

Abstract
The targeting of telomerase and telomere maintenance mechanisms represents a promising therapeutic approach for various types of cancer. In this work, we designed a new protocol to screen for and rank the efficacy of compounds specifically targeting telomeres and telomerase. This approach used two isogenic cell lines containing a circular human artificial chromosome (HAC, lacking telomeres) and a linear HAC (containing telomeres) marked with the EGFP transgene: compounds that target telomerase or telomeres should preferentially induce loss of the linear HAC but not the circular HAC. Our assay allowed quantification of chromosome loss by routine flow cytometry. We applied this dual-HAC assay to rank a set of known and newly developed compounds, including G-quadruplex (G4) ligands. Among the latter group, two compounds -Cu-ttpy and Pt-ttpy- induced a high rate of linear HAC loss with no significant effect on the mitotic stability of a circular HAC. Analysis of the mitotic phenotypes induced by these drugs revealed an elevated rate of chromatin bridges in late mitosis and cytokinesis as well as UFB (Ultrafine Bridges). Chromosome loss after Pt-ttpy or Cu-ttpy treatment correlated with the induction of telomere-associated DNA damage. Overall, this platform enables identification and ranking of compounds that greatly increase chromosome mis-segregation rates as a result of telomere dysfunction and may expedite the development of new therapeutic strategies for cancer treatment.
Keywords: chromosome instability, CIN, human artificial chromosome, HAC, telomerase inhibitors
Introduction
Aneuploidy or an abnormal chromosome number is a common feature of many cancers and is often accompanied by an elevated rate of chromosome instability (CIN) (1). Gain or loss of entire chromosomes leads to changes in gene copy number and expression levels. Recent findings point to that experimentally induced CIN may be a barrier to tumor growth that can be exploited therapeutically (2–5). However, drugs that increase CIN beyond the therapeutic threshold are currently limited. Developing novel strategies to screen the compounds specifically modulating CIN and to exploit the fitness cost associated with excessive aneuploidy is important for the successful treatment of cancer.
Telomeres - protective caps at the ends of human chromosomes – are potential targets for increasing CIN. Telomeres shorten with each successive cell division in normal cells whereas in tumors they are continuously elongated by telomerase reverse transcriptase (hTERT). Telomerase is overexpressed in 80–95% of cancers and is present at very low levels in normal cells. Because telomerase plays a key role in cancer cell proliferation it may serve as a useful target for anticancer therapeutics. Inhibition of telomerase in telomerase-positive tumors could potentially lead to a decrease of telomere length resulting in CIN, cell senescence and apoptosis.
Several strategies have been developed for telomerase inhibition based on targeting either hTERT or its RNA subunit with small molecule inhibitors or antisense oligonucleotides (6). However, interest in their use for therapeutic approaches is diminished by the observation that significant effects on tumor growth were obtained only after long-term drug administration required for telomeres to reach a critical length. Such long-term telomerase inhibition has the potential to select for cells expressing the alternative lengthening of telomeres (ALT) pathway in which chromosomal ends are maintained by a recombination-based mechanism (7).
The telomeric structure itself offers a potential target for telomere-binding compounds in short-term treatments (8). Thus, G4 ligands, a class of molecules that are able to interact with G-quadruplex (abbreviated G4) structures formed by the G-rich overhang of telomeric DNA have recently received considerable attention (9–11). G4-stabilizing molecules such as Telomestatin, Braco-19, 360A (PDC) and RHPS4 can reduce cancer growth by rending telomeres dysfunctional. In contrast to hTERT inhibitors, G4 ligands represent a good example of a multimodal class of drugs, able even after a short treatment to simultaneously affect multiple targets participating in several distinct pathways (including the complex mechanism of gene expression). This simplifies the treatment modalities, improving the selectivity against cancer cells and avoiding the selection of ALT cells. Until now there has been no reliable method to quantify the effect of different G4 ligands on chromosome instability.
In our previous work, we developed a quantitative assay to measure CIN in response to cell treatment by different compounds (12). This assay was based on the use of a circular human artificial chromosome (HAC) constructed in our lab (13). The HAC has previously been used for the efficient and regulated expression of genes of interest (14) and kinetochore studies (15,16,17). It contains centromeric repeats that form a functional centromere/kinetochore allowing its stable inheritance as a nonessential chromosome, albeit with a loss rate roughly 10x that of the native chromosomes (15). In order to develop a quantitative (“loss of signal”) assay for chromosome mis-segregation, we used a modified HAC carrying a constitutively expressed EGFP transgene (12). Cells that inherit the HAC display green fluorescence, while cells lacking the HAC do not. This allows the measurement of HAC loss rate by flow cytometry, providing a quick and efficient way to screen hundreds of drugs and identify those affecting chromosome mis-segregation. The assay was successfully used to rank different anticancer drugs according to their effects on chromosome transmission (18). More recently, this HAC-based assay was adapted for high-throughput screening of chemical libraries using a fluorescence microplate reader to identify compounds that elevate chromosome mis-segregation and drive lethal aneuploidy (19). In the modified assay, cells carry the EGFP transgene integrated in the genome and the HAC carries a constitutively expressed shRNA against EGFP. Cells display green fluorescence only after loss of the HAC (“gain of signal” assay). Using both HAC-based systems we have identified new and potentially less toxic agents that selectively elevate CIN in cancer cells.
In the present work, we further modified our HAC-based screening protocol to identify compounds specifically targeting telomeres or telomerase. This protocol is based on the use of two isogenic cell lines expressing the EGFP transgene: one carrying a circular HAC lacking telomeres and the other carrying a linear HAC with telomeres. We hypothesized that compounds specifically inhibiting telomerase or other telomere functions would induce loss of the linear HAC but not the circular HAC. Our screen included known telomerase inhibitors as well as a set of known and newly developed G4 ligands. Among this last group, Pt-ttpy and Cu-ttpy (20,21) induced the highest rate of loss of the linear HAC.
Identification of new compounds that greatly increase chromosome mis-segregation rates as a result of telomere dysfunction may expedite the development of new therapeutic strategies for cancer.
Materials and Methods
Cell lines and culture
The human fibrosarcoma (HT1080; ATCC® CCL-121™), human colon carcinoma (HCT116; (ATCC® CCL-247™) and human osteosarcoma (U2OS; ATCC® HTB-96™) cell lines were obtained from the American Type Culture Collection and were authenticated both morphologically and by short tandem repeat analysis. All cell lines were tested regularly to confirm lack of mycoplasma infection with mycoplasma detection kit PlasmoTest from InvivoGen.
The human fibrosarcoma HT1080 cells (telomerase positive) harboring either alphoidtetO-HAC-EGFP or 21ΔqHAC-EGFP were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% (v/v) tet system-approved fetal bovine serum (FBS, Clontech Laboratories, Inc.) at 37°C in 5% CO2. Human colon carcinoma HCT116 cells (telomerase positive) were cultured in McCoy’s 5A medium supplemented with 10% FBS at 37oC and 5% CO2. Human osteosarcoma U2OS cells (telomerase negative) were cultured in DMEM supplemented with 10% FBS at 37oC and 5% CO2.
For chromosome instability experiments, HT1080 cells were grown in blasticidin-containing medium to prevent HAC loss prior to treatment with the drugs being tested (both linear and circular HACs contain the BS marker). After drug treatment, the cells were cultured in a non-selective medium to allow HAC loss, i.e. under conditions when the cells that have lost a HAC are able to grow. For mitotic abnormality experiments, the cells were not exposed to blasticidin because the experiments were carried out in HT1080, HCT116 and U2OS cell lines not containing any HAC.
Flow cytometry
Analysis of EGFP expression was performed on a FACS Calibur instrument (BD Biosciences) using CellQuest acquisition software and analyzed statistically with FlowJo software. The cells were harvested by trypsin-treatment. Intensities of fluorescence were determined by flow cytometry. A minimum of 4 × 104 cells was analyzed for each cell sample.
Compounds and treatments
23 different compounds were used in our experiments (Supplementary Table S1). Our experiment protocol was as follows. HT1080 cells containing a EGFP-HAC were maintained on blasticidin selection to select for the presence of the HAC. Approximately 1 × 105 cells were cultured either in the presence or absence of blasticidin selection in parallel with a third culture that was exposed to the agent under examination to test its effect on EGFP-HAC segregation. The compound concentration applied for measuring chromosome instability was adjusted to the LC50 level for each compound (determined using a proliferation assay described below). Concentrations of compounds and lengths of treatment are presented in Supplementary Table S2. After treatment, the compound was removed by performing three consecutive medium washes and the cells were subsequently grown without blasticidin selection for 1–14 days. At the end of the experiment, cells were collected and analyzed by flow cytometry to detect the proportion of cells that retain EGFP fluorescence. This served as a measure of EGFP-HAC stability following compound treatment. For each compound, the experiments on measuring EGFP-HAC loss were carried out in triplicate. The results were reproducible and the std were small (for example, Pt-ttpy: SD±0.9%; MST-312: SD±1.1%).
To study mitotic abnormalities induced by Pt-ttpy and MST-312 in the telomerase positive HT1080 and HCT116 and telomerase negative U2OS cell lines, we used much lower concentrations of the drugs as at the LC50 only very small numbers of mitotic cells were seen. For these experiments, the cells were treated by noncytotoxic drugs concentrations as a dose based on published data and proven experimentally for our cell lines (see Supplementary Table S2).
Calculation of the rate of spontaneous HAC loss and after compound treatment
In our study, we determined the normal rate of spontaneous HAC mis-segregation (RNormal) in the host cell line HT1080 using the formula,; where P0 is the percentage of EGFP(+) cells at the start of the experiment as determined by FACS. These cells were cultured under HAC selection conditions using blaticidin. PNormal is the percentage EGFP(+) cells after culturing without HAC selection (no blasticidin) for a duration of t1. In this study t1 was 14 days. n1 is the number of cell doublings that occurs during culturing without blasticidin selection. The doubling time of HT1080 under normal growth conditions is approximately 18 hours. The number of cell divisions (n) is calculated by (t / host cell doubling time).
Once (RNormal) was obtained, the rate of HAC loss induced by drug treatment (RDrug) is determined using the formula, . As before, P0 represents the percentage of EGFP(+) cells at the start of the experiment, cultured under HAC selection condition. PTreated is the percentage of EGFP(+) cells at the end of the drug treatment experiment with a duration of (t2 + t3), where t2 is the duration of drug treatment and t3 is the duration of culturing after the drug is removed. (t2 + t3) was 14 days in this study. n2 is the number of cell doublings that occurs during drug treatment, while n3 is the number of cell doublings that occurs during the culturing without selection after drug treatment.
In the present study, the duration of most drug treatments was less than the duration of a single cell cycle of HT1080 (t2 <18 hr). We made the assumption that any significant increase in HAC loss occurs only during the first mitotic division after wash-out of the drug (n2 = 1). Thus n3 = (14d / 18hr - 1). The algorithm we used is valid between the ranges R = 0 to 1. R values large than 1 indicate that the assumptions made in this model are incorrect. The assumption of synchronous growth in the model means that the estimated mis-segregation rate is lower than real values. As the spontaneous rate of HAC mis-segregation (RNormal) was found to be low, this algorithm is relatively insensitive to the number of cell divisions that occurs post drug treatment (12).
Cell viability test for measuring HAC loss in response to drug treatment
For each compound LC50 was determined using a MTS tetrazolium cell viability assay according to the manufacturer’s instructions (CellTiter 96 AQueous Assay reagent; Promega). Briefly, the CellTiter 96 AQueous One Solution Reagent was added to each well and incubated at 37°C for 3 hr. Cell proliferation was determined by measuring the absorbance at 490 nm using a microtiter plate reader (Molecular Devices, Sunnyvale, CA). The LC50 was obtained from the MTS viability curves using GraphPad Prism 5. Experiments were carried out in triplicate.
FISH analysis for the circular HAC
The presence of the circular HAC in an autonomous form was confirmed by FISH analysis as previously described (22). HT1080 cells containing the HAC were grown in DMEM medium to 70–80% confluence. Metaphase cells were obtained by adding colcemid (Gibco) to a final concentration of 0.05 μg/ml and incubating overnight. Media was aspirated, and the plate washed with 1x PBS. Cells were detached from the plate by 0.25% Trypsin, washed off with DMEM, pelleted and resuspended in 10 ml of 50 mM KCl hypotonic solution for 30 min at 37°C. Cells were fixed by three washes of fixative solution (75% acetic acid, 25% methanol). Between washes, cells were pelleted by centrifugation at 900 rpm for 4 min. Metaphase cells were evenly spread on a microscope slide and the fixative solution evaporated over boiling water. Dry slides were rehydrated with 1xPBS for 15 min and fixed in 4% formaldehyde-1x PBS for 2 min, followed by three 5 min 1xPBS washes and ethanol series dehydration. PNA (peptide nucleic acid) labeled probes used were telomere (CCCTAA)3-Cy3) (PerSeptive Biosystems, Inc.) and tetO-alphoid array (FITC-OO-ACCACTCCCTATCAG) (Panagene, South Korea). Ten nanomol of each PNA probe was mixed with hybridization buffer and applied to the slide, followed by denaturation at 800C for 3 min. Slides were hybridized for 2 hours at room temperature in the dark. Slides were washed twice in 70% formamide, 10 mM Tris pH 7.2, 0.1% BSA, followed by three washes with 1xTBS, 0.08% Tween-20. Slides were dehydrated gradually with a series of 70%, 90% and 100% ethanol washes and mounted (Vectorshield with DAPI). Images were captured using a Zeiss Microscope (Axiophot) equipped with a cooled-charge-coupled device (CCD) camera (Cool SNAP HQ, Photometric) and analyzed by IP lab software (Signal Analytics). The PNA-DNA hybrid probes demonstrated a high hybridization efficiency and staining.
FISH analysis for the linear HAC
Slides with metaphases were rehydrated with 1xPBS for 15 min at RT, fixed with 4% Paraformaldehyde (PFA), prepared on 1xPBS, for 2 min and washed 3 times with 1xPBS for 5 min. Then slides were gradually dehydrated at RT for 5 min each: 70% EtOH, 90% EtOH, 100% EtOH and then dried. Hybridization mix (10 mM Tris-HCl, pH7.4; 70% Formamide; 5% Dextran sulfate; 10 ng PNA-TRITC-telomere (Panagen); 10 ng Biotin-labeled-EGFF sequence in the linear HAC was applied onto each dehydrated slide in 20 uL of volume and covered with a coverslip. Slides were next denaturated at 80°C on heating table for 3 min in the dark and then incubated 2–6 hours at RT in the dark. Cover glasses were then removed and slides washed 2 times in washing Solution I (70% Formamide/10 mM Tris-HCl pH 7.4/0.1% BSA) for 15 min, 3 times in washing Solution II (20 mM Tris-HCl pH 7.4; 136 mM NaCl; 0.08% Tween) for 5 min, and briefly rinsed once in PBS. For biotin detection, 60 μL of Avidin-FITC solution (Sigma) was applied, covered with a 24×60 mm coverslip and incubated for 40 min at 37°C in a moist chamber in the dark. To amplify the signal, slides were washed in 4x SSC/0.1% Tween-20 three times for 2 min at 45°C, after which 60 μl of anti-avidin solution (Sigma, 1:200 dilution in 4xSSC/0.1 Tween-20) was applied, covered with 24×60 mm coverslip and incubated for 40 min at 37°C in a moist chamber in the dark. Next, a second round of detection was performed in the same manner. Then slides were gradually dehydrated at RT for 5 min each: 70% EtOH, 90% EtOH, 100% EtOH and dried. Slides were mounted in Vectashield mounting media, containing DAPI (Vector Laboratories). Images were captured and analyzed using a DeltaVision microscopy imaging system and software in the CRC, LRBGE Fluorescence Imaging Facility (NIH).
Immunofluorescence
For Figures 1 and 8: treated cells were fixed in 4% PFA in PBS for 15 min at RT. Cells were rinsed two times quickly with PBS followed by incubation for 15 min with a last PBS wash at RT. 200 ml of 5% BSA in PBS-TT (PBS containing 0.5% Tween-20, 0.1% Triton X-100) were added to the washed cells and incubated for 30 min in a humid chamber. Cells were rinsed once in PBS-T (PBS, containing 0.1% Tween-20) for 5 min. 200 μl of γHistone2AX antibody (Abcam, Cat. No. # 05–636, mouse origin, dilution 1:500) and TRF2 antibody (Santa Cruz, Cat. No. # sc-9143, rabbit origin, dilution 1:200) in 1% BSA in PBS-TT were added for 2 hr at RT in the humid chamber. The samples were washed three times for 5 min in PBS-T. 200 μl of secondary antibodies (goat-anti-mouse Alexa 488, dilution 1:500, Life Technology A11029; goat-anti-rabbit Alexa 555, dilution 1:500, Life Technology A21428) were applied at RT in the humid chamber for 1 hr. The samples were washed three times in PBS-T for 5 min. The samples were counterstained with DAPI and mounted with mounting media (ProLong™ Diamond Antifade Mountant with DAPI, Life Technology, P36962). Samples were analyzed using DeltaVision Microscopic System at the CRC, LRBGE Fluorescence Imaging Facility (NIH). For each compound, at least 120 nuclei were analyzed.
Figure 1.
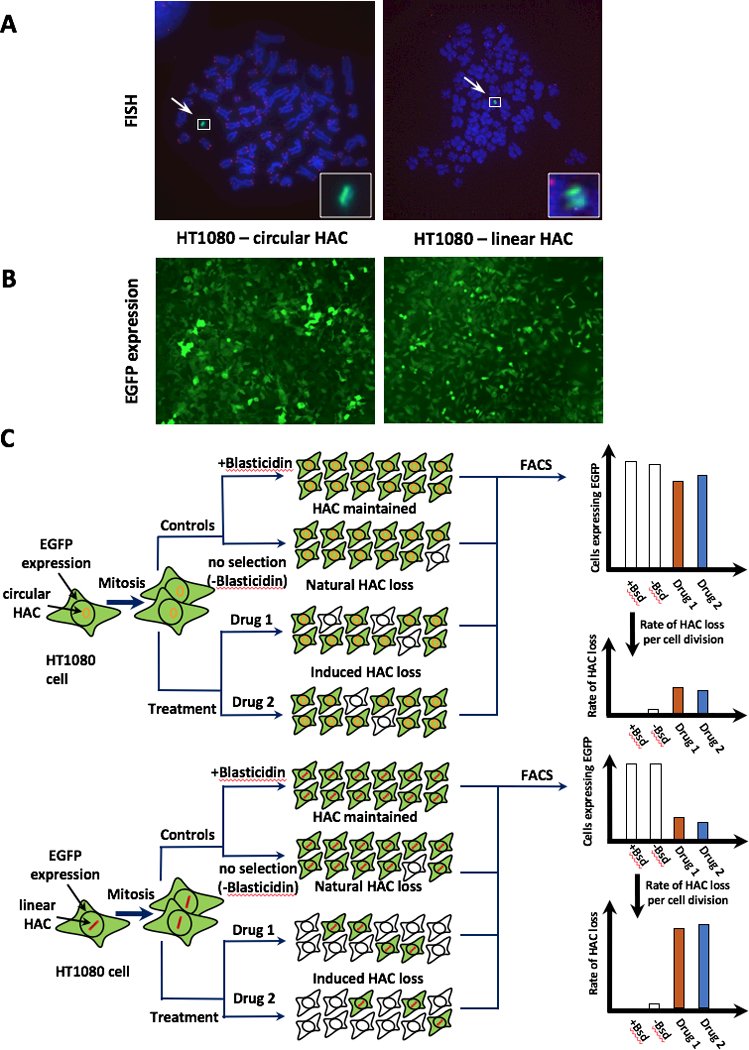
Scheme of an assay for measuring chromosome instability (CIN) based on the use of a linear HAC versus a circular HAC, both containing the EGFP transgene. A, FISH analysis of the HACs in HT1080 cells. The circular HAC was visualized using PNA probes for telomeres and for the vector part of the HAC as previously described (22,24). The linear HAC was visualized using the PNA probes designed in this work (see Materials and Methods for details). Chromosomal DNA was counterstained with DAPI. The HACs are indicated by arrowhead. B, Fluorescence images of cells carrying the EGFP-containing HACs. C, Scheme of an assay for measuring CIN based on the use of linear versus circular HACs, both containing the EGFP transgene. Cells that inherit any of these HACs display green fluorescence, while cells that lack them do not. Because both HACs are pretty stable during cell division, it is expected that the control cells should display uniform green fluorescence while there will be a mixture of EGFP positive and negative cells in the drug-treated population if the drug is specific to telomeres or telomerase (a linear HAC). The actual percentage of cells carrying EGFP-HAC can be measured by FACS as previously described (18).
For Figures 4, 5, 6, 7 and Supplementary Figures S5, S6, S7, S8, S9, S10, S11, S12, S13 and S14: after drug treatment, the cells were rinsed in PBS and then fixed in 4% paraformaldehyde in PBS. Cells were then permeabilised and blocked before incubation with primary antibodies. Primary antibodies used in this study include: mouse monoclonal anti α-tubulin (SIGMA), rabbit polyclonal anti-Survivin (NOVUS), rabbit polyclonal anti-Aurora B (Abcam), rabbit polyclonal anti-INCENP (Cell Signalling), mouse monoclonal antibody anti-Borealin (MBL), rabbit polyclonal anti-phosphorylated Histone3 Ser10 (Millipore), mouse monoclonal anti-LAP2 (BD Biosciences), mouse monoclonal anti-phosphorylated Gamma H2A.X Ser139 (Abcam). Fluorophore-conjugated secondary antibodies were purchased from Jackson Immunoresearch. Samples were counterstained with DAPI and mounted in Vectashield. Microscope images were acquired on a DeltaVision Core system (Applied Precision) using an inverted Olympus IX-71 stand, with an Olympus UPlanSApo ×100 oil immersion objective [numerical aperture (NA) 1.4] and a LED light source. The camera (Photometrics Cool Snap HQ), shutter and stage were controlled by SoftWorx 5.5.0 (Applied Precision). Z-series were collected with a spacing of 0.2 μm, and image stacks were subsequently deconvolved using SoftWorx. Projected Z-sections were exported as TIFF files into Adobe Photoshop.
Figure 4.
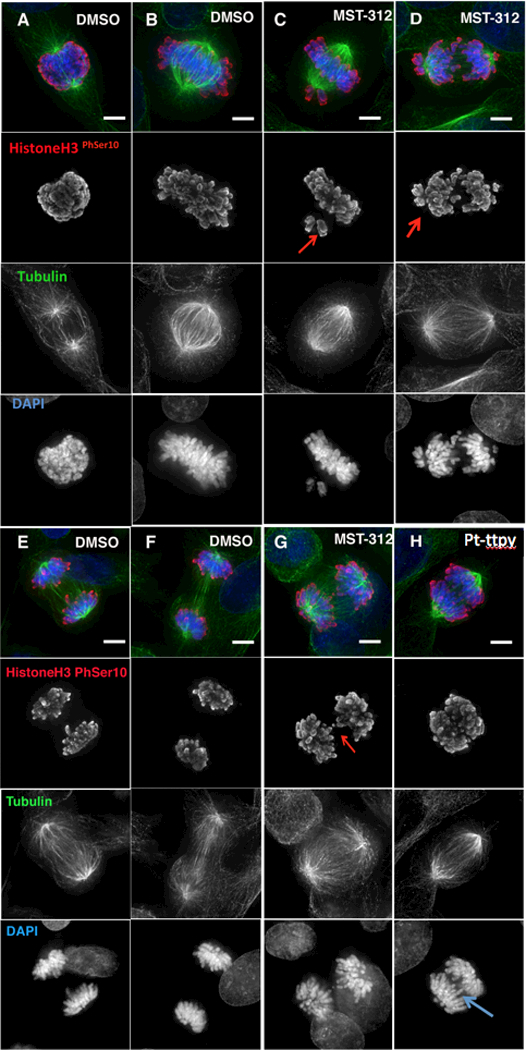
Analysis of mitotic phenotypes induced by MST-312 and Pt-ttpy treatment of HT1080 cells. Cells in different stages of mitosis were stained with antibodies against phosphorylated HistoneH3 (Ser10) –a mitotic marker (in red) and α-tubulin (in green). A-B, and E-F, DMSO-treated control cells. C-D, MST-312-treated cells show chromosome alignment defects. Red arrows point at uncongressed chromosomes. G-H, Chromatin bridges appear in both MST-312- and Pt-ttpy-treated cells. Red/blue arrows point at chromatin bridges.
Figure 5.
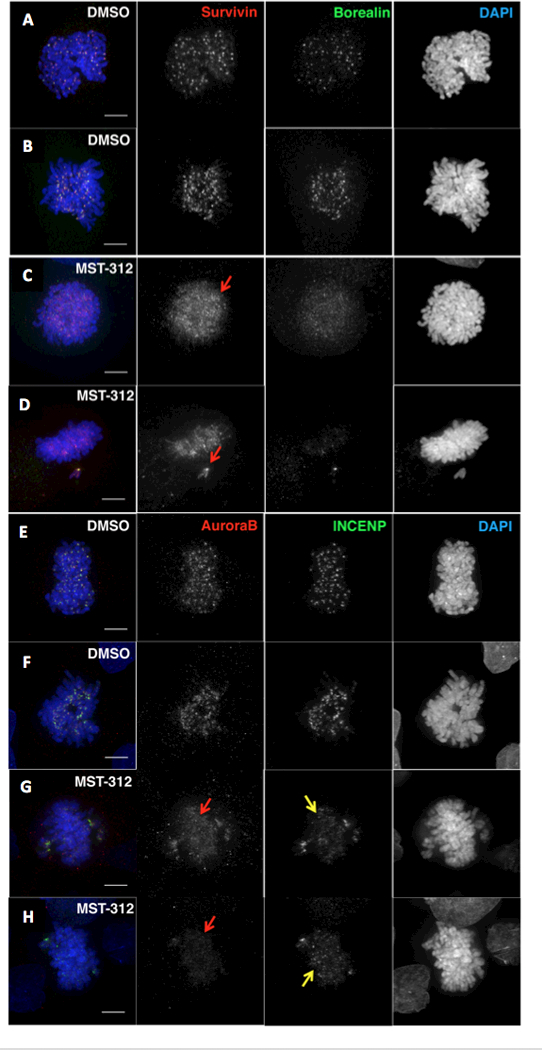
Analysis of the level and localization of the Chromosomal Passenger Complex (CPC) in early mitosis in MST-312-treated HT1080 cells. A-D, Cells were stained with antibodies against Survivin (in red) and Borealin (in green). Localization of Survivin/Borealin in (A-B) control cells (DMSO) and (C-D) MST-312-treated cells in early mitosis. Red arrows point to examples where the proteins are dispersed along chromatin in treated cells. E-H, Cells were stained with antibodies against Aurora B (in red) and INCENP (in green). Localization of Aurora B and INCENP in (E-F) control cells (DMSO) and (G-H) MST-312-treated cells in early mitosis. In drug-treated cells the levels of all four CPC components are lower and the proteins appears to be dispersed along chromosome arms (red/yellow arrows).
Figure 6.
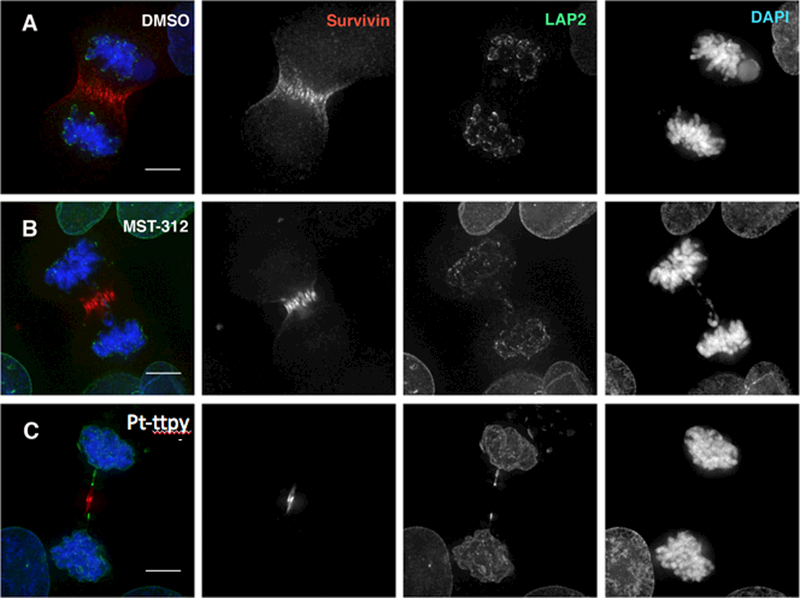
MST-312 and Pt-ttpy treatment induces chromatin bridges in late mitosis in HT1080 cells. Cells in mitosis were stained with antibodies against LAP2 (in green) and α-Survivin (in red). A, Control cells (DMSO); B, MST-312-treated cells. C, Pt-ttpy-treated cells.
Figure 7.
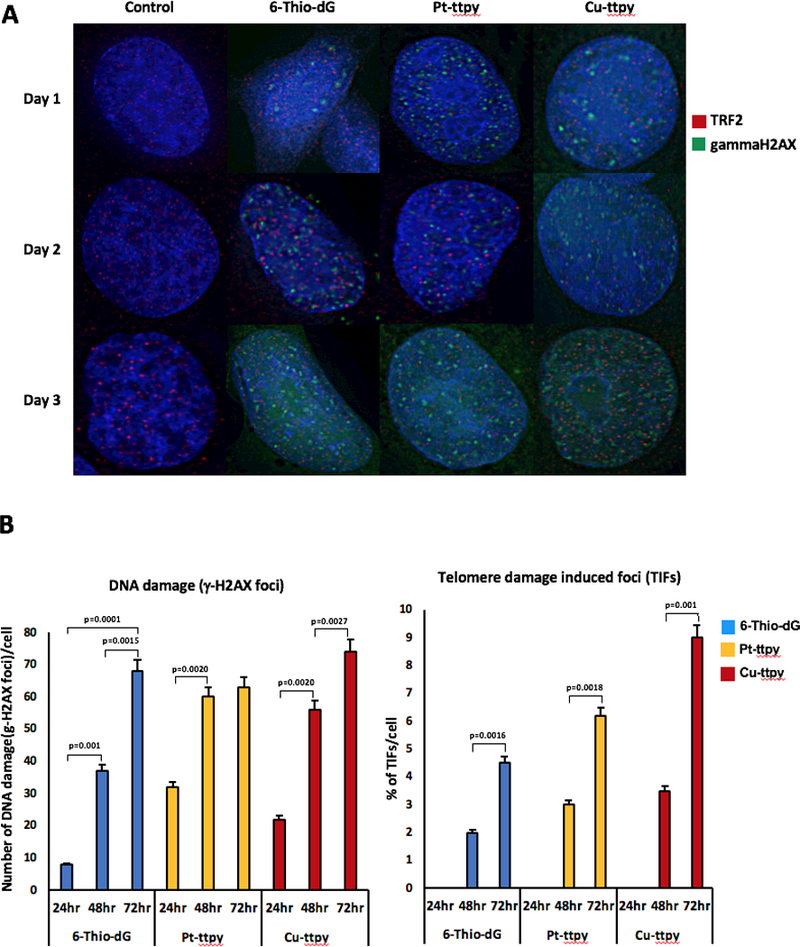
Co-localization of γHistone2AX foci with the telomeric protein TRF2. A, Example of immuno-staining of the cells treated with telomere-binding drugs 6-Thio-dG, Pt-ttpy and Cu-ttpy. Green signals – γHistone2AX staining. Red signals – TRF2 as a marker for telomeres localization. Accumulation of γHistone2AX foci occurred at day 3 in all cases. Co-localization of green and red signals indicates for the presence of double-strand breaks (DSBs) in the telomeric sequences (for details see Materials and Methods). B, A statistical effect of co-localization of γHistone2AX foci and TRF2 protein. A statistical effect was determined at day 3. Left panel shows the number of γHistone2AX foci in the cells treated with the drugs after 24, 48 and 72 hrs. Right panel shows the percentage of γHistone2AX foci present in the telomeric sequences. For each sample at least 120 nuclei were analyzed. Statistically significant (Fisher exact test: p-value; 2-tailed) results are indicated on the figure with square brackets.
Trichostatin A treatment
Aliquots of ∼2.4 × 106 HT1080 cells carrying a circular alphoidtetO-HAC or a linear 21ΔqHAC both carrying EGFP were incubated in 2 ml of non-selective medium containing 100 ng/ml of Trichostatin A (TSA) (Wako) for 24 hr in the presence of doxycycline. Then cell samples were collected and analyzed by FACS.
Western blotting
For Western blot analysis, HT1080 lysates were subjected to electrophoresis on SDS-PAGE followed by transfer to Hybond membrane (Amersham Biosciences, NJ) according to the manufacturer’s instructions. Concentrations for blot analysis of anti-hTERT as well as secondary antibodies varied according to the manufacturer’s recommendation (Santa Cruz Biotehnology, Jackson ImmunoResearch, West Grove, PA). The blot was developed with an ECL system (Amersham Biosciences).
Results
Experimental design for the identification of compounds specifically targeting telomeres and telomerase
To identify compounds specifically targeting telomeres or telomerase, we developed an assay based on the use of two different human artificial chromosomes (HACs) – Figure 1. Both HACs are of approximately the same size (5 Mb) (23,24) but one of them is circular while the other is linear and has telomeres (Fig. 1A). Both HACs have functional kinetochores and are maintained as non-essential extra chromosomes, replicating and segregating like normal chromosomes in human cells. The rate of spontaneous loss (no treatment) was approximately the same for circular and linear HACs (loss rate = 0.0024 and 0.0054, correspondingly). Both HACs carry the same EGFP (enhanced green fluorescence protein) transgene flanked by HS4 insulators that is stably expressed from the HACs. Cells that inherit the HAC display green fluorescence, while cells that lack it do not (Fig. 1B). After drug treatment, there are two expected outcomes: either there is no change in the percentage of EGFP-expressing cells (no effect on HAC stability) or, if the compound induces chromosome segregation errors, there is an increase in the percentage of EGFP-negative cells (Fig. 1C). Control untreated cells containing either of these HACs show uniform green fluorescence. The actual percentage of cells carrying EGFP-HAC can be measured by FACS as previously described (18).
The circular HAC used in our experiments is the alphoidtetO-HAC (15), which was engineered from a synthetic alphoid DNA array and was previously used to screen for drugs inducing chromosome instability (CIN) (18). The linear 21ΔqHAC was engineered by truncation of human chromosome 21 using telomere containing plasmids (25). These HACs possess several useful and similar features: 1) they have a well-defined architecture (24,26); 2) they are present episomally, independently of the host chromosomes (13,14,23,27); 3) they are mitotically stable both in human cell lines and mice, but slightly less stable than natural human chromosomes, so that the system is sensitized (15,25,28–32); 4) EGFP expression from the HACs is stable (33,34). No epigenetic silencing of EGFP was observed after 24 months of continuous culturing of HAC-containing human HT1080 cells under blasticidin selection (33,34). Importantly for these experiments, HT1080 cells containing either the linear or circular HAC express human telomerase (hTERT) (Supplementary Figure S1). The experimental design comparing segregation fidelity of the circular and linear HACs reported here can be used not only to identify new compounds that specifically target telomeres or telomerase but also to rank those compounds according to their effect on the segregation of the linear HAC
Compounds used in this study
Initially we chose several groups of known anti-telomerase compounds. They included antisense oligonucleotides that target the RNA template of human telomerase (hTERT), a set of small molecule telomerase inhibitors that block the catalytic activity of the enzyme and compounds that block the access of telomerase to telomeres, among them G-quadruplex (G4) stabilizers. These compounds can bind to and stabilize the secondary DNA structures formed by genomic G-rich sequences such as telomeres. This blocks telomerase access to telomeres, uncapping them and mimicking ssDNA overhang exposure. Thus, these agents are good candidates to increase chromosome instability. In this study, we used a panel of G-quadruplex stabilizers belonging to various chemical classes: the bisquinolinium series comprised of Phen-DC3, Phen-DC6, PDC and their functional derivatives Phen-DC3-C4Bn, PDC-C4Bn, Phen-DC3 Bisalk1; the styryl derivatives series PhenDV, PhenDV-An and Bisphenyl-Vpy; the metal complexes series CuBisQ, Pt-ttpy and Cu-ttpy; and a series of benchmark compounds of diverse structures MMQ3, TrisQ, TMPyP4, BRACO19, PIPER and Pyridostatin (Supplementary Table S1). A negative control CN1 that has no G4-binding ability was also included in the analysis. The compounds effecting telomerase were GRN163L, BIBR1532, MST-312 and 6-Thio-dG (Supplementary Table S1). Chemical structures of most of the compounds have been published previously (see references in Supplementary Table S1). The structures and synthesis of four G4-stabilizers developed recently in our group, Phen-DC3 Bisalk1, Phen-DC3C4Bn, PhenDMA and Bisphenyl-VPy, are shown in Supplementary Figure S2A-D. The structures of previously developed components, Pt-ttpy and Cu-ttpy, are shown in Supplementary Figure S2E-F.
We previously demonstrated in other screens that the highest rate of drug-induced HAC loss occurs at the compound’s LC50 (12). Higher concentrations of drugs killed more cells but did not increase the rate of HAC loss (12,18). In contrast, treatment at lower concentrations induced either no or lower frequency HAC loss. Because the LC50 provides a useful parameter to normalize the results from different drugs/compounds we determined LC50 values for all compounds, except the specific telomerase inhibitor GRN163L, i.e. conditions under which HT1080 cell viability was around 50%. For GRN163L we could not determine the LC50 because even the highest concentration of this drug did not lead to detectable cell death. Thus, for GRN163L we used concentrations recommended in the literature (10,35). The complete list of compounds used in this work is presented in Supplementary Table S1. The results of LC50 determinations are shown in Supplementary Table S2.
Verification of the system: effect of GRN163L on mitotic stability of circular and linear HACs
Drugs that induce CIN via mechanisms that do not involve telomeres/telomerase should affect HAC stability independently of the presence or absence of telomeres. In our previous work using a circular HAC to rank a set of anticancer drugs according to their effect on chromosome instability (18) significant effects were observed after treatment with Taxol (microtubule stabilizing agent), LMP400 and Camptothecin [specific inhibitors of topoisomerase 1 (TOP1)], Olaparib (PARP inhibitor), Cisplatin and Gemcitabine (DNA damage compounds). In the present control experiments, the rate of loss in response to drug treatment was indistinguishable between linear and circular HACs (Supplementary Figure S3A). Notably no increase in green cells was observed when the histone deacetylase inhibitor TSA (trichostatin A) was added to the culture (Supplementary Figure S3B), indicating that these inhibitors do not induce transgene silencing but rather induces a high rate of linear HAC loss.
We next investigated the effect on the stability of circular vs linear HAC of one of the most specific telomerase inhibitors described to date, imetelstat or GRN163L. This compound consists of a 13-mer oligonucleotide N3’-P5’ thio-phosphoramidate covalently attached to a C16 (palmitoyl) lipid moiety. It directly binds to the RNA component of telomerase (hTERT) with very high affinity in the active site of the telomerase enzyme (10,36). GRN163L has been shown to inhibit telomerase in a wide range of human solid tumor cells, including lung, breast, prostate liver, brain and also in hematological malignancies, including multiple myeloma and lymphoma (10,35).
Cells carrying either a linear or a circular HAC, were treated with increasing concentrations of GRN163L. Treatments were carried out for 3 days, as shorter treatment times do not result in a significant shortage of the telomeres and chromosome instability (10,35). As expected, treatment with increasing concentrations of GRN163L resulted in a specific concentration-dependent destabilization of the linear HAC (Fig. 2A). Importantly, the mitotic stability of the circular HAC was not affected under the same conditions. This proof of principle experiment indicated that our dual-HAC system allows to identify new compounds specifically targeting telomeres or telomerase.
Figure 2.
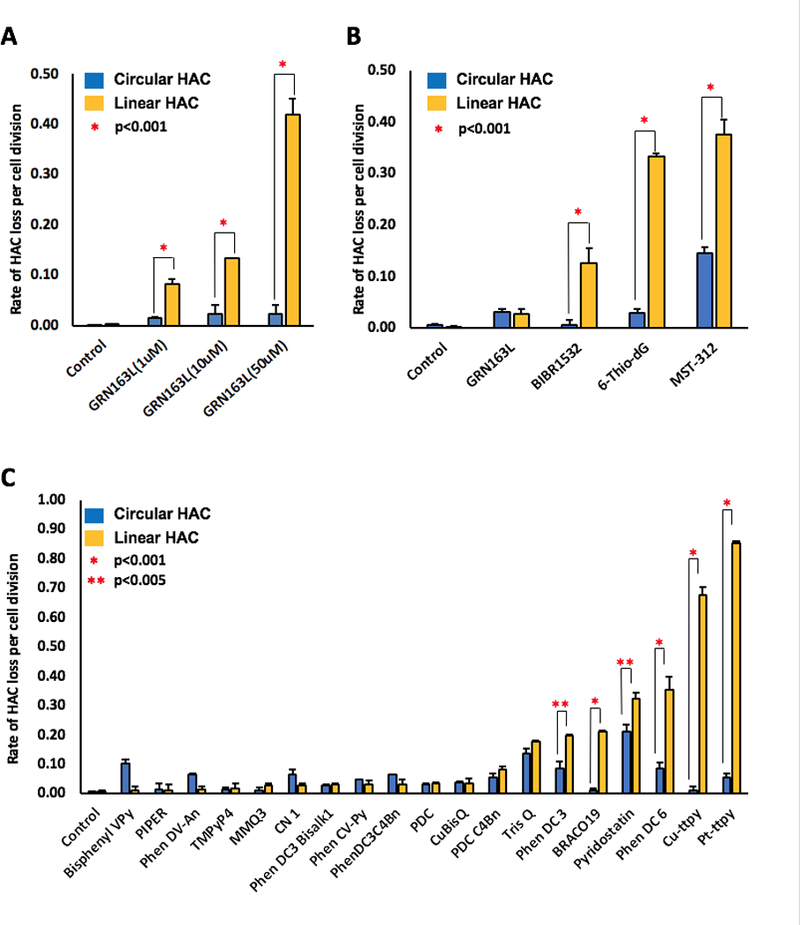
Effect of telomerase and telomeres inhibitors on a linear and a circular HAC mis-segregation rate. A, Effect of the telomerase inhibitor GRN163L, that directly binds with high affinity and specificity to the RNA component of telomerase (hTERT), on the stability of a linear and a circular HAC. HT1080 cells containing either a linear or a circular HAC were treated by GRN163L at concentrations of 1 μM, 10 μM or 50 μM for 3 days. A specific concentration-dependent destabilization of the linear HAC was observed. B, Effect of telomerase and telomeres inhibitors, 6-Thio-dG, BIBR1532, GRN163L and MST-312, on a linear versus a circular HAC mis-segregation rate. HT1080 cells containing either a linear or a circular HAC were treated with the compounds at the concentrations corresponding to their LC50 (Supplementary Table S2) overnight. MST-312 was the strongest CIN inducer (approximately 40-fold increase of loss rate of a linear HAC). C, Effect of 19 G-Quadruplex ligands that may recognize telomere repeats on a linear and a circular HAC mis-segregation rate. The highest rate of the linear HAC loss was observed after treatment by Cu-ttpy and Pt-ttpy (almost 70- and 90-fold increase compared to the control, correspondingly). *stands for <0.001 (done by t-test). ** stands for p<0.005 (done by t-test).
6-Thio-dG and MST-312 have the highest effects on the linear HAC mis-segregation rate
Four known telomerase and telomere inhibitors, 6-Thio-dG (36), BIBR1532 (37), GRN163L (10,35), and MST-312 (38), were included in the analysis. The LC50 of these compounds in HT1080 cells is shown in Supplementary Table S2. HT1080 cells carrying either a linear or a circular EGFP-HAC were treated with each compound overnight. Figure 2B summarizes the results on HAC stability. Treatment with BIBR1532 increased the rate of loss of the linear HAC approximately 15 times compared to control untreated cells. The two other inhibitors tested, 6-Thio-dG and MST-312, also resulted in a significant increase of the rate of loss of the linear HAC, with the highest effect observed after treatment with MST-312 (approximately a 40-fold increase). Interestingly, this inhibitor also affected the stability of the circular HAC (approximately a 15-fold increase). Overnight treatment by GRN163L at 50 μM only slightly and equally affected stability of linear and circular HACs despite the fact that much lower concentrations of the same compound (1 μM and 10 μM) but longer treatment (3 days) specifically and significantly increased the rate of loss of only the linear HAC (Fig. 2A).
To summarize, the dual-HAC assay allowed us to rank the strongest and most specific inhibitors of telomerase and telomeres according to their effects on chromosome stability. Two inhibitors in particular, 6-Thio-dG and MST-312, caused a significant increase of the rate of loss of the linear HAC after one day of treatment. 6-Thio-dG appeared to be the best among the compounds analyzed, as it exhibited both the highest and most specific effect on linear HAC stability (Fig. 2B).
Analysis of G-quadruplex ligands revealed two compounds, Pt-ttpy and Cu-ttpy, with significant effects on mitotic transmission of the linear HAC
Next, we investigated how structurally diverse G-quadruplex ligands affect chromosome stability. HT1080 cells carrying the circular or the linear EGFP-HAC were treated overnight with 19 different G4 ligands: Phen-DC3, Phen-DC6, TrisQ, Phen-DC3C4Bn, Phen-DV-Py, Phen-DV-An, CN 1, Phen-DC3 Bisalk1, PDC, PDC C4Bn, CuBisQ, Pt-ttpy, Cu-ttpy, Bisphenyl-Vpy, MMQ3, TMPyP4, BRACO19, PIPER and Pyridostatin. (LC50 values of these compounds are shown in Supplementary Table S2.) Figure 2C illustrates the effect of these compounds on the rate of HAC loss. As seen, no significant increase in loss of the circular HAC was detected with most of the analyzed compounds. Eight compounds, PDC C4Bn, TrisQ, Phen-DC3, BRACO19, Pyridostatin, Phen-DC6, Cu-ttpy, and Pt-ttpy, increased the rate of linear HAC loss. After treatment by Cu-ttpy and Pt-ttpy this rate was almost 70- and 90-fold higher compared to that observed in control untreated cells (Fig. 2C).
In summary, our dual-HAC assay allowed us to rank 19 G-quadruplex ligands according to their effects on chromosome stability. The two compounds, Cu-ttpy and Pt-ttpy, exhibited the highest effects on stability of the linear but not the circular HAC. This makes them potentially interesting candidates for future development as therapeutic agents, and therefore we decided to investigate further their mechanism of action in cancer cells.
Pt-ttpy and MST-312 treatments produce defects in mitosis
Pt-ttpy and Cu-ttpy are metallo-organic complexes of a similar structure containing a tolylterpyridine moiety (ttpy) coordinated with either copper (Cu 2+) or platinum (Pt 2+) cation (Supplementary Figure S2E-F) (20,21). To explore the mechanism by which they disturb chromosome transmission, we have performed the first detailed analysis of the effect of Pt-ttpy in mitosis. In parallel, we analyzed the effect of the telomerase inhibitor MST-312, a compound that also induces a high rate of instability in our assay for the linear HAC (Fig. 2B). In these experiments, the cells were treated by noncytotoxic drugs concentrations as a dose based on published data and proven experimentally for our cell lines (see Supplementary Table S2). Preliminary studies of MST-312 have revealed some mitotic abnormalities, but still many questions remained unanswered in this regard (38–40). To rule out cell-line specific phenotypes, all experiments were carried out in two different telomerase-expressing cell lines, HT1080 (Supplementary Figure S1) and HCT116 (41).
Pt-ttpy- and MST-312-treated HT1080 and HCT116 cells were stained with antibodies against phosphorylated HistoneH3 (Ser10) – a mitosis-specific modification- and against α-tubulin to visualise the mitotic spindles (Fig. 3A-D and Fig. 4A-H; Supplementary Figures S4A-F and S5A-I). MST-312-treatment of HT1080 cells caused a significant decrease of the mitotic index (the fraction of cells in a population undergoing mitosis) (Fig. 3A), associated with a significant increase in the proportion of mitotic cells in prometaphase and a decrease of the proportion of cells in cytokinesis (Fig. 3B). Further quantification revealed a significant increase in mitotic abnormalities in MST-312-treated cells (Fig. 3C). These included defects in chromosome alignment (Fig. 4C-D) and chromatin bridges in late mitosis (Fig. 4G). Quantification of chromatin bridges is of special interest because they can form when telomeres of sister chromatids fuse and fail to completely segregate into the respective daughter cells (42). In contrast, Pt-ttpy treatment of HT1080 cells caused no significant decrease in the mitotic index (Fig. 3A) or total percentage of mitotic abnormalities (Fig. 3C). Notably, Pt-ttpy also induced chromatin bridges (Fig. 4H).
Figure 3.
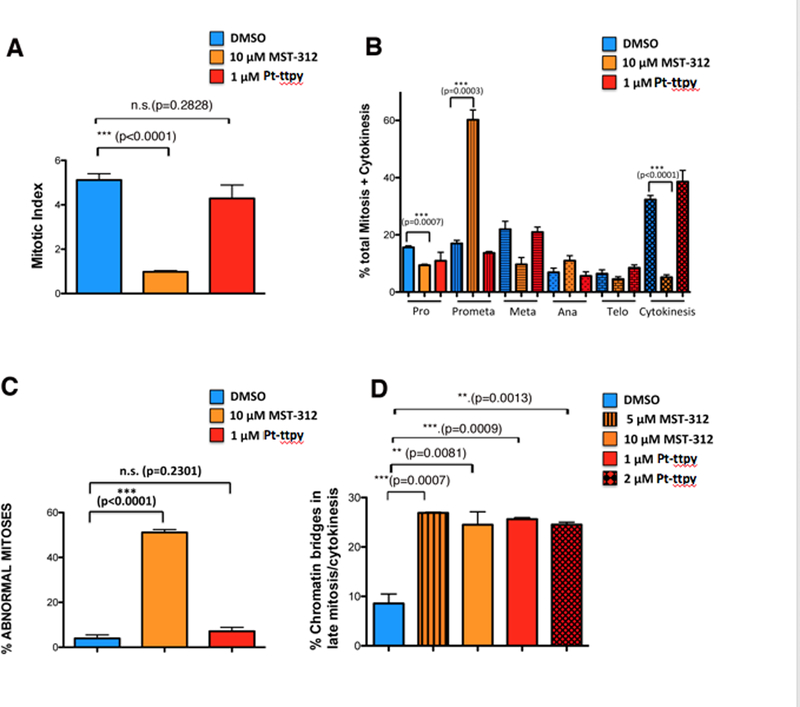
Quantification of defects in mitosis upon MST-312 and Pt-ttpy treatment of HT1080 cells. A, Mitotic index (a ratio between the number of cells in a population undergoing mitosis to the total number of cells) upon MST-312 or Pt-ttpy treatment. Three independent experiments were performed for each drug and more than 1,000 cells (n) were screened. B, Distribution of cells in different stages of mitosis upon MST-312 or Pt-ttpy treatment. Three independent experiments were performed for each drug and more than 100 mitotic cells (n) were screened. C, The percentage of cells exhibiting defects in mitosis upon MST-312 or Pt-ttpy treatment. D, The percentage of cells presenting chromatin bridges in late mitosis upon MST-312 (5 μM, 10 μM) or Pt-ttpy (1 μM, 2 μM) treatment, quantified using LAP2 as a marker. In all experiments bars compare results of untreated cells (DMSO) with drug-treated cells. Relevant results from statistical analysis (t tests) are shown; error bars correspond to SD; N.S.= not significant.
HTC116-treated cells showed similar phenotypes to HT1080-treated cells (Supplementary Figures S4A-F and S5A-I) but in addition we observed an increased frequency of spindle pole defects (Supplementary Figure S5E-F). It thus appears that the spindle phenotypes are specific to the HCT116 cell line. We have observed similar effects previously in this cell line when studying the effect of other drugs treatments (Carmena, unpublished results). In contrast, the MST-312-induced chromosome misalignment phenotype was observed in both cell lines and is consistent with the increase in the frequency of prometaphases.
MST-312 treatment affects the localization and function of the Chromosomal Passenger Complex (CPC)
The defects in chromosome alignment shown by the MST-312-treated cells are reminiscent of the phenotypes caused by defects in CPC function. Aurora B kinase (the enzymatically active component of the CPC) destabilizes incorrect kinetochore-microtubule attachments and, therefore, promotes correct and timely chromosome bi-orientation (43,44).
Levels and localization of the CPC components were analysed in HT1080 (Fig. 5) and HCT116 cells (Supplemental Figure S6). Levels of Survivin were reduced in MST-312-treated cells and its localization was abnormal in early mitosis (Fig. 5A-D and Supplementary Figure S6A-C). Similar results were obtained when studying the distribution of the other CPC components, Borealin (Fig. 5A-D), INCENP and Aurora B kinase (Fig. 5E-H and Supplementary Figure S6D-E). To assess whether this had an effect on Aurora B activity, we stained MST-312-treated cells with an antibody against the activated form of the kinase (anti-Aurora B phospho-T232) (Supplementary Figure S7A-H). This revealed that the level of the active Aurora B was reduced in early mitosis. Thus, MST-312 affects - either directly or indirectly - the function of Aurora B kinase in both HT1080 and HCT116 cells. This is likely to be the cause of the defects in chromosome alignment and consequent delay in prometaphase observed in drug-treated cells.
Pt-ttpy treatment did not result in chromosome alignment defects as described above (Fig. 4; Supplementary Figure S5). As known, MST-312 inhibits telomerase activity (45) while Pt-ttpy binds to G-quadruplexes, hindering telomerase access to telomeres (but not affecting telomerase activity) (20,21). Thus, one possible explanation is that telomerase activity is somehow required for Aurora B/CPC function, so this would not be altered after treatment with Pt-ttpy, as telomerase remains active. Another possible explanation is that inhibition of Aurora B kinase is an off-target effect of MST-312.
To test this possibility, we used U2OS, an ALT cell line that does not express telomerase (46). MST-312-treated U2OS cells showed the chromosome misalignment phenotype [Supplementary Figures S8A-F (DMSO); Supplementary Figures S8A’-F‘ (MST-312) and Supplementary Figure S9A-B] suggesting that indeed Aurora B kinase inhibition is likely an off-target, telomerase-independent effect of MST-312. This effect possibly contributed to the elevated rate of chromosome instability detected in our original screen and could also explain why this compound has also an effect on segregation of the circular HAC. (Fig. 2A).
Pt-ttpy and MST-312 both induce chromatin bridges and UFBs (Ultrafine Bridges) in mitosis
Our analysis of mitosis in drug-treated cells revealed the presence of chromatin bridges in late mitosis and cytokinesis (see Fig. 4 and Supplementary Figure S5). To carry out a detailed quantification of the frequency of chromosome bridges in late mitosis in HT1080 and HCT116 cells treated with Pt-ttpy and MST-312, we used an antibody against Survivin and LAP2, a nuclear envelope protein that decorates chromatin bridges. This works because during late anaphase and telophase LAP2 and other nuclear envelope components decorate the decondensing chromatin while it is still in the process of moving polewards. With LAP2 antibody, bridges are readily visible, even when they are too fine to be visualised by DAPI staining (Fig. 6A-C and Supplementary Figure S10A-E). These experiments revealed that our previous analysis had underestimated the number of chromatin bridges in late mitosis (anaphase to cytokinesis). As described above, we observed a statistically significant increase in the percentage of late mitotic cells showing chromatin bridges in cells treated with either Pt-ttpy or MST-312 (Fig. 3D; Supplementary Figure S4F). A similar analysis in telomerase-negative U2OS cells showed no significant increase in chromatin bridges (Supplementary Figure S9B). We, therefore, concluded that this phenotype is likely to be a consequence of the disruption of telomerase activity and/or telomere function.
We also analysed the formation of Ultrafine Bridges (UFBs) following Pt-ttpy and MST-312 treatment. HCT116-treated cells (Supplementary Figure S11A-C) were stained with antibodies against Blooms Syndrome protein (BLM) and against phosphorylated histone H2AX (γHistone2AX). UFBs were increased in anaphase/telophase after either treatment (Supplementary Figures S11D). The similar results were obtained in HT1080-treated cells (Supplementary Figure S12A-D). Importantly, no significant increase in the frequency of UFBs was observed in the telomerase-negative U2OS cells (Supplementary Figure S13A-D).
Pt-ttpy-treated cells show an increased number of double-stranded breaks (DSBs) that colocalise with telomeric markers
To begin to investigate the mechanism(s) by which drug treatment resulted in chromosome loss, we stained double-stranded breaks (DSBs) in interphase drug-treated cells with an antibody against phosphorylated γH2AX. The number of DSBs was increased after treatment with either Pt-ttpy or MST-312 (Supplementary Figure S14A-F). To determine whether DSBs are associated with telomeric sequences, we examined co-localization of γH2AX foci with the telomeric protein TRF2 (telomeric repeat binding factor 2) (Fig. 7A). Because the nucleoside analog 6-Thio-dG is known to be incorporated into de novo-synthesized telomeres and cause DNA breaks (36,47) it was used as a control in this experiment. In addition, we included in this analysis Cu-ttpy, a compound with a structure similar to Pt-ttpy. Immunostaining with antibodies against TRF2 and γH2AX was carried out every 24 hours for 3 days (see Materials and Methods). A statistically significant co-localization of γH2AX foci and TRF2 protein was observed at day 3 for 6-Thio-dG (4.5%), Pt-ttpy (6.2%), and Cu-ttpy (9%) (Fig. 7B). Note that telomeres represent only ~1/6,000 of the total human genome. Therefore, if the γH2AX foci were distributed evenly along the chromosomes, at telomeres we would expect to observe only 0.016% colocalization. As shown above, Pt-ttpy also induces additional DSBs outside of telomeres. This is consistent with the description of non-telomeric targets for G4-stabilizers in the promoter sequences of some oncogenes and other G-rich genomic regions (21,48,49). G-quadruplex-forming genomic sequences, similar to telomeres, represent natural replication fork barriers.
To summarize all of the above experiments, chromosome loss after Pt-ttpy treatment may be due to the induction of DSBs that are predominantly localized at telomeres. Such telomere damage could explain the increased formation of chromosome bridges and UFBs in the drug treated cells.
Discussion
As telomerase is constitutively expressed in many human cancers, telomerase-targeting therapy has been considered to be a potentially promising approach for cancer treatment (6,50–52). However, a limited number of chemical compounds that target telomerase or telomeres have been identified and only some of these are in clinical trials. Moreover, protocols that allow quantitative comparison of the efficiency and specificity of these compounds were lacking.
To address this point, we developed a novel assay allowing comparison of libraries of compounds for their ability to induce telomere dysfunction leading to chromosome loss. Even transient telomere dysfunction can induce chromosomal instability (CIN) in human cells (53). Therefore, the activity of each compound can be evaluated based on its effect on CIN. To quantify this effect, we used two isogenic cell lines with linear and circular HACs carrying the EGFP color marker. Specific destabilization of the linear HAC (containing telomeres) in response to drug treatment is consistent with specific targeting of telomeres or telomerase. Conversely, destabilization of both linear and circular HACs suggests that a compound has off-target effects in addition to its effect on telomerase. We initially verified our dual-HAC system using GRN163L, a well-characterized and highly specific inhibitor of telomerase. We found that this experimental design can be used to rank known and newly developed compounds targeting telomerase or telomeres.
In this study, the dual-HAC assay was applied to analyze a set of known and newly developed compounds targeting telomerase or telomeres, including G4 ligands. For each drug, the rate of HAC loss was quantified and within each analyzed group, the compounds were ranked according to their HAC-destabilizing potency. Analysis of four well-known telomerase inhibitors revealed the nucleoside analogue 6-Thio-dG (36,47) as the highest and the most specific drug with respect to its effect on the linear HAC stability in this assay. It was quite unexpected that this drug, which may be incorporated into DNA strands during replication, induced a more specific effect on linear HAC stability than other analyzed telomerase inhibitors such as MST-312. Furthermore, our studies of MST-312 action in ALT cells that lack active telomerase suggested that this drug has other targets that are important for chromosome segregation, possibly including the Chromosomal Passenger Complex.
Ranking of 19 G-quadruplex (G4) ligands revealed that two recently developed compounds, Cu-ttpy and Pt-ttpy, exhibited the highest rate of linear HAC mis-segregation, i.e. a 70 and 90-fold increase compared to the controls, respectively. Our results suggest that these two compounds or their derivatives may be useful for a new telomere-addressed anticancer approach, by exacerbating the CIN phenotype in cancer cells. One possible mechanism that might explain chromosome loss after Pt-ttpy and Cu-ttpy treatment is an increased number of double-stranded breaks (DSBs) at or near telomeres. Such telomere damage may lead to the formation of chromosome bridges and UFBs that we observed in anaphase/telophase and could ultimately result in chromosome mis-segregation. Alternatively, treatment with these agents might interfere with telomere replication, leading to UFBs formation and subsequent DNA damage. It is worth noting that we cannot exclude additional mechaisms of action for these G4 ligands such as transcriptional inactivation of genes important for chromosome segregation or induction of chromosomal DSBs as previously was described for other G4 stabilizers (21,48,49). Indeed, other authors have reported that non-specific induction of DNA damage enhances telomeric dysfunction induced by G quadruplex-stabilizing agents (54).
Among other possible applications of the dual-HAC system described here is analysis of the genetic control of the alternative lengthening of telomeres (ALT) pathway in telomerase-negative cells. The ALT pathway is specific for cancer cells (7). Until now the development of ALT-targeted therapy has been challenging as the identification of proteins specific to ALT has proven elusive: all enzymes thus far shown to play any role in ALT are also critical for normal cellular functions. Transfer of circular and linear HACs into telomerase-negative cells might be used to identify genes or compounds that disturb the ALT pathway (for example, by siRNA gene depletion).
To summarize, this study describes a novel approach that may be applied to the quantitative analysis of known and novel compounds used in cancer therapy to determine their ability and potency to specifically target telomeres or telomerase. The identification of compounds that selectively inactivate telomere replication and interfere with cell proliferation could lay the foundation for new treatment strategies for cancer.
Supplementary Material
Acknowledgements
The authors are grateful to Dr. Kazuki from Institute of Regenerative Medicine and Biofunction, Tottori University for providing information about 21ΔqHAC. The authors are also grateful to Dr. Shay from University of Texas, Southwestern Medical Center for providing us GRN163L and 6-Thio-dG and fruitful discussion. The authors also would like to thank the CRC, LRBGE Fluorescence Imaging Facility (NIH) and personally Drs. Karpova and Dr. McNally for instructions, consultations and help with the usage of a DeltaVision microscopy imaging system.
Financial Support
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, USA (V. Larionov, N. Kouprina and Y. Pommier; grant number BC 006150), a Wellcome Trust Principal Research Fellowship (W. C. Earnshaw; grant number 073915), the Grand-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, Science and Technology of Japan (H. Masumoto; grant numbers 16H04747 and 16H01414) and the Kazusa DNA Research Institute Foundation (H. Masumoto).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol 2010;20:R285–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colombo R, Moll J. Targeting aneuploid cancer cells. Expert Opin Ther Targets 2011;15:595–608 [DOI] [PubMed] [Google Scholar]
- 3.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011;333:1895–8 [DOI] [PubMed] [Google Scholar]
- 4.Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci U S A 2013;110:E4134–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swanton C, Nicke B, Schuett M, Eklund AC, Ng C, Li Q, et al. Chromosomal instability determines taxane response. Proc Natl Acad Sci U S A 2009;106:8671–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruden M, Puri N. Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev 2013;39:444–56 [DOI] [PubMed] [Google Scholar]
- 7.Sobinoff AP, Pickett HA. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet 2017;33:921–32 [DOI] [PubMed] [Google Scholar]
- 8.Arndt GM, MacKenzie KL. New prospects for targeting telomerase beyond the telomere. Nat Rev Cancer 2016;16:508–24 [DOI] [PubMed] [Google Scholar]
- 9.Crees Z, Girard J, Rios Z, Botting GM, Harrington K, Shearrow C, et al. Oligonucleotides and G-quadruplex stabilizers: targeting telomeres and telomerase in cancer therapy. Curr Pharm Des 2014;20:6422–37 [DOI] [PubMed] [Google Scholar]
- 10.Man RJ, Chen LW, Zhu HL. Telomerase inhibitors: a patent review (2010–2015). Expert Opin Ther Pat 2016;26:679–88 [DOI] [PubMed] [Google Scholar]
- 11.Neidle S. Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J 2010;277:1118–25 [DOI] [PubMed] [Google Scholar]
- 12.Lee HS, Lee NC, Grimes BR, Samoshkin A, Kononenko AV, Bansal R, et al. A new assay for measuring chromosome instability (CIN) and identification of drugs that elevate CIN in cancer cells. BMC Cancer 2013;13:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouprina N, Earnshaw WC, Masumoto H, Larionov V. A new generation of human artificial chromosomes for functional genomics and gene therapy. Cell Mol Life Sci 2013;70:1135–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kouprina N, Tomilin AN, Masumoto H, Earnshaw WC, Larionov V. Human artificial chromosome-based gene delivery vectors for biomedicine and biotechnology. Expert Opin Drug Deliv 2014;11:517–35 [DOI] [PubMed] [Google Scholar]
- 15.Nakano M, Cardinale S, Noskov VN, Gassmann R, Vagnarelli P, Kandels-Lewis S, et al. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev Cell 2008;14:507–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergmann JH, Martins NM, Larionov V, Masumoto H, Earnshaw WC. HACking the centromere chromatin code: insights from human artificial chromosomes. Chromosome Res 2012;20:505–19 [DOI] [PubMed] [Google Scholar]
- 17.Molina O, Kouprina N, Masumoto H, Larionov V, Earnshaw WC. Using human artificial chromosomes to study centromere assembly and function. Chromosoma 2017;126:559–75 [DOI] [PubMed] [Google Scholar]
- 18.Lee HS, Lee NC, Kouprina N, Kim JH, Kagansky A, Bates S, et al. Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res 2016;76:902–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JH, Lee HS, Lee NC, Goncharov NV, Kumeiko V, Masumoto H, et al. Development of a novel HAC-based “gain of signal” quantitative assay for measuring chromosome instability (CIN) in cancer cells. Oncotarget 2016;7:14841–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertrand H, Monchaud D, De Cian A, Guillot R, Mergny JL, Teulade-Fichou MP. The importance of metal geometry in the recognition of G-quadruplex-DNA by metal-terpyridine complexes. Org Biomol Chem 2007;5:2555–9 [DOI] [PubMed] [Google Scholar]
- 21.Trajkovski M, Morel E, Hamon F, Bombard S, Teulade-Fichou MP, Plavec J. Interactions of Pt-ttpy with G-Quadruplexes Originating from Promoter Region of the c-myc Gene Deciphered by NMR and Gel Electrophoresis Analysis. Chemistry 2015;21:7798–807 [DOI] [PubMed] [Google Scholar]
- 22.Erliandri I, Fu H, Nakano M, Kim JH, Miga KH, Liskovykh M, et al. Replication of alpha-satellite DNA arrays in endogenous human centromeric regions and in human artificial chromosome. Nucleic Acids Res 2014;42:11502–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kazuki Y, Oshimura M. Human artificial chromosomes for gene delivery and the development of animal models. Mol Ther 2011;19:1591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kouprina N, Samoshkin A, Erliandri I, Nakano M, Lee HS, Fu H, et al. Organization of synthetic alphoid DNA array in human artificial chromosome (HAC) with a conditional centromere. ACS Synth Biol 2012;1:590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katoh M, Ayabe F, Norikane S, Okada T, Masumoto H, Horike S, et al. Construction of a novel human artificial chromosome vector for gene delivery. Biochem Biophys Res Commun 2004;321:280–90 [DOI] [PubMed] [Google Scholar]
- 26.Kazuki Y, Hoshiya H, Takiguchi M, Abe S, Iida Y, Osaki M, et al. Refined human artificial chromosome vectors for gene therapy and animal transgenesis. Gene Ther 2011;18:384–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oshimura M, Uno N, Kazuki Y, Katoh M, Inoue T. A pathway from chromosome transfer to engineering resulting in human and mouse artificial chromosomes for a variety of applications to bio-medical challenges. Chromosome Res 2015;23:111–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoshiya H, Kazuki Y, Abe S, Takiguchi M, Kajitani N, Watanabe Y, et al. A highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4 Mb entire human dystrophin gene. Mol Ther 2009;17:309–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kazuki Y, Kobayashi K, Aueviriyavit S, Oshima T, Kuroiwa Y, Tsukazaki Y, et al. Trans-chromosomic mice containing a human CYP3A cluster for prediction of xenobiotic metabolism in humans. Hum Mol Genet 2013;22:578–92 [DOI] [PubMed] [Google Scholar]
- 30.Kim JH, Kononenko A, Erliandri I, Kim TA, Nakano M, Iida Y, et al. Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proc Natl Acad Sci U S A 2011;108:20048–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liskovykh M, Ponomartsev S, Popova E, Bader M, Kouprina N, Larionov V, et al. Stable maintenance of de novo assembled human artificial chromosomes in embryonic stem cells and their differentiated progeny in mice. Cell Cycle 2015;14:1268–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tedesco FS, Gerli MF, Perani L, Benedetti S, Ungaro F, Cassano M, et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Transl Med 2012;4:140ra89. [DOI] [PubMed] [Google Scholar]
- 33.Iida Y, Kim JH, Kazuki Y, Hoshiya H, Takiguchi M, Hayashi M, et al. Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Res 2010;17:293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otsuki A, Tahimic CG, Tomimatsu N, Katoh M, Chen DJ, Kurimasa A, et al. Construction of a novel expression system on a human artificial chromosome. Biochem Biophys Res Commun 2005;329:1018–25 [DOI] [PubMed] [Google Scholar]
- 35.Roth A, Harley CB, Baerlocher GM. Imetelstat (GRN163L)--telomerase-based cancer therapy. Recent Results Cancer Res 2010;184:221–34 [DOI] [PubMed] [Google Scholar]
- 36.Mender I, Gryaznov S, Dikmen ZG, Wright WE, Shay JW. Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2’-deoxyguanosine. Cancer Discov 2015;5:82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Daly H, Kull M, Zimmermann S, Pantic M, Waller CF, Martens UM.Selective cytotoxicity and telomere damage in leukemia cells using the telomerase inhibitor BIBR1532. Blood 2005;105:1742–9 [DOI] [PubMed] [Google Scholar]
- 38.Gurung RL, Lim SN, Low GK, Hande MP. MST-312 Alters Telomere Dynamics, Gene Expression Profiles and Growth in Human Breast Cancer Cells. J Nutrigenet Nutrigenomics 2014;7:283–98 [DOI] [PubMed] [Google Scholar]
- 39.Jin Y, Stewenius Y, Lindgren D, Frigyesi A, Calcagnile O, Jonson T, et al. Distinct mitotic segregation errors mediate chromosomal instability in aggressive urothelial cancers. Clin Cancer Res 2007;13:1703–12 [DOI] [PubMed] [Google Scholar]
- 40.Lundberg G, Rosengren AH, Hakanson U, Stewenius H, Jin Y, Stewenius Y, et al. Binomial mitotic segregation of MYCN-carrying double minutes in neuroblastoma illustrates the role of randomness in oncogene amplification. PLoS One 2008;3:e3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saha A, Shree Padhi S, Roy S, Banerjee B. HCT116 colonospheres shows elevated expression of hTERT and beta-catenin protein - a short report. J Stem Cells 2014;9:243–51 [PubMed] [Google Scholar]
- 42.de Lange T. How telomeres solve the end-protection problem. Science 2009;326:948–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 2012;13:789–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krenn V, Musacchio A. The Aurora B Kinase in Chromosome Bi-Orientation and Spindle Checkpoint Signaling. Front Oncol 2015;5:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seimiya H, Oh-hara T, Suzuki T, Naasani I, Shimazaki T, Tsuchiya K, et al. Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991. Mol Cancer Ther 2002;1:657–65 [PubMed] [Google Scholar]
- 46.Hu L, Wu QQ, Wang WB, Jiang HG, Yang L, Liu Y, et al. Suppression of Ku80 correlates with radiosensitivity and telomere shortening in the U2OS telomerase-negative osteosarcoma cell line. Asian Pac J Cancer Prev 2013;14:795–9 [DOI] [PubMed] [Google Scholar]
- 47.Mender I, Gryaznov S, Shay JW. A novel telomerase substrate precursor rapidly induces telomere dysfunction in telomerase positive cancer cells but not telomerase silent normal cells. Oncoscience 2015;2:693–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov 2011;10:261–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zimmer J, Tacconi EMC, Folio C, Badie S, Porru M, Klare K, et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol Cell 2016;61:449–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berardinelli F, Coluzzi E, Sgura A, Antoccia A. Targeting telomerase and telomeres to enhance ionizing radiation effects in in vitro and in vivo cancer models. Mutat Res 2017;773:204–19 [DOI] [PubMed] [Google Scholar]
- 51.Mocellin S, Pooley KA, Nitti D. Telomerase and the search for the end of cancer. Trends Mol Med 2013;19:125–33 [DOI] [PubMed] [Google Scholar]
- 52.Xu Y, Goldkorn A. Telomere and Telomerase Therapeutics in Cancer. Genes (Basel) 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meena J, Rudolph KL, Günes C Telomere Dysfunction, Chromosomal Instability and Cancer. Recent Results Cancer Res. 2015; 200:61–79. [DOI] [PubMed] [Google Scholar]
- 54.Merle P, Gueugneau M, Teulade-Fichou MP, Muller-Barthelemy M, Amiard S, Chautard E, et al. Highly efficient radiosensitization of human glioblastoma and lung cancer cells by a G-quadruplex DNA binding compound. Sci Rep 2015;5:16255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.