

Abstract
Purpose:
Clinically available BH3 mimetic drugs targeting BCLXL and/or BCL2 (navitoclax and venetoclax, respectively) are effective in some hematological malignancies, but have limited efficacy in solid tumors. This study aimed to identify combination therapies that exploit clinical BH3 mimetics for prostate cancer (PCa).
Experimental Design:
PCa cells or xenografts were treated with BH3 mimetics as single agents or in combination with other agents, and effects on MCL1 and apoptosis were assessed. MCL1 was also targeted directly using RNAi, CRISPR, or an MCL1 specific BH3 mimetic, S63845.
Results:
We initially found that MCL1 depletion or inhibition markedly sensitized PCa cells to apoptosis mediated by navitoclax, but not venetoclax, in vitro and in vivo, indicating that they are primed to undergo apoptosis and protected by MCL1 and BCLXL. Small molecule EGFR kinase inhibitors (erlotinib, lapatinib) also dramatically sensitized to navitoclax-mediated apoptosis, and this was associated with markedly increased proteasome-dependent degradation of MCL1. This increased MCL1 degradation appeared to be through a novel mechanism as it was not dependent upon GSK3β-mediated phosphorylation and subsequent ubiquitylation by the ubiquitin ligases βTRCP and FBW7, or through other previously identified MCL1 ubiquitin ligases or deubiquitinases. Inhibitors targeting additional kinases (cabozantinib and sorafenib) similarly caused GSK3β-independent MCL1 degradation, and in combination with navitoclax drove apoptosis in vitro and in vivo.
Conclusions:
These results show that PCa cells are primed to undergo apoptosis, and that co-targeting BCLXL and MCL1, directly or indirectly through agents that increase MCL1 degradation, can induce dramatic apoptotic responses.
Keywords: prostate cancer, MCL1, BH3 mimetics, apoptosis, navitoclax
Introduction
The standard treatment for metastatic prostate cancer (PCa) is androgen deprivation therapy (ADT, medical or surgical castration) to suppress activity of the androgen receptor (AR), but tumors invariably recur (castration-resistant prostate cancer, CRPC). Many will respond to agents that further suppress androgen synthesis or to AR antagonists (abiraterone and enzalutamide, respectively) (1,2), but most men relapse within 1–2 years and these relapses appear to be driven by multiple AR dependent and independent mechanisms (3). Further responses may be obtained with additional agents including taxanes, immunotherapy (Sipuleucel-T), radium-223, or with PARP inhibitors in tumors with DNA damage response defects (4), but these responses are generally partial and not durable. Therefore, there is a critical need for more effective PCa therapies.
BH3-mimetic agents are an extremely promising class of drugs that act by binding to anti-apoptotic BCL2 family members (5). This blocks their binding to pro-apoptotic BH3 only proteins such as BIM and their ability to neutralize BAX/BAK, thereby enhancing apoptosis. ABT-737 (6) and ABT-263 (navitoclax, orally bioavailable analogue of ABT-737) (7) are BH3-mimetics that bind to BCL2, BCLXL, and BCLW, but not MCL1. Navitoclax has single-agent activity in hematological malignancies (8), but causes thrombocytopenia due to BCLXL inhibition. A more selective agent that just blocks BCL2 (ABT-199, venetoclax), and thereby spares platelets, is similarly active and is now FDA approved for chronic lymphocytic leukemia (9,10). In contrast to hematological malignancies, these currently clinically available BH3-mimetics have limited single agent activity in most solid tumors (11). One basis for this relative resistance may be ineffective blockade of MCL1, as high levels of MCL1 are associated with resistance to these BH3 mimetics in several solid tumors (11–16). Preclinical studies have indicated that navitoclax may be efficacious in some solid tumors when used in combination with other agents through a variety of mechanisms, including decreasing MCL1 expression by transcriptional, translational, or post-translational mechanisms (11,16–21).
MCL1 expression can be stimulated by a variety of growth factors acting through JAK/STAT and other pathways, and downregulated by a series of miRNAs (22–24). MCL1 mRNA translation can be regulated by eIF2a and mTORC1, and thereby suppressed by inhibition of the PI3K/AKT/mTOR pathway (25,26). MCL1 protein also undergoes rapid turnover through the ubiquitin-proteasome system (24,27). The major ubiquitin ligases known to target MCL1 are the HECT domain E3 ligase HUWE1 (MULE) (28,29), and the RING-type E3 ligases FBW7 and βTRCP (30–32). HUWE1 has a BH3 domain that mediates its binding to the N-terminus of MCL1, and its ubiquitylation of MCL1 appears to be constitutive. In contrast, MCL1 ubiquitylation by FBW7 and βTRCP is dependent on GSK3β-mediated phosphorylation at sites in an MCL1 C-terminal phosphodegron (S155 and S159) (33). This is preceded by a priming phosphorylation at T163 that can be mediated by JNK, but other kinases may also target this site and can stabilize MCL1 in some contexts where GSK3β is not active (34,35).
The objective of this study was to determine whether ABT-737/263 could be exploited in PCa to lower the apoptotic threshold and sensitize tumors to other agents. We found that MCL1 depletion (by RNAi or CRISPR) or inhibition with an MCL1-specific BH3 mimetic, in combination with ABT-737/263 treatment, induced a rapid apoptotic response in PCa cells in vitro and in vivo, indicating that they are primed to undergo apoptosis and protected by MCL1 and BCL2/BCLXL. We further found that multiple receptor tyrosine kinase (RTK) inhibitors could rapidly and dramatically enhance MCL1 protein degradation, and drive apoptosis in combination with ABT-737/263 in vitro and in vivo. Interestingly, while RTK inhibitors have been reported to enhance GSK3β-mediated MCL1 degradation (21,36–38), we found that MCL1 degradation in response to RTK inhibition was independent of GSK3β and the downstream ubiquitin ligases FBW7 and βTRCP. We also confirmed that HUWE1 was a mediator of basal MCL1 degradation, and identified TRIM21 as a novel MCL1 ubiquitin ligase, but found that neither of these mediated MCL1 degradation in response to RTK inhibition. These results show that PCa cells are primed to undergo apoptosis, and that co-targeting BCLXL and MCL1 (directly or indirectly through agents that increase its degradation) can induce dramatic apoptotic responses.
Materials and Methods
Cell culture and Matrigel based colony formation assays
LNCaP, C4–2, PC3 and RV1 cells were obtained from ATCC and cultured in RPM1640 medium with 10% FBS and penicillin-streptomycin (100 IU/ml). VCaP, MDA-MB-468, MCF7 (from ATCC) and A549 (kindly provided by Dr. Susumu Kobayashi, BIDMC) were cultured in DMEM medium with 10% FBS and penicillin-streptomycin (100 IU/ml). Cell identify was confirmed by STR analysis, and Mycoplasma testing was negative. For most immunoblotting or RT-PCR experiments, cells were grown to 50%−60% confluence in 10% FBS containing medium for 1 day and then treated with indicated drugs. Transfections were carried out using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instruction. For colony formation assays, approximately 4,000 cells were seeded onto chamber slides (LAB-TEK) coated with growth factor-reduced Matrigel (BD Biosciences), and then cultured in RPMI1640 medium replenished with 2% FBS and 2% Matrigel. The medium was replaced every 4 days. ABT-737 was from Selleck Chemicals. ABT-263 was kindly provided by AbbVie Inc. S63845 was from MedChemExpress.
Primary culture of patient-derived xenografts (PDXs)
LuCaP 35CR and LuCaP 70CR PDX tumors were obtained from University of Washington and passaged in castrated male SCID mice (Taconic Laboratories) (39). The harvested tumors were minced in trypsin, and cells were seeded in DMEM medium with 10% FBS, 2% Matrigel, and penicillin-streptomycin (100 IU/ml) for 1 day, followed by drug treatment.
Immunoblotting
Cells were lysed in RIPA buffer (Pierce) supplemented with protease and phosphatase inhibitor cocktails (Thermo Scientific). Blots were incubated with anti-MCL1 (1:1000, Cell Signaling), anti-phopho-MCL1 Ser159 (1:1000, Abcam), anti-phopho-MCL1 Thr163 (1:1000, Cell Signaling), anti-cleaved caspase 3 (CC3) (1:250, Cell Signaling), anti-β-actin (1:10000, Abcam), anti-vinculin (1:20000, Sigma-Aldrich), anti-BCL2 (1:500, Cell Signaling), anti-BCLXL (1:1000, Cell Signaling), anti-BIM (1:1000, Cell Signaling), anti-phospho-AKT Thr308 (1:1000, Cell Signaling), anti-phospho-AKT Ser473 (1:1000, Cell Signaling), anti-phopho-S6 Ser235/236 (1:3000, Cell Signaling), anti-phopho-ERK1/2 Thr202/Tyr204 (1:1000, Cell Signaling), anti-p21 (1:1000, Santa Cruz Biotechnology), anti-p27 (1:1000, Cell Signaling), anti-p53 (1:1000, Santa Cruz Biotechnology), anti-c-MYC (1:1000, Santa Cruz Biotechnology), anti-β-catenin (1:1000, Cell Signaling), anti-phospho-EGFR Tyr845 (1:500, Cell Signaling), anti-phospho-EGFR Tyr1068 (1:500, Cell Signaling), anti-EGFR (1:1000, Cell Signaling), anti-HA (1:1000, Cell Signaling), anti-TRIM21 (1:1000, Bethyl Laboratories), or anti-USP9X (1:1000, Bethyl Laboratories), and then with 1:5000 of anti-rabbit or anti-mouse secondary antibodies (Promega).
RT-PCR
Quantitative real-time RT-PCR amplification was performed on RNA extracted from cells using RNeasy Mini kit (Qiagen). RNA (50 ng) was used for each reaction, and the results were normalized by co-amplification of GAPDH. Reactions were performed on an ABI Prism 7700 Sequence Detection System using TaqMan one-step RT-PCR reagents. Primer mix for MCL1 (Hs01050896_m1) and GAPDH was purchased from Thermo Fisher Scientific.
Flow cytometry
Cells were immunostained for Annexin V and Propidium Iodide using Annexin V Apoptosis Detection Kit with PI (Biolegend) following the manufacturer’s instructions. Flow cytometry was performed with FACSCalibur (Becton-Dickinson) and the data were analyzed using FlowJo software (Tree Star).
Caspase 3/7 luciferase reporter assays
Caspase-Glo 3/7 assay kit (Promega) was used to evaluate caspase 3 activity according to the manufacturer’s manual. Briefly, following drug treatment and cell lysis, a luminogenic caspase-3/7 substrate containing the tetrapeptide sequence DEVD was added. Luminescence generated by caspase cleavage and subsequent luciferase reaction was measured by a luminometer in triplicate samples.
Formalin fixed paraffin embedded (FFPE) tissue immunohistochemistry (IHC)
IHC staining was performed using Vectastain Elite ABC HRP kit (Vector Laboratory). Tissue slides were deparaffinized, antigen retrieved and then immunostained for cleaved caspase 3 (1:1000; Cell Signaling). DAB HRP substrate kit (Vector) was used to visualize the signal.
RNA interference
For transient silencing of MCL1, EGFR, MULE, TRIM21, βTRCP, FBW7, USP9X, or USP24, LNCaP cells were transfected with target siRNA and then analyzed 48 to 72 hours later. All pooled siRNAs including control siRNA were purchased from GE Dharmacon. For stable silencing of MCL1, LNCaP cells were transfected with 5 different MCL1 target shRNAs (#1; TRCN0000196390, #2; TRCN0000196914, #3; TRCN0000197024, #4; TRCN0000199070, #5; TRCN0000199377, Sigma-Aldrich) respectively or non-targeting shRNA control (Sigma-Aldrich) and were selected with puromycin. For doxycycline-inducible shRNA-mediated knockdown of MCL1, a single-stranded oligonucleotide encoding MCL1 target shRNA and its complement (sense, 5’-CCGGCCTAGTTTATCACCAATAATCTCGAGATTATTGGTGATAAACTAGGTTTTT-3’) was synthesized. The oligonucleotide sense and antisense pair were annealed and were inserted into a tet-on pLKO vector (40). 293 cells were co-transfected with control or shRNA-containing tet-on pLKO vector, VSVG, and dR8.91 for 48 hours, and then LNCaP cells were infected with the produced lentiviral supernatants and were selected with puromycin.
Generation of MCL1 knockout cell line
LNCaP cells were co-transfected with MCL1 CRISPR/Cas9 KO and MCL1 HDR plasmid (pool of 3 guide RNAs, sc-400079) at a ratio of 1:1. Cells were then selected with 2 µg/ml of puromycin for two weeks. The selective medium was replaced every 2–3 days. The single clones were picked and checked for MCL1 expression. Control CRISPR/Cas9 plasmid (sc-418922) was used as a negative control. All plasmids were from Santa Cruz Biotechnology, Inc.
Coimmunoprecipitation combined with mass spectrometry (IP-MS)
Plasmids encoding HA-tagged MCL1 wild-type (WT) and mutant (3A; S155A, S159A, T163A) were kindly provided by Dr. Wenyi Wei (BIDMC). For IP-MS, LNCaP cells stably overexpressing HA-MCL1-WT were treated with erlotinib or lapatinib in combination with proteasome inhibitors for 4 hours, the cell lysate was subject to immunoprecipitation using anti-HA-conjugated agarose beads (Sigma-Aldrich) and then eluted with HA peptide (Sigma-Aldrich). The eluted proteins were analyzed by microcapillary reversed-phase (C18) liquid chromatography-tandem mass spectrometry (LC-MS/MS) as previously reported (41). MS/MS data were searched against the Uniprot Human protein database (version 20151209 containing 21,024 entries) using Mascot 2.5.1 (Matrix Science), and data analysis was performed using the Scaffold 4.4.8 software (Proteome Software). Peptides and modified peptides were accepted if they passed a 1% FDR threshold.
Xenograft experiments
Xenografts were established in the flanks of male nude mice (Taconic) by subcutaneous inoculation of ~3 million of indicated cells mixed with 50% Matrigel (BD Bioscience). When the tumors reached the indicated volumes, treatments were initiated. Growth was then either monitored by caliper measurements or tumor samples were collected for protein analysis or IHC staining. All animal experiments were approved by Beth Israel Deaconess Medical Center’s Institutional Animal Care and Use Committee and were performed in accordance with institutional and national guidelines.
Statistical analysis
Significance of difference between 2 groups was determined by 2-tailed Student’s t test using R software (version 3.3.2). For comparison of dose-response curves of MCL1 protein after erlotinib treatment between EGFR knockdown and the control, normalized MCL1 protein (MCL1/Actin) data were analyzed by paired-T test with Bonferroni correction to compare replicate means at each dose. Statistical significance was accepted at p < 0.05.
Results
MCL1 or BCLXL are sufficient to prevent apoptosis in PCa cells
We initially asked whether PCa cells were primed to undergo apoptosis and were protected by BCL2, BCLXL, and/or MCL1. Treatment with ABT-737 to antagonize BCL2 and BCLXL did not cause apoptosis in LNCaP or PC3 cells (Fig. 1A). Similarly, depleting MCL1 with siRNA did not stimulate apoptosis. However, the combination of MCL1 depletion by siRNA and ABT-737 treatment caused a dramatic apoptotic response within 4 hours (Fig. 1A). We then examined LNCaP cells stably expressing a series of MCL1 shRNA. ABT-737 again caused rapid apoptosis in the two lines in which MCL1 was most markedly depleted (Fig. 1B). We next generated LNCaP cells with doxycycline regulated expression of the most effective shRNA (shMCL1–1) (Fig. S1). ABT-737 had minimal effect in the absence of doxycycline, but induced a rapid and marked apoptotic response in cells that were pretreated with doxycycline to deplete MCL1 (Fig. 1C). Finally, we used Cas9/CRISPR with three different guide RNAs to delete MCL1. Consistent with the RNAi results, there was a dramatic apoptotic response to ABT-263 (navitoclax) in each of three MCL1-depleted lines (Fig. 1D). Significantly, while ABT-737 and −263 induced marked apoptosis in MCL1 depleted cells, ABT-199 (venetoclax, which selectively inactivates BCL2) was not effective (Fig. 1E). Together these data show that these PCa cells are primed to undergo apoptosis, and that MCL1 or BCLXL are sufficient to prevent this apoptosis.
Fig. 1. MCL1 Depletion Sensitizes BH3 Mimetics to Induce Apoptosis in Prostate Cancer Cells in vitro.
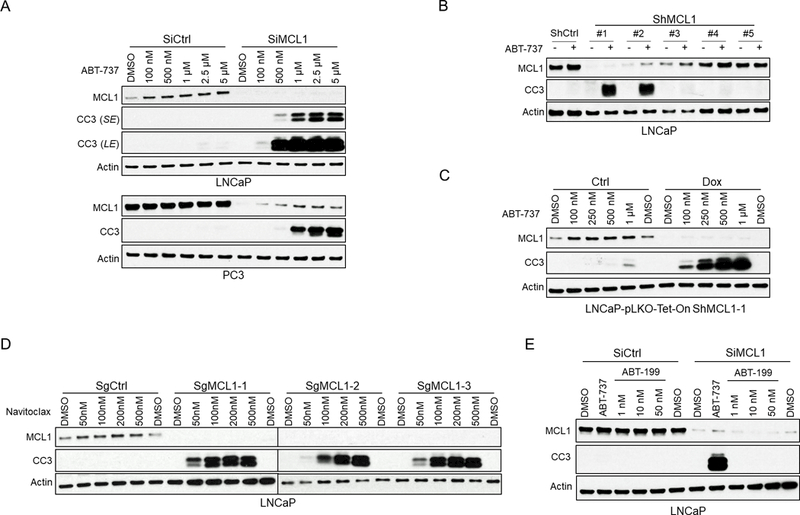
(A) LNCaP or PC3 cells transfected with MCL1 siRNA or non-target control siRNA were treated with ABT-737 (0 – 5 μM) for 4 hours. Total cell lysates were immunoblotted for indicated proteins. Apoptosis induction was detected with cleaved caspase 3 (CC3) signal. SE, short exposure; LE, long exposure. (B) LNCaP cells stably expressing 5 distinct MCL1 lentiviral shRNAs or a non-target shRNA were treated with ABT-737 (500 nM) for 4 hours, followed by immunoblotting. (C) LNCaP cells stably expressing doxycycline-inducible lentiviral MCL1 shRNA were treated with ABT-737 for 4 hours with and without 48 hours of doxycycline (DOX) pre-incubation. (D) Three MCL1 deficient LNCaP subclones generated with CAS9/CRISPR and independent guide RNAs (SgMCL1) and 1 negative control clone (non-specific guide RNA, SgCtrl) were treated with ABT-263 for 4 hours. (E) LNCaP cells were transfected with MCL1 siRNA or non-target siRNA and then treated with ABT-737 (500 nM) or ABT-199 (venetoclax, BCL2 selective inhibitor), and harvested 48 hours later.
Both MCL1 and BCLXL mRNA, but not BCL2 mRNA, are increased in primary PCa relative to normal prostate epithelium, suggesting these are similarly acting to suppress apoptosis in vivo (Fig. S2A-C). MCL1 protein is also increased in a series of PCa cell lines and PDXs relative to levels in a normal prostate epithelial cell line (RWPE-1 cells), while levels of BCLXL and BCL2 in the PCa cells relative to RWPE-1 were more variable (Fig. S2D). To assess priming for apoptosis in vivo, we established xenografts with LNCaP cells expressing doxycycline inducible MCL1 shRNA. When the xenografts reached ~400 mm3, mice were treated for 3 days with doxycycline or normal diet, followed by 3 days of ABT-263 (50 mg/kg/day by i.p. injection) or vehicle control (12.5% DMSO/PEG-400), and tumors were harvested at ~6 hours after the final dose. Immunoblotting of protein lysates (Fig. 2A) and immunohistochemistry (Fig. 2B) indicated that there was synergy between MCL1 depletion and ABT-263 in driving apoptosis. We also generated xenografts from LNCaP cells with Cas9/CRISPR-mediated deletion of MCL1 (sgMCL1–1 from Fig. 1D). Consistent with the in vitro results, these xenografts grew well in vivo with low levels of apoptosis (Fig. 2C and D, controls). In contrast, 3 days of treatment with ABT-263 led to dramatic tumor regression in both small (~100 mm3) and larger (~400 mm3) tumors (Fig. 2C and Fig. S3). This was associated with a marked increase in apoptosis, as assessed at 6 hours after the third dose of ABT-263 (Fig. 2D).
Fig. 2. MCL1 Depletion Sensitizes BH3 Mimetics to Induce Apoptosis in Prostate Cancer Xenografts.
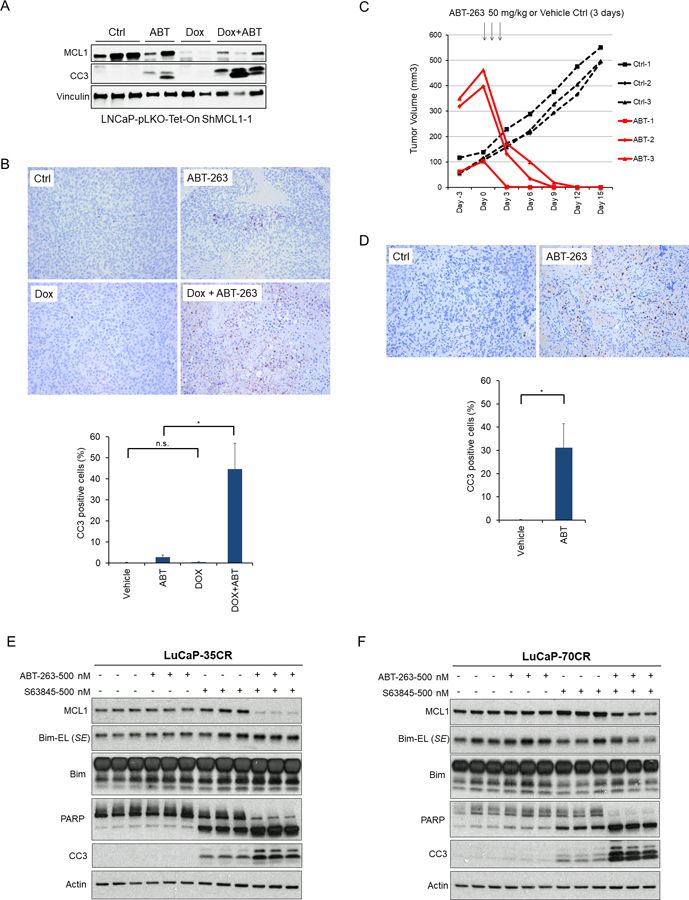
(A) Nude mice bearing LNCaP xenografts (~400 mm3) that inducibly express MCL1 shRNA were fed with doxycycline containing diets or normal diet for 3 days and then i.p. injected with ABT-263 (50 mg/kg/day) or vehicle control (12.5% DMSO/PEG-400) once daily for consecutive 3 days. Tumors were harvested 6 hours after the last injection and proteins were analyzed by western blot. CC3, cleaved caspase 3. (B) Representative images of immunohistochemical staining for CC3 at each treatment group in Figure 2A. Ratios of CC3 positive to total cells were quantified in at least 3 high power fields (mean ± SEM). (C) LNCaP-SgMCL-1 (Cas9/CRISPR MCL1 depletion) xenografts bearing mice were i.p. injected with ABT-263 (n=3, 50 mg/kg) or vehicle control (n=3, 12.5% DMSO/PEG-400) once daily for consecutive 3 days, and tumor volumes were monitored. Arrows indicate the timepoint of injection (started at Day 0 and finished at Day 2). (D) Representative images of immunohistochemical staining for CC3 in MCL1 depleted tumors that were untreated (Ctrl) or harvested 6 hours after third treatment with ABT-263. Ratio of CC3 positive to total cells were quantified (mean ± SEM). (E) Primary LuCaP35CR and (F) LuCaP70CR cultures were treated with ABT-263 alone, S63845 alone, or combination for 4 hours in 2D culture. The cell lysates then were immunoblotted for indicated proteins. SE, short exposure.
To determine whether these observations could be extended to additional PCa models, we examined primary cultures from the LuCaP 35CR and LuCaP 70CR castration-resistant patient derived xenografts (PDXs) (39). Consistent with the LNCaP and PC3 cells, ABT-263 had no clear effect in either PDX (Fig. 2E, F). In contrast, 4 hour treatment with S63845, an MCL1 selective BH3 mimetic (42), caused an apoptotic response in both models as assessed by both CC3 and PARP cleavage. However, this apoptosis was markedly increased by combined treatment with S63845 and ABT-263. It should be noted that while there was a decrease in MCL1 in response to the combination therapy, this is likely secondary to caspase activation and subsequent caspase mediated MCL1 degradation.
EGFR inhibition synergizes with ABT-737 in driving apoptosis by decreasing MCL1
In parallel studies we screened in vitro a small series of drugs in combination with ABT-737 to identify potentially effective combination therapies. The dual EGFR/ERBB2 inhibitor lapatinib in combination with ABT-737 was particularly effective at decreasing LNCaP cell growth (Fig. 3A) and spheroid formation (Fig. 3B). Annexin V staining showed that ABT-737 or lapatinib alone did not have clear effects on apoptosis, while the combination for as little as 4 hours markedly increased the number of apoptotic cells (Fig. 3C and Fig. S4A). Consistent with this result, luciferase assays for activated caspase 3/7 showed that ABT-737 for 4 hours caused modest caspase activation, but this was markedly enhanced by lapatinib (with levels being comparable to those induced by staurosporine) (Fig. 3D).
Fig. 3. Dual EGFR/ERBB2 Inhibition Sensitizes Prostate Cancer Cells to BH3 Mimetic-induced Apoptosis via Downregulating MCL1.
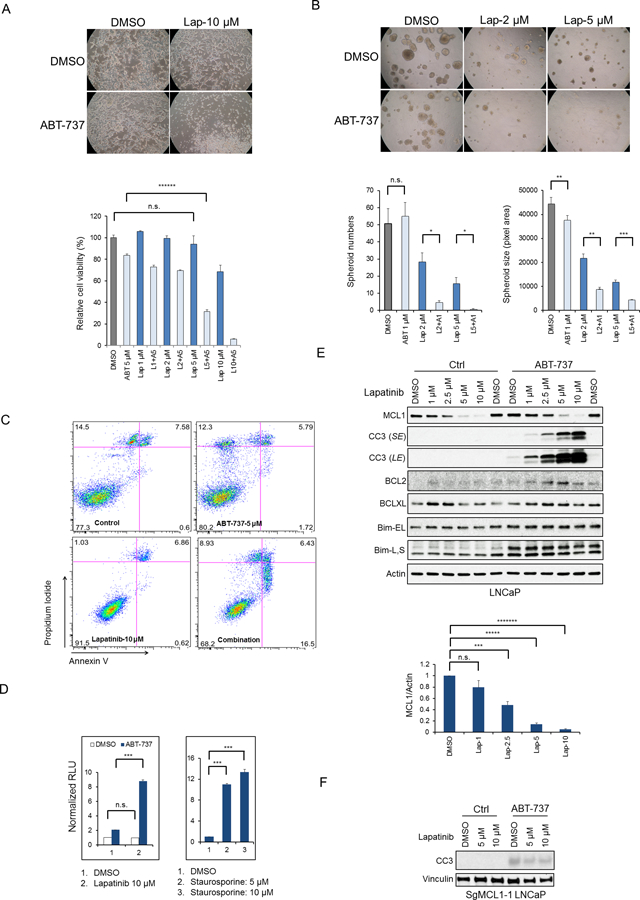
(A) LNCaP cells were cultured 3 days in the presence of DMSO, ABT-737, lapatinib, or the combination, and pictures were taken (upper panel) and relative cell viabilities were measured (bottom panel). Data shown are mean ± SEM (n=6). n.s., not significant; ******, p < 1×10−6. (B) LNCaP cells were grown in Matrigel based 3D culture matrix for 7 days, followed by indicated treatments on day 7 and day 10. Pictures were taken 3 days after the last treatment. The spheroid size was quantified in pixel area using ImageJ. The numbers of spheroids above certain size (7500 pixel area) were counted. n.s., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001. (C) LNCaP cells treated with indicated drugs for 4 hours were stained with propidium iodide and annexin V antibody and then analysed by flow cytometry. Representative data from 2 experiments are shown, and both are summarized in Fig. S4A. (D) LNCaP cells in each treatment group were analysed for caspase-3/7 activities using luciferase reproter assay. All data were normalized to control (DMSO) group and shown as mean± SEM. n.s., not significant; ***, p < 0.001; RLU, relative light unit. (E) LNCaP cells were treated with lapatinib (0 – 10 μM) with and without ABT-737 (5 μM) for 4 hours. MCL1 bands were measured by imageJ and normalized to actin (bottom panel). CC3, cleaved caspase 3; SE, short exposure; LE, long exposure; Bim-EL, extra large form of Bim; Bim-L, large form of Bim; Bim-S, small form of Bim; n.s., not significant; ***, p < 0.001; *****, p < 1×10−5; *******, p < 1×10−7. (F) LNCaP cells with CAS9/CRISPR mediated MCL1 depletion were treated with lapatinib alone or in the presence of ABT-737 (500 nM) for 4 hours. Representative data from 2 experiments are shown.
Significantly, immunoblotting showed that 4 hours treatment with lapatinib also markedly decreased MCL1 levels (Fig. 3E). Moreover, this decrease in MCL1 correlated with induction of apoptosis by ABT-737, suggesting that lapatinib was acting by decreasing MCL1. Consistent with this hypothesis, lapatinib did not substantially decrease the levels of other anti-apoptotic proteins or increase the expression of pro-apoptotic proteins BH3-only proteins including BIM (which is increased by EGFR inhibition in lung cancer cells driven by mutant EGFR) (43,44) (Fig. 3E). Interestingly, ABT-737 did increase BIM protein, and particularly the BIM-L form, which presumably reflects redistribution of BIM from BCL2/BCLXL to other sites where it is more stable. Moreover, lapatinib did not further enhance ABT-737-mediated apoptosis in cells with Cas9/CRISPR mediated MCL1 loss (Fig. 3F). Lapatinib similarly decreased MCL1 and synergized with ABT-737 in driving apoptosis in additional PCa cell lines (C4–2, VCaP, PC3, and RV1) (Fig. S4B) and in breast and lung cancer cell lines (Fig. S4C, D).
Previous studies in cell lines with amplified, overexpressed or mutated EGFR have found that EGFR inhibition can decrease MCL1 or increase BIM, and can synergize with ABT-737/263 in driving apoptosis (18,43,44). To assess the role of EGFR versus ERBB2 inhibition by lapatinib in LNCaP cells (which do not have EGFR or ERBB2 amplification or mutations), we compared effects of selective inhibitors. Both erlotinib (EGFR inhibitor) and TAK165 (ERBB2 inhibitor) could decrease MCL1 and synergize with ABT-737 in inducing apoptosis, with erlotinib being more effective and with the combination being as effective as lapatinib (Fig. 4A and Fig. S5A). Similar results were obtained when we used a luciferase substrate to assess caspase 3/7 activation (Fig. 4B). One prominent pathway for ERBB2 signaling is through ERBB3 phosphorylation and subsequent PI3K/AKT pathway activation, and previous studies indicate that PI3K activation can enhance MCL1 mRNA translation via mTORC1 (11,20,45). Consistent with these previous studies, a PI3K inhibitor (BKM120) decreased MCL1 and synergized with ABT-737 in driving caspase 3/7 activation (Fig. 4C, D). However, lapatinib treatment did not have a marked effect on AKT activation, suggesting its effects on MCL1 are not substantially mediated via AKT/mTORC1 signaling (Fig. 4E). Moreover, treatment with a direct AKT inhibitor or with rapamycin caused only a modest decrease in MCL1 and no clear synergy with ABT-737 in driving apoptosis (Fig. 4E, F), further indicating that AKT-independent mechanisms are contributing to the effects of lapatinib and PI3K inhibition on MCL1. Finally, treatment with a MEK inhibitor (U0126) similarly did not decrease MCL1 or synergize with ABT-737, indicating that EGFR and ERBB2 inhibitors are suppressing MCL1 by an ERK-independent mechanism (Fig. 4E and S5B).
Fig. 4. EGFR/ERBB2 Inhibitors Decrease MCL1 through Blocking EGFR and ERBB2 Independent of AKT/mTOR and ERK Pathways.
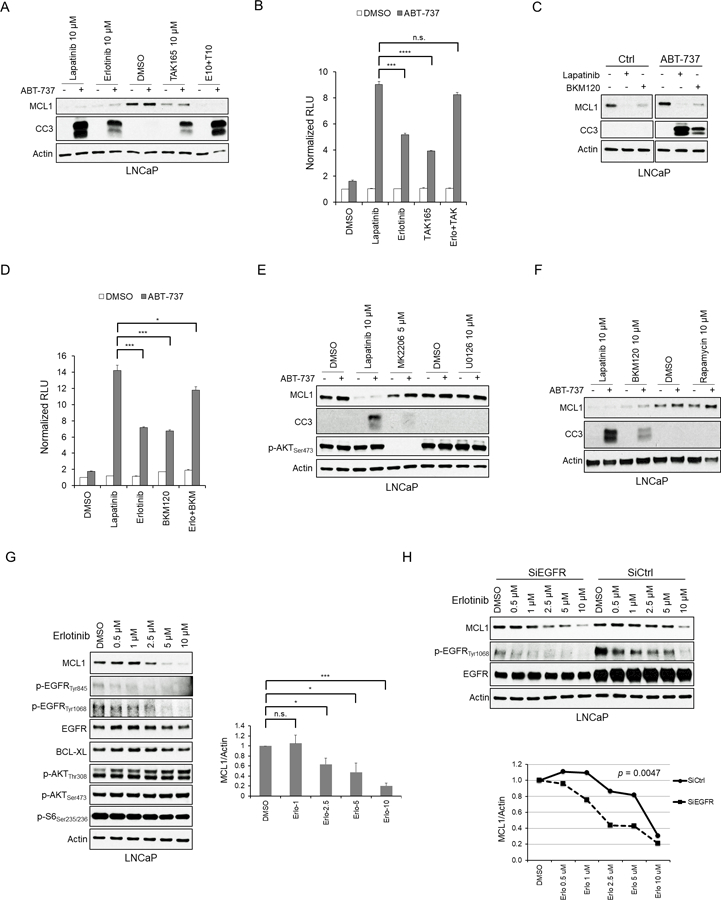
(A) LNCaP cells were treated with DMSO, lapatinib, erlotinib (EGFR inhibitor), TAK165 (ERBB2 inhibitor) and the combination (erlotinib plus TAK165) with or without ABT-737 (5 μM) for 5 hours, and then harvested, followed by western blot analysis. Results are representative of 3 experiments, which are summarized with respect to MCL1 in Fig. S5A. CC3, cleaved caspase 3. (B) LNCaP cells treated as in (A) were applied to Caspase-3/7 activity luciferase reporter assay. Data represents the fold-change of relative light units (RLU) relative to control (DMSO) group. The data shown are mean ± SEM (n=3). n.s., not significant; ***, p < 0.001; ****, p < 0.0001. (C) LNCaP cells were treated for 4 hours with DMSO, lapatinib (10 μM) and BKM120 (PI3K inhibitor, 10 μM), with or without ABT-737 (5 μM). Representative data of 3 experiments are shown, with a summary in Fig. 5SA. (D) LNCaP cells were treated with DMSO, lapatinib (10 μM), erlotinib (10 μM), BKM120 (10 μM), and the combination (erlotinib plus BKM120) with or without ABT-737 (5 μM) for 4 hours. Caspase-3/7 activities in each group (n=3) were examined as in (B). *, p < 0.05; ***, p < 0.001. (E) LNCaP cells were treated with lapatinib (10 μM), MK2206 (AKT inhibitor, 5 μM) and UO126 (MEK inhibitor) with or without ABT-737 (5 μM) for 4 hours. Representative data of 3 experiments are shown, and suumarized for MCL1 in Fig. S5A. (F) LNCaP cells were treated with lapatinib (10 μM), BKM120 (10 μM) or rapamycin (mTORC1 inhibitor, 10 μM) with or without ABT-737 (5 μM) for 4 hours. Representative data from 3 experiments are shown. (G) LNCaP cells were treated with erlotinib (0 – 10 μM) for 4 hours, followed by Western blot. MCL1 bands were measured by imageJ and normalized to actin (bottom panel). n.s., not significant; *, p < 0.05; ***, p < 0.001. (H) LNCaP cells were transfected with EGFR siRNA or non-target siRNA for 72 hours and then treated with erlotinib for 4 hours (left panel). MCL1 bands were quantified by ImageJ and normalized to actin (right panel). Statistical difference of dose-response curves of MCL1 protein levels between EGFR knockdown and control is shown.
To further examine the role of EGFR, we assessed the effects of erlotinib over a range of concentrations. Significantly, we again found that erlotinib could markedly decrease MCL1 expression, and there was a correlation between decreases in MCL1 protein and EGFR phosphorylation, supporting an on-target effect of the drug (Fig. 4G). As expected, there were no clear effects of erlotinib on AKT activation. Comparable effects on MCL1 were observed with another selective EGFR inhibitor (gefitinib) and with another dual EGFR/ERBB2 inhibitor (afatinib) (Fig. S5C). We then examined the effects of decreasing EGFR with siRNA. While EGFR downregulation did not clearly decrease MCL1 levels, it shifted to the left the dose response curve for erlotinib (Fig. 4H). Moreover, immunoblotting for pEGFR again indicated that high-level suppression of EGFR activity was required to decrease MCL1. Together these findings indicate that the effects of lapatinib on MCL1 are mediated both by EGFR and ERBB2, with the ERBB2 effects being mediated at least in part through PI3K. Consistent with this conclusion, combination treatment with erlotinib plus BKM120 sensitized to ABT-737 to a similar degree as lapatinib (see Fig. 4D). Moreover, the results show that high-level repression of EGFR activity is required to markedly decrease MCL1 expression.
EGFR inhibition increases MCL1 protein degradation by GSK3β-independent mechanism
Lapatinib did not decrease MCL1 mRNA, indicating a posttranscriptional mechanism for decreasing MCL1 (Fig. S6A). Significantly, proteasome inhibition (with MG115 and MG132) prevented the decrease in MCL1 in response to lapatinib and erlotinib, indicating that both drugs are acting primarily by increasing MCL1 degradation (Fig. 5A, B, results quantified in Fig. S6B, C). Consistent with this conclusion, using cycloheximide to block new protein synthesis we found that lapatinib decreased MCL1 protein half-life from ~45 minutes to ~15 minutes (Fig. S6D). One major pathway regulating MCL1 degradation is a priming phosphorylation at Thr163 followed by GSK3β-mediated phosphorylation of Ser159 and Ser155 (33), and subsequent ubiquitylation by βTRCP and FBW7 (30,31). However, while treatment with a GSK3β inhibitor (LY2090314) increased MYC and β-catenin, it did not prevent the erlotinib-mediated decrease in MCL1 (Fig. 5C, results quantified in Fig. S7A).
Fig. 5. EGFR Inhibition Increases Proteasome-dependent Degradation of MCL1.
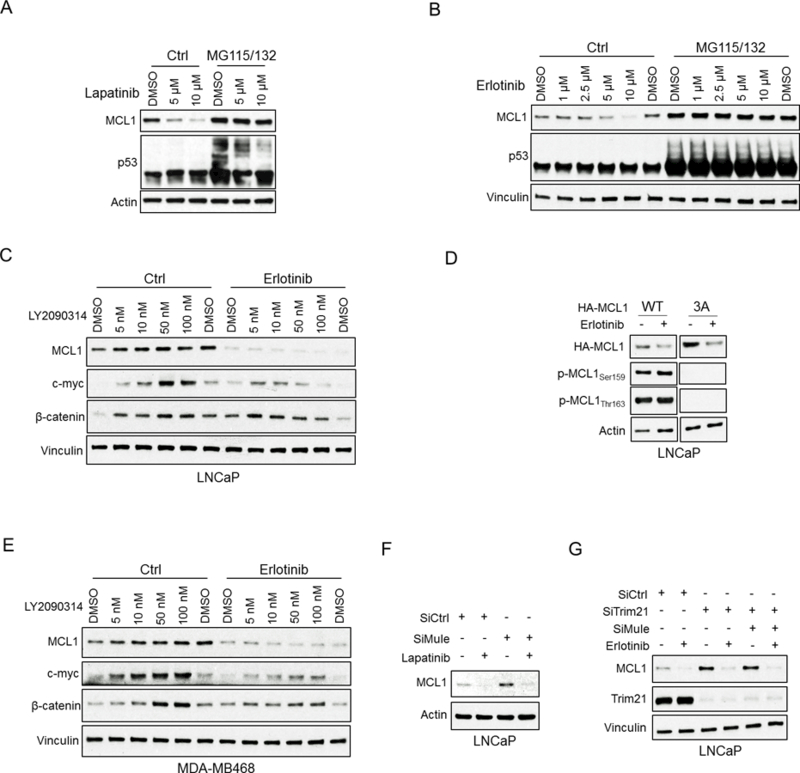
(A and B) LNCaP cells were pretreated with MG115 (10 μM) and MG132 (10 μM) for 30 min, followed by treatment with lapatinib (10 μM) (A) or erlotinib (10 μM) (B) for 4 hours. Efficacy of proteasome block was confirmed by blotting for p53. Results are quantified in Fig. S6B,C. Data shown in A-G are representative of 3 experiments. (C) LNCaP cells were pretreated with GSK3β inhibitor LY2090314 for 1 hour, followed by treatment with erlotinib (10 μM) for 4 hours. Blotting for c-Myc and β-catenin was carried out as a positive control to confirm suppression of GSK3β activity. (D) LNCaP cells were transfected with HA-tagged MCL1 wild-type or 3A (S155A, S159A, T163A) mutant for 24 hours and then treated with DMSO or erlotinib (10 μM) for 4 hours. Lystates were then blotted for HA tagged MCL1, phospho-MCL1 at S159 or T163. (E) MDA-MB468 cells (breast cancer cell line) were pretreated with LY2090314 for 1 hour, followed by treatment with erlotinib (10 μM) for 4 hours. (F) LNCaP cells were transfected with Mule siRNA or non-target siRNA for 48 hours and then treated with DMSO and lapatinib (10 μM) for 4 hours. (G) LNCaP cells were transfected with Trim21 siRNA, double (Trim21 and Mule) siRNA, or non-target siRNA for 48 hours and then treated with DMSO or erlotinib (10 μM) for 4 hours.
Treatment with another GSK3β inhibitor (SB216763) similarly failed to prevent the erlotinib-mediated decrease in MCL1 (Fig. S7B). Moreover, immunoblotting for MCL1 pThr163 or pSer159 showed that phosphorylation at these sites was not increased by erlotinib (Fig. 5D). In addition, threonine or serine to alanine mutations at T163, S159, and S155 (3A) did not prevent the effects of erlotinib (Fig. 5D). GSK3β inhibition similarly did not block the effects of erlotinib in MDA-MB468 breast cancer cells (Fig. 5E) or in additional cell lines examined (RV1, MCF7, or A549 cells) (Fig. S8A,B,C). Consistent with these findings, siRNA targeting βTRCP and FBW7 did not increase MCL1 or prevent the lapatinib-mediated decrease in its expression (Fig. S8D).
To identify additional ubiquitin ligases or deubiquitinases (DUBs) that may regulate MCL1 degradation in response to EGFR inhibition, we carried out large scale immunopurifications of MCL1 from cells treated with erlotinib or lapatinib in combination with proteasome inhibitors (MG115 and MG132), followed by LC-MS/MS. Amongst the MCL1-associated proteins we identified were HUWE1 (MULE), a cullin-independent ubiquitin ligase that has been shown previously to target MCL1 (28,29) (Table S1). We also identified TRIM21, which has ubiquitin ligase activity but has not previously been linked to MCL1 (46,47). Significantly, HUWE1 siRNA increased basal MCL1 expression, but did not prevent the decrease in response to lapatinib (Fig. 5F). TRIM21 siRNA also increased MCL1, but alone or combined with HUWE1 siRNA similarly failed to block the effects of erlotinib (Fig. 5G). The LC-MS/MS did not identify any DUBs associated with MCL1. Nonetheless, as USP9X and USP24 have been reported to act as DUBs for MCL1 (48), we also treated with USP9X and USP24 siRNA, and found that this did not decrease MCL1 protein (Fig. S8E). Finally, depletion of REGγ, which is a mediator of ubiquitin-independent proteasomal degradation, did not prevent MCL1 degradation in response to erlotinib (Fig. S8F). Together these data indicate that the effects of EGFR inhibition are mediated by activation of a novel ubiquitin ligase, or potentially by inactivation of a novel MCL1 DUB.
MCL1 degradation can be enhanced by multiple tyrosine kinase inhibitors
Suppression of other tyrosine kinases with the multi-kinase inhibitor cabozantinib at 1.0 – 2.5 μM also rapidly decreased MCL1 protein, and this effect could be blocked by proteasome inhibition (although decreases at higher concentrations may occur by a proteasome-independent mechanism) (Fig. 6A). This decrease in MCL1 correlated with increased apoptosis in response to ABT-737 (Fig. 6B). The multi-kinase inhibitor sorafenib also markedly decreased MCL1, and this could be partially rescued by proteasome inhibition (Fig. 6C, effects of cabozantinib and sorafenib on MCL1 are quantified in Fig. S9A). Previous studies indicate that sorafenib may also suppress MCL1 transcription through an NFkB or STAT3 pathway (23,49), or increase its degradation by an ERK/PIN1 pathway (38). Significantly, combined treatment with relatively lower concentrations of erlotinib, cabozantinib, and sorafenib (500 nM each) caused a proteasome-dependent decrease in MCL1 (Fig. 6C) and rapid induction of apoptosis in combination with ABT-737 (Fig. 6D). These results indicate that multiple RTKs may feed into a common downstream pathway for stabilizing MCL1. Similarly to EGFR inhibition, the decreases of MCL1 in response to cabozantinib and sorafenib were independent of GSK3β, as they were not prevented by LY2090314 (Supplementary Fig. S9B,C).
Fig. 6. Inhibition of Multiple Receptor Tyrosine Kinases Enhances MCL1 Degradation and Synergizes with BH3 Mimetics in vitro and in vivo.
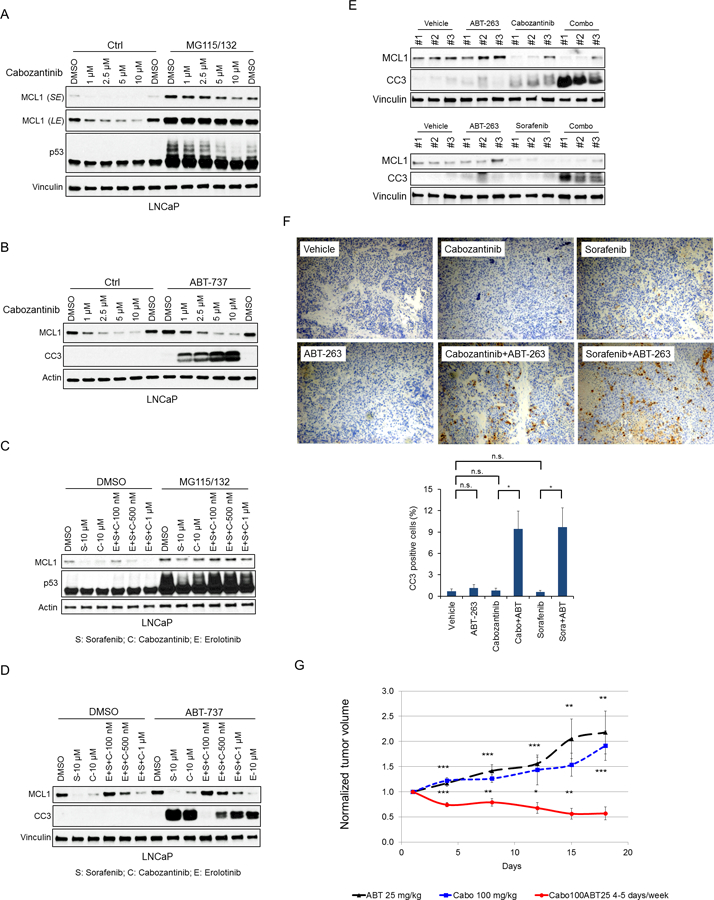
(A) LNCaP cells were pretreated with MG115 (10 μM) and MG132 (10 μM) for 30 min, followed by treatment with cabozantinib for 4 hours. p53 was immunoblotted as a positive control for proteasome inhibition. SE, short exposure; LE, long exposure. (B) LNCaP cells were treated with cabozantinib with or without ABT-737 (5 μM) for 4 hours. CC3, cleaved caspase 3. (C) LNCaP cells were pretreated with MG115 (10 μM) and MG132 (10 μM) for 30 min, followed by treatment with sorafenib (S), cabozantinib (C), and/or erlotinib (E), each at the indicated concentrations, for 4 hours. Data in A-D are representative data of 3 experiments, and effects on MCL1 are quantified in Fig. S9A. (D) LNCaP cells were treated with sorafenib, cabozantinib, and/or erlotinib with or without ABT-737 (5 μM) for 4 hours. (E) LNCaP xenografts bearing mice were i.p. injected with vehicle, ABT-263 (50 mg/kg), cabozantinib (50 mg/kg), sorafenib (30 mg/kg), or the combination (cabozantinib or sorafenib plus ABT-263) once daily for consecutive 3 days. Tumors were harvested 6 hours after the last treatment, and portions (E) were snap frozen for protein analysis and portions (F) were formalin fixed and paraffin embedded for IHC. Representative immunohistochemical stainings for cleaved caspase 3 were shown. Ratios of CC3 positive to total cells were quantified in high power fields from at least 3 different areas. The data shown are mean ± SEM. n.s., not significant; *, p < 0.05. (G) Nude mice bearing LNCaP xenografts were i.p. injected with cabozantinib (100 mg/kg, n=6), ABT-263 (25 mg/kg, n=3) alone or combination (n=8) once daily for 5 days every week, and tumor volumes were normalized to day 0. The data shown are mean ± SEM. Tumor volume fold change at each time point were compared between combination treatment (cabozantinib and ABT-263) and ABT-263 or cabozantinib monotherapy. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine whether RTK inhibitors could decrease MCL1 levels in vivo, and whether this could drive apoptosis in combination with ABT-263, we treated a series of subcutaneous PCa xenografts. We initially used an implantable microdevice with multiple reservoirs that releases microdoses of single agents or drug combinations into spatially distinct regions of the tumor (50). Reservoirs were loaded with a series of kinase inhibitors alone or in combination with ABT-263, and inserted into xenografts generated from PC3 or VCaP PCa cells. Tumors were then removed after 3 days and apoptosis was assessed by IHC for cleaved caspase 3. The combination of lapatinib plus ABT-263 induced more apoptosis than either agent alone in both xenografts (Supplementary Fig. S10A, C). ABT-263 combined with BKM120, MK2206, or dinaciclib were similarly more effective than single agents in PC3, with the latter combination being most effective (consistent with previous data and presumably related to CDK9 inhibition and decreased MCL1 mRNA synthesis). Representative images of cleaved caspase 3 staining in PC3 and VCaP xenografts are also shown (Supplementary Fig. S10B, D).
We next carried out systemic treatments in subcutaneous LNCaP xenografts. Mice bearing LNCaP xenografts were i.p. injected with vehicle, ABT-263 (50 mg/kg), cabozantinib (50 mg/kg), sorafenib (30 mg/kg), or the combination (cabozantinib or sorafenib plus ABT-263) once daily for consecutive 3 days, and tumors were harvested 6 hours after the last treatment. As shown by immunoblotting, the single agent treatment with cabozantinib or sorafenib decreased MCL1 protein levels (Fig. 6E). Moreover, in combination the ABT-263, this was associated with increased apoptosis. Similar results were obtained by IHC for cleaved caspase 3 (Fig. 6F). Finally, to determine whether combination therapy could induce tumor regression, we established another series of LNCaP xenografts. When xenografts reached ~250 mm3, mice were randomized to treatment with cabozantinib (100 mg/kg), ABT-263 (25 mg/kg), or the combination by i.p. injection once daily for 5 days every week. The combination therapy, but not either agent alone, resulted in tumor regression (Fig. 6G).
Discussion
The BH3 mimetic drugs currently in the clinic (navitoclax and venetoclax, respectively targeting BCL2/BCLXL or BCL2) have efficacy in hematological malignancy, but have had limited single-agent efficacy in solid tumors (8,10,11,51). However, preclinical studies in solid tumor models have shown that navitoclax may be synergistic with a number of agents acting by a variety of mechanisms, including upregulation of pro-apoptotic BH3-only proteins such as BIM and NOXA, or downregulation of MCL1 (11,16–21). We found that apoptosis could be rapidly induced in PCa cells by depletion or inhibition of MCL1 in combination with ABT-737/263. We further found that RTK inhibitors, including erlotinib, sorafenib, and cabozantinib, were synergistic with ABT-737/263 in driving apoptosis, and this was associated with a rapid and dramatic increase in MCL1 protein degradation. Previous studies have found synergy between ABT-737/263 and erlotinib that was mediated by increased BIM (43,44), and synergy with sorafenib that was mediated by increased GSK3β-dependent MCL1 degradation (23). In contrast, we found that MCL1 degradation in response to RTK inhibitors was independent of GSK3β in LNCaP and additional epithelial cancer cell lines, and was independent of the downstream ubiquitin ligases FBW7 and βTRCP (30,31,33). These results show that PCa cells are primed to undergo apoptosis, and that co-targeting BCLXL and MCL1 (directly or indirectly through agents that decrease MCL1 synthesis or increase its degradation) can induce dramatic apoptotic responses.
The RAS/RAF/MAPK and PI3K/AKT/mTOR pathways are activated downstream of RTKs and clearly can impact responses to navitoclax by multiple mechanisms. As noted above, in previous studies erlotinib was found to have synergy with navitoclax through increased BIM (43,44). This was observed in cells with mutant or amplified EGFR and downstream RAS/RAF/MAPK pathway activation, and is consistent with ERK being a negative regulator of BIM transcription. Significantly, a phase I clinical trial of navitoclax combined with erlotinib in advanced solid tumors failed to show evidence of efficacy (52). However, EGFR status was not assessed and effects of erlotinib on BIM or MCL1 were not determined. PI3K pathway inhibition may enhance apoptosis through multiple mechanisms, and particularly may decrease MCL1 translation through suppression of mTORC1 and increased MCL1 degradation through activation of GSK3β (25,26). However, the effects we observed of EGFR inhibition on MCL1 appear to be independent of MAPK and PI3K signaling.
To identify potentially novel mechanisms driving MCL1 degradation in response to EGFR inhibition, we immunopurified MCL1 from cells treated with erlotinib or lapatinib in the presence of a proteasome inhibitor, followed by LC-MS/MS. This analysis identified several proteins known to interact with MCL1, including BIM and the ubiquitin ligase HUWE1 (MULE), and a ubiquitin ligase not previously linked to MCL1 (TRIM21) (46,47). Significantly, siRNA against HUWE1 and TRIM21 increased basal levels of MCL1, indicating that these ubiquitin ligases targeted MCL1. However, MCL1 degradation in response to EGFR inhibition was not impaired, indicating that this response is not mediated through HUWE1 or TRIM21.
Although the precise molecular mechanism driving MCL1 degradation in response to RTK inhibition remains to be identified, these studies support further exploration of therapeutic strategies based on synergistic combinations with navitoclax in PCa and other solid tumors. Moreover, as effective combination therapies would induce apoptosis rather than just suppress growth, it is likely that intermittent schedules will be effective and could possibly mitigate dose limiting myelosuppression or other toxicities. It also should be noted that selective MCL1 inhibitors are currently in development and show great promise in preclinical studies (42,51,53). Our data suggest that combination therapy with small molecules targeting MCL1 and BCLXL may be very effective, and a recent study suggests that toxicity may be manageable as most normal tissues may be relatively resistant due to lower levels of BAX and BAK (54). Nonetheless, the potential toxicity associated with global MCL1 antagonism in combination with navitoclax may be circumvented by therapies with agents such as kinase inhibitors that may more selectively target MCL1 in tumor cells, and may also provide an apoptotic stimulus through inhibition PI3K, MAPK, or other signaling pathways. In any case, the availability of MCL1 inhibitors will provide new opportunities to discover efficacious combinations that may be effective in definable subsets of tumors.
Supplementary Material
Translational Relevance.
BH3 mimetic drugs in the clinic targeting BCL2 (venetoclax) or both BCLXL and BCL2 (navitoclax) have limited efficacy in solid tumors including prostate cancer (PCa). This study demonstrates that PCa cells are primed to undergo apoptosis, that BCLXL and MCL1 have redundant functions in preventing apoptosis, and that therapeutic strategies jointly targeting BCL2/BCLXL and MCL1 may be highly effective. One straightforward approach, if not limited by toxicity, may be to combine BCL2/BCLXL (or selective BCLXL inhibitors when available) with an MCL1 inhibitor. An alternative approach supported by this study is to target MCL1 indirectly through agents that decrease its synthesis or enhance its degradation. Specifically, this study finds that MCL1 degradation can be markedly enhanced by receptor tyrosine kinase inhibitors, and thereby provides a rationale for trials that combine these agents with navitoclax or other BH3 mimetics targeting BCLXL/BCL2.
Acknowledgments
We thank Drs. Wenyi Wei and Susumu Kobayashi (BIDMC) for helpful discussions and reagents, Dr. John Asara (BIDMC) for guidance on mass spectrometry, and AbbVie for providing navitoclax.
Financial Support: This work was supported by NIH grants P01 CA163227 (S.P. Balk, E. Corey), P50 CA090381 (S.P. Balk), R33 CA223904 (O. Jonas), the Bridge Project (partnership between the Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center) (S.P. Balk, O. Jonas), a Research Fellowship Award from Gunma University Hospital (S. Arai), and a Career Enhancement Award from P50CA090381 (S. Chen).
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368(2):138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371(5):424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014;33(22):2815–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med 2015;373(18):1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov 2017;16(4):273–84. [DOI] [PubMed] [Google Scholar]
- 6.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005;435(7042):677–81. [DOI] [PubMed] [Google Scholar]
- 7.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008;68(9):3421–8. [DOI] [PubMed] [Google Scholar]
- 8.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 2012;30(5):488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 2014;4(3):362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med 2016;374(4):311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faber AC, Farago AF, Costa C, Dastur A, Gomez-Caraballo M, Robbins R, et al. Assessment of ABT-263 activity across a cancer cell line collection leads to a potent combination therapy for small-cell lung cancer. Proc Natl Acad Sci U S A 2015;112(11):E1288–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006;10(5):389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006;10(5):375–88. [DOI] [PubMed] [Google Scholar]
- 14.Santer FR, Erb HH, Oh SJ, Handle F, Feiersinger GE, Luef B, et al. Mechanistic rationale for MCL1 inhibition during androgen deprivation therapy. Oncotarget 2015;6(8):6105–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams MM, Lee L, Hicks DJ, Joly MM, Elion D, Rahman B, et al. Key Survival Factor, Mcl-1, Correlates with Sensitivity to Combined Bcl-2/Bcl-xL Blockade. Mol Cancer Res 2017;15(3):259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao Y, Nimmer P, Sheppard GS, Bruncko M, Hessler P, Lu X, et al. MCL-1 Is a Key Determinant of Breast Cancer Cell Survival: Validation of MCL-1 Dependency Utilizing a Highly Selective Small Molecule Inhibitor. Mol Cancer Ther 2015;14(8):1837–47. [DOI] [PubMed] [Google Scholar]
- 17.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis 2015;6:e1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Jin S, Abraham V, Huang X, Liu B, Mitten MJ, et al. The Bcl-2/Bcl-X(L)/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol Cancer Ther 2011;10(12):2340–9. [DOI] [PubMed] [Google Scholar]
- 19.Modugno M, Banfi P, Gasparri F, Borzilleri R, Carter P, Cornelius L, et al. Mcl-1 antagonism is a potential therapeutic strategy in a subset of solid cancers. Exp Cell Res 2015;332(2):267–77. [DOI] [PubMed] [Google Scholar]
- 20.Anderson GR, Wardell SE, Cakir M, Crawford L, Leeds JC, Nussbaum DP, et al. PIK3CA mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci Transl Med 2016;8(369):369ra175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong J, Wang P, Tan S, Chen D, Nikolovska-Coleska Z, Zou F, et al. Mcl-1 degradation is required for targeted therapeutics to eradicate colon cancer cells. Cancer Res 2017;77(9):2512–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett 2010;584(14):2981–9. [DOI] [PubMed] [Google Scholar]
- 23.Abdulghani J, Allen JE, Dicker DT, Liu YY, Goldenberg D, Smith CD, et al. Sorafenib sensitizes solid tumors to Apo2L/TRAIL and Apo2L/TRAIL receptor agonist antibodies by the Jak2-Stat3-Mcl1 axis. PLoS One 2013;8(9):e75414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ertel F, Nguyen M, Roulston A, Shore GC. Programming cancer cells for high expression levels of Mcl1. EMBO Rep 2013;14(4):328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A 2008;105(31):10853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fritsch RM, Schneider G, Saur D, Scheibel M, Schmid RM. Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J Biol Chem 2007;282(31):22551–62. [DOI] [PubMed] [Google Scholar]
- 27.Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells 2014;3(2):418–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005;121(7):1085–95. [DOI] [PubMed] [Google Scholar]
- 29.Warr MR, Acoca S, Liu Z, Germain M, Watson M, Blanchette M, et al. BH3-ligand regulates access of MCL-1 to its E3 ligase. FEBS Lett 2005;579(25):5603–8. [DOI] [PubMed] [Google Scholar]
- 30.Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 2007;27(11):4006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011;471(7336):104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011;471(7336):110–4. [DOI] [PubMed] [Google Scholar]
- 33.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006;21(6):749–60. [DOI] [PubMed] [Google Scholar]
- 34.Morel C, Carlson SM, White FM, Davis RJ. Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol Cell Biol 2009;29(14):3845–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nifoussi SK, Vrana JA, Domina AM, De Biasio A, Gui J, Gregory MA, et al. Thr 163 phosphorylation causes Mcl-1 stabilization when degradation is independent of the adjacent GSK3-targeted phosphodegron, promoting drug resistance in cancer. PLoS One 2012;7(10):e47060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elgendy M, Abdel-Aziz AK, Renne SL, Bornaghi V, Procopio G, Colecchia M, et al. Dual modulation of MCL-1 and mTOR determines the response to sunitinib. J Clin Invest 2017;127(1):153–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang R, Xia L, Gabrilove J, Waxman S, Jing Y. Sorafenib Inhibition of Mcl-1 Accelerates ATRA-Induced Apoptosis in Differentiation-Responsive AML Cells. Clin Cancer Res 2016;22(5):1211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao Y, et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res 2008;68(15):6109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Higano CS, et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017;77(6):654–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chin YR, Yuan X, Balk SP, Toker A. PTEN-deficient tumors depend on AKT2 for maintenance and survival. Cancer Discov 2014;4(8):942–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breitkopf SB, Yuan M, Helenius KP, Lyssiotis CA, Asara JM. Triomics Analysis of Imatinib-Treated Myeloma Cells Connects Kinase Inhibition to RNA Processing and Decreased Lipid Biosynthesis. Anal Chem 2015;87(21):10995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016;538(7626):477–82. [DOI] [PubMed] [Google Scholar]
- 43.Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med 2007;4(10):e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med 2007;4(10):1681–89; discussion 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov 2014;4(1):42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat Immunol 2013;14(2):172–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z, et al. TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis. Mol Cell 2016;61(5):720–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010;463(7277):103–7. [DOI] [PubMed] [Google Scholar]
- 49.Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 2007;12(1):66–80. [DOI] [PubMed] [Google Scholar]
- 50.Jonas O, Landry HM, Fuller JE, Santini JT Jr., Baselga J, Tepper RI, et al. An implantable microdevice to perform high-throughput in vivo drug sensitivity testing in tumors. Sci Transl Med 2015;7(284):284ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov 2017;16(4):273–284. [DOI] [PubMed] [Google Scholar]
- 52.Tolcher AW, LoRusso P, Arzt J, Busman TA, Lian G, Rudersdorf NS, et al. Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with erlotinib in patients with advanced solid tumors. Cancer Chemother Pharmacol 2015;76(5):1025–32. [DOI] [PubMed] [Google Scholar]
- 53.Akcay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, et al. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol 2016;12(11):931–36. [DOI] [PubMed] [Google Scholar]
- 54.Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 2017;31(1):142–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.