchange data (Figure S15), 2D 1H-Nz-1H ZZ exchange data (25 °C, pH 9.2, Figure S16A), and 3D 1H-15N-1H NOESY-HSQC data (17 °C, pH 9.2). Overall, the amide shifts of the His–Fe–His conformation of Fe(II) PPDME GlbN-A at pH 9.2 and PP GlbN-A at pH 7.1 are similar. In contrast, the His–Fe–Lys conformation of Fe(II) PPDME GlbN-A gives rise to a distinct 1H-15N correlation map. The annotated 2D 1H-Nz-1H ZZ exchange spectrum shown in Figure S16A identifies 32 resolved correlations between amide resonances in the His–Fe–His and His–Fe–Lys forms. Some amides, such as that of Lys42, were assigned only in the His–Fe–His conformation and showed strong magnetization transfer to the water frequency. The alkaline pH necessary to populate the His–Fe–Lys form permitted only partial 1H-15N assignments. Solvent exchange also confounded the measurement of conformational exchange rates using 1H-Nz-15N ZZ data. However, the chemical shift separations detected between the two conformations in the 1H-Nz-15N ZZ and 1H-Nz-1H ZZ spectra are consistent with the apparent rate constants determined for the lysine side chain with EXSY data. The His–Fe–Lys resonances observed by 1H-15N HSQC respond to pH (not shown) and pressure (Figure S13) in concert with the upfield Lys42 peaks, supporting a global rather than local conformational change on ligand switching.
Structural Consequences of Lys42 Ligation.
A coarse picture of the structural properties of the His–Fe–Lys conformation of Fe(II) PPDME GlbN-A can be gleaned from the NMR data by comparison to the His–Fe–His conformation. In Figure 8, backbone amide CSPs are colored on the His–Fe–His GlbN-A structure according to their magnitude (values are shown in Figure S16B). The subset of amides detectable at alkaline pH forms a pattern by which the F, G, and H helices are relatively unperturbed. The largest changes in amide chemical shift are observed for the B, C, and E helices. In addition, many resonances from the heme and several side chains on both sides of the heme experience measurable shifts (> 0.2 ppm, Table S2). The proximal histidine and adjacent residues are among those affected. These data, although distorted by the large and anisotropic effect of the porphyrin ring current, demonstrate a structural rearrangement of the heme cavity.
Figure 8.

The 1H-15N backbone CSPs obtained by comparison of the His–Fe–His and His–Fe–Lys forms of Fe(II) PPDME GlbN-A (pH 9.8) depicted on the His–Fe–His structure (PDB ID 4MAX). Blue (CSP < 0.95 ppm), gold (0.95 ppm < CSP < 1.40 ppm) and red (1.40 ppm < CSP). The average CSP is 0.50 ppm and the standard deviation is 0.45 (Figure S16). Backbone amides in grey could not be assigned at this alkaline pH.
Additional features of the His–Fe–Lys conformation were derived from the 1H-1H NOESY data. A series of characteristic dipolar contacts are recognized in both His–Fe–His and His–Fe–Lys states; these stem from the proximity of Thr80 and His83 (forming a resilient hydrogen-bonded cap);58 interactions between Val121 and His70; contacts between Phe84 and His117; and packing of Phe61 against the heme 8-CH3. When Lys42 or His46 acts as a heme ligand, each exhibits dipolar contact with Phe35, and common heme-protein contacts are observed for Phe35 in both conformations. Of note are the effects observed between Phe50, the heme group, and either His46 (His–Fe–His state) or the axial lysine (His–Fe–Lys state) (Figure 9). When His46 is ligated to the iron, the imidazole Cε1H is oriented toward Phe50 on the α-meso side of the heme; in this conformation, the distance between Phe50 and Lys42 side chains is ~14 Å (Figure 9A). Figure 9C presents a portion of the 1H-1H NOESY map showing contacts made by the CδH2 and CεH2 protons of coordinated Lys42 with Phe50 in addition to Phe35 and the heme meso protons. In the alkaline conformation, Phe50 apparently moves “below” pyrrole B as the E-helix reorients, making new contacts with the heme and the axial lysine. Analogous to the environment surrounding the Lys53 distal ligand in C. reinhardtii THB1,6, 53 the proximity of coordinated Lys42 with Phe50 and Phe35, along with the presence of Phe21 and Tyr22 on the B-helix, may produce a relatively hydrophobic heme distal pocket in which the lysine NζH2 group is protected from rapid exchange with solvent and observable by NMR spectroscopy. Interestingly, the constellation of NOEs detected in the His–Fe–Lys conformation (Figure 9D) differs from that expected of both the His–Fe–His (Figure 9A) and His–Fe–CN (Figure 9) states, illustrating the plasticity of the GlbN heme pocket.
Figure 9.

Structural changes in Fe(II) PPDME GlbN-A associated with axial lysine ligation. Key residues are shown in (A) the His–Fe–His and (B) the His–Fe–CN X-ray structures. (C) Portion of a NOESY spectrum (pH* 9.6, 99% 2H2O, 17 °C, 80 ms mixing time) illustrating Lys42 cross peaks in the His–Fe–Lys conformation. (D) The network of observed NOEs (red dashed lines) does not correspond to either known structure (A and B) and requires a reorganization of the E helix.
Lys42 Ligation and PTM.
The conformational changes required for lysine ligation in Fe(II) GlbN-A raise two questions. First, is heme–protein cross-linking necessary to stabilize the His–Fe–Lys state, and second, does the non-native conformation influence the rate of the PTM? NMR signals characteristic of bound lysine are detected in Fe(II) PPDME H117A GlbN and in freshly reduced, high pH Fe(II) PPDME GlbN samples, prior to PTM (data not shown). We conclude that covalent attachment of the heme to the protein is not required for the alternative ligation state.
At pH 9.2, formation of the His117–heme covalent linkage, which requires protonation of the vinyl group, is sufficiently slow (2.3 ± 0.1 × 10−3 s−1) that the reaction can be observed optically by manual mixing of the unmodified ferric protein with DT.33 The His–Fe–His/His–Fe–Lys ligand exchange occurs much faster (~20 s−1) and essentially establishes a condition of rapid pre-equilibrium. The ability of Fe(II) PPDME GlbN to undergo the PTM was inspected by manual mixing at pH 9.2 (Figures S17 and S18). A 2-fold reduction in the apparent rate constant for PTM formation is observed (1.0 ± 0.1 × 10−3 s−1), which suggests that ligand exchange retards the reaction. Control experiments carried out with PPDME K42L GlbN and PP K42L GlbN return rate constants of 1.7 ± 0.1 × 10−3 s−1, and 2.7 ± 0.1 × 10−3 s−1, respectively, both close to the wild-type value. Only the protein exhibiting ligand exchange has a clearly decelerated rate constant, by a factor approximately consistent with the pre-equilibrium factor. This may be linked to the conformational difference between the two ligation schemes. However, the effect is small, and the variations observed in the control experiments highlight the sensitivity of the rate constant to multiple factors.
DISCUSSION
Composition of Alkaline Samples and Limitations to their Characterization.
The high pH conditions required to populate the His–Fe–Lys complex impose severe restrictions on the extent of structural and kinetic analyses that can be performed. Backbone amide exchange rates are accelerated, and many useful NMR spectroscopic handles are therefore obliterated. Furthermore, in PPDME GlbN samples, hydrolysis of the heme dimethyl esters occurs over the course of NMR data acquisition. Although the hydrolysis rate is slower in the ferrous state than the ferric state and allows for the collection of multidimensional data sets, the complexity borne out by the presence of multiple exchanging species in evolving amounts and the limited number of spectral probes prevented a detailed description of the His–Fe–Lys complex. With these limitations in mind, we derive the following points.
Lysine versus Histidine as Axial Ligands.
The competition between the two nitrogenous ligands for axial ligation on the distal side requires an explanation. In particular, Lys42 appears capable of displacing His46 as the distal ligand in the ferrous (Figures 3 and 4) but not the ferric (Figure S7) oxidation state of the heme iron. The strength of an iron–ligand coordination bond opposed to a histidine axial ligand is difficult to measure experimentally in part because the pentacoordinate state of Figure 5 is often inaccessible.59 However, equilibrium binding studies of nitrogenous ligands to ferric heme peptide N-acetylmicroperoxidase-8 found that imidazole and propylamine were equally apt at displacing bound water.60 Taking this observation to be applicable to Fe(III) GlbN, the sole persistence of His–Fe–His coordination at alkaline pH may then represent entatic control61 exerted by the protein scaffold in maintaining the native ligand set.
Theoretical calculations estimate that the heme Fe(III)–imidazole Fe–N bond is about 30 kJ/mol stronger than the Fe(II)–imidazole Fe–N bond.59 In addition, studies of sequential histidine ligation in His–Fe–His model peptides have demonstrated that the second histidine binds ferrous iron with lower affinity than the first, whereas the opposite is true in the ferric oxidation state.62 Displacement of the iron from the porphyrin plane and steric occlusion by the pyrrole nitrogens destabilizes ferrous bis-histidine ligation. This “face strain” was invoked in discussing the heightened flexibility of ferrous GlbN as pictured by 15N relaxation data.35 The weaker histidine ligation and increased flexibility of GlbN-A in the ferrous state likely contribute to the ability of Lys42 to displace His46 as the distal iron ligand. Dedicated studies will be needed to reconcile the results of different model systems and clarify the energetics of histidine and lysine ligand exchange in both iron oxidation states.
Effect of Propionate Esterification.
At alkaline pH (≥ 8) the heme propionate groups are expected to be deprotonated and negatively charged.63 The structure of the His–Fe–His conformation (Figure 1) suggests that an electrostatic interaction exists between the charged Lys42 and the 6-propionate. In the Fe(II) PP GlbN-A complex the His–Fe–His conformation is fully populated at pH 9.2,33 and the His–Fe–Lys conformation is present only as a minor form at pH 11 (~20%, Figure 4C). This result indicates that strongly basic conditions are required to disrupt the Lys42-heme 6-propionate interaction. Upon propionate esterification, however, the His–Fe–Lys form is populated to a significant degree at pH 9 (~20%) and reaches a maximum of ~60% at pH 11 (Figure 6). The heme modification must therefore lower the pKa of some ionizable group, the most likely candidate being Lys42. It appears that the interaction between the 6-propionate and Lys42 prevents this residue from displacing His46 at pH lower than ~10 and contributes to the stability of the His–Fe–His complex. This is in contrast to the proposed role of the heme propionates in the alkaline transition of ferric cytochrome c, deprotonation of which may promote lysine coordination.64–66 In this protein, the heme propionates are not exposed to solvent and do not interact strongly with lysines, which contributes to the distinct behavior.
Structural Perturbation and Flexibility of the E-Helix.
The CSP and NOE results lead to limited conclusions about the conformational rearrangement required for lysine ligation. Much of the protein undergoes some perturbation, including many of the residues composing the heme binding pocket. Not surprisingly, the largest manifestations are detected on the distal side and in particular the E helix (Figure 8). The observed contacts between Lys42 and Phe50 in the His–Fe–Lys conformation appear to require some change in secondary structure (Figure 9). One model consistent with the data involves loss of helical content for at minimum the first ~5 residues (40 through 44).
The plausibility of a local structural distortion can be assessed with known properties of GlbN. At neutral pH, the backbone amides composing the N-terminal portion of the E helix of Fe(III) PP GlbN (residues 40–46) have low protection factors, becoming undetectable within a few minutes after dissolution of the protein into 2H2O.34 The modest local stability for this region of the protein is consistent with 15N relaxation measurements,35 which show increased millisecond motions on the distal side of Fe(II) PP GlbN-A relative to the rest of the protein. This is enhanced for the carbon monoxy adduct of Fe(II) PP GlbN-A, in which the first turn of the E helix has elevated R2 values. Heightened flexibility in the ferrous, compared to ferric, GlbN-A extends throughout the protein backbone whether in the bis-histidine state or with exogenous ligand bound. In amide HSQC spectra of Fe(II) PPDME GlbN-A acquired at basic pH, we were unable to detect most of the signals arising from the N-terminal region of helix E, likely because of rapid hydrogen exchange. These observations agree with the notion that residues 40–44 are prone to unfolding.
Lysine as a Heme Ligand and Ligand Switching.
Coordination of a lysine residue requires a neutral amino group, that is, an energetic expenditure to lower the pKa of that group to the physiological range. In GlbN, the His–Fe–Lys state is significantly populated at pH values much higher than neutral, which suggests that per se the alternative ligation mode is unlikely to have functional significance. Likewise, the artificial PPDME prosthetic group used to populate the His–Fe–Lys conformation has no direct biological relevance. However, both high pH and propionate esterification reveal an energetically low-lying conformation that GlbN and relatives can sample and perhaps adopt when interacting with a binding partner.
It is interesting to note that, in approximately 51% of TrHb1s, a histidine occupies the same position as His46 (referred to as E10 by sequence analogy to the myoglobin notation, but topologically equivalent to E7 in that protein). Whether this residue is an axial ligand in all instances of TrHb1 is uncertain. It is likely that coordination occurs with different stability and population of the five-coordinate intermediate shown in Figure 5.67 Approximately 30% of TrHb1s have a lysine at the analogous position as Lys42 (E6) and 35% of those having a histidine at E10 have lysine at E6 (additional statistics are presented in Table S3). Synechocystis GlbN illustrates a second example of His/Lys ligand swapping (Figure 4D). Given the statistics, we expect that switching occurs in many TrHb1s, with a range of apparent pKa values. In a survey of b heme proteins regardless of the fold, lysine is found to interact with a heme propionate only in 10% of tabulated instances. Arginine, in contrast, is present in 38% of the instances.68 The enrichment of lysine at the edge of the heme cavity in GlbN relatives may signify a specific functional requirement related to iron coordination.
The structure of murine neuroglobin, a bis-histidine globin of the same lineage as myoglobin, shows an interaction between Lys67 (E10, three residues beyond the distal His E7) and a heme propionate group that is disrupted upon CO binding.22 When the distal histidine is replaced with a leucine residue, Lys67 is able to coordinate the ferrous heme with an apparent pKa of 10.69 Similarly, a bis-histidine Arabidopsis hemoglobin has recently been shown to use Lys69 (E10) as a ferrous heme ligand when the distal histidine (His66, E7) is replaced.70 In contrast, the H46L variant of GlbN-A appears predominantly pentacoordinate in the ferrous state even at alkaline pH (Figure S19). As no lysine coordination is detected, these results probably reflect the effect of a leucine at the distal position, either favoring the pentacoordinate state or interfering with a potential His–Fe–Lys conformation.
An Intriguing Parallel to Cytochrome c.
Distal ligand replacement has gained considerable attention over the years for its importance in sensing and signaling.71,72 Various triggers have been identified, for example a redox state change in E. coli direct oxygen sensor (EcDos),73 binding of CO to the transcription factor CooA,74 and changes in pressure in cytochromes P420 and P450.75 A timely example of pH-triggered ligand switching is provided by cytochrome c, in which structural changes related to deligation of the axial Met80 allow the cytochrome to transform itself from an electron carrier to a peroxidase and act as an early signal in apoptosis.76–79 There is now some evidence that His–Fe–Lys conformers detected through alkaline transitions contribute to the complex relationship between structure and function in the cytochrome.80,81
The response to pH exhibited by Fe(II) PPDME GlbN-A is reminiscent of the Fe(III) cytochrome Met-to-Lys switch, but with some differences. The cytochrome transition involves replacement of the axial Met80, a “softer” ligand than His46 in GlbN. Lys73 and Lys79, however, are unable to displace Met80 in the ferrous oxidation state82 despite the relatively weak Fe–Met bond.59 This implies a significantly reduced stability of lysine ligation in the ferrous state, consistent with the low reduction potential of the alkaline form.82 The K73H variant undergoes a His–Fe–His to His–Fe–Lys transition in the ferric state,83 whereas GlbN-A does not seem to depart from the His–Fe–His state when oxidized, highlighting the role of the protein scaffold in ligand selection. As an additional common feature, both GlbN-A and yeast iso-1-cytochrome c exhibit heightened backbone motions on the μs–ms timescale in the oxidation state capable of ligand switching.35,84,85 Residual dipolar coupling and 1H-15N NOE measurements on alkaline K79A cytochrome c are consistent with increased ps–ns motions in the ferric His–Fe–Lys form relative to the His–Fe–Met form.86 However, human cytochrome c displays greater rigidity in the ferric oxidation state87,88 while still undergoing the alkaline transition, albeit with a higher pKa than the yeast protein.89 Thus, the connection between local and global backbone dynamics and ligand switching, which occurs on the ms–s timescale, remains to be established. The full consequences of lysine ligation in GlbN-A are unknown, but it is clear that such scheme is made possible by the low local stability of several structural elements and imparts a different conformation beyond the edge of the heme cavity. The inability of the ferric state to undergo the same distal ligand transition as the ferrous state raises an interesting avenue to a redox sensing mechanism.
CONCLUSION
The heme propionates have been implicated in adjusting multiple holoprotein properties.90–94 In this study, we have shown that electrostatic interactions along the heme periphery play a part in determining the coordination state of the cyanobacterial TrHb1, GlbN. For residues such as lysine, that can both form salt bridges and act as potential heme ligands, interaction with the propionates may effectively compete with iron coordination. Perhaps more importantly, we characterized a ligand switching process occurring on the sub-second time scale in one of two common oxidation states and leading to a stable conformation not captured in the existing structural models. These results exemplify well the malleability of the GlbN heme distal pocket and expand our view of the conformational landscape available to TrHb1s. Studies of other proteins with similar lysine–heme interactions are in progress to explore the generality of this mode of electrostatic control.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Belinda Wenke, Emily Adney, and Lukas Gilevicius for preparation of Synechocystis GlbN, K42L GlbN and wild-type GlbN. This work was supported by the National Science Foundation grant MCB-1330488 to JTJL and National Institutes of Health grant T32 GM080189 (DBN).
Funding Sources: This work was supported by the National Science Foundation grant MCB-1330488 to JTJL and National Institutes of Health grant T32 GM080189 (DBN).
Abbreviations:
- 1D
one-dimensional
- 2D
two-dimensional
- 3D
three-dimensional
- CSP
chemical shift perturbation
- DEAE
diethylaminoethyl
- DMSO
dimethyl sulfoxide
- DT
sodium dithionite
- EXSY
exchange spectroscopy
- Fe PP
iron-protoporphyrin IX
- Fe PPDME
iron-protorporphyrin IX dimethylester
- GlbN
Synechococcus sp. PCC 7002 hemoglobin
- GlbN-A
GlbN with His117–heme covalent attachment
- Hb
hemoglobin
- HSQC
heteronuclear single quantum coherence
- NOE
nuclear Overhauser effect
- PDB
Protein Data Bank
- pH*
pH uncorrected for isotope effect
- PTM
post-translational modification
- RNS
reactive nitrogen species
- THB1
hemoglobin 1 from Chlamydomonas reinhardtii
- TrHb1
Group I truncated hemoglobin
Footnotes
ASSOCIATED CONTENT
Supporting Information
Figures S1 to S19, Tables S1 to S3, and the derivation of the apparent pKa equation.
References
- (1).Kepp KP (2017) Heme: From quantum spin crossover to oxygen manager of life. Coord. Chem. Rev 344, 363–374. [Google Scholar]
- 2).Rydberg P, Sigfridsson E, and Ryde U (2004) On the role of the axial ligand in heme proteins: a theoretical study. J. Biol. Inorg. Chem 9, 203–223. [DOI] [PubMed] [Google Scholar]
- 3).Gibney BR (2016) Equilibrium studies of designed metalloproteins. Methods Enzymol 580, 417–438. [DOI] [PubMed] [Google Scholar]
- 4).Koder RL, Anderson JLR, Solomon LA, Reddy KS, Moser CC, and Dutton PL (2009) Design and engineering of an O2 transport protein. Nature 458, 305–U64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Johnson EA, Rice SL, Preimesberger MR, Nye DB, Gilevicius L, Wenke BB, Brown JM, Witman GB, and Lecomte JTJ (2014) Characterization of THB1, a Chlamydomonas reinhardtii truncated hemoglobin: linkage to nitrogen metabolism and identification of lysine as the distal heme ligand. Biochemistry 53, 4573–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Rice SL, Boucher LE, Schlessman JL, Preimesberger MR, Bosch J, and Lecomte JTJ (2015) Structure of Chlamydomonas reinhardtii THB1, a group 1 truncated hemoglobin with a rare histidine-lysine heme ligation. Acta Crystallogr. F Struct. Biol. Commun 71, 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Teh AH, Saito JA, Najimudin N, and Alam M (2015) Open and Lys-His hexacoordinated closed structures of a globin with swapped proximal and distal sites. Sci. Rep 5, 11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Ilcu L, Rother W, Birke J, Brausemann A, Einsle O, and Jendrossek D (2017) Structural and functional analysis of latex clearing protein (Lcp) provides insight into the enzymatic cleavage of rubber. Sci. Rep 7, 6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Trent JT, Hvitved A, and Hargrove MS (2001) A model for ligand binding to hexacoordinate hemoglobins. Biochemistry 40, 6155–6163. [DOI] [PubMed] [Google Scholar]
- 10).Simonneaux G, and Bondon A (2005) Mechanism of electron transfer in heme proteins and models: the NMR approach. Chem. Rev 105, 2627–2646. [DOI] [PubMed] [Google Scholar]
- 11).Kiger L, Tilleman L, Geuens E, Hoogewijs D, Lechauve C, Moens L, Dewilde S, and Marden MC (2011) Electron transfer function versus oxygen delivery: A comparative study for several hexacoordinated globins across the animal kingdom. PLoS ONE 6, e20478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).De Henau S, Tilleman L, Vangheel M, Luyckx E, Trashin S, Pauwels M, Germani F, Vlaeminck C, Vanfleteren JR, Bert W, Pesce A, Nardini M, Bolognesi M, De Wael K, Moens L, Dewilde S, and Braeckman BP (2015) A redox signalling globin is essential for reproduction in Caenorhabditis elegans. Nat Commun 6, 8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).He BY, Schulz CE, and Li JF (2015) Synthesis and characterization of a modified “picket fence” porphyrin complex - stronger π bonding interactions between Fe(II) and axial ligands. Dalton Trans 44, 13651–13661. [DOI] [PubMed] [Google Scholar]
- 14).Marques HM (2007) Insights into porphyrin chemistry provided by the microperoxidases, the haempeptides derived from cytochrome c. Dalton Trans 0, 4371–4385. [DOI] [PubMed] [Google Scholar]
- 15).Du J, Perera R, and Dawson JH (2011) Alkylamine-ligated H93G myoglobin cavity mutant: a model system for endogenous lysine and terminal amine ligation in heme proteins such as nitrite reductase and cytochrome f. Inorg. Chem 50, 1242–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Reedy CJ, and Gibney BR (2004) Heme protein assemblies. Chem. Rev 104, 617–649. [DOI] [PubMed] [Google Scholar]
- 17).Lu Y, Berry SM, and Pfister TD (2001) Engineering novel metalloproteins: Design of metal-binding sites into native protein scaffolds. Chem. Rev 101, 3047–3080. [DOI] [PubMed] [Google Scholar]
- 18).Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, and Pecoraro VL (2014) Protein Design: Toward Functional Metalloenzymes. Chem. Rev 114, 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Nastri F, Chino M, Maglio O, Bhagi-Damodaran A, Lu Y, and Lombardi A (2016) Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev 45, 5020–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Farid TA, Kodali G, Solomon LA, Lichtenstein BR, Sheehan MM, Fry BA, Bialas C, Ennist NM, Siedlecki JA, Zhao Z, Stetz MA, Valentine KG, Anderson JLR, Wand AJ, Discher BM, Moser CC, and Dutton PL (2013) Elementary tetrahelical protein design for diverse oxidoreductase functions. Nat. Chem. Biol 9, 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Makino M, Sawai H, Shiro Y, and Sugimoto H (2011) Crystal structure of the carbon monoxide complex of human cytoglobin. Proteins 79, 1143–1153. [DOI] [PubMed] [Google Scholar]
- 22).Vallone B, Nienhaus K, Matthes A, Brunori M, and Nienhaus GU (2004) The structure of carbonmonoxy neuroglobin reveals a heme-sliding mechanism for control of ligand affinity. Proc. Natl. Acad. Sci. U. S. A 101, 17351–17356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).de Sanctis D, Ascenzi P, Bocedi A, Dewilde S, Burmester T, Hankeln T, Moens L, and Bolognesi M (2006) Cyanide binding and heme cavity conformational transitions in Drosophila melanogaster hexacoordinate hemoglobin. Biochemistry 45, 10054–10061. [DOI] [PubMed] [Google Scholar]
- 24).Rosell FI, Ferrer JC, and Mauk AG (1998) Proton-linked protein conformational switching: Definition of the alkaline conformational transition of yeast iso-1-ferricytochrome c. J. Am. Chem. Soc 120, 11234–11245. [Google Scholar]
- 25).Ferrer JC, Guillemette JG, Bogumil R, Inglis SC, Smith M, and Mauk AG (1993) Identification of Lys79 as an iron ligand in one form of alkaline yeast iso-1-ferricytochrome c. J. Am. Chem. Soc 115, 7507–7508. [Google Scholar]
- 26).Pearce LL, Gartner AL, Smith M, and Mauk AG (1989) Mutation-induced perturbation of the cytochrome c alkaline transition. Biochemistry 28, 3152–3156. [DOI] [PubMed] [Google Scholar]
- 27).Josephs TM, Liptak MD, Hughes G, Lo A, Smith RM, Wilbanks SM, Bren KL, and Ledgerwood EC (2013) Conformational change and human cytochrome c function: mutation of residue 41 modulates caspase activation and destabilizes Met-80 coordination. J. Biol. Inorg. Chem 18, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Nall BT, Zuniga EH, White TB, Wood LC, and Ramdas L (1989) Replacement of a conserved proline and the alkaline conformational change in iso-2-cytochrome c. Biochemistry 28, 9834–9839. [DOI] [PubMed] [Google Scholar]
- 29).Goldes ME, Jeakins-Cooley ME, McClelland LJ, Mou TC, and Bowler BE (2016) Disruption of a hydrogen bond network in human versus spider monkey cytochrome c affects heme crevice stability. J. Inorg. Biochem 158, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Wenke BB, Lecomte JTJ, Heroux A, and Schlessman JL (2014) The 2/2 hemoglobin from the cyanobacterium Synechococcus sp. PCC 7002 with covalently attached heme: comparison of X-ray and NMR structures. Proteins 82, 528–534. [DOI] [PubMed] [Google Scholar]
- 31).Scott NL, Falzone CJ, Vuletich DA, Zhao J, Bryant DA, and Lecomte JTJ (2002) The hemoglobin of the cyanobacterium Synechococcus sp. PCC 7002: Evidence for hexacoordination and covalent adduct formation in the ferric recombinant protein. Biochemistry 41, 6902–6910. [DOI] [PubMed] [Google Scholar]
- 32).Nothnagel HJ, Preimesberger MR, Pond MP, Winer BY, Adney EM, and Lecomte JTJ (2011) Chemical reactivity of Synechococcus sp. PCC 7002 and Synechocystis sp. PCC 6803 hemoglobins: covalent heme attachment and bishistidine coordination. J. Biol. Inorg. Chem 16, 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Preimesberger MR, Pond MP, Majumdar A, and Lecomte JTJ (2012) Electron self-exchange and self-amplified posttranslational modification in the hemoglobins from Synechocystis sp. PCC 6803 and Synechococcus sp. PCC 7002. J. Biol. Inorg. Chem 17, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Vuletich DA, Falzone CJ, and Lecomte JTJ (2006) Structural and dynamic repercussions of heme binding and heme-protein cross-linking in Synechococcus sp. PCC 7002 hemoglobin. Biochemistry 45, 14075–14084. [DOI] [PubMed] [Google Scholar]
- 35).Pond MP, Majumdar A, and Lecomte JTJ (2012) Influence of heme post-translational modification and distal ligation on the backbone dynamics of a monomeric hemoglobin. Biochemistry 51, 5733–5747. [DOI] [PubMed] [Google Scholar]
- 36).Rice SL, Preimesberger MR, Johnson EA, and Lecomte JTJ (2014) Introduction of a covalent histidine-heme linkage in a hemoglobin: A promising tool for heme protein engineering. J. Inorg. Biochem 141, 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Scott NL, and Lecomte JTJ (2000) Cloning, expression, purification, and preliminary characterization of a putative hemoglobin from the cyanobacterium Synechocystis sp. PCC 6803. Protein Sci 9, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Teale FWJ (1959) Cleavage of heme-protein link by acid methylethylketone. Biochim. Biophys. Acta 35, 543. [DOI] [PubMed] [Google Scholar]
- 39).Neya S, Nagai M, Nagatomo S, Hoshino T, Yoneda T, and Kawaguchi AT (2016) Utility of heme analogues to intentionally modify heme-globin interactions in myoglobin. Biochim. Biophys. Acta 1857, 582–588. [DOI] [PubMed] [Google Scholar]
- 40).Hendler RW, and Shrager RI (1994) Deconvolutions based on singular value decomposition and the pseudoinverse: a guide for beginners. J. Biochem. Biophys. Methods 28, 1–33. [DOI] [PubMed] [Google Scholar]
- 41).Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, and Sykes BD (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140. [DOI] [PubMed] [Google Scholar]
- 42).Jeener J, Meier BH, Bachmann P, and Ernst RR (1979) Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys 71, 4546–4553. [Google Scholar]
- 43).Farrow NA, Zhang O, Forman-Kay JD, and Kay LE (1994) A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J. Biomol. NMR 4, 727–734. [DOI] [PubMed] [Google Scholar]
- 44).Delory GE, and King EJ (1945) A sodium carbonate-bicarbonate buffer for alkaline phosphatases. Biochem. J 39, 245. [PubMed] [Google Scholar]
- 45).Otting G, Liepinsh E, and Wüthrich K (1993) Disulfide bond isomerization in BPTI and BPTI(G36S): an NMR study of correlated mobility in proteins. Biochemistry 32, 3571–3582. [DOI] [PubMed] [Google Scholar]
- 46).Hardman CH, Broadhurst RW, Raine ARC, Grasser KD, Thomas JO, and Laue ED (1995) Structure of the A-domain of HMG1 and its interaction with DNA as studied by heteronuclear three- and four-dimensional NMR spectroscopy. Biochemistry 34, 16596–16607. [DOI] [PubMed] [Google Scholar]
- 47).Pond MP, Vuletich DA, Falzone CJ, Majumdar A, and Lecomte JTJ (2009) 1H, 15N, and 13C resonance assignments of the 2/2 hemoglobin from the cyanobacterium Synechococcus sp. PCC 7002 in the ferric bis-histidine state. Biomol. NMR Assign 3, 211–214. [DOI] [PubMed] [Google Scholar]
- 48).Scott NL, Xu Y, Shen G, Vuletich DA, Falzone CJ, Li Z, Ludwig M, Pond MP, Preimesberger MR, Bryant DA, and Lecomte JTJ (2010) Functional and structural characterization of the 2/2 hemoglobin from Synechococcus sp. PCC 7002. Biochemistry 49, 7000–7011. [DOI] [PubMed] [Google Scholar]
- 49).Preimesberger MR, Johnson EA, Nye DB, and Lecomte JTJ (2017) Covalent attachment of the heme to Synechococcus hemoglobin alters its reactivity toward nitric oxide. J. Inorg. Biochem 177, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50).Pesce A, Couture M, Dewilde S, Guertin M, Yamauchi K, Ascenzi P, Moens L, and Bolognesi M (2000) A novel two-over-two α-helical sandwich fold is characteristic of the truncated hemoglobin family. EMBO J 19, 2424–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51).Banci L, Bertini I, Branchini BR, Hajieva P, Spyroulias GA, and Turano P (2001) Dimethyl propionate ester heme-containing cytochrome b5: structure and stability. J. Biol. Inorg. Chem 6, 490–503. [DOI] [PubMed] [Google Scholar]
- 52).Antonini E, and Brunori M (1970) Hemoglobin. Annu. Rev. Biochem 39, 977–1042. [DOI] [PubMed] [Google Scholar]
- 53).Preimesberger MR, Majumdar A, and Lecomte JTJ, (2017) The dynamics of lysine as a heme axial ligand: NMR analysis of the Chlamydomonas reinhardtii hemoglobin THB1. Biochemistry 56, 551–569. [DOI] [PubMed] [Google Scholar]
- 54).Ubbink M, Campos AP, Teixeira M, Hunt NI, Hill HA, and Canters GW (1994) Characterization of mutant Met100Lys of cytochrome c-550 from Thiobacillus versutus with lysine-histidine heme ligation. Biochemistry 33, 10051–10059. [DOI] [PubMed] [Google Scholar]
- 55).Davis LA, Schejter A, and Hess GP (1974) Alkaline isomerization of oxidized cytochrome c. Equilibrium and kinetic measurements. J. Biol. Chem 249, 2624–2632. [PubMed] [Google Scholar]
- 56).Tsuda M, Shirotani I, Minomura S, and Terayama Y (1976) Effect of pressure on dissociation of weak acids in aqueous buffers. B. Chem. Soc. Jpn 49, 2952–2955. [Google Scholar]
- 57).Markley J (1975) Observation of histidine residues in proteins by means of nuclear magnetic resonance spectroscopy. Acc. Chem. Res 8, 70–80. [Google Scholar]
- 58).Preimesberger MR, Majumdar A, Rice SL, Que L, and Lecomte JTJ (2015) Helix-capping histidines: Diversity of N-H…N hydrogen bond strength revealed by 2hJNN scalar couplings. Biochemistry 54, 6896–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59).Kroll T, Hadt RG, Wilson SA, Lundberg M, Yan JJ, Weng TC, Sokaras D, Alonso-Mori R, Casa D, Upton MH, Hedman B, Hodgson KO, and Solomon EI (2014) Resonant inelastic X-ray scattering on ferrous and ferric bis-imidazole porphyrin and cytochrome c: Nature and role of the axial methionine-Fe bond. J. Am. Chem. Soc 136, 18087–18099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60).Marques HM, Munro OQ, Munro T, de Wet M, and Vashi PR (1999) Coordination of N-donor ligands by the monomeric ferric porphyrin N-acetylmicroperoxidase-8. Inorg. Chem 38, 2312–2319. [Google Scholar]
- 61).Vallee BL, and Williams RJ (1968) Metalloenzymes: the entatic nature of their active sites. Proc. Natl. Acad. Sci. U. S. A 59, 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62).Cowley AB, Kennedy ML, Silchenko S, Lukat-Rodgers GS, Rodgers KR, and Benson DR (2006) Insight into heme protein redox potential control and functional aspects of six-coordinate ligand-sensing heme proteins from studies of synthetic heme peptides. Inorg. Chem 45, 9985–10001. [DOI] [PubMed] [Google Scholar]
- 63).Das DK, and Medhi OK (1998) The role of heme propionate in controlling the redox potential of heme: Square wave voltammetry of protoporphyrinato IX iron(III) in aqueous surfactant micelles. J. Inorg. Biochem 70, 83–90. [DOI] [PubMed] [Google Scholar]
- 64).Tonge P, Moore GR, and Wharton CW (1989) Fourier-transform infra-red studies of the alkaline isomerization of mitochondrial cytochrome c and the ionization of carboxylic-acids. Biochem. J 258, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65).Hartshorn RT, and Moore GR (1989) A denaturation-induced proton-uptake study of horse ferricytochrome c. Biochem. J 258, 595–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66).Gu J, Shin D-W, and Pletneva EV (2017) Remote perturbations in tertiary contacts trigger ligation of lysine to the heme iron in cytochrome c. Biochemistry 56, 2950–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67).Kakar S, Hoffman FG, Storz JF, Fabian M, and Hargrove MS (2010) Structure and reactivity of hexacoordinate hemoglobins. Biophys. Chem 152, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68).Schneider S, Marles-Wright J, Sharp KH, and Paoli M (2007) Diversity and conservation of interactions for binding heme in b-type heme proteins. Nat. Prod. Rep 24, 621–630. [DOI] [PubMed] [Google Scholar]
- 69).Nienhaus K, Kriegl JM, and Nienhaus GU (2004) Structural dynamics in the active site of murine neuroglobin and its effects on ligand binding. J. Biol. Chem 279, 22944–22952. [DOI] [PubMed] [Google Scholar]
- 70).Kumar N, Astegno A, Chen J, Giorgetti A, and Dominici P (2016) Residues in the distal heme pocket of Arabidopsis non-symbiotic hemoglobins: Implication for nitrite reductase activity. Int. J. Mol. Sci 17, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71).Martínková M, Kitanishi K, and Shimizu T (2013) Heme-based globin-coupled oxygen sensors: Linking oxygen binding to functional regulation of diguanylate cyclase, histidine kinase, and methyl-accepting chemotaxis. J. Biol. Chem 288, 27702–27711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72).Gilles-Gonzalez MA, and Gonzalez G (2005) Heme-based sensors: defining characteristics, recent developments, and regulatory hypotheses. J. Inorg. Biochem 99, 1–22. [DOI] [PubMed] [Google Scholar]
- 73).Kurokawa H, Lee DS, Watanabe M, Sagami I, Mikami B, Raman CS, and Shimizu T (2004) A redox-controlled molecular switch revealed by the crystal structure of a bacterial heme PAS sensor. J. Biol. Chem 279, 20186–20193. [DOI] [PubMed] [Google Scholar]
- 74).Lanzilotta WN, Schuller DJ, Thorsteinsson MV, Kerby RL, Roberts GP, and Poulos TL (2000) Structure of the CO sensing transcription activator CooA. Nat. Struct. Biol 7, 876–880. [DOI] [PubMed] [Google Scholar]
- 75).Sun YH, Zeng WQ, Benabbas A, Ye X, Denisov I, Sligar SG, Du J, Dawson JH, and Champion PM (2013) Investigations of heme Ligation and ligand switching in cytochromes P450 and P420. Biochemistry 52, 5941–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76).McClelland LJ, Mou TC, Jeakins-Cooley ME, Sprang SR, and Bowler BE (2014) Structure of a mitochondrial cytochrome c conformer competent for peroxidase activity. Proc. Natl. Acad. Sci. U. S. A 111, 6648–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77).Kagan VE, Tyurin VA, Jiang JF, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou MM, Di P, Svistunenko DA, Kurnikov IV, and Borisenko GG (2005) Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Res 1, 223–232. [DOI] [PubMed] [Google Scholar]
- 78).Bren KL, and Raven EL (2017) Locked and loaded for apoptosis. Science 356, 1236–1236. [DOI] [PubMed] [Google Scholar]
- 79).Mara MW, Hadt RG, Reinhard ME, Kroll T, Lim H, Hartsock RW, Alonso-Mori R, Chollet M, Glownia JM, Nelson S, Sokaras D, Kunnus K, Hodgson KO, Hedman B, Bergmann U, Gaffney KJ, and Solomon EI (2017) Metalloprotein entatic control of ligand-metal bonds quantified by ultrafast x-ray spectroscopy. Science 356, 1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80).Amacher JF, Zhong F, Lisi GP, Zhu MQ, Alden SL, Hoke KR, Madden DR, and Pletneva EV (2015) A compact structure of cytochrome c trapped in a lysine-ligated state: loop refolding and functional implications of a conformational switch. J. Am. Chem. Soc 137, 8435–8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81).Yin V, Shaw GS, and Konermann L (2017) Cytochrome c as a peroxidase: Activation of the pre-catalytic native state by H2O2-induced covalent modifications. J. Am. Chem. Soc 44, 15701–15709. [DOI] [PubMed] [Google Scholar]
- 82).Barker PD, and Mauk AG (1992) pH-Linked conformational regulation of a metalloprotein oxidation-reduction equilibrium: electrochemical analysis of the alkaline form of cytochrome c. J. Am. Chem. Soc 114, 3619–3624. [Google Scholar]
- 83).Nelson CJ, and Bowler BE (2000) pH dependence of formation of a partially unfolded state of a Lys 73 -> His variant of iso-1-cytochrome c: Implications for the alkaline conformational transition of cytochrome c. Biochemistry 39, 13584–13594. [DOI] [PubMed] [Google Scholar]
- 84).Fetrow JS, and Baxter SM (1999) Assignment of 15N chemical shifts and 15N relaxation measurements for oxidized and reduced iso-1-cytochrome c. Biochemistry 38, 4480–4492. [DOI] [PubMed] [Google Scholar]
- 85).Barker PD, Bertini I, Del Conte R, Ferguson SJ, Hajieva P, Tomlinson E, Turano P, and Viezzoli MS (2001) A further clue to understanding the mobility of mitochondrial yeast cytochrome c: a 15N T1ρ investigation of the oxidized and reduced species. Eur. J. Biochem 268, 4468–4476. [DOI] [PubMed] [Google Scholar]
- 86).Assfalg M, Bertini I, Turano P, Mauk AG, Winkler JR, and Gray HB (2003) 15N-1H Residual dipolar coupling analysis of native and alkaline-K79A Saccharomyces cerevisiae cytochrome c. Biophys. J 84, 3917–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87).Sakamoto K, Kamiya M, Uchida T, Kawano K, and Ishimori K (2010) Redox-controlled backbone dynamics of human cytochrome c revealed by 15N NMR relaxation measurements. Biochem. Biophys. Res. Commun 398, 231–236. [DOI] [PubMed] [Google Scholar]
- 88).Imai M, Saio T, Kumeta H, Uchida T, Inagaki F, and Ishimori K (2016) Investigation of the redox-dependent modulation of structure and dynamics in human cytochrome c. Biochem. Biophys. Res. Commun 469, 978–984. [DOI] [PubMed] [Google Scholar]
- 89).Ying T, Zhong F, Xie J, Feng Y, Wang ZH, Huang ZX, and Tan X (2009) Evolutionary alkaline transition in human cytochrome c. J. Bioenerg. Biomembr 41, 251–257. [DOI] [PubMed] [Google Scholar]
- 90).Andrew CR, Petrova ON, Lamarre I, Lambry J-C, Rappaport F, and Negrerie M (2016) The dynamics behind the affinity: Controlling heme-gas affinity via geminate recombination and heme propionate conformation in the NO carrier cytochrome c’. ACS Chem. Biol 11, 3191–3201. [DOI] [PubMed] [Google Scholar]
- 91).Bhagi-Damodaran A, Petrik ID, Marshall NM, Robinson H, and Lu Y (2014) Systematic tuning of heme redox potentials and its effects on O2 reduction rates in a designed oxidase in myoglobin. J. Am. Chem. Soc 136, 11882–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92).Brändén G, Brändén M, Schmidt B, Mills DA, Ferguson-Miller S, and Brzezinski P (2005) The protonation state of a heme propionate controls electron transfer in cytochrome c oxidase. Biochemistry 44, 10466–10474. [DOI] [PubMed] [Google Scholar]
- 93).Warren JJ, and Mayer JM (2011) Proton-coupled electron transfer reactions at a heme-propionate in an iron-protoporphyrin-IX model compound. J. Am. Chem. Soc 133, 8544–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94).Mauk MR, Mauk AG, Weber PC, and Matthew JB (1986) Electrostatic analysis of the interaction of cytochrome c with native and dimethyl ester heme substituted cytochrome b5. Biochemistry 25, 7085–7091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.