

SUMMARY
Cells respond to temperature stress via up- and down-regulation of hundreds of genes. This process is thought to be regulated by the heat shock factor HSF1, which controls the release of RNAPII from promoter-proximal pausing. Here we analyze the events taking place in hESCs upstream of RNAPII release. We find that temperature stress results in the activation or decommissioning of thousands of enhancers. This process involves alterations in the occupancy of transcription factors HSF1, AP-1, NANOG, KLF4, and OCT4, accompanied by nucleosome remodeling by BRG1 and changes in H3K27ac. Furthermore, redistribution of RAD21 and CTCF results in the formation and disassembly of interactions mediated by these two proteins. These alterations tether/untether enhancers to their cognate promoters or refashion insulated neighborhoods, thus transforming the landscape of enhancer-promoter interactions. Details of the 3D interactome remodeling process support loop extrusion initiating at random sites as a mechanism for the establishment of CTCF/cohesin loops.
Keywords: Cohesin, CTCF, AP-1, loop extrusion, pluripotency, stem cell, heat shock, enhancer
Graphical Abstract
Lyu et al show that AP-1 and pluripotency factors play a critical role in the temperature stress response in hESCs. Recruitment of these factors to regulatory sequences is accompanied by changes in chromatin 3D organization mediated by CTCF and cohesin, which regulate the establishment of new enhancer-promoter interaction via a randomly-initiated loop extrusion mechanism.
INTRODUCTION
Studies in Drosophila and mammals have shown that cells respond to temperature stress by reprograming their transcriptome, involving the silencing of hundreds of previously active genes and the activation of a number of previously silenced genes (Mahat et al., 2016). The molecular processes involved in this response are comparable to the changes in gene expression that take place during cell differentiation but the response to temperature stress occurs in a very short time period, facilitating the analysis of the temporal events involved in transcription activation and repression of a large group of genes. Changes in gene expression in response to stress are controlled in part by the transcription factor HSF1 in vertebrates, which forms homotrimers or heterotrimers with HSF2 in response to thermal or oxidative stress (Trinklein et al., 2004). In Drosophila, activation of transcription of Hsp genes involves recruitment of HSF and release of RNAPII into productive elongation, a process that has been analyzed in detail by John Lis and colleagues (Fuda et al., 2009). However, not all genes containing HSFs in Drosophila or mammals are induced by temperature stress, and not all genes induced after heat shock contain HSFs bound in the proximity of the activated genes (Mahat et al., 2016), suggesting that the process is more complex, requiring the involvement of other transcription factors and distant regulatory elements that may loop to the promoter to control the release of RNAPII.
The genome of mammalian cells is organized in the three-dimensional nuclear space into higher-order structures, created in part by interactions between distant sites containing CTCF and the cohesin complex (Phillips-Cremins et al., 2013; Tang et al., 2015). These interactions regulate contacts among enhancers and promoters located far apart in the linear genome. In addition, CTCF loops establish insulated neighborhoods to dampen interactions between regulatory sequences located on either side of the loop (Hnisz et al., 2016). Loops between CTCF sites are formed preferentially between genomic regions with convergent CTCF motifs (de Wit et al., 2015; Guo et al., 2015; Rao et al., 2014). To explain their tendency to interact when in a convergent head-to-head orientation, it has been proposed that CTCF loops are formed via an extrusion mechanism involving cohesin (Alipour and Marko, 2012; Fudenberg et al., 2016; Nasmyth, 2001; Nichols and Corces, 2015; Sanborn et al., 2015). This extrusion process could be initiated at CTCF sites, cohesin loading sites, or randomly. In embryonic stem cell (ESCs), evidence suggests that pluripotency factors such as KLF4 and NANOG also contribute to the three-dimensional organization of the ESC genome (de Wit et al., 2013).
Here we explore the ability of the human embryonic stem cell (hESC) 3D nucleome to mediate the dramatic changes in transcription elicited by temperature stress. We find that decommissioning and activation of enhancers correlate with nucleosome remodeling and changes in the occupancy of pluripotency factors and AP-1. Furthermore, we find that activation of transcription by these enhancers involves reorganization of enhancer-promoter contacts that correlates with changes in the occupancy of CTCF and cohesin to enable or confine interactions between these regulatory sequences. Analysis of cohesin-mediated interactions obtained by HiChIP allows us to visualize intermediate and final steps in the formation of new loops that we interpret as evidence of a randomly initiated extrusion process in the establishment of new contacts between regulatory sequences. The results offer a comprehensive and detailed view of how the hESC genome coordinates a complex transcriptional response to environmental stress.
RESULTS
Temperature stress induces global changes in transcription in hESCs
To gain insights into the changes in gene expression that accompany the response of hESCs to temperature stress, we incubated H9 hESCs at 43 °C for 60 min and then examined alterations in nascent transcription using EU-seq (Table S1) (Paulsen et al., 2014). Many genes (n=2506) are upregulated by at least 2-fold in response to temperature stress with respect to the normal growing temperature, and 1610 are downregulated (Figures 1A and1B). To examine the mechanisms underlying these changes in gene expression, we first performed ChIP-seq in control and temperature-stressed H9 hESCs using antibodies to H3K4me1, H3K4me3, H3K27ac, H3K27me3, and RNAPII. We also performed ChIP-seq with antibodies to CTCF, RAD21, and ZNF143, which co-localizes extensively with CTCF and cohesin (Bailey et al., 2015), AP-1, the pluripotency factors NANOG, OCT4, and KLF4, and the PRC1 complex protein RING1B (Table S1). To precisely define the location of enhancers in H9 hESCs, we performed ATAC-seq using the FastATAC procedure (Corces et al., 2016) and separated reads in the<115 bp range from those in the 180–247 bp range in order to independently map transcription factor binding sites (Tn5 hypersensitive sites, THSSs) and nucleosomes, respectively (Table S1) (Schep et al., 2015). We defined enhancers by identifying sites containing ATAC-seq THSSs, H3K4me1 and various levels of H3K27ac but lacking H3K4me3. We then compared enhancers between control and temperature-stressed cells based on the levels of H3K27ac as a measureof enhancer activity. First, we identified sites in the genome where levels of H3K27ac are altered by at least 3-fold (p≤0.05) between control and heat shocked cells. Using this approach, we identified 16,942 enhancers that are enriched by at least 3-fold in H3K27ac in control cells and 5,839 enriched by at least 3-fold in heat shocked cells (Figure 1C). Of the rest of the 61,192 enhancers identified, 5,647 show a statistically significant but less than 3-fold alteration in the levels of H3K27ac and were not considered in subsequent analyses.
Figure 1. Alterations of transcription in response to temperature stress correlate with changes of transcription factors at enhancers.

(A) MAplot of nascent transcript FPKM values for all Refseq annotated genes determined by EU-seq. Pink dots: Heat shock (HS) over control (Ctrl) fold change > 2 (p<0.01) in FPKM; cyan dots: Ctrl over HS fold change > 2 (p<0.01) in FPKM; gray dots: fold change < 2 or p>=0.01.
(B) Heatmaps showing nascent transcript FPKM values determined by EU-seq of up- and down-regulated genes in Ctrl and HS (left). Log2 fold change in FPKM values of HS over Ctrl in the same order (right).
(C) Active H3K27ac enhancers that undergo more than 3-fold changes (p < 0.05) in H3K27ac ChIP-seq signal between Ctrl and HS are highlighted in scatter plots as red and blue dots. Equal and 2-fold changes are marked by black and olive dashed lines.
(D) Heatmaps showing changes induced after temperature stress for H3K27ac, H3K4me1, HSF1, ATAC-seq THSSs, KLF4, c-FOS (AP-1), and RAD21, at HS gained enhancers defined in figure 1C based on ChIP-seq signals ±1 kb of enhancer summit (upper panels). Changes of the same features at Ctrl lost enhancers are shown in the lower panels. RPM: reads per million per 50 bp bin.
(E) Analysis of transcription factor motifs found at enhancers activated after temperature stress. Example of a motif corresponding to the KLF4 pluripotency factor. Average profiles of ATAC-seq signal (<115 bp) at KLF4 motifs show gain of footprints after temperature stress.
(F) Bar graph showing percentages of all active enhancers present in temperature-stressed cells (HS all aE) and enhancers that become active after temperature stress (HS gained aE) overlapping top enriched TF motifs (AP-1: JUN and FOS; KLF4; OCT4::SOX2; CTCF; ZNF143; YY1) and two HS related TFs (SRF and HSF1) (top). Same analysis on enhancers lost after temperature stress (Ctrl lost aE and Ctrl all aE) (bottom). aE: active enhancers in Ctrl or HS.
(G) Average profiles of ChIP-seq signals for FOS, CTCF, ZNF143, RAD21, NIPBL, and WAPL at HSF1+ and HSF1- HS-gained enhancers. RPMPCG: reads per million per 50 bp covered bin. Shading indicates 95% confidence intervals estimated by non-parametric bootstrapping.
(H) Average profiles of ChIP-seq signals for FOS, CTCF, ZNF143, RAD21, NIPBL, and WAPL at enhancers that become decommissioned after temperature stress. RPMPCG: reads per million per 50 bp covered bin.
Enhancers altered in response to temperature stress and identified as described above contain H3K4me1 but lack H3K4me3. However, highly transcribed enhancers have been shown to also contain high levels of H3K4me3 (Henriques et al., 2018). To determine whether this class of enhancers are also involved in the heat shock response, we identified non-TSS sequences containing ATAC-seq THSSs, H3K4me3, and various levels of H3K27ac. Using these criteria, we identified 7,576 HS-gained and 11,232 Ctrl-lost enhancers. Levels of H3K4me3 do not change significantly at these enhancers whereas RAD21 and pluripotency factors increase or decrease in response to temperature stress (Figure S1A). As a comparison, levels of H3K4me3 are low in H3K4me1 enhancers and they do not change in response to temperature stress (Figure S1B)
AP-1 and pluripotency factors are recruited to enhancers activated upon temperature stress
Enhancers that become active or decommissioned in response to temperature stress based on changes in H3K27ac maintain levels of H3K4me1 (Figure 1D). To understand the relationship between activation or decommissioning of enhancers and possible binding of the HSF1 transcription factor, we performed ChIP-seq with antibodies to this protein in control and temperature-stressed cells. We find that enhancers that become inactive after temperature stress lack HSF1 in both control and stressed cells, whereas those that become active can be divided into two clusters. Approximately 11% of enhancers activated after temperature stress contain low levels of HSF1 in control cells, but the amount of this protein increases dramatically after heat shock (Figure 1D, cluster 1). This set of enhancers are enriched in the HSF1 motif. Cluster 2 lacks HSF1 in control cells but the levels increase slightly after heat shock, and these enhancers lack HSF1 motifs (Figure 1D). Interestingly, although the amount of HSF1 in cluster 2 gained enhancers increases only slightly, the THSS ATAC-seq signal increases dramatically, suggesting that other transcription factors bind to these enhancers in temperature-stressed H9 cells (Figure 1D).
To gain further insights into the mechanisms by which enhancers are activated and decommissioned after temperature stress, we attempted to identify putative transcription factors involved in this process by examining binding motifs present at the submits of THSSs. We used ATAC-seq subnucleosomal reads to map footprints at control-lost and heat shock-gained enhancers using Wellington and HINT (Gusmao et al., 2016; Piper et al., 2013). Results show that enhancers activated after heat shock have weak footprints in control cells but show strong footprints, indicative of bound proteins, in temperature-stressed cells. On the other hand, enhancers lost after temperature stress show a concomitant loss of footprint signal in cells exposed to high temperature. This is illustrated in Figure 1E for KLF4, whose binding motif is among the top enriched sequences at enhancers activated after temperature stress, and for other transcription factors in Figure S1C. Enriched motifs at these enhancers include those for AP-1 components JUN and FOS, KLF4, OCT4, and SOX2 (Figure 1F). Motifs for the architectural proteins CTCF, ZNF143, and YY1 are also enriched but the heat shock transcription factor HSF1 is one of the least prevalent motifs at these enhancers. These observations support the idea that activation of most enhancers during heat shock may involve binding of transcription factors other than HSF1. To confirm this hypothesis, we used ChIP-seq data for pluripotency factors and examined their enrichment at these enhancer sequences. A large number of ChIP-seq peaks for the pluripotency factors KLF4, NANOG, and OCT4 are present at active enhancers in control cells and are lost at enhancers that become decommissioned after heat shock. These same pluripotency factors are enriched in heat shock specific enhancers that were not active in control cells (Figures 1D, S1D and S1E). We also performed ChIP-seq with antibodies to FOS in control and heat shocked H9 cells (Figure S1F). Results show an enrichment of this protein at enhancers that become active, and a depletion at enhancers that become decommissioned, after temperature stress (Figures 1D, 1G, and 1H). A similar change in distribution can be observed for other proteins related to enhancer function, including CTCF, RAD21, ZNF143, NIPBL, and WAPL (Figures 1G and1H). AP-1 is depleted in control cells from enhancers that will become active after heat shock, and it becomes enriched at these activated enhancers after temperature stress (Figure 1G). Furthermore, AP-1 is enriched in control cells at enhancers that will become inactive after heat shock but is lost from these enhancers when they become decommissioned (Figure 1H). These results suggest a critical role for AP-1 in enhancer dynamics during temperature stress.
Activation and decommissioning of enhancers in response to temperature stress involves nucleosome remodeling by BRG1
Loss and recruitment of transcription factors to enhancers during the stress response may be accompanied by changes in the organization of the 10-nm chromatin fiber. Analysis of ATAC-seq mono-nucleosome reads indicates that a subset of heat shock gained enhancers show two well-positioned nucleosomes flanking a strong THSSs after temperature-stress, suggesting binding of transcription factors and repositioning of nucleosomes (cluster 1 in Figures 2A and2B). The other half of these enhancers show one positioned nucleosome either at the THSSs where the transcription factor is presumably bound (cluster 3) or at either side of this site (cluster 2). The opposite can be seen for active enhancers in control cells that become inactive after heat shock (Figures 2A and2B). It has been previously shown that HSF1 interacts in vitro with BRG1, which is the ATPase subunit of SWI/SNF in human cells (Sullivan et al., 2001). To test a possible involvement of BRG1 in remodeling nucleosomes at enhancers involved in the transcriptional response to temperature stress, we performed ChIP-seq with antibodies to this protein (Table S1). Results indicate that BRG1 is already present at low levels in control cells at enhancers that will become active after temperature stress, but BRG1 levels increase dramatically at these enhances after heat shock (Figures 2C and2D). In addition, BRG1 is present in control cells at enhancers that will become decommissioned after temperature stress, which happens concomitantly with a decrease in this protein (Figures 2C and2D). To further test a possible role for nucleosome remodeling by SWI/SNF in the activation and decommissioning of enhancers, we used CRISPR to make a small deletion in exon 1 of the BRG1 gene. We obtained a clone trans-heterozygous for two different alleles of BRG1, one containing a 34 bp deletion and a second one caused by an 8 bp deletion, both of which result in a reading frame shift (Figure S2A) and complete absence of BRG1 protein (Figure S2B). We then performed ATAC-seq on the BRG1 −/− cells (Table S1) and compared the results with those obtained in wildtype cells. Results indicate that control-lost and heat shock-gained enhancers lack positioned nucleosomes around the location of THSSs in both conditions (Figures 2A and2B). Interestingly, the THSSs signal in control cells at enhancers that become activated after heat shock appears the same in wild type and BRG1 −/− cells, suggesting that the presence of transcription factors bound to these enhancers in control cells is not altered by the lack of BRG1 (Figure 2B). However, the increase in THSSs signal at these enhancers in temperature-stressed cells is much higher in wildtype than in BRG1 −/− cells, suggesting that nucleosome remodeling by SWI/SNF is required to attain full occupancy of these enhancers by transcription factors (Figures 2B).
Figure 2. Alterations in enhancer activity induced by temperature stress involve nucleosome remodeling by BRG1.

(A) Repositioning of nucleosomes flanking HS gained enhancers observed in control H9 cells after temperature stress, based on ATAC-seq nucleosomal (180~247bp) read signal at enhancer summits ±1 kb (top). BRG1−/− cells fail to remodel nucleosomes at the same enhancers in after heat shock. Enhancers that become inactive after temperature stress lose positioned nucleosomes in control H9 cells based on ATAC-seq nucleosomal read signal at enhancer summits ±1 kb (bottom). BRG1−/− cells lack well positioned nucleosomes at enhancers lost after temperature stress even in control cells. Subtraction heatmaps included for comparison. RPMPCG: reads per million per covered region in 50 bp bin.
(B) Average profiles of ATAC-seq THSSs and nucleosomes on HS gained and lost enhancers in H9 cells in Ctrl and HS (top) show repositioning of nucleosomes, which is impaired in BRG1−/−H9 cells. Clusters derived by K-means clustering of nucleosomal reads. RPMPCG: reads per million per covered region in 50 bp bin
(C) Distribution of BRG1 ChIP-seq signal at enhancers gained (top) or lost (bottom) after temperature stress; anchors are in the same order as (D). Subtraction heatmaps included. RPMPCG: reads per million per covered region in 50 bp bin
(D) Average profiles of BRG1 ChIP-seq signals on the same anchors as (B). RPMPCG: reads per million per covered region in 50 bp bin
(E) Distribution of H3K27ac ChIP-seq signal at enhancers gained (top) or lost (bottom) in control and temperature stressed wild type and BRG1 KO cells; anchors are in the same order as (B). RPMPCG: reads per million per covered region in 50 bp bin.
Activation or decommissioning of enhancers during the response of hESCs to temperature stress is also accompanied by changes in H3K27ac. To examine whether these changes take place up- or down-stream of transcription factor recruitment and nucleosome remodeling, we performed ChIP-seq with antibodies to H3K27ac in wildtype and BRG1 −/− cells. Results show that enhancers normally activated after heat shock fail to accumulate increased levels of H3K27ac in the absence of BRG1, suggesting that acetylation of H3K27 takes place after nucleosome remodeling by SWI/SNF (Figure 2E).
The cohesin-mediated 3D interactome of H9 hESCs
To explore the relationship between the observed changes in transcription and enhancer function in response to temperature stress and possible alterations in 3D genome organization, we first examined the ground state of the 3D interactome in H9 hESCs growing at 37 °C using HiChIP (Mumbach et al., 2016). Since cohesin is involved in many of the interactions between enhancers and promoters we first performed HiChIP using antibodies to RAD21. We obtained 4 biological replicates with high correlation in raw signals (Pearson cor >= 0.97) (Figure S2C, Table S2) and in significant interactions (Figure S2E). We therefore pooled all 3 replicates for subsequent analyses and determined significant interactions taking place at more than 20 kb, requiring an FDR≤0.01 and PET number ≥10. Using this approach, we obtained 97,351 significant interactions mediated by RAD21 in H9 hESCs with a resolution of 1 kb. CTCF motif orientation bias in RAD21-mediated loops identified by HiChIP is similar to that observed by CTCF Chia-PET (Figures S2F and S2G), with ca 70% loops taking place between CTCF sites in convergent orientation (F-R) and less than 30% of the loops between motifs oriented in tandem (F-F or R-R).
Visualization of data obtained in control cells at 1 kb resolution shows that RAD21 HiChIP captures chromatin loops between CTCF/cohesin sites at a higher signal-to-noise ratio than Hi-C and reveals fine structures that appear as uniform interactions in Hi-C maps (Figure 3A). Analysis of all 97,351 significant interactions mediated by RAD21 in control H9 hESCs indicates that approximately 30% of loops take place between sites co-occupied by CTCF and RAD21 (Figures 3B and S2H). Approximately 78% of all loops detected by RAD21 HiChIP contain this protein in both anchors and an additional 22% contain RAD21 in one anchor (Figures 3B and S2H and S3A). Of the loops containing RAD21 at both anchors and CTCF at only one, the second anchor contains either an enhancer or a promoter, and these loops extend over distances of up to 1 Mb (Figure S3B). RAD21 loops containing CTCF at both anchors not always have a recognizable CTCF motif, which could be due to a deviation from the consensus or recruitment of CTCF by a different DNA binding protein (Figure S3C). Sites in the genome with a recognizable CTCF motif and bound by CTCF protein contain the motif in a convergent forward-reverse orientation or tandem forward-forward or reverse-reverse orientation, with the former mediating more frequent interactions and over longer distances (Figure S3D). RAD21 loops with tandemly oriented CTCF motifs exhibit active chromatin features such as H3K27ac, ATAC-seq accessibility, and active nascent transcription, suggesting that these loops primarily represent enhancer-promoter (E-P) interactions in the same direction as CTCF motifs (Figures S3E and S3F). Other RAD21-mediated loops lack CTCF and are associated with active regulatory elements containing RNAPII and/or H3K27ac. Loop anchors are adjacent to actively transcribed genes based on results from EU-seq (Figure S3E; see specific example in Figure 3A).
Figure 3. High resolution analysis of cohesin-mediated interactions in H9 hESCs.

(A) RAD21 HiChIP captures CTCF loops at higher signal-to-noise ratio than Hi-C. Juicebox views of RAD21 HiChIP on an example region (chr12: 7,689,000–7,810,000). Lower panels depict arc views of significant interactions from RAD21 HiChIP, IGV views of RAD21, CTCF, H3K27ac, and RNAPII ChIP-seq, and nascent transcripts obtained by EU-seq. RAD21 loops lacking CTCF mediate contacts between enhancers and promoters. Red arrows: forward CTCF motifs; green arrows: reverse CTCF motifs. HiChIP data are read-normalized.
(B-F) E: enhancer, P: promoter. C: CTCF binding sites. aE: active enhancers; iE: primed enhancers; pE: poised enhancers; aP: active promoters; pP: poised promoters.
(B) Quantification of protein occupancy on E-P loop anchors from RAD21 HiChIP shows most E-P loops contain NANOG in both anchors; OCT4, KLF4, CTCF, ZNF143, AP-1 and RNAPII in at least one anchor, but RING1B is depleted in both anchors (left). Clustering of E-P loops by protein co-occupancy on both/one/neither loop anchors shows that E-P loops with RNAPII on only one anchor are enriched in NANOG and CTCF/ZNF143 on the other anchors.
(C) Pipeline for obtaining significant interactions from RAD21 HiChIP in H9 cells (left). Bar plot shows 70% of RAD21 loops take place between regulatory elements and CTCF binding sites (middle). Of these loops, E-E, E-P loops are most frequent (right). Circle size is scaled with E/P/C number, stick thickness is scaled with loop number.
(D) Summary of different categories of E-P loops shows that most RAD21 mediated loops take place between active enhancers and promoters. n is number of loops observed in each category. Color scale: median contact strengths based on RAD21 HiChIP. Statistical significance p values from random permutation test of distinct types of E-P loop numbers over genome background are also shown.
(E) Summary of number of active promoters interacting with active, primed and poised enhancers, respectively and same analysis on poised promoters. ** p<0.005 by random permutation test.
(F) Average profile of ChIP-seq signal on all promoters (left columns) and enhancers (right columns) in H9 cells. RING1B profiles based on ChIP-seq are shown as a control of chromatin states of enhancers and promoters. RPM: reads per million per 50 bp coverage.
See also Figures S2 and S3, and Table S2
To further explore the significance of RAD21-mediated interactions in the context of the state of different regulatory sequences, we subdivided these sequences into active enhancers (aE) if they contain H3K4me1 and H3K27ac, poised enhancers (pE) if they contain H3K4me1 and H3K27me3, and primed (iE) if they contain H3K4me1 but lack H3K27ac (Shlyueva et al., 2014). A very large fraction of RAD21 loops contain NANOG in both anchors (Figure 3B, left panel). Clustering of all interactions detected in RAD21 HiChIP in the context of the location of different proteins gives a detailed view of their co-occupancy at the anchors of RAD21 loops (Figure 3B, right panel). Many interactions mediated by RAD21 take place between active enhancers and promoters (Figure 3C). However, a substantial number of RAD21 loops mediate contacts between active enhancers and poised promoters before transcription activation, and ca 2,000 RAD21 loops form interactions between promoters of transcriptionally active genes and primed enhancers that lack H3K27ac (Figure 3D). The same data analyzed from a different perspective indicates that active and inactive enhancers contact ca 4,500 and ca 1,000 active promoters, respectively, whereas the number of poised promoters contacted by the same two types of enhancers is below 300 (Figure 3E). RAD21 and CTCF are enriched in loop anchors containing active and poised promoters with respect to the same sequences when not present in loop anchors. These two proteins are also enriched in loops formed by active and primed, but not poised, enhancers with respect to the same sequences when not present in loops (Figure 3F). KLF4 (Figure 3F), OCT4, NANOG, and ZNF143 (Figure S2I) are enriched at active and primed enhancers when present in loops but also significantly enriched in active enhancers that do not form RAD21-mediated loops. These observations suggest that enhancers can show signs of activation i.e. Tn5 accessibility, RNAPII occupancy and TF binding whether they establish contacts with a promoter or not. These observations set the stage to now investigate the basis for the transcriptional changes taking place in H9 hESCs in response to temperature stress and their relationship to alterations in 3D organization.
RAD21-mediated interactions between enhancers and promoters in temperature-stressed hESCs
To study changes in the 3D interactome during temperature stress, we next performed HiChIP using RAD21 antibodies in H9 cells subjected to heat shock. We obtained 4 highly correlated replicates (Figure S2D) and pooled the data for subsequent analysis (Table S2). Significant interactions were called using Mango (Figure S2E). We then examined alterations in RAD21-mediated interactions around genes found to be up- or down-regulated in response to temperature stress. As an example, the TMEM158 gene, which is down-regulated after heat shock, contacts a RAD21 site adjacent to an active enhancer containing H3K27ac in control cells. After heat shock, levels of RAD21 decrease slightly and the interaction identified by Mango is lost, while levels of H3K27ac at the adjacent enhancer decrease dramatically and nascent transcription of the TMEM158 gene is markedly reduced (Figure 4A). In contrast, HES1, an up-regulated gene, does not have significant interactions with distant sequences in control cells. After temperature stress, the HES1 gene undergoes strong interactions with a presumed enhancer where the levels of H3K27ac increase dramatically but the levels of RAD21 remain the same (Figure 4B). In both examples, activation of transcription correlates with changes in histone modifications and the formation of a loop between the enhancer and promoter. To explore the generality of these observations, we examined RAD21-mediated interactions that are enriched in either control or temperature stress conditions by at least −2-fold in PET number and FDR cut-off of 0.1 (Figure 4C). Results show that RAD21 loops enriched in control cells are mainly involved in mediating interactions among enhancers whose H3K27ac levels decrease, and promoters that become transcriptionally inactive, after temperature stress. RAD21 loops enriched in heat shocked cells mediate interactions among enhancers enriched in H3K27ac and promoters of genes that become transcriptionally active during temperature stress (Figure S4B). Taken together, the results suggest a correlation between increased gene transcription, higher levels of H3K27ac at enhancers and promoters, and more frequent interactions among these sequences in heat shocked cells, with the opposite being true for genes down-regulated after temperature stress.
Figure 4. RAD21-mediates interactions between enhancers and promoters in temperature-stressed hESCs.

(A) Juicebox view of read-normalized Ctrl and HS RAD21 HiChIP in an example region (chr3: 45,217,312 – 45,341,523) containing the HS-repressed gene TMEM158, which forms a significant loop (blue arc) with an H3K27ac-containing enhancer adjacent to a RAD21 site based on ChIP-seq (tracks below) in Ctrl. Loss of RAD21 and H3K27ac at this enhancer correlates with loss of loops and transcription from TMEM158 in HS. Orange vertical stripe highlights TMEM158 genic region; blue vertical stripe highlights region containing enhancer whose interaction is lost after temperature stress.
(B) Juicebox view of read-normalized Ctrl and HS RAD21 HiChIP on an example region (chr3:194,050,118 – 194,142,554) containing the HS-induced gene HES1, which forms a new induced loop (red arc) with a nearby HS gained RAD21 site on an H3K27ac enhancer. Orange vertical stripe highlights HES1 genic region; blue vertical stripe highlights its gained interacting enhancers in HS.
(C) HS induced loss and gain of RAD21 mediated loops (significance -fdr <0.1 and fold change >2) observed in RAD21 HiChIP are shown in MAplot as red and blue dots. Equal and 2-fold changes between Ctrl and HS are marked as yellow and blue dashed lines.
(D) Contact frequency changes at temperature stress-induced or decommissioned enhancers correlate with occupancy changes in pluripotency factors, AP-1, and HSF1. HS dynamic enhancers are divided by contact frequency log2FC between Ctrl and HS based on RAD21 HiChIP, and protein occupancy log2FC in each group are averaged and compared between Ctrl and HS based on ChIP-seq signal. Black arrows indicate increase of HS over Ctrl log2FC in contact frequency in HiChIP.
(E) Transcription changes of genes interacting with enhancers induced or decommissioned after temperature stress correlate with occupancy changes in pluripotency factors, AP-1, and HSF1at these enhancers. Target genes of dynamic enhancers were determined using Rad21 HiChIP and divided into ten groups according to transcriptional changes measured as log2 between Ctrl and HS based on EU-seq (y-axis). Protein occupancy at these enhancers in each group are averaged and compared as the log2FC between Ctrl and HS based on ChIP-seq signal (x-axis).
(F) Heatmaps comparing changes in virtual 4C view of Rad21 HiChIP and H3K4me1 HiChIP signals between all HS-induced gene promoters and HS gained activated enhancers (top) and HS-repressed gene promoters and HS decommissioned enhancers (bottom). Viewpoint: promoters. Dynamic enhancers are ranked by distance from upstream to downstream of promoters. Only interactions within 500 kb are plotted. Norm enrich score: VC_SQRT normalized enrichment score.
Changes in transcription factor occupancy underlie alterations in enhancer activity in response to temperature stress
To further explore the involvement of AP-1, pluripotency, and HSF1 transcription factors in gene expression after temperature stress, we examined changes in interactions at enhancers occupied by these factors. We examined changes in contact strength, defined as the normalized enrichment score of contact frequency (see STAR methods) of RAD21 HiChIP describing a specific interaction, at sites occupied by each of the factors described above. We find a strong correlation between the increased contact strength obtained from RAD21 HiChIP and the level of occupancy of pluripotency factors, AP-1, or HSF1, determined from ChIP-seq (Figure 4D). These results suggest that the amount of a transcription factor recruited to a specific enhancer correlates with the frequency or strength of interactions mediated by this enhancer. Furthermore, we find a strong positive correlation between the increase in transcription factor occupancy at enhancers that become activated after temperature stress and upregulation of expression of genes contacted by these enhancers (Figure 4E).
To further explore the role of enhancer-promoter contacts in the activation of transcription of genes induced by temperature stress, we performed HiChIP using antibodies to H3K4me1 (see Table S2 for details on read number and quality control steps), since the levels of this modification remain constant at enhancers during the heat shock response (Figure 1D, S1A, and S1B). An example of H3K4me1-mediated interactions in the regions surrounding the TMEM58 and HES1 genes is shown in Figures S4D and S4E; these are the same regions shown in Figures 4A and4B displaying RAD21interactions. We find that H3K4me1-mediated interactions between control-lost and heat shock-gained enhancers and their target genes decrease or increase, respectively, following the same pattern as RAD21-mediated interactions (Figure S4C). Analysis of RAD21 and H3K4me1-mediated interactions between promoters of up or downregulated genes and enhancers gained or lost after heat shock, respectively, shows that enhancer activation correlates with an increase in contacts with promoters, whereas enhancer decommissioning is accompanied by a decrease of interactions with target promoters (Figure 4F). A similar analysis of changes in ChIP-seq signals indicates that, whereas H3K27ac levels increase at HS-gained and decrease at Ctrl-lost enhancers, levels of H3K4me1 remain the same (Figure S4F). These results support the conclusion that changes in transcription are accompanied by changes in interactions between activated or decommissioned enhancers and promoters. Similarly, interactions mediated by enhancers containing high levels of H3K4me3 (Figure S1A) also increase at HS-gained and decrease at Ctrl-lost enhancers based on analyses of RAD21 and H3K4me1 HiChIP data (Figure S4G).
Inhibition of HSF1 function interferes with changes in 3D chromatin organization in temperature stressed hESCs
To examine the role of HSF1 in the stress response, we first test whether it is required for the alteration of 3D interactions mediated by RAD21. To this end, we obtained two RAD21 HiChIP replicates (Table S2) in heat shocked H9 hESCs treated with a combination of the kinase inhibitors SP600125, U0126, and SB239063 (HSKI), which inhibit phosphorylation of HSF1 and abrogate the heat shock response (Byun et al., 2013). In the presence of the kinase inhibitors, interactions mediated by enhancers that become decommissioned decrease slightly after temperature stress (Figure S5A, top), whereas novel interactions involving activated enhancers fail to form (Figure S5A, bottom). We then examined the generality of this observation by analyzing the enrichment of all RAD21-mediated interactions in a 200 kb region around RAD21 loop anchors present at enhancers in control and temperature stress cells in the absence and presence of the inhibitor. In the presence of the kinase inhibitors, there is a depletion of heat shock-enriched RAD21 loops, suggesting a failure to undergo changes in 3D architecture in response to temperature stress in the absence of functional HSF1 (Figures 5A and S5B). The depletion of RAD21-mediated interactions takes place at sites with both high or low levels of HSF1 (Figure S5C). However, loops that normally remain stable after temperature stress are not affected by the presence of kinase inhibitors (Figures 5A). These observations also suggest that the alterations in RAD21-mediated interactions after heat shock are not a consequence of the change in temperature per se, since they fail to occur when HSF1 is not phosphorylated, despite the temperature increase. Interestingly, control-enriched cohesin loops still decrease after heat shock in the presence of inhibitor, as they do in control (Figures 5A, S5B, and S5D). This agrees with results showing that downregulation of transcription after temperature stress occurs by mechanisms independent of HSF1 (Mahat et al., 2016).
Figure 5. Pluripotency factors participate in 3D interactions during the transcriptional response to temperature stress in hESCs.

(A) Comparison of RAD21 HiChIP in control (Ctrl), temperature stressed (HS), and temperature stressed in the presence of kinase inhibitors (HSKI) cells. APA metaplots derived from RAD21 HiChIP show that inhibition of HSF1 phosphorylation by kinase inhibitors reverses the temperature stress-induced loss and gain of RAD21-mediated loops. Color scale: blue < white < red. Ctrl or HS enriched RAD21 loop anchors ± 100 kb were used for analysis.
(B-H) SE: super-enhancers; TE: typical enhancers; PD: polycomb domains. (B) Browser view of H3K4me1, RAD21, and OCT4 HiChIP in an example region (chr14: 89.4–89.65 Mb) containing two SEs, together with tracks showing ChIP-seq signals for H3K27ac, OCT4, RAD21, CTCF and ZNF143. Reads are normalized between different HiChIP samples for visualization. Yellow vertical stripes highlight SEs; blue vertical stripes highlight other enhancers. Black circles highlight interactions between SEs; blue circles highlight interactions between SE and TE.
(C) Browser view of H3K4me1, RAD21 and OCT4 HiChIP on a region adjacent to the HOXD locus (chr2: 171.5–176.5 Mb) containing three PDs, together with tracks showing ChIP-seq signals for H3K27ac, OCT4, RAD21, CTCF and ZNF143. Reads are normalized between different HiChIP samples for visualization. Blue vertical stripes highlight PDs. Black circles highlight interactions between SEs; blue circles highlight interactions between an SE and a TE.
(D) Analysis of OCT4, NANOG and KLF4 HiChIP results. APA metaplots show that HiChIP data for this pluripotency factors recapitulate loss of cohesin loops associated with Ctrl lost enhancers (upper row) and gain of cohesin loops associated with HS gained enhancers (lower row) in temperature-stressed cells. Color scale: blue < white < red. Loop anchors ± 100 kb were used for analysis.
(E) Heatmaps comparing OCT4 HiChIP contact strengths in different categories of enhancer-promoter loops. Abbreviations are the same as in Figure 2D.
(F) Heatmaps comparing RAD21, OCT4, and H3K4me1 HiChIP contact strengths between PD, SE and TE. HiChIP reads are normalized as inputs for analysis.
(G) Subtraction heatmaps of RAD21, OCT4, and H3K4me1 HiChIP contact strengths between PD, SE and TE. Temperature stress induces dramatic increase of contact strength between SE and PD, which is better captured by OCT4 HiChIP (left) and H3K4me1 HiChIP (right).
(H) Boxplot comparing transcription increase of genes gaining SE versus genes gaining TE in HS. Gain of enhancers based on HiChIP differential loops. Increase of transcription calculated by subtraction of HS FPKM minus Ctrl FPKM based on EU-seq. p value obtained by Wilcoxon test.
Pluripotency factors mediate 3D interactions during the transcriptional response to temperature stress in hESCs
To examine the role of pluripotency factors in the reorganization of enhancer-promoter interactions during the heat shock response we performed HiChIP with antibodies to NANOG, OCT4, and KLF4 (Table S2). At genomic regions where they colocalize, HiChIP results for RAD21, H3K4me1 and pluripotency factors uncover the same interactions between various types of regulatory sequences. An example of interactions between two super-enhancers located in the FOXN3 gene is shown in Figure 5B, where sites containing OCT4, RAD21, and H3K4me1 in control cells can be seen to interact in each of the three datasets. Similarly, the HOXD locus, which is repressed in hESCs by the Polycomb complex but is occupied by pluripotency factors, can be seen to interact with other Pc domains in the RAD21 and OCT4 HiChIP datasets (Figure 5C). Similar results were obtained using NANOG and KLF4 HiChIP experiments (Figures S5E and S5F). When we compare the enrichment of loop anchors mediating interactions between sequences containing pluripotency factors at control-enriched RAD21 loops, we find that interactions between genomic sites for NANOG, OCT4, and KLF4, are enriched at the same anchors as control-specific RAD21 interactions, and that they decrease at these sites in cells undergoing temperature stress (Figure 5D). The opposite is the case for heat shock-enriched RAD21 loops, which are depleted of pluripotency factor-mediated interactions in control cells but become enriched in temperature stressed cells (Figure 5D). Analysis of enhancer-promoter interactions identified by OCT4 HiChIP indicates that this protein is enriched in interactions between poised promoters and active enhancers in control cells (Figure 5E). Furthermore, OCT4 is also enriched in interactions between Polycomb domains and between super-enhancers in control cells (Figure 5F). These interactions are also enriched in H3K4me1 HiChIP (Figure 5F). In H9 hESCs undergoing temperature stress, OCT4-mediated interactions are also highly enriched for contacts between Polycomb domains and super-enhancers (Figure 5G). Interestingly, the expression of heat shock induced genes contacting super-enhancers is higher than that of genes contacting normal enhancers, suggesting that pluripotency factors at super-enhancers play a distinct functional role by facilitating interactions with the promoters of genes that need to be transcribed at high levels in temperature-stressed cells (Figure 5H).
Changes of insulated neighborhoods in hESCs in response to temperature stress
CTCF occupancy is altered at activated and decommissioned enhancers concomitantly with the up- or down-regulation of gene expression in response to temperature stress (Figures S1C and S1D). CTCF can affect transcription by either facilitating interactions between regulatory sequences and their cognate promoters or by forming loops that establish insulated neighborhoods (Hnisz et al., 2016). To analyze this aspect of CTCF function, we first identified all CTCF sites in the genome located between enhancers and promoters whose interactions are altered by heat shock. To this end, we performed CTCF ChIP-Nexus (Table S1) (He et al., 2015b) to precisely map the location and orientation of CTCF sites in the genome of control and heat shocked H9 hESCs. Quantitative analysis of changes in CTCF sites using MAnorm (Shao et al., 2012) shows the existence of 808 CTCF sites that significantly decrease, and 1140 sites that significantly increase, in heat shocked versus control cells by more than 2-fold (Figure S6A). Heatmaps showing differences in CTCF binding at these sites are shown in Figure 6A. However, levels of RAD21 at these sites remain the same between the two conditions (Figure S6B). To explore potential mechanisms by which CTCF is recruited to new sites in the genome of temperature stressed cells, we plotted subnucleosomal- and nucleosomal-size reads from ATAC-seq experiments with respect to the CTCF motif at sites gained during the response to temperature stress. We find that sites in the genome where CTCF is recruited after heat shock are already flanked by well positioned nucleosomes in control cells (Figure S6C). Cells depleted of BRG1 lack positioned nucleosomes around these sites, and fail to recruit CTCF after temperature stress (Figure S6C). We then performed HiChIP using antibodies to CTCF in control and heat shocked cells (Table S2). An example of changes in CTCF-mediated interactions is shown in Figure S6D, where one interaction decreases and a second one increases after heat shock. A representative example of the similarities between RAD21 and CTCF HiChIP is displayed in Figure S6E.
Figure 6. Changes in CTCF occupancy during temperature stress regulate enhancer-promoter interactions.

(A) Heatmaps comparing CTCF ChIP-seq signals in control and temperature-stressed cells at Ctrl-enriched CTCF sites between HS gained aE-P loops (top). Same analysis at HS-enriched CTCF sites between HS lost aE-P loops (bottom). CTCF summits ± 1 kb are analyzed. Subtraction of HS minus Ctrl signals also included. RPM: reads per million per 50 bp bin.
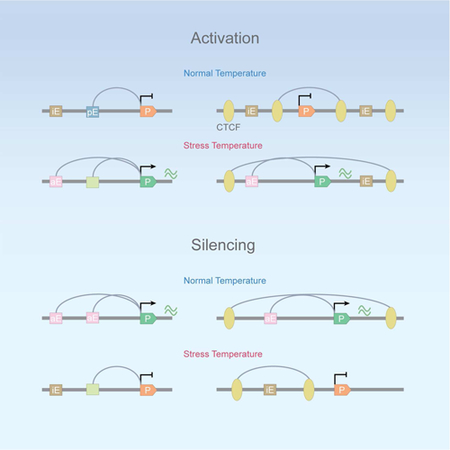
(B) Diagram showing HS-induced genes gain HS activated enhancers when CTCF loops insulating them from nearby enhancers are lost due to loss of CTCF binding on both anchors, and larger CTCF loops are formed (top). CTCF sites were identified from CTCF ChIP-Nexus in control and temperature-stressed cells. CTCF loops identified by RAD21 and CTCF HiChIP. sE, inactive enhancers.
(C) Metaplots of HS over Ctrl log2 FC in CTCF HiChIP showing loss of CTCF loops insulating HS-induced genes and their enhancers and gain of larger CTCF loops in HS as shown in Figure 7B. Similar effects observed in RAD21 HiChIP. CTCF motifs are listed for all four anchors to show that bigger CTCF loops form at motifs in F-R convergent orientation. Black circles highlight lost and gained CTCF loops. Dashed rectangles in RAD21 HiChIP indicate higher E-P interactions outside Ctrl CTCF loops in HS.
(D) Genes repressed after heat shock loose contacts with enhancers when CTCF loops containing the genes and their regulatory sequences are lost, and smaller CTCF loops insulating genes from their enhancers are formed due to increase of CTCF levels at both anchors. CTCF sites were identified from CTCF ChIP-Nexus in control and temperature-stressed cells. CTCF loops identified by RAD21 and CTCF HiChIP. sE, inactive enhancers.
(E) Metaplots of HS over Ctrl log2 FC in CTCF HiChIP showing gain of CTCF loops insulating HS-induced genes and their enhancers and loss of larger CTCF loops enclosing the gene and regulatory sequences in HS as shown in Figure 7B (bottom). Similar effects observed in RAD21 HiChIP. CTCF motifs are listed for all four anchors. Black circles highlight lost and gained CTCF loops. Dashed rectangles in RAD21 HiChIP indicate lower E-P interactions outside Ctrl CTCF loops in HS.
(F) Diagram showing HS-induced changes in CTCF loops accompanied by changes in CTCF occupancy at only one anchor of CTCF loops. HS-lost CTCF binding at one anchor to allow E-P loops in temperature-stressed cells. sE, inactive enhancers.
(G) Metaplots of HS over Ctrl log2 FC in CTCF HiChIP to visualize CTCF loop rearrangements as shown in Figure 7E (top). Similar changes are observed in RAD21 HiChIP. CTCF motifs are listed for all three anchors. Blue and red arrowheads highlight lost and gained CTCF loops.
(H) Diagram showing HS-induced changes in CTCF loops accompanied by changes in CTCF occupancy at only one anchor of CTCF loops. HS-gained CTCF binding at one anchor to insulate E-P loops after heat shock. sE, inactive enhancers.
(I) Metaplots of HS over Ctrl log2 FC in CTCF HiChIP to visualize CTCF loop rearrangements as shown in Figure 7E (bottom). Similar changes are observed in RAD21 HiChIP. CTCF motifs are listed for all three anchors. Blue and red arrowheads highlight lost and gained CTCF loops.
We first used CTCF HiChIP data to examine changes in CTCF contacts in the regions surrounding up- and down-regulated genes. We find that interactions between CTCF anchors present at the bases of loops containing downregulated genes increase after heat shock whereas loops containing upregulated genes show a decrease in interactions between their anchors (Figure S6F). Loops in these two categories can be further sub-divided based on the location of enhancers and promoters with respect to the loop anchors. One group involves cases in which CTCF sites with differential occupancy between control and heat shock cells are located between enhancers and promoters of genes whose expression is altered. In the case of genes whose transcription is up-regulated by temperature stress, the amount of CTCF at these sites is significantly reduced after heat shock (Figure 6B). This arrangement suggests that the enhancer and promoter of control-specific genes are in different CTCF loops in control cells, whereas these two sequences are present in one larger loop creating an insulated neighborhood in heat shocked cells that allows expression of the gene (Figure 6B). To test this hypothesis, we examined all CTCF loops containing either an enhancer or a promoter of a gene up-regulated by heat shock and we determined alterations in interactions between the CTCF anchors of these loops during the heat shock response. We find that interactions between these anchors decrease after heat shock, whereas interactions between more distant flanking CTCF sites increase (Figure 6C). The same pattern can be observed when using RAD21 HiChIP data (Figure 6C, middle panel). These results suggest that CTCF sites present in control cells may insulate enhancer-promoter interactions, and the insulating effect decreases in heat shocked cells because of decreased CTCF levels, resulting in the up-regulation of transcription. It has been hypothesized that formation of new CTCF loops involves cohesin-mediated loop extrusion that requires loading of cohesin by NIPBL (Schwarzer et al., 2017). To test whether this is the case during the heat shock response, we used a lentivirus vector to downregulate the expression of NIPBL (Figure S6G). We then performed CTCF HiChIP in siNIPBL cells (Table S2). We find that new loops encompassing the active enhancers and target genes are not formed in cells lacking NIPBL, supporting the requirement of continuous cohesin loading for the establishment of these new interactions (Figure 6C, bottom). We then examined genes expressed in control cells and down-regulated after heat shock. In this case, CTCF occupancy at sites located between active enhancers and promoters in control cells increases after heat shock (Figure 6D). Consequently, CTCF-mediated interactions between the two anchors now flanking the enhancers or the promoters increases after heat shock, resulting in the formation of loops that separate the enhancer from the promoter and an increase of insulation strength in temperature-stressed cells (Figure 6E). These interaction changes can also be seen in RAD21 HiChIP data (Figure 6E, middle panel), and the new loops also fail to form in cells depleted of NIPBL (Figure 6E, bottom).
The second group of genes whose expression is altered during heat shock because of differential CTCF occupancy displays a different arrangement of enhancers and promoters relative to CTCF sites and their interactions. In this group, the enhancers or promoters of heat shock up-regulated genes are located inside a loop in control cells, whereas the promoters or enhancers, respectively, are located outside this loop (Figure 6F). During temperature stress, occupancy at one of the two CTCF site anchors decreases, whereas occupancy at a distal site increases. This results in the decrease of interactions mediated by the original anchor and an increase of contacts by the new anchor, resulting in the formation of a larger loop that now encloses the enhancer and promoter of the gene (Figure 6G). The same outcome can be observed in RAD21 HiChIP data (figure 6G, middle panel), but new loops fail to form in cells in which NIPBL is down-regulated (Figure 6G, bottom). On the other hand, genes whose expression decreases after heat shock have their enhancers and promoters within a large CTCF loop in control cells (Figure 6H). After heat shock, occupancy at an internal CTCF site increases, leading to the formation of a new, smaller loop that separates the enhancer from the promotor (Figure 6I). This result can be observed using both CTCF and RAD21 HiChIP data, and these new loops also fail to form in cells depleted of NIPBL (Figure 6I, bottom).
To confirm the effect of changes in CTCF loops on enhancer-promoter interactions, we used RAD21 and H3K4me1 HiChIP data. Loss of CTCF occupancy results in increased interactions between sequences located inside and outside of the original CTCF loops formed between anchors arranged in a convergent forward-reverse orientation (Figure 7A, left panel; see arrowheads pointing to red signal). This is also the case when only one of the CTCF sites is lost (tail site) from a pair of sites arranged in a forward-forward or reverse-reverse orientations (Figure 7A, middle and right panels). The same results can be observed using H3K4me1 HiChIP (Figure 7B). Depletion of NIPBL, which results in the absence of RAD21-mediated loops, also causes the disappearance of enhancer-promoter interactions observed in H3K4me1 HiChIP (Figure 7C). These results suggest that orchestration of the transcriptional changes taking place in response to temperature stress requires CTCF to perform a role as insulator to allow or inhibit contacts between enhancers and their cognate promoters in addition to its role as a facilitator of enhancer promoter interactions
Figure 7. Changes in CTCF- and RAD21-mediated contacts suggests loop extrusion as a mechanism for forming new interactions.

(A) Metaplots of HS over Ctrl log2 FC of RAD21 HiChIP signal showing HS-induced increase in interactions escaping lost CTCF loops on F-R, F- F and R-R anchors.
(B) Metaplots of HS over Ctrl log2 FC of H3K4me1 HiChIP signal showing HS-induced increase in interactions escaping lost CTCF loops on F-R, F- F and R-R anchors.
(C) NIPBL knockdown impairs rearrangements of H3K4me1 interactions found in (B) based on H3K4me1 HiChIP in siNIPBL cells.
(D) Metaplots of RAD21 HiChIP on forward-reverse (F-R, top) and forward-forward (F-F, middle) CTCF loops showing extrusion lines in the same direction as CTCF motifs. The extrusion lines from the left F anchors of F-F loops continue after passing the right F anchor, whereas the line from left F anchors of F-R loops is greatly reduced after passing the right R anchor. Subtraction of FR from the FF metaplots more clearly shows the differences in the extrusion lines between these two types of loops, indicating the preferential pause of the cohesin complex at F-R sites.
(E) (Top) Metaplot of actual RAD21 HiChIP data on all F-R CTCF loops showing increase of signal on extrusion lines from the left F anchor to the right R anchor. Regions between F and R anchors were scaled to 10 bins. A: Forward CTCF anchor. B-F: non-CTCF bins inside loops used as control random extrusion start sites in Figure 7F.
(Middle) Metaplot of simulated RAD21 HiChIP signals based on the assumption that RAD21 starts extrusion from random sites inside F-R CTCF loops, showing increased signal on the extrusion lines from the left F to the right R anchor. Red dashed line indicates the same line in Figure 7G.
(Bottom) Metaplot of simulated RAD21 HiChIP signals based on the assumption that RAD21 starts extrusion from CTCF sites of F-R CTCF loops. Results show constant signal on the extrusion lines from the left F to the right R anchors. Green dashed line indicates the same line in Figure 7G.
(F) Slopes of signals of actual Hi-C enrichment on lines starting at CTCF forward motif and non-CTCF anchors A, B, C, D, E, F shown in Figure 7E (top). Same colors under A-F in Figure 7E (top) diagrams were used for lines starting at A-F locus.
(G) Slopes of signals from actual and simulated Hi-C enrichment on lines starting at CTCF forward motif. Red dotted lines were labeled in Figure 7E (middle) representing slopes anchoring the CTCF site in random-starting simulation matrix, green dotted lines were labeled in Figure 7E (bottom) representing slopes anchoring the CTCF site in CTCF-starting simulation matrix.
Analysis of changes in CTCF- and RAD21-mediated contacts suggests random loop extrusion as a mechanism for forming new interactions
Establishment of interactions between CTCF sites are thought to require a cohesin-mediated loop extrusion mechanism. We tested whether evidence for loop extrusion can be obtained from the RAD21 and CTCF HiChIP data. RAD21 HiChIP shows an intense “line” of interactions leading up to the signal representing interactions between the loop anchors (see arrowheads in Figure S7A). These represent interactions between the CTCF anchor and every locus interior to the loop domain, which is likely indicative of the HiChIP capturing extrusion events at different stages in different cells of the population. We further examined the line representing these interactions, hereafter called extrusion lines, by categorizing CTCF loops into those occurring in forward-reverse, forward-forward, or reverse-reverse orientation. When CTCF sites are in forward-reverse orientation, the extrusion lines converge at the point representing interactions between the left (forward oriented) and right (reverse oriented) CTCF anchors (Figure 7D and S7A). When the two CTCF sites are in forward-forward or reverse-reverse orientation, the extrusion line no longer converges as markedly, but predominately follows the orientation of CTCF (Figure 7D and S7A). Interestingly, a line of weaker signal is oriented opposite to the direction of the CTCF motif, indicating that some amount of extrusion can bypass CTCF motifs in the reverse convergent orientation, which is likely how non-convergent CTCF loops form. In comparing these loop categories, we also noticed a stronger signal past the loop in the forward-forward orientation, indicating that forward-forward loops represent weaker boundaries, allowing extrusion to continue past the loop anchor better than forward-reverse loops (Figure 7D bottom panel).
It has been proposed that loop extrusion starts at CTCF sites (Nichols and Corces, 2015) or randomly along chromatin, stopping at properly oriented CTCF sites (Fudenberg et al., 2016). The RAD21 HiChIP data could suggest that extrusion starts at CTCF sites, based on the extrusion line starting at each CTCF anchor. To differentiate between these two possibilities, we compared RAD21 and CTCF HiChIP data and found that loops are stronger in CTCF HiChIP, while extrusion lines are stronger in RAD21 HiChIP data (Figure S7B). This indicates that, while RAD21 HiChIP detects stages of loop extrusion, CTCF HiChIP preferentially pulls down completely extruded loops. Interestingly, RAD21 HiChIP shows stronger interaction signal in the interior of the loop domain (Figure S7B) which agrees with random extrusion start sites as described by Fudenberg et al. To further differentiate between the two extrusion models, based on random-starts or CTCF-starts, we created a simple simulation of the interaction signal in each case. In the model where extrusion starts at CTCF sites, all interactions captured at each extrusion time point will lie at the CTCF anchor on one side (Figure S7C), which would contribute to an intense and uniformly constant signal along the extrusion line (Figure 7E, bottom). In contrast, the random-start model predicts that loop extrusion begins at a random point inside the domain and therefore the interaction signal will not always be at CTCF anchors (Figure S7D). After the extrusion complex reaches CTCF on one side, the interaction signal will be seen at CTCF anchors (Figure S7D). Given a population of random started extrusion complexes, signal at the extrusion line should gain intensity as we move away from the diagonal leading up to the loop (Figure 7E middle panel). We examined actual RAD21 HiChiP data and saw that the extrusion line signal gains intensity away from the diagonal as predicted by the random-start model (Figure 7E, top). To quantify this, we calculated the slope of the theoretical extrusion line which runs from the left CTCF anchor (corresponding to bin A in Figure 7E) to the right CTCF anchor. This interaction signal increases as we move away from the diagonal closer to the other anchor (Figure 7F blue line). We compared this slope to the slope of bins that are internal to CTCF loops and found no such increases (Figure 7F, see also Figure 7E bins B-F). We then compared the experimentally derived slope (Figure 7G blue line) to the slope provided by the random-start model (Figure 7G red dashes, corresponding to Figure 7E red dashes) and found high concordance between the two. In contrast, when we plot the slope of the CTCF-start model (Figure 7G green dashes, corresponding to Figure 7E green dashes), we see no concordance. The upward slope of the extrusion line and its concordance with the random-start model indicates that loop-extrusion likely starts randomly within loop domains.
DISCUSSION
The response of cells to environmental stresses takes place over a short time scale, involves changes in the expression of a few thousand genes, and thus can serve as a paradigm to study mechanisms by which cells mount global transcriptional changes of genes located throughout the genome. Results presented here suggest that enhancers that will become active upon temperature stress are either in an inactive primed or poised state in hESCs. Their activation involves an increase in transcription factor binding to enhancers detected by ATAC-seq. Correlated with this event, we also observe nucleosome remodeling involving the BRG1 ATP-dependent chromatin remodeling complex followed by an increase in H3K27ac. Motif enrichment analyses suggest that enhancers that become active after heat shock are enriched for binding sites for AP-1, NANOG, KLF4, OCT4, and HSF1. Analysis of ChIP-seq data confirms the presence low levels of these proteins in control cells at enhancers that will become activated in response to temperature stress. Activation of these enhancers after temperature elevation involves increased recruitment, whereas decommissioning of active enhancers correlates with lower levels of pluripotency factors and AP-1. Contrary to the dramatic changes in the levels of various transcription factors at decommissioned or activated enhancers, levels of cohesin remain relatively unchanged. This observation can be explained if cohesin is not directly recruited to enhancers but rather its occurrence at these sequences is a consequence of its temporary presence during the extrusion process, which takes place whether the enhancer is active or not. In the case of genes down or upregulated after temperature stress, it appears that the levels of transcription factors at enhancers directly correlate with the frequency of interactions mediated by RAD21, H3K4me1, or pluripotency factors, which in turn correlate with the level of nascent transcription. The direct correlation between these three features may suggest that large protein complexes at enhancers and promoters slow down the cohesin extrusion process, leading to increased enhancer-promoter interactions and higher frequency of transcription bursting.
In addition to facilitating enhancer-promoter interactions, cohesin, together with CTCF, plays a role in forming insulated neighborhoods that separate enhancers and promoters located within the loop from regulatory interactions with nearby sequences located outside the loop (Hnisz et al., 2016). We observe up- and down-regulation of CTCF occupancy at hundreds of sites located adjacent to, but not within, enhancers and promoters. Changes in interactions at these sites in response to temperature stress serve to relocate regulatory sequences out or in new insulated neighborhoods. Upregulation of transcription in response to temperature stress involves the disruption of loops, which separate either enhancers or promoters from their cognate sequences in control cells, and formation of larger loops to create insulated neighborhoods that enclose the enhancer and promoter within the same loop. Down-regulation of transcription after temperature stress involves the opposite process. These results suggest that the orchestration of changes taking place in response to temperature stress is a complex process that involves not only activation of enhancers and tethering these enhancers to their cognate promoters via cohesin loops but also the assembly/disassembly of interactions with the purpose of allowing or precluding, rather than directing, new contacts among regulatory sequences. The mechanisms by which these new interactions are established are not well understood. To explain the directionality of CTCF-mediated interactions, it has been proposed that CTCF/cohesin loops form by an extrusion mechanism whereby cohesin loads either at random or specific sites and progresses extruding a loop until it finds CTCF sites. Results from our experiments agree with a loop extrusion mechanism in which the extrusion process initiates at random locations in the loop.
Results presented here suggest that genome-wide alterations of the transcriptome require a precisely choreographed series of events involving activation and decommissioning of enhancers, processes that may be initiated by the binding or dismissal of cell-type specific transcription factors. These processes are accompanied by changes in 3D interactions mediated by cohesin with or without CTCF in order to tether enhancers to their cognate promoters or to foster the formation or disassembly of insulated neighborhood to modulate interactions between enhancers and their cognate promoters.
STAR*METHODS
Contact for Reagent and Resource Sharing For information regarding resources and reagents, please contact the Lead Contact, Victor Corces (vgcorces@gmail.com).
Experimental Model and Subject Details
Cell culture and heat shock treatment
H9 human embryonic stem cells were obtained from WiCell. Cells were cultured in STEMPRO hESC SFM (Thermo Fisher) on cultureware coated with Geltrex Matrix (Thermo Fisher) at 37°C under 5% CO2. Medium was changed every day. Conditioned SFM medium from 80% confluent cultured H9 cells was filtered through a 0.45 μm sterilized filter and stored at 4°C for heat shock experiments. For heat shock treatment, 80% confluent H9 cells were changed into 42°C prewarmed conditioned SFM medium and transferred into a 42°C incubator for 1 h.
Generation of BRG1 knockout H9 hESCs
CRISPR was used to derive BRG1 knockout H9 hESCs. sgRNA was designed to target the first exon of the BRG1 gene by using the on-line tool at http://crispr.mit.edu:8079/guides/5081480023640509. sgRNA sequence is (ATCTGGTGCATGTTGTCCTG)AGG. iCas9 H9 hESCs was gifted by Dr. Danwei Huangfu and obtained as described (Gonzalez et al., 2014). 24 h before transfection of sgRNA, Doxycycline was added to cells to induce Cas9 expression. sgRNA was delivered by lipofectamine3000. 48 h after transfection, genomic PCR and T7EI digestion as well as RFLP assays were applied to the whole cell population to determine the INDEL efficiency. The remaining cells were digested to single cells with TRYPLE and seeded at 2000 cells per 10-cm dish for expansion of single cell colonies. Genomic PCR and RFPL assays were done on single cell colonies 3–4 weeks later to identify mutations in both alleles. PCR products of colonies showing mutations in both alleles from RFLP digestion were sub-cloned into the pJET vector (ThermoFisher) for Sanger sequencing of both alleles. Cells with INDELs leading to frame shifts in both alleles were analyzed for BRG1 protein level by Western blot to ensure knock out efficiency.
NIPBL knockdown
To knock down NIPBL, shRNA lenti-viral particles (sc-75921-V, Santa Cruz) were incubated with H9 hESCs for 24 h. Puromycin was added to culture media 48 h after infection to select cells with stable expression of the shRNA. Knock down efficiency was determined by Western blot when collecting cells for HiChIP experiments.
Method Details
In-situ Hi-C
We generated in-situ Hi-C libraries from H9 cells grown at 37°C (Ctrl) or 42°C (HS) using HindIII restriction enzyme following the protocol as described (Rao et al., 2014). Briefly, 5 million H9 cells for each condition were crosslinked with 1% formaldehyde, quenched with glycine, washed with PBS, and permeabilized to obtain intact nuclei. Nuclear DNA was then digested with HindIII, the 5’-overhangs were filled with biotinylated dCTPs and dA/dT/dGTPs to make blunt-end fragments, which were then ligated, reverse-crosslinked, and purified by standard DNA ethanol precipitation. Purified DNA was sonicated to 200–500 bp small fragments and captured with streptavidin beads. Standard Illumina TruSeq library preparation steps including end-repairing, A-tailing, and ligation with universal adaptors were all performed on beads, washing twice in TWB between each step. DNA on the beads was PCR amplified with barcoded primers using KAPA SYBR FAST qPCR Master Mix for 5~12 PCR cycles to obtain enough DNA for sequencing. Generated libraries were paired-end sequenced on Illumina HiSeq2500 v4. Two sets of biological replicates were generated for Ctrl and HS conditions. Replicates were combined after ensuring high correlation.
Nascent transcript sequencing using EU-seq
EU-seq was modified from Bru-seq (Paulsen et al., 2014) to increase efficiency. eU was used instead of BrU to label nascent RNA in live H9 cells. For both Ctrl and HS cells, cells were grown to 80% confluency in one 15-cm culture plate and labeled with 2 mM eU in 12 ml conditioned medium. For heat shock, eU was added to the medium 30 min after the beginning of HS treatment, at which time eU was also added to Ctrl H9 cells. After adding eU, both Ctrl and HS cells were labeled for 30 min at 37°C and 42 °C respectively to ensure equal labeling time. Medium was then removed and RNA was extracted using TRIZOL reagent and isolated by standard protocols. Isolated RNA was converted by click-chemistry using Click-iT Nascent RNA Capture Kit (Thermo Fisher) into Biotin-U labeled RNA. Converted RNA was heated at 80°C for 10 min and then immediately put on ice, followed by capturing biotin-U labeled RNA with streptavidin beads. After washing the beads several times with 0.1% BSA in DEPC-PBS, bio-RNA was eluted in 40 μl of DEPC H2O by heating the beads at 95°C for 10 min and store d at −80°C. 250 ng of Bio-RNA was reverse transcribed usi ng Superscript II to synthesize first strand cDNA, followed by purification with AMpure RNAclean beads. Second strand cDNA was synthesized with DNA Polymerase I to obtain double stranded cDNA, which was purified using AMpure beads and used to make Illumina TruSeq libraries following standard protocols, including end-repairing, A-tailing, adapter ligation, and PCR amplification and barcoding using KAPA SYBR FAST qPCR Master Mix.
HiChIP
HiChIP libraries were prepared for Rad21, CTCF, OCT4, NANOG, KLF4 and H3K4me1 in Ctrl and HS treated H9 ES cells as described (Mumbach et al., 2016) with some modifications. For each HiChIP, 107 cells grown to 80% confluency at 37°C and 42°C res pectively were crosslinked in 1% formaldehyde for 10 min at room temperature, after which 0.2 M glycine was added for 5 min to quench the reaction. PBS washed cells were pelleted in a 2 ml Eppendorf tube and resuspended in 1500 μl cold Hi-C lysis buffer (10 mM Tris-HCl pH8, 10 mM NaCl, 0.2% Igepal CA-630, and 1× Protease Inhibitor Roche 11873580001) and incubated on ice for 15 min. Nuclei were pelleted at 2500 rcf for 5 min at 4°C, washed once in 1500 μl cold Hi-C lysis buffer and pelleted at 2500 rcf for 5 min at 4°C. N uclei pellets were then resuspended in 150 μl 0.5% SDS, and incubated for 5 min at 65°C; after wh ich we added 450 μl of water and 75 μl of 10% Triton X-100, then incubated samples for 15 min at 37°C. 75 μl of 10× DpnII buffer and 300 U of DpnII (NEB R0543) were added and samples were digested overnight at 37°C with rotation. After digestion, samples were incubated at 65°C for 10 min to inactivate DpnII, and reactions were cooled to room temperature. Biotin fill-in was done with 67.5 μl of water, 4.5 μl each of 10 mM dTTP, dATP, and dGTP, 45 μl of 1 mM biotin-16-dCTP (Jena Bioscience JBSNU-809-BIO16), and 24 μl of 5 U/μl DNA polymerase I Large (Klenow) fragment (NEB M210). This reaction was placed at 37°C for 1 h, after which samples were ligated for 4 h at room temperature with addition of 1,989 μl H2O, 360 μl 10× NEB T4 DNA Ligase buffer, 300 μl 10% Triton X-100, 36 μl 10 mg/ml BSA, and 15 μl 400 U/ μl T4 DNA Ligase (NEB M0202). Following ligation, nuclei were pelleted and resuspended in 1.5 ml cold Nuclei Lysis Buffer (50 mM Tris-HCl pH 9, 10 mM EDTA, 1% SDS, and 1× Protease Inhibitors) with incubation on ice for 20 min. After incubation, lysates were sonicated using a Bioruptor to approximately 200–500 bp fragments, then 75 ul of 20% tritonX-100 was added to quench SDS. Lysates were centrifuged at 15,000 rpm for 20 min at 4 °C to remove cell deb ris and supernatant was transferred into a new 1.5 ml tube for immunoprecipitation. After a preclear step with 10 μl IgG coated Protein A or G magnetic beads for 1 hr, supernatant was incubated with antibody coated beads overnight at 4 °C for immunoprecipitation. After IP, samples wer e washed 3× with Low Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA pH 8, 20 mM Tris-HCl pH 8, 150 mM NaCl), 2× with High Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA pH 8, 20 mM Tris-HCl pH 8, 500 mM NaCl), 2× with LiCl Buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 0.25 M LiCl, 1% Igepal CA-630, 1% DOC), and 1× with TE buffer. DNA was eluted using 400 μl IP elution buffer (0.1 M NaHCO3, 1% SDS) for 20 min at 65°C and transferred to a new tube. Eluates were reverse crosslinked at 65 °C for 2 h in 20 μl 5 M NaCl, 8 μ l 0.5 M EDTA and 16 μl 1 M Tris-HCl pH8, followed by 8 μl Proteinase K digestion at 55°C for 1 h. Ethanol precipitated DNA was resuspended in 300 μl 10 mM Tris-Cl pH 8.0. DNA was incubated with TWB (5mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20) washed 2× in Binding Buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl), and incubated with streptavidin beads at room temperature for 15 min with rotation. Afterwards, samples were washed 2× in TWB and the standard Hi-C library preparation protocol was performed as previously. Generated libraries were paired-end sequenced on an Illumina HiSeq2500 v4.
For Rad21 HiChIP, we generated three biological replicates in each condition with DpnII and another biological replicate with MboI in each condition. For CTCF HiChIP, we generated three biological replicates for Ctrl and two biological replicates in HS. All other HiChIP have two biological replicates in each condition except KLF4 in heat shock. We combined biological replicates for most analysis to obtain higher resolution contact matrices.
ChIP-Seq
ChIP-seq experiments in control and temperature-stressed H9 hESCs were carried out as described (Li et al., 2015). After removal of medium, cells were crosslinked in 1% formaldehyde in PBS at room temperature for 10 min and quenched with glycine. PBS rinsed cell pellets were flash frozen in liquid nitrogen and stored at −80°C or continue with cell and nuclear lysis steps. Nuclear lysates were precleared with protein A/G beads followed by incubation with proper antibodies. After washing with high salt buffer, LiCl buffer, and TE, chromatin was eluted and reverse-crosslinked. Purified DNA was ethanol precipitated followed by Illumina Truseq library preparation. Libraries for Illumina sequencing were constructed using the following standard protocol. Fragment ends were repaired using the NEBNext End Repair Module and adenosine was added at the 3’ ends of fragments using Klenow fragment (3’ to 5’ exo minus, New England Biolabs), universal adaptors were ligated to the A-tailed DNA fragments at room temperature for 1 h with T4 DNA ligase (New England Biolabs) and amplified with Illumina barcoded primers using KAPA SYBR FAST qPCR Master Mix for 5~12 PCR cycles to obtain enough DNA for sequencing. Generated libraries were paired-end sequenced on Illumina HiSeq2500 v4.
ChIP-Nexus with Rad21 and CTCF antibodies were performed as described previously (He et al., 2015b).
ATAC-seq
Fast-ATAC-seq was performed as described (Corces et al., 2016) with some modifications. For each ATAC-seq reaction, H9 cells grown to 80% confluency in one well of a 96-well plate in Ctrl or HS conditions were used. After removing medium from Ctrl or HS cells, cells were incubated with Tn5 transposase mixture containing 0.01% digitonin for 20 min with occasional shaking. Following incubation, genomic DNA was isolated with the Qiagen Minelute Kit and directly used for PCR amplification with 2× KAPA HiFi mix and 1 μM indexed primers using the following PCR conditions: 72°C for 5 min; 98°C for 30 s; and 10–1 1 cycles at 98°C for 10 s, 63°C for 30 s, and 72°C for 1 min. The amplified libraries from both F ast-ATAC were cleaned with AMpure XP beads at a 1.1× ratio to avoid losing low molecular weight fragments and paired-end sequenced on an Illumina HiSeq2500 v4 instrument.
Quantification and Statistical Analysis
HiChIP data processing
Paired-end reads from HiChIP experiments were aligned to the human hg38 reference genome using Juicer (Rao et al., 2014). After PCR duplicates and low-quality reads were removed, high-quality reads were assigned to MboI restriction fragments, filtered for valid interaction PETs, and used to generate binned contact matrix hic files. For visualization and further analysis of HiChIP interaction signals, Vanilla coverage square root (VCsqrt) normalized signal for the interaction matrices were derived using the Juicebox tools dump command. Mango (Phanstiel et al., 2015) was used to call significant interactions with default parameters and FDR cutoff at 0.01 and PETs >=10. HiCCUPS (Rao et al., 2014) was used to call CTCF loops in Rad21 HiChIP for the analysis shown in Figure S3. Mango loops were visualized with the WashU Epigenome browser to obtain arc views of significant loops.
Virtual 4C analysis of HiChIP data
To derive the HiChIP virtual 4C line plots, promoters were used as viewpoints, VCsqrt normalized observed over expected (O/E) contact matrices binned into 10 kb fragments were generated using Juicebox dump tools and used to compute contact frequencies between other genomic bins with each viewpoint. Viewpoints were ranked by distances between promoters and interacting dynamic enhancers from upstream to downstream. For clarity of the results, only interactions within 500 kb up/down-stream were plotted. Virtual 4C signals were analyzed with in house python scripts. Briefly, virtual 4C was performed by taking the HiChIP VCsqrt normalized observed over expected (O/E) contact frequency of each bin interacting with the anchor. To account for intensities at various resolutions, individual bins at high resolution were combined using a sliding window congruent to the resolution step, then the average signal across combined bins was calculated for each resolution step examined until reaching the lowest specified resolution. Final normalized enriched scores shown in Figure 4 were generated by calculating the average intensity of each bin across all resolutions from 10 kb to 500 kb, and plotted using java treeview.
APA metaplot analysis
Aggregate peak analysis (APA) metaplots and scores were generated as described (Rao et al., 2014) using 10 kb resolution contact matrices. To measure the enrichment of loops over the local background and normalize for different loop distances and protein occupancy bias, the VCsqrt normalized observed over expected (O/E) contact frequency of pixels of loops as well as the surrounding pixels up to 10 bins away in both x and y directions, i.e. 210 kb*210 kb local contact matrices, were collected. To generate the aggregate heatmaps, the median O/E for each position of all 210 kb*210 kb contact matrices for a set of loops were calculated and plotted using the heatmap.2 R package. APA scores were determined by dividing the center pixel value by the mean value of the 25 (5*5) pixels in the lower right section of the APA plot.
Multiple anchor metaplots
Multiple anchor metaplots shown in Figure 6 were obtained at 10 kb resolution and the distance between anchors was scaled to 10 equal bins. For three CTCF anchors, the anchors were oriented such that the stable anchors are the first ones on the left. To compare libraries from cells subjected to different treatments, the observed interaction matrices were normalized between samples by random picking to obtain equal numbers of PETs for each library. VCsqrt Normalization was then applied to all contact matrices. To compare HiChIP aggregate signal changes between different samples on distinct anchor sets, subtraction or log2 fold changes of treatment versus Ctrl were calculated for each loop separately and then summarized by taking the median values of all anchors for visualization.
Calling of differential loops in HiChIP data
For comparison between different HiChIP libraries, the following normalization steps were taken. 1) PETs of each library were randomly picked to match the size of the library with lowest numbers of PETs; 2) distance normalized by observed over expected; 3) VCsqrt normalized to balance protein occupancy bias. The normalized matrices were used to call differential loops between conditions by the following procedures. 1) Mango loops from both conditions were combined; 2) normalized contact frequencies in both conditions were combined pairwise on all resulting combined loops in step 1); 3) pairwise interaction frequencies from step 2) were used as input in the edgeR R package to identify condition-enriched significant differential loops (FDR cutoff < 0.1 and fold change >2).
Overlapping of ChIP-Seq peaks with HiChIP loop anchors
For analyzing overlaps of ChIP-Seq peaks and loop anchors, Mango loop anchors ± 5kb were used to overlap with ChIP-Seq peaks using the bedtools intersect function. To obtain the expected overlapping ratio, ChIP-Seq peaks were shuffled 1000 times and the same analysis was repeated. Significant p values were derived by numbers of times when observed < expected happens divided by 1000.
ChIP-Seq data processing
All reads were mapped to unique genomic regions using Bowtie and the hg38 genome. PCR duplicates were removed manually. Bedtools genomeCoverage function was used to derive bedgraph files for further analysis. To compare changes in ChIP-Seq signals, libraries were normalized by random picking to obtain the same numbers of reads. Normalized reads were used to derive bedgraph files for comparison in IGV. MACS2 was used to call peaks using default parameters with IgG as control. Differential peaks of H3K27ac and RAD21 were found using the edgeR (Robinson et al., 2010) R package at p<0.05, fold change >=3. CTCF differential peaks were obtained using Manorm (Shao et al., 2012) at p<0.05.
Promoter definition
Refseq genes TSS ± 1 kb were defined as promoters. HS and Ctrl enriched promoters correspond to promoters of HS-induced and HS-repressed genes (shown in Figure 1), respectively.
Enhancer definition
Enhancers were defined by using H3K4me1 peaks without H3K4me3 but overlapping ATAC-seq THSSs peaks, TSS±1b was excluded. Among these enhancers, those overlapping H3K27ac peaks were taken as active enhancers, those overlapping with H3K27me3 peaks were taken as poised enhancers, those overlapping with neither H3K27ac nor H3K27me3 peaks were considered primed enhancers.
Differential enhancers were found using the edgeR R package based on H3K27ac ChIP-Seq signals with a p value cutoff at 0.05 and more than 3-fold changes in either condition.
Super enhancer definition
Super enhancers were identified using ROSE (Whyte et al., 2013) with H3K27ac ChIP-Seq signal as input using default parameters.
Polycomb domain identification
To identify Polycomb domains, 10 kb bins with a 1 kb sliding window were used to calculate windows with RPKM > 10 in RING1B ChIP-Seq. Windows selected were merged.
Heatmaps and average profiles of ChIP-Seq and clustering
For deriving heatmaps of ChIP-Seq signal, anchors plus flanking regions were binned equally to get a blank matrix (anchors × bins). To compare between samples, reads from the same antibody analysis were normalized by random pick. Normalized read pairs were mapped to each genomic bin with the bedtools intersect function to obtain reads counts in each bin for the whole matrix. To normalize for sequencing depth, values in the matrix were divided by library sizes in millions to obtain reads per million per covered bin (RPMPCG or RPM), which was then visualized with Java treeview to derive heatmaps. To obtain average profiles of the ChIP-Seq data, mean values of bins at the same distances from anchors were calculated and plotted. Clustering of ChIP-Seq heatmaps was done using Cluster3 on center ± 3 bins signals of appropriate heatmaps. K-means clustering was used.
ATAC-seq data processing
All reads were mapped with Bowtie2 and PCR duplicates were removed by Picard tools with in house python script pipeline. After this, reads were split into two ranges based on sizes: THSSs (transposases hypersensitive sites, bound by TFs, <115bp) and mono-nucleosomes (180~247bp). To obtain the exact positioning of nucleosomes, DANPOS (Chen et al., 2013) was used to derive the nucleosomal signals genome wide by dpos function using 180~247 bp fragments as input and 115 bp fragments as background using -p 1 -a 1 -jd 20 -u 0 -m 1. Reads were normalized between samples before running DANPOS. THSSs bedgraphs were made using the bedtools genomeCoverage function.
Heatmaps and average profile of ATAC-seq THSSs and nucleosomal signals were derived in the same way as ChIP-Seq data. Clustering of ATAC-seq heatmaps was done using Cluster3 with K-means clustering.
EU-seq data processing
All reads were process as described for Bru-Seq (Paulsen et al., 2014). First, all sequence reads were mapped to the human ribosomal DNA complete repeating unit (U13369.1) using Bowtie to obtain non-rRNA reads, then those non-rRNA reads were mapped to the human reference genome assembly hg38 using TopHat2 and default parameters. Only uniquely mapped reads with up to two read segment mismatches were allowed to split between exons in RefSeq transcript annotation, but de novo splice junction calling was not performed, since nascent RNA reads are mainly intronic. Picard was used to remove PCR duplicates. Finally, fragments were normalized between Ctrl and HS for obtaining bedgraph files at fragments per million (FPM) for visualization in IGV. Cufflinks was used to calculate gene FPKM values and cuffdiff was used to calculate p-values for differentially expressed genes. Genes with more than 2-fold changes in FPKM values and p <0.01 in cuffdiff were considered Ctrl or HS enriched genes.
Percentage of sites overlapping motifs
Significant motifs of ChIP-Seq peaks and differential loop anchors were found by MEME. Exact motifs sequences were scanned using FIMO and the JASPAR_CORE_2016_vertebrates database against a set of peaks or anchors to obtain the overlapping percentages.
TFs footprint analysis
To analyze the footprints of TFs in ATAC-seq data, motifs on a set of peaks or loop anchors were used as anchors for running dnase_average_profile.py scripts of the Wellington program in ATAC-seq mode. To compare between Ctrl and HS samples, read-normalized ATAC-seq THSSs (<115 bp) fragments were used as input to obtain the footprint average profiles. The footprint p values of all motifs on a set of peaks or anchors were derived using the wellington_footprints.py scripts of the Wellington program in ATAC-seq mode on read-normalized ATAC-seq THSSs (<115 bp) fragments.
Analysis of CTCF motif orientation
CTCF motifs were downloaded from the JASPER database. To find the CTCF motif orientation at loop anchors, loops with only one CTCF binding site overlapping one CTCF motif or multiple CTCF motifs in the same orientation were kept for analysis. The percentage of all four possible pairwise combinations of motifs were calculated on the loop anchors.
For analysis of CTCF motif orientation at loop anchors, the number of forward motifs was divided by the number of reverse motifs and log2 transformed. For three CTCF anchors, since the anchors were oriented such that the stable anchors are the first ones on the left, the CTCF motif orientations were also inverted if the three anchors coordinates were in reverse decreasing order in the genome. Therefore, the CTCF orientation on the plot of three anchors are relative to the expansion or shrinking orientation of CTCF loops.
Quantile change analysis
Changes of Eigenvector or contact frequencies were ranked by values and divided into ten groups. The ChIP-Seq signals changes in each group were then averaged and plotted using the heatmap.2 R package.
Contact enrichment and contact strength between genomic regions
To measure the enrichment of contacts between genomic regions, pairwise anchors were random shuffled 100 times to generate 100 random pairwise anchors with comparable distance distribution to the experimental set. The significance p value was determined by counting the number of random control sets with more contacts than the experimental set and divided by the number of experiments (100).
Simulation of loop extrusion
To simulate the extrusion complex starting at CTCF sites, an 11*11 empty matrix was generated, where bin0 indicates F anchor and bin11 indicates R anchor. As the diagram in Figure S7C shows, we start assigning contact counts to the left anchor at bin0 and to the right anchor at bin1, and then keep assigning counts to the left anchor on bin0 and the right anchor on bin2, bin3…bin11, then a loop is formed using the forward CTCF as starting point. In the other direction, we performed the same analysis using the reverse CTCF motif as starting point, so that the right anchor was constantly bin11, and left anchors were gradually decreased from bin10 to bin0. This process was repeated 1000 times. After averaging these two types of matrices, the final simulation matrix was derived.
To simulate the extrusion complex starting at random sites between two convergent F-R CTCF motifs, an 11*11 empty matrix was generated, where bin0 indicates the F anchor and bin11 indicates the R anchor. As the diagram in Figure S7D shows, we generated a random bin number bin(n) as a start of extrusion and assigned this bin as i and j in the matrix[i,j]. Each extrusion step was simulated by matrix[i−1,j+1] with a read added at each step. When i reached a CTCF site on the left, it was left constant while j was increased by matrix[i,j+1]. We generated the random starting bin number n by 50 times iterations and all matrices are summed up. These processes are repeated 1000 times. After averaging 1000 matrices above, the final simulation matrix is derived.
Slopes of Hi-C contact enrichment for locus A-F in Figure 7F were determined by off the diagonal signal determined by iterating through matrix[i, o]/counts[i,i], where o ranges between 1 and 8 as shown in Figure 7G. This was done for the CTCF anchor A, as well as for anchors interior to the domain (B – F).
Supplementary Material
HIGHLIGHTS.
hESCs respond to temperature stress by altering the expression of hundreds of genes
Alteration of transcription correlates with changes of AP-1 and pluripotency factors
CTCF/cohesin changes result in new loops that regulate enhancer-promoter interactions
HiChIP data for CTCF and cohesin support loop formation by random extrusion
ACKNOWLEGMENTS
We would like to thank the Genomic Services Lab at the HudsonAlpha Institute for Biotechnology, and specially Drs. Angela Jones and Braden Boone, for their help in performing Illumina sequencing of samples. This work was supported by U.S. Public Health Service Award R01 GM035463 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
ChIP-seq, ATAC-seq, Hi-C, HiChIP and EU-seq data are available from NCBI’s Gene Expression Omnibus (GEO) under accession number GSE105028.
DECLARATION OF INTERESTS
The authors declare no competing interests
REFERENCES
- Alipour E, and Marko JF (2012). Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research 40, 11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ay F, Bailey TL, and Noble WS (2014). Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res 24, 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SD, Zhang X, Desai K, Aid M, Corradin O, Cowper-Sal Lari R, Akhtar-Zaidi B, Scacheri PC, Haibe-Kains B, and Lupien M (2015). ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nature communications 2, 6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Williams N, Misleh C, and Li WW (2006). MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34, W369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun K, Kim TK, Oh J, Bayarsaikhan E, Kim D, Lee MY, Pack CG, Hwang D, and Lee B (2013). Heat shock instructs hESCs to exit from the self-renewal program through negative regulation of OCT4 by SAPK/JNK and HSF1 pathway. Stem cell research 11, 1323–1334. [DOI] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, and Li W (2013). DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Bouwman BA, Zhu Y, Klous P, Splinter E, Verstegen MJ, Krijger PH, Festuccia N, Nora EP, Welling M, et al. (2013). The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature 501, 227–231. [DOI] [PubMed] [Google Scholar]
- de Wit E, Vos ESM, Holwerda SJB, Valdes-Quezada C, Verstegen MJAM, Teunissen H, Splinter E, Wijchers PJ, Krijger PHL, and de Laat W (2015). CTCF Binding Polarity Determines Chromatin Looping. Molecular Cell 60, 676–684. [DOI] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016a). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Systems 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016b). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuda NJ, Ardehali MB, and Lis JT (2009). Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Reports 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F, Zhu Z, Shi ZD, Lelli K, Verma N, Li QV, and Huangfu D (2014). An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell 15, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, et al. (2015). CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 162, 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusmao EG, Allhoff M, Zenke M, and Costa IG (2016). Analysis of computational footprinting methods for DNase sequencing experiments. Nat Methods 13, 303–309. [DOI] [PubMed] [Google Scholar]
- He C, Zhang MQ, and Wang X (2015a). MICC: an R package for identifying chromatin interactions from ChIA-PET data. Bioinformatics (Oxford, England) 31, 3832–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Johnston J, and Zeitlinger J (2015b). ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat Biotechnol 33, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, and Adelman K (2018). Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 32, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Day DS, and Young RA (2016). Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 167, 1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Dadon Daniel B., Powell Benjamin E., Fan Zi P., Borges-Rivera D, Shachar S, Weintraub Abraham S., Hnisz D, Pegoraro G, Lee Tong I., et al. (2016). 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 18, 262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lyu X, Hou C, Takenaka N, Nguyen HQ, Ong C-T, Cubeñas-Potts C, Hu M, Lei EP, Bosco G, et al. (2015). Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Molecular Cell 58, 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T (2014). Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol 1150, 81–95. [DOI] [PubMed] [Google Scholar]
- Mahat DB, Salamanca HH, Duarte FM, Danko CG, and Lis JT (2016). Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol Cell 62, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K (2001). Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annual Review of Genetics 35, 673–745. [DOI] [PubMed] [Google Scholar]
- Nichols MH, and Corces VG (2015). A CTCF Code for 3D Genome Architecture. Cell 162, 703–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE, and Ljungman M (2014). Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 67, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Boyle AP, Heidari N, and Snyder MP (2015). Mango: a bias-correcting ChIA-PET analysis pipeline. Bioinformatics 31, 3092–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper J, Elze MC, Cauchy P, Cockerill PN, Bonifer C, and Ott S (2013). Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res 41, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014). BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Current protocols in bioinformatics 47, 11 12.1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SSP, Huang S-C, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, and Greenleaf WJ (2015). Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res 25, 1757–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, C, H. H, Mirny L, et al. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan GC, Orkin SH, and Waxman DJ (2012). MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, and Stark A (2014). Transcriptional enhancers: from properties to genome-wide predictions. Nature reviews. Genetics 15, 272–286. [DOI] [PubMed] [Google Scholar]
- Sullivan EK, Weirich CS, Guyon JR, Sif S, and Kingston RE (2001). Transcriptional activation domains of human heat shock factor 1 recruit human SWI/SNF. Mol Cell Biol 21, 5826–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo Oscar J., Li X, Zheng M, Zhu Jacqueline J., Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. (2015). CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163, 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinklein ND, Murray JI, Hartman SJ, Botstein D, and Myers RM (2004). The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Molecular biology of the cell 15, 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, Yang L, and Chen LL (2016). The Biogenesis of Nascent Circular RNAs. Cell Rep 15, 611–624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.