

Abstract
Background and Purpose:
Tobacco cigarette smoking is considered to be a strong risk factor for intracranial aneurysmal rupture. Nicotine is a major biologically-active constituent of tobacco products. Nicotine’s interactions with vascular cell nicotinic acetylcholine receptors containing α7 subunits (α7*-nAChR) are thought to promote local inflammation and sustained angiogenesis. In this study, using a mouse intracranial aneurysm model, we assessed potential contributions of nicotine exposure and activation of α7*-nAChR to the development of aneurysmal rupture.
Methods:
Intracranial aneurysms were induced by a combination of deoxycorticosterone-salt induced hypertension and a single elastase injection into cerebrospinal fluid in mice.
Results:
Exposure to nicotine or an α7*-nAChR-selective agonist significantly increased aneurysm rupture rate. Co-exposure to an α7*-nAChR antagonist abolished nicotine’s deleterious effect. Additionally, nicotine’s promotion of aneurysm rupture was absent in smooth muscle cell-specific α7*-nAChR subunit knockout mice, but not in mice lacking α7*-nAChR on endothelial cells or macrophages. Nicotine treatment increased the mRNA levels of vascular endothelial growth factor, platelet-derived growth factor-B, and inflammatory cytokines. α7*-nAChR antagonist reversed nicotine-induced up-regulation of these growth factors and cytokines.
Conclusion:
Our findings indicate that nicotine exposure promotes aneurysmal rupture through actions on vascular smooth muscle cell α7*-nAChR.
Keywords: Nicotine, alpha7 nicotinic acetylcholine receptor, angiogenesis, intracranial aneurysm, models, animal
Introduction
Unruptured intracranial aneurysms are common, and 1 to 5% of the population may harbor an unruptured intracranial aneurysm.1 Despite advances in management, subarachnoid hemorrhage from aneurysmal rupture still has high morbidity and mortality.1 Tobacco contains over 7,000 different chemicals, including nicotine, and tobacco product use is associated with various cardiovascular and cerebrovascular diseases.2 Tobacco cigarette smoking is an independent risk factor for intracranial aneurysmal rupture, and the rupture risk in current smokers is three times higher than that of non-smokers.3
Nicotinic acetylcholine receptors (nAChR) are the biological targets of nicotine and mediate natural chemical signaling by acetylcholine.4–6 nAChRs are diverse members of the cys loop family of neurotransmitter-gated ion channels. In mammals, nAChRs are composed as pentamers of different combinations of sixteen, genetically-distinct, transmembrane protein subunits. Each nAChR subtype is defined by its subunit combination and isoforms of those subtypes are further distinguished based on subunit ratios and arrangements.7 The accepted nomenclature for nAChR subtypes is based on known or inferred subunit combination and employs an “*” to indicate that there are known or potential assembly partners in addition to the subunit(s) specified.8
Each nAChR subtype or isoform has a unique distribution across cell types, organs and organ regions, and even subcellularly as well as its own pharmacological profile and responsiveness and sensitivity to nicotine and acetylcholine. Vascular cells including endothelial cells and smooth muscle cells express the nicotinic acetylcholine receptors containing α7 subunits (α7*-nAChR).9, 10 Nicotine’s interaction with α7*-nAChR on vascular cells promotes inflammation and angiogenesis,2, 10, 11 biological processes that are important for the development, growth, and rupture of intracranial aneurysms.12–14 We hypothesized that nicotine contributes to the pathophysiology of intracranial aneurysm through the activation of α7*-nAChR in the vascular wall. Therefore, in this study, using a mouse model of intracranial aneurysm,13–15 we studied potential contribution of nicotine exposure to aneurysmal rupture and roles for those effects of α7*-nAChR. Furthermore, utilizing cell-type specific nAChR α7 subunit knockout mice, we defined a cell type involved in nicotine’s effects on aneurysmal rupture.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. Experiments were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee. We obtained α7nAChRflox/flox (α7f/f) mice16, α7nAChR knockout (α7nAChR KO) mice17, mice expressing Cre recombinase under the control of the myeloid-specific lysosome M (LysM) promoter (LysMCre18), the transgelin (smooth muscle protein 22-alpha) promoter (SM22Cre19), and the endothelial-specific receptor tyrosine kinase (Tie2) promoter (Tie2Cre20) from Jackson Laboratory (Bar Harbor, Maine). We used LysMCre, SM22Cre and Tie2Cre negative α7f/f mice as control and LysMCre, SM22Cre, and Tie2Cre positive α7f/f mice as myeloid-, smooth muscle cell-, and endothelial cell-specific α7nAChR knockout mice. We used 9-week-old male C57BL/6J. To induce aneurysm formation, we combined systemic hypertension and a single injection of elastase (17.5mU) into the cerebrospinal fluid.13–15, 21 To induce systemic hypertension, we used deoxycorticosterone acetate (DOCA)-salt-induced hypertension.
Nicotine (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in saline and administrated subcutaneously thorough Alzet osmotic minipumps (2004, Durect Corporation, Cupertino, CA) (5 mg/kg/day22). α7nAChR agonist (AR-R17779, 5mg/kg in 5%DMSO) and α7nAChR antagonist (5mg/kg in saline) were injected intraperitoneally once a day.23 Vehicle control groups received saline or 5% DMSO. All drugs or vehicle were administered from one week before elastase injection, and the treatments were continued for 4 weeks.
To detect aneurysmal rupture, two blinded observers performed daily neurological examinations as previously described.14 Mice were euthanized when they developed neurological symptoms (score, 1–5). Asymptomatic mice were euthanized 21 days after aneurysm induction as previously described.14 The brain samples were perfused with phosphate-buffered saline, followed by a gelatin-containing blue dye to visualize cerebral arteries. Aneurysms were defined as a localized outward bulging of the vascular wall, whose diameter was greater than the parent artery diameter. Two observers who were blinded to the treatments and mouse genotypes conducted a daily neurological examination and assessed subarachnoid hemorrhage and aneurysm formations. A detailed Methods section is available in the online-only Data Supplement.
Statistical Analysis
We used the Fisher’s exact test to analyze the incidence of aneurysms (number of mice with any aneurysms [ruptured or unruptured]/total number of mice) and rupture rate (number of mice with ruptured aneurysms/number of mice with any aneurysms). Mice that did not show aneurysm formation were excluded from the calculation of the rupture rate. As an exploratory analysis, the survival analysis was performed using the log-rank test. Levels of mRNA and blood pressure were analyzed by two-way ANOVA, followed by Turkey-Kramer post hoc test. Statistical significance was accepted at P < 0.05. Quantitative results were expressed as mean ± SD.
Results
Nicotine exposure promotes aneurysmal rupture in a mouse model of intracranial aneurysm.
As a first step, we assessed effects of nicotine exposure on the formation and rupture of intracranial aneurysms. There was no significant difference in the overall incidence of aneurysms between the vehicle and nicotine treatment groups (68% versus 76%; n = 22 versus n = 25; Figure 1A). However, nicotine treatment significantly increased the aneurysm rupture rate (Figure 1A; vehicle versus nicotine: 46% versus 89%; P < 0.01). Mice treated with nicotine had significantly worse symptom-free survival compared to vehicle (Kaplan-Meier log-rank P < 0.05; Figure 1A). Figures 1B, 1C, and 1D show normal cerebral arteries, an unruptured aneurysm from a mouse that was asymptomatic throughout the experimental period, and a ruptured aneurysm with subarachnoid hemorrhage from a mouse that became symptomatic 8 days after aneurysm induction, respectively. There were hemosiderin deposits on the brain surface along the Circle of Willis due to the blood leakage from the needle track of the elastase injection. There was no significant difference in blood pressure between two groups at any time points (Table I in Online Data Supplement).
Figure 1.
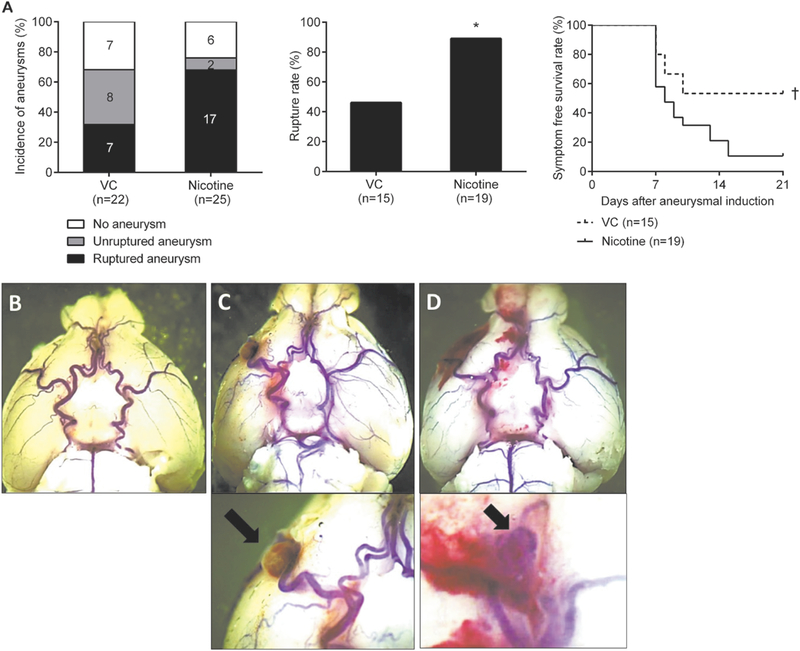
A, Nicotine exposure promotes the development of aneurysm rupture in male wild-type mice. Representative pictures of intracranial aneurysm. B, No aneurysm. C, Unruptured aneurysm. D, Ruptured aneurysm. VC indicates vehicle control. *P < 0.01 vs VC and †P < 0.05 vs VC.
Effects of α7*-nAChR agonist or antagonist exposure on the development of intracranial aneurysm rupture
Since nicotine increased the rate of aneurysmal rupture, we then investigated whether activation of α7*-nAChR mediates nicotine’s effects utilizing an α7*-nAChR-selective agonist, AR-R17779. There was no significant difference in the incidence of aneurysm formations between vehicle or α7*-nAChR agonist treatment groups (62% versus 72%; n = 16 versus n = 18; Figure 2A). However, the α7*-nAChR agonist significantly increased aneurysmal rupture (Figure 2A; 40% versus 92%; P < 0.05). Mice treated with the α7*-nAChR agonist had significantly worse symptom-free survival compared to vehicle (Kaplan-Meier log-rank P < 0.05; Figure 2A). There was no significant difference in blood pressure, aneurysm location, or aneurysm size between two groups (Table I-II in Online Data Supplement).
Figure 2.
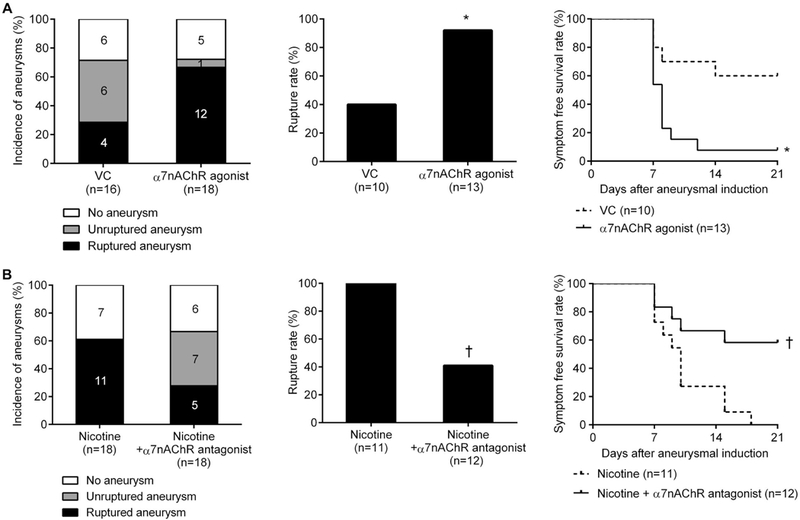
A, An α7*-nicotinic acetylcholine receptor (α7*-nAChR) agonist (AR-R17779) promotes aneurysm rupture in male wild-type mice. B, Co-exposure to nicotine plus an α7*-nAChR antagonist (methyllycaconitine) abolishes aneurysm rupture in male wild-type mice. VC indicates vehicle control. *P < 0.05 vs VC and †P < 0.01 vs VC.
Next, to test whether α7*-nAChR activation mediates nicotine’s promotion of aneurysmal rupture, we compared the rupture rate between mice treated with nicotine alone and those treated with nicotine plus an α7*-nAChR-selective antagonist, methyllycaconitine. There was no significant difference in the incidence of aneurysm formations between nicotine and nicotine + α7*-nAChR-selective antagonist treated groups (66% versus 77%; n = 18 versus n = 18; Figure 2B). However, the α7*-nAChR antagonist effectively abolished nicotine’s promotion of aneurysmal rupture (Figure 2B; nicotine versus nicotine + antagonist: 100% versus 42%; P < 0.01). Mice treated with nicotine plus the α7*-nAChR antagonist had significantly better symptom-free survival compared to those exposed to nicotine alone (Kaplan-Meier log-rank P < 0.05; Figure 2B). α7nAChR antagonist alone did not have any significant effect on the rupture rate (42% versus 38%, n = 12 versus 13, P = 1.00). There was no significant difference in blood pressure among three groups (Table I in Online Data Supplement).
Aneurysmal rupture in global nAChR α7 subunit knockout mice
To further confirm that nicotine exposure promotes aneurysmal rupture through α7*-nAChR, we compared effects of nicotine treatment between nAChR α7 subunit knockout (α7 KO) mice and wild-type littermates.
There was no significant difference in the overall incidence of aneurysms (Figure 3A) and blood pressure (Table I in Online Data Supplement) in among four groups. However, consistent with results presented in Figure 1, nicotine treatment significantly increased aneurysmal rupture compared to the vehicle treatment in the wild-type mice (Figure 3B; vehicle versus nicotine: 46% versus 84%; P < 0.01). More importantly, the effect of nicotine exposure on aneurysmal rupture was abolished in α7 KO mice (Figure 3B; wild-type + nicotine versus α7 KO + nicotine: 84% versus 27%; P < 0.05). In α7 KO mice, nicotine did not affect the rupture rate (Figure 3B; vehicle versus nicotine: 36% versus 27%; P = 1.00).
Figure 3.
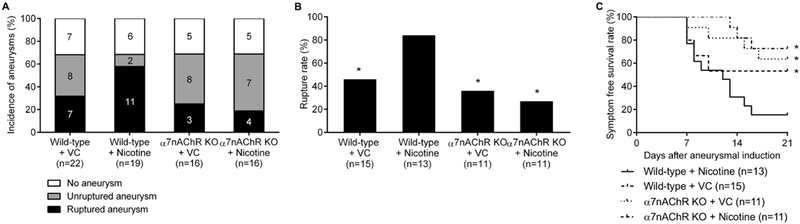
Nicotine’s promotion of aneurysm rupture requires α7*-nicotinic acetylcholine receptors. A, Incidence of aneurysms. B, Rupture rate. C, Symptom-free curve (Kaplan-Meier analysis curve). VC and α7 KO indicates vehicle control and nAChR α7 subunit knockout. *P < 0.05 vs WT + nicotine.
Nicotine promotes the expression of growth factors and inflammation in the cerebral arteries.
Previous studies have shown that the activation of α7*-nAChR in vascular cells promotes sustained angiogenesis and inflammation through the release of angiogenic factors.2, 10, 11 Therefore, to explore the mechanisms by which nicotine promotes aneurysmal rupture, we used real-time RT-PCR to determine levels of mRNA for angiogenic factors and inflammatory cytokines in cerebral arteries in the vehicle, nicotine, and nicotine + α7*-nAChR antagonist-treated groups.
Expression of vascular endothelial growth factor (VEGF) and platelet-derived growth factor-B (PDGF-B) in cerebral arteries were significantly higher in the nicotine treatment group than in the vehicle group (n=7 and 6; VEGF: 1.0 ± 0.3 versus 1.6 ± 0.2; PDGF-B: 1.0 ± 0.3 versus 1.6 ± 0.3, all P < 0.01; Figure 4A). Moreover, the presence of an α7*-nAChR antagonist reduced nicotine-mediated elevation of VEGF and PDGF-B (n=7 and 6; VEGF: 1.6 ± 0.2 versus 0.8 ± 0.3; PDGF-B: 1.6 ± 0.3 versus 0.9 ± 0.1, all P < 0.01; Figure 4A). Meanwhile, expression of transforming growth factor-β (TGF-β) was significantly lower in the nicotine group than in the vehicle group (n=7 and 6; TGF-β: 1.0 ± 0.4 versus 0.5 ± 0.1, all P < 0.05; Figure 4A). α7*-nAChR antagonist exposure did not normalize levels of TGF-β mRNA in nicotine-treated mice. There was no difference in fibroblast growth factor-2 (FGF2) or hepatocyte growth factor (HGF) levels across groups.
Figure 4.
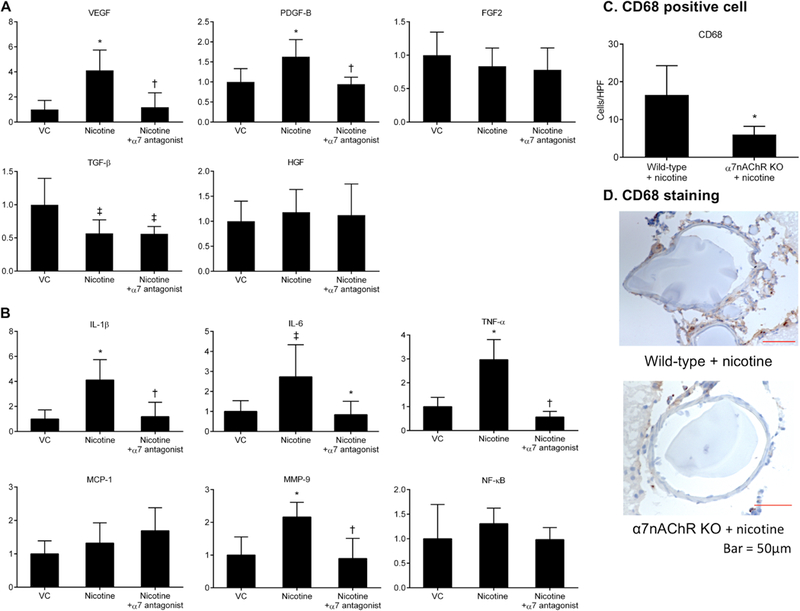
A, Effects of nicotine exposure on mRNA levels of growth factors. B, Effects of nicotine treatment on mRNA levels of inflammatory cytokines. *P < 0.01 vs VC, †P < 0.01 vs nicotine, and ‡P < 0.05 vs VC (two-way ANOVA). C, Effects of α7*-nicotinic acetylcholine receptor activation by nicotine on the infiltration of macrophages. CD68 positive cell numbers were significantly lower in the collected tissue from nicotine-treated α7 knockout (α7nAChRKO) mice than in nicotine-treated wild-type mice. *P < 0.01 vs wild-type + nicotine. D, Representative immunohistochemistry images comparing nicotine-treated α7 KO mice with nicotine-treated wild-type mice. HPF indicates high-power field. VC indicates vehicle control; α7 antagonist, α7*-nAChR antagonist.
mRNA levels of interleukin-6 (IL-6), IL-1β, tumor necrosis factor-α (TNF-α), and matrix metalloproteinase-9 (MMP-9) in cerebral arteries were significantly higher in the nicotine group than in the vehicle group (n = 7 and 6; IL-6: 1.0 ± 0.5 versus 2.9 ± 1.8; IL-1β: 1.0 ± 0.7 versus 4.1 ± 1.7; TNF-α: 1.0 ± 0.3 versus 2.9 ± 0.7; MMP-9; 1.0 ± 0.5 versus 2.1 ± 0.4, IL-1b, MMP-9, and TNF-α P < 0.01, IL-6 P < 0.05; Figure 4B). Furthermore, addition of an α7*-nAChR antagonist blocked nicotine exposure-induced increases in IL-6, IL-1β, TNF-α, and MMP-9 (n = 6; IL-6: 0.5 ± 0.3 versus 2.9 ± 1.8; IL-1β: 1.1 ± 1.1 versus 4.1 ± 1.7; TNF-α: 0.5 ± 0.2 versus 2.9 ± 0.7; MMP-9: 0.8 ± 0.6 versus 2.1 ± 0.4, all P < 0.01; Figure 4B). There was no difference in monocyte chemoattractant protein-1 or NF-κB across groups.
Roles of α7*-nAChR in nicotine-induced inflammation of cerebral arteries were further examined by assessing macrophage infiltration in cerebral arteries. CD68 positive cell density was significantly lower in cerebral arteries from nicotine-treated α7 KO mice (n = 6) than in those from nicotine-treated wild-type mice (n = 6; CD68: 6.0 ± 2.1 versus 16.5 ± 7.7, P < 0.01; Figure 4C). Figure 4D shows representative staining of cerebral arteries for CD68.
Nicotine promotes aneurysmal rupture via the activation of VSMC α7*-nAChR.
To explore the cell type that mediates deleterious effects of nicotine-induced α7*-nAChR activation, we compared the incidence of aneurysmal ruptures among vascular smooth muscle cell (VSMC)-specific α7 KO mice (α7f/fSM22Cre+), myeloid lineage cell (macrophages, monocytes, and granulocytes)-specific α7 KO mice (α7f/fLysMCre+), endothelial cell-specific α7 KO mice (α7f/fTie2Cre+), and their corresponding control littermates (α7f/f).
As shown in Figure 5, there was no difference in the rupture rate between α7f/fLysMCre+ mice treated with nicotine and their nicotine-treated control littermates (Figure 5B; α7f/f + nicotine versus α7f/fLysMCre+ + nicotine: 92% versus 87%; P = 1.00). However, nicotine’s promotion of aneurysm rupture was abolished in mice lacking α7*-nAChR on vascular smooth muscle cells (α7f/fSM22Cre+) (Figure 5A; α7f/f + nicotine versus α7f/fSM22Cre+ + nicotine: 91% versus 22%; P < 0.01). α7f/fSM22Cre+ mice treated with nicotine had significantly improved symptom-free survival compared with α7f/f mice treated with nicotine (P < 0.05; Figure 5A). Meanwhile, there was no difference in the rupture between α7f/fTie2Cre+ treated with nicotine and their control littermates treated with nicotine (Figure 5C; α7f/f + nicotine versus α7f/fTie2Cre+ + nicotine: 81% versus 72%; P = 1.00). There was no significant difference in the overall incidence of aneurysm formation (Figure 5) or blood pressure (Table I in Online Data Supplement) between any of three cell type-specific α7 KO mice and their corresponding control littermates.
Figure 5.
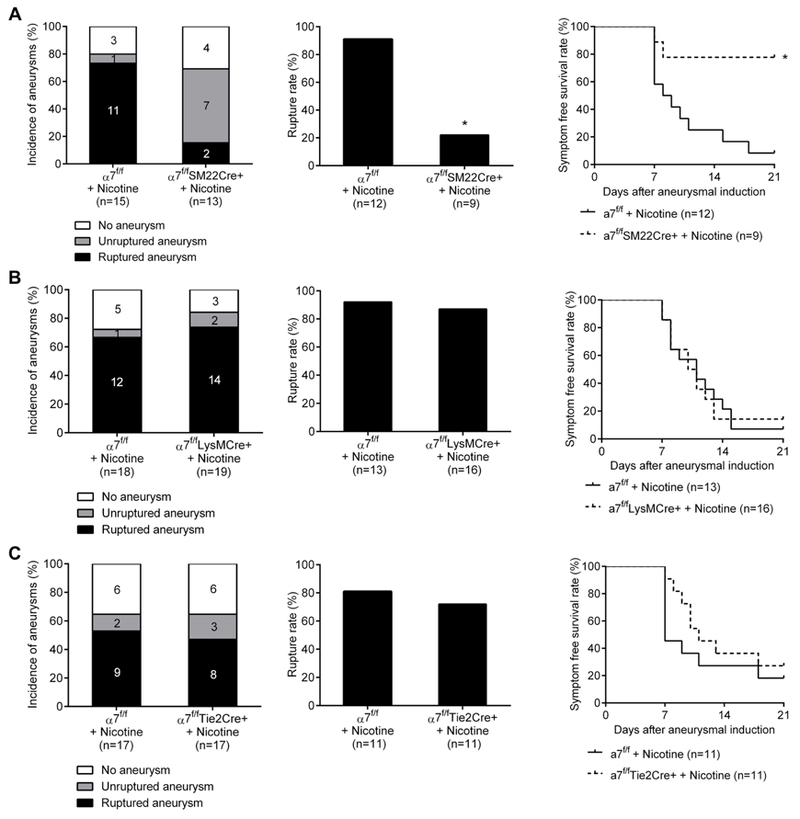
Nicotine’s promotion of aneurysm rupture requires α7*-nicotinic acetylcholine receptors (α7*-nAChR) expressed on smooth muscle cells. A, α7f/f SM22Cre+ treated with nicotine and α7f/f treated with nicotine. B, α7f/f LysMCre+ treated with nicotine and α7f/f treated with nicotine. C, α7f/f Tie2Cre+ treated with nicotine and α7f/f treated with nicotine. *P < 0.01 vs α7f/f + Nicotine.
Consistent with results presented in Figure 4A, mRNA levels for VEGF and PDGF-B in cerebral arteries were significantly lower in α7f/fSM22Cre+ mice treated with nicotine than in α7f/f mice treated with nicotine (n=5; VEGF: 1.0 ± 0.1 versus 0.3 ± 0.1; PDGF-B: 1.0 ± 0.2 versus 0.4 ± 0.1, all P < 0.01; Figure 6). Moreover, expression of FGF2 was significantly lower in α7f/fSM22Cre+ mice treated with nicotine than in α7f/f mice treated with nicotine (n=5; FGF2: 1.0 ± 0.1 versus 0.4 ± 0.1, P < 0.01; Figure 6). There was no difference in TGF-β and HGF expression between these groups.
Figure 6.
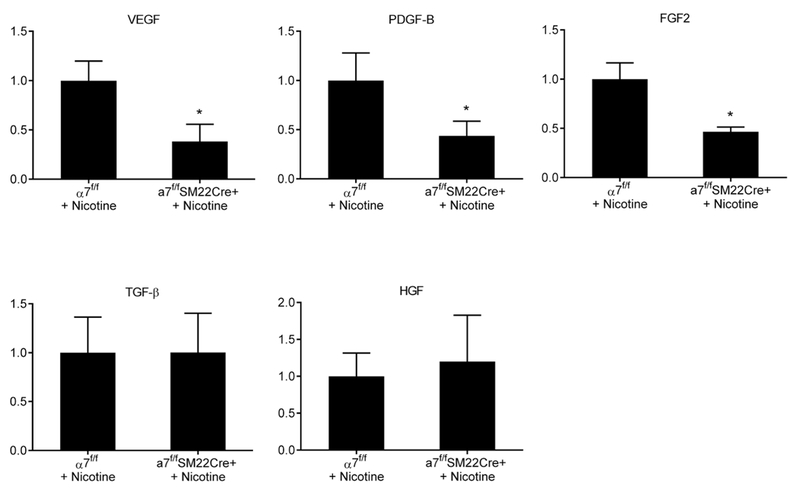
A, Effect of VSMC α7*-nAChR modulation on mRNA levels of growth factors. Expression of VEGF, PDGF-B, and FGF2 in cerebral arteries was significantly lower in α7f/fSM22Cre+ mice treated with nicotine than in α7f/f mice treated with nicotine. *P < 0.01 vs α7f/f + nicotine.
Discussion
Epidemiological studies have repeatedly shown a positive association between tobacco cigarette smoking and aneurysmal rupture (i.e., aneurysmal subarachnoid hemorrhage). However, underlying mechanisms are not well understood, nor is the specific role played by nicotine itself as opposed to other constituents of tobacco and of cigarette smoke. Our data indicate that nicotine exposure alone increases rates of aneurysm rupture in a mouse model and that this occurs through activation of vascular smooth muscle cell α7*-nAChR. These observations are supported by studies showing that α7*-nAChR-selective antagonism or global knockout of nAChR α7 subunits abolishes the deleterious effect of nicotine exposure on aneurysmal rupture. Moreover, α7 KO specific to VSMCs, but not in inflammatory cells such as macrophages and granulocytes, abolished nicotine’s promotion of aneurysmal rupture.
Although stimulation of α7*-nAChR of macrophages and lymphocytes exerts anti-inflammatory effects,24 vascular cell α7*-nAChR have been shown to mediate VSMC proliferation, angiogenesis, and vascular inflammation.2, 10, 11 Roles of α7nAChRs in VSMCs have been extensively studied in the cell culture settings. Stimulation of α7*-nAChR by nicotine can directly increase migration and proliferation of VSMCs in vitro.25, 26 In addition, nicotine’s interactions with 7*-nAChR may indirectly affect the migration and proliferation of vascular and inflammatory cells through the release of cytokines.10, 26, 27 7*-nAChR mediate nicotine-induced production and release of angiogenic growth factors, including PDGF-BB and VEGF, from vascular cells.10, 26, 27 PDGF-BB and VEGF, while causing smooth muscle cell proliferation and sustained angiogenesis, promote the release of inflammatory cell attractants into the vascular wall.27 Expression of VEGF and PDGF-BB in ruptured aneurysms is reported to be higher than those in unruptured aneurysms in humans.28 Inflammatory cytokines such as TNF-α and IL-1 secreted by inflammatory cells stimulate the release of growth factors from endothelial cells and VSMCs.29, 30 In our study, nicotine treatment increased expression of VEGF, PDGF-B, and inflammatory cytokines through 7*-nAChR. Both human and animal studies suggest that sustained inflammation and angiogenesis play key roles in the growth and rupture of intracranial aneurysms.28, 31, 32 Nicotine may promote aneurysm rupture by directly and indirectly causing excessive vascular remodeling and inflammation through the activation of 7*-nAChR.
Interestingly, nicotine reduced mRNA levels of TGF-β in our model, but an α7*-nAChR antagonist did prevent this effect of nicotine. Other nAChR subtypes, such as α9*- or α4β2-nAChR may regulate TGF-β signaling instead.33 As TGF-β can protect against pathological vascular remodeling,34, 35 α7*-nAChR-independent down-regulation of TGF-β by nicotine may further destabilize vascular wall and promote rupture of intracranial aneurysms.
In this study, we did not detect a significant effect of nicotine on the incidence of aneurysm formations. In this model, aneurysmal formations are induced, while aneurysmal ruptures occur spontaneously. As the incidence of aneurysm formation in this model was already as high as 70% in the control mice when 17.5 milli-units of elastase was used, it may be difficult to reliably detect the further increase in the incidence of aneurysm in the nicotine-treated mice. In addition, it may take a long-term exposure to nicotine to promote the formation of aneurysms. Alternatively, the mechanism for aneurysm formation may be different from the mechanism for aneurysmal rupture. Different doses of nicotine or exposure length may be required for the formation and rupture of intracranial aneurysms.
Different nAChR subtypes can have opposing effects.10 For example, inhibition or knockdown of α1*-nAChR is reported to reduce aortic plaque development.2, 36 α4β2-nAChR is reported to have anti-inflammatory effects in vitro.10, 33 While it is possible that other nAChR subtypes may contribute to different aspects of the pathophysiology of intracranial aneurysms, our data indicate a critical role of α7*-nAChR on vascular smooth muscle cells in mediating nicotine’s effects. Further studies using other nAChR subtype-specific knockout mice or inhibitors will elucidate the roles of other nAChRs in the pathophysiology of intracranial aneurysm.
Different doses of nicotine can lead to activation or desensitization of nAChR function and do so at different concentration ranges for different nAChR subtypes.37 The dose of nicotine used in this study approximates levels previously reported in heavy smokers.22, 38 This dose has been widely used to study deleterious effects of nicotine in various animal models.22, 38 However, future studies could investigate whether low doses of nicotine experienced as second-hand smoke affect intracranial aneurysmal rupture.
Finally, as an initial step to study roles of nicotine and α7*-nAChR in the pathophysiology of intracranial aneurysms, we used only male mice in this study. Our previous studies showed that the incidence of aneurysm formation and rupture rates are higher in ovariectomized female mice than male mice and sham-ovariectomized female mice, indicating protective effects of estrogen against the formation and rupture of intracranial aneurysms.21, 39 The baseline differences in the incidence of aneurysm formations and aneurysmal ruptures among male, non-ovariectomized female, and ovariectomized female mice make the inclusion and comparison of these groups would make the experimental design far more complex and highly expansive. While this proof-of-concept study started with experiments using male animals, roles of sex differences and sex steroids should be carefully examined in future studies.
Conclusions
This study using a mouse model of intracranial aneurysm shows that nicotine exposure increases aneurysmal rupture through actions on α7*-nAChR on VSMCs and via promotion of sustained angiogenesis and inflammation. α7*-nAChR may serve as a potential therapeutic target for the prevention of intracranial aneurysmal rupture.
Supplementary Material
Acknowledgment
Sources of Funding
The project was supported by grant number R01NS055876 (TH) and R01NS082280 (TH) from the National Institute of Neurological Disorders and Stroke (NIH/NINDS), Brain Aneurysm Foundation (TK, DK), and Barrow Neurological Foundation (RJL, MTL, TH). The authors have no personal, financial, or institutional interest in any of the drugs, materials, or devices described in this article.
The authors would like to thank Ms. Cindy Giljames and Barrow Neurological Institute Neuroscience Publications for the production of Visual Abstract.
Footnotes
Disclosures
None.
Contributor Information
Yoshinobu Kamio, Department of Neurosurgery, Barrow Aneurysm and AVM Research Center, Barrow Neurological Institute, Phoenix, AZ.
Takeshi Miyamoto, Department of Neurosurgery and Neurobiology, Barrow Neurological Institute, Phoenix, AZ.
Tetsuro Kimura, Department of Neurosurgery and Neurobiology, Barrow Aneurysm and AVM Research Center, Barrow Neurological Institute, Phoenix, AZ.
Kazuha Mitsui, Department of Anesthesia and Perioperative Care, University of California, San Francisco.
Hajime Furukawa, Department of Anesthesia and Perioperative Care, University of California, San Francisco.
Dingding Zhang, Department of Anesthesia and Perioperative Care, University of California, San Francisco.
Kimihiko Yokosuka, Department of Anesthesia and Perioperative Care, University of California, San Francisco.
Masaaki Korai, Department of Anesthesia and Perioperative Care, University of California, San Francisco.
Daisuke Kudo, Department of Neurosurgery and Neurobiology, Barrow Aneurysm and AVM Research Center, Barrow Neurological Institute, Phoenix, AZ.
Ronald J. Lukas, Department of Neurobiology, Barrow Aneurysm and AVM Research Center, Barrow Neurological Institute, Phoenix, AZ.
Michael T. Lawton, Department of Neurosurgery, Barrow Aneurysm and AVM Research Center, Barrow Neurological Institute, Phoenix, AZ.
Tomoki Hashimoto, Department of Neurosurgery and Neurobiology, Barrow Aneurysm and AVM Research Center, Barrow Neurological Institute, Phoenix, AZ.
References
- 1.Connolly ES Jr., Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: A guideline for healthcare professionals from the american heart association/american stroke association. Stroke 2012;43:1711–1737 [DOI] [PubMed] [Google Scholar]
- 2.Lee J, Cooke JP. Nicotine and pathological angiogenesis. Life Sci 2012;91:1058–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Can A, Castro VM, Ozdemir YH, Dagen S, Yu S, Dligach D, et al. Association of intracranial aneurysm rupture with smoking duration, intensity, and cessation. Neurology 2017;89:1408–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: Structural revelations, target identifications, and therapeutic inspirations. J Med Chem 2005;48:4705–4745 [DOI] [PubMed] [Google Scholar]
- 5.Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov 2009;8:733–750 [DOI] [PubMed] [Google Scholar]
- 6.Lukas RJ, Bencherif M. Recent developments in nicotinic acetylcholine receptor biology. Arias HR, ed. Biological and biophysical aspects of ligand-gated ion channel receptor superfamilies, 2006. Kerala, India: Research Signpost; 2006:27–59. [Google Scholar]
- 7.Lucero LM, Weltzin MM, Eaton JB, Cooper JF, Lindstrom JM, Lukas RJ, et al. Differential alpha4(+)/(−)beta2 agonist-binding site contributions to alpha4beta2 nicotinic acetylcholine receptor function within and between isoforms. J Biol Chem 2016;291:2444–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, et al. International union of pharmacology. Xx. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 1999;51:397–401 [PubMed] [Google Scholar]
- 9.Li DJ, Huang F, Ni M, Fu H, Zhang LS, Shen FM. Alpha7 nicotinic acetylcholine receptor relieves angiotensin ii-induced senescence in vascular smooth muscle cells by raising nicotinamide adenine dinucleotide-dependent sirt1 activity. Arterioscler Thromb Vasc Biol 2016;36:1566–1576 [DOI] [PubMed] [Google Scholar]
- 10.Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, et al. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med 2001;7:833–839 [DOI] [PubMed] [Google Scholar]
- 11.Heeschen C, Weis M, Aicher A, Dimmeler S, Cooke JP. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J Clin Invest 2002;110:527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frosen J, Tulamo R, Paetau A, Laaksamo E, Korja M, Laakso A, et al. Saccular intracranial aneurysm: Pathology and mechanisms. Acta Neuropathol 2012;123:773–786 [DOI] [PubMed] [Google Scholar]
- 13.Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, et al. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 2011;42:173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makino H, Tada Y, Wada K, Liang EI, Chang M, Mobashery S, et al. Pharmacological stabilization of intracranial aneurysms in mice: A feasibility study. Stroke 2012;43:2450–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nuki Y, Tsou TL, Kurihara C, Kanematsu M, Kanematsu Y, Hashimoto T. Elastase-induced intracranial aneurysms in hypertensive mice. Hypertension 2009;54:1337–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernandez CM, Cortez I, Gu Z, Colon-Saez JO, Lamb PW, Wakamiya M, et al. Research tool: Validation of floxed alpha7 nicotinic acetylcholine receptor conditional knockout mice using in vitro and in vivo approaches. J Physiol 2014;592:3201–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, et al. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci 1997;17:9165–9171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using lysmcre mice. Transgenic Res 1999;8:265–277 [DOI] [PubMed] [Google Scholar]
- 19.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, et al. Smooth muscle-selective deletion of guanylyl cyclase-a prevents the acute but not chronic effects of anp on blood pressure. Proc Natl Acad Sci U S A 2002;99:7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev Biol 2001;230:230–242 [DOI] [PubMed] [Google Scholar]
- 21.Tada Y, Wada K, Shimada K, Makino H, Liang EI, Murakami S, et al. Estrogen protects against intracranial aneurysm rupture in ovariectomized mice. Hypertension 2014;63:1339–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, et al. Activation of amp-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med 2012;18:902–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis AS, Mineur YS, Smith PH, Cahuzac ELM, Picciotto MR. Modulation of aggressive behavior in mice by nicotinic receptor subtypes. Biochem Pharmacol 2015;97:488–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384–388 [DOI] [PubMed] [Google Scholar]
- 25.Li S, Zhao T, Xin H, Ye LH, Zhang X, Tanaka H, et al. Nicotinic acetylcholine receptor alpha7 subunit mediates migration of vascular smooth muscle cells toward nicotine. Journal of pharmacological sciences 2004;94:334–338 [DOI] [PubMed] [Google Scholar]
- 26.Cucina A, Fuso A, Coluccia P, Cavallaro A. Nicotine inhibits apoptosis and stimulates proliferation in aortic smooth muscle cells through a functional nicotinic acetylcholine receptor. J Surg Res 2008;150:227–235 [DOI] [PubMed] [Google Scholar]
- 27.Naldini A, Leali D, Pucci A, Morena E, Carraro F, Nico B, et al. Cutting edge: Il-1beta mediates the proangiogenic activity of osteopontin-activated human monocytes. J Immunol 2006;177:4267–4270 [DOI] [PubMed] [Google Scholar]
- 28.Frosen J, Piippo A, Paetau A, Kangasniemi M, Niemela M, Hernesniemi J, et al. Growth factor receptor expression and remodeling of saccular cerebral artery aneurysm walls: Implications for biological therapy preventing rupture. Neurosurgery 2006;58:534–541; discussion 534–541 [DOI] [PubMed] [Google Scholar]
- 29.Chidlow JH Jr., Shukla D, Grisham MB, Kevil CG. Pathogenic angiogenesis in ibd and experimental colitis: New ideas and therapeutic avenues. Am J Physiol Gastrointest Liver Physiol 2007;293:G5–G18 [DOI] [PubMed] [Google Scholar]
- 30.Deban L, Correale C, Vetrano S, Malesci A, Danese S. Multiple pathogenic roles of microvasculature in inflammatory bowel disease: A jack of all trades. Am J Pathol 2008;172:1457–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke 1999;30:1396–1401 [DOI] [PubMed] [Google Scholar]
- 32.Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery 1999;45:1137–1146; discussion 1146–1137 [DOI] [PubMed] [Google Scholar]
- 33.Wu JC, Chruscinski A, De Jesus Perez VA, Singh H, Pitsiouni M, Rabinovitch M, et al. Cholinergic modulation of angiogenesis: Role of the 7 nicotinic acetylcholine receptor. Journal of cellular biochemistry 2009;108:433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, et al. Tgf-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin ii-infused mice. J Clin Invest 2010;120:422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angelov SN, Hu JH, Wei H, Airhart N, Shi M, Dichek DA. Tgf-beta (transforming growth factor-beta) signaling protects the thoracic and abdominal aorta from angiotensin ii-induced pathology by distinct mechanisms. Arterioscler Thromb Vasc Biol 2017;37:2102–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang G, Marshall AL, Thomas AL, Kernan KA, Su Y, LeBoeuf RC, et al. In vivo knockdown of nicotinic acetylcholine receptor alpha1 diminishes aortic atherosclerosis. Atherosclerosis 2011;215:34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newhouse P, Kellar K, Aisen P, White H, Wesnes K, Coderre E, et al. Nicotine treatment of mild cognitive impairment: A 6-month double-blind pilot clinical trial. Neurology 2012;78:91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maegdefessel L, Azuma J, Toh R, Deng A, Merk DR, Raiesdana A, et al. Microrna-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci Transl Med 2012;4:122ra122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tada Y, Makino H, Furukawa H, Shimada K, Wada K, Liang EI, et al. Roles of estrogen in the formation of intracranial aneurysms in ovariectomized female mice. Neurosurgery 2014;75:690–695; discussion 695 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.