

Abstract
The metastatic cascade is a complex process that requires cancer cells to survive despite conditions of high physiologic stress. Previously a cooperation between the glucocorticoid receptor (GR) and hypoxia inducible factors (HIFs) was reported as a point of convergence for host and cellular stress signaling. These studies indicated p38 MAPK-dependent phosphorylation of GR on Ser134 and subsequent p-GR/HIF-dependent induction of breast tumor kinase (PTK6/Brk), as a mediator of aggressive cancer phenotypes. Herein, p-Ser134 GR was quantified in human primary breast tumors (n=281) and the levels of p-GR were increased in triple negative breast cancer (TNBC) relative to luminal breast cancer. Brk was robustly induced following exposure of TNBC model systems to chemotherapeutic agents (Taxol or 5-FU) and growth in suspension (ultra-low attachment (ULA). Notably, both Taxol and ULA resulted in upregulation of the Aryl hydrocarbon receptor (AhR), a known mediator of cancer pro-survival phenotypes. Mechanistically, AhR and GR co-purified and following chemotherapy and ULA, these factors assembled at the Brk promoter and induced Brk expression in a HIF-dependent manner. Further, Brk expression was upregulated in Taxol-resistant breast cancer (MCF-7) models. Ultimately, Brk was critical for TNBC cell proliferation and survival during Taxol treatment and in the context of ULA as well as for basal cancer cell migration, acquired biological phenotypes that enable cancer cells to successfully complete the metastatic cascade. These studies nominate AhR as a p-GR binding partner and reveal ways to target epigenetic events such as adaptive and stress-induced acquisition of cancer skill sets required for metastatic cancer spread.
INTRODUCTION
Cancer cell metastasis is a complicated, multi-step process (1). Key rate-limiting steps include the survival of the cancer cells while in the circulation and upon colonization of distant metastatic sites. Multiple steps of the metastatic cascade require cancer cells to resist anoikis, the process through which normal epithelial cells undergo programmed cell death when detached from the basement membrane (2). An emerging mediator of cell survival in suspension environments is the aryl-hydrocarbon receptor (AhR), a member of the basic-helix-loop-helix (bHLH) Per-ARNT-Sim (PAS) superfamily of transcriptional factors (3), that includes the Hypoxia-Inducible Factor (HIF) family, which we previously showed is required for inducible breast tumor kinase (Brk) expression (4). Whereas the HIFs are stabilized and activated by decreasing oxygen tensions, AhR is activated by a diverse group of ligands, including polycyclic aromatic hydrocarbons (PAH), natural plant flavonoids and indoles, and metabolites of the tryptophan pathway. Once ligand-activated, AhR heterodimerizes with aryl hydrocarbon receptor nuclear translocator (ARNT), also known as HIF-1beta, in order to regulate expression of target genes. AhR is required for normal mammary gland development (5,6) and increased expression of AhR and AhR target genes have been found in multiple cancer types, including breast tumors (7). In immortalized normal mammary epithelial cells, high AhR expression confers increased cell invasion, migration, and proliferation (8).
Brk, also known as PTK6, is a soluble tyrosine kinase distantly related to the c-Src family of oncogenic protein kinases. Brk is overexpressed in ~86% of breast tumors and has been found to be mislocalized to the membrane in transformed mammary epithelial cells (9,10). Numerous growth factor receptors, including MET, EGF receptor, and HER2, activate Brk signaling upstream of Rac1 ((11,12) and reviewed in (13)). Once activated, Brk mediates aggressive phenotypes in breast cancer cells, many of which are critical for steps in the metastatic process, including resistance to anoikis (14), anchorage-independent growth (15), modulation of EMT markers (16), and growth factor-induced cell migration (11,17).
We previously demonstrated upregulation of Brk expression in triple negative breast cancer (TNBC) cells in response to physiologic stress stimuli mediated by hypoxia-inducible factors, HIF-1α and HIF-2α (4), principal mediators of transcriptional responses to physiologic stress stimuli (18). HIFs and HIF gene signatures are highly expressed in TNBC (19–21); overexpression of HIF-1α in breast tumors predicts a higher risk of metastasis and relapse of disease (22,23). Notably, 15-40% of TNBC express high levels of the glucocorticoid receptor (GR), which mediates the biological effects of glucocorticoid (GC) signaling (24,25). GR/GCs are emerging critical modulators of epithelial cell survival and resistance to chemotherapy-induced cell death in solid tumors (25–30). Interestingly, in ER-negative breast tumors, GR expression predicts increased risk of metastasis and decreased overall survival (25,26,31). Our previous studies demonstrated that GR cooperates with HIFs to induce Brk expression in TNBC models following diverse physiologic stress stimuli (ROS, hypoxia, nutrient starvation) in addition to hormonal (i.e. GC driven) cues (32).
As breast cancer patients typically receive high doses of GCs just prior to chemotherapy treatment in order alleviate adverse side-effects (33), understanding the unintended consequences of this GC treatment is critical for optimizing patient care. Herein we investigated the concept that combined GC and chemotherapy treatment, itself a potent mediator of HIF induction (4,32) may unintentionally activate the stress-induced p-GR/HIF-dependent transcriptional program that includes Brk, ultimately leading to increased cancer cell survival coupled to increased metastatic potential.
MATERIALS AND METHODS
Cell Culture and CRISPR-Cas9 Gene Editing
MDA-MB-231 cell lines were a gift from Dr. Roland Wenger and were cultured as previously described (4). Cells used to generate CRISPR-mediated gene sub-lines were authenticated April 27, 2017 by the University of Arizona Genetics Core and results were compared with the ATCC short-tandem repeat (STR) database. To knockout HIF1A and HIF2A by CRISPR/Cas9 “nickase” technology, paired guide RNAs (gRNA) were designed to have zero off target effects using the MIT algorithm (www.crispr.mit.edu) (Supplementary Table 1).
Each gRNA to target exon 1 of HIF1A was individually cloned into the pX462-puromycin vector, which expresses Cas9n (AddGene). pX462 was also modified to express hygromycin instead of puromycin and the HIF2A guides were individually cloned into pX462-hygromycin. MDA-MB-231 cells obtained from ATCC were transfected with either pX462-puromycin vector alone (empty vector control) or 1:1 ratios of pX462-puro-HIF1A gRNAs using FuGene HD followed by selection in puromycin. A clonal line was selected in which HIF1A was undetectable by western blotting (clone 1-2), which was then transfected with 1:1 ratios of pX462-hygromycin-HIF2A gRNAs and pools of clones selected by hygromycin to generate a HIF double knockout (DKO) line. Knockdown was confirmed by western blot (Supp. Figure 5). U2OS WT-GR and S134A-GR cells (previously described (34)) were cultured in DMEM/F12 50:50 with 10% FBS, 1% P/S, 1% glutamax, .2mg/mL Hygromycin, and 200μg/mL G418. Generation of Customized Guide RNA Expression Construct for Brk/PTK6 knock out: In order to knockout the expression of Brk/PTK6 in MDA-MB-231 cells, an anti-sense sgRNA targeting exon 2 of Brk was cloned into a CRISPR/Cas9-GFP expression vector (PX458). Briefly, a sense oligo 5′-CACCGCCGACGCACAGCTTCCGAG-3′ and an anti-sense oligo 5′-AAACCTCGGAAGCTGTGCGTCGGC-3′, containing four base-pair overhangs compatible with PX458 BbsI restriction enzyme digestion sites, were annealed and ligated into PX458. Correct incorporation of this sgRNA sequence was confirmed by Sanger sequencing (Genewiz). MDA-MB-231 Transfections and screening for Brk knockout clones: The PX458-PTK6-exon 2 sgRNA plasmid was electroporated into MDA-MB-231 cells using a Neon electroporator (Invitrogen), and allowed to recover for two days. GFP positive cells were then collected by FACS sorting, expanded and sub-cloned by limited dilution into 96 well plates. Single cell clones were identified and expanded further into 24 well plates for genomic DNA collection and PCR screening. In order to identify CRISPR/Cas9 edited cells, two primers that span exon 2 of Brk, Brk exon 2 ScrF1 5′- GACATTCAGGGCGTCTGG-3′ and Brk exon 2 ScrR1 5′- TGGAGAGGAGCCCATGT-3′ were used to produce a 414 bp amplicon by PCR. Resulting amplicons from individual clones were further cloned into pCR4-TOPO vector using a TOPO TA cloning kit (Invitrogen). Plasmid DNA was isolated from individual transformants and sequenced by Sanger sequencing (Genewiz). Clones with indels were tested for Brk knockout by western blot. Cells were maintained in 5% CO2 at normoxia (20% O2). Cells were cultured in DMEM with 10% FBS and 1% pen/strep. For experiments containing dexamethasone treatment conditions, cells were plated and 24 hours washed twice with 1X PBS before being starved in phenol red free media with 10% dextran charcoal stripped serum (DCC) and 1% pen/strep. After 18 hours, cells were treated with ethanol vehicle or 10μM dexamethasone (treatment times are noted in figure legends).
Forced suspension culture
MDA-MB-231 cells were plated for 24 or 48 hours in Corning Ultra-low attachment (ULA) dishes (catalog #3262). ULA experiments were performed using cells suspended in 1% methyl cellulose to prevent aggregation. In order to achieve similar densities at time of harvest, cells were plated in 10cm2 dishes at 3×106 (attached) or 4×106 (ULA) for Western blot experiments and in 6-well plates at 3×105 (attached) or 5×105 (ULA). After 48 hours in ULA culture, cells were pelleted and rinsed with 1X PBS several times to remove methyl cellulose. Cells were then re-plated in adherent dishes for 2 hours, trypsinized and then counted with trypan blue to determine viability.
Tissue Microarray
A luminal breast cancer tissue microarray (TMA) was generated by the University of Minnesota Histology and Immunohistochemistry Laboratory from 209 de-identified breast cancer patient samples (represented by 1719 individual tissue spots). From this set, 209 tumor samples contained four different pathological regions were independently included in the array: invasive, inflammatory, DCIS, and adjacent-normal-like (normal) tissue within tumor-containing tissue. Patient and tumor characteristics were extracted from pathological reports and used for analysis.
A TNBC TMA was generated by the UT Southwestern Pathology Laboratory from 72 patients with high-grade primary invasive TNBC. This study was approved by the UT Southwestern University Institutional Review Board (no. STU032011-117). Pathologists reviewed breast carcinoma cases at Zale Lipshy University Hospital, Dallas, TX, from 2004 to 2011. The diagnosis of TNBC was established by immunohistochemical analysis and confirmed by human epithelial growth factor receptor 2 (HER2) fluorescence in situ hybridization assay. The criteria for determining triple negativity were based on immunohistochemical staining and image quantitation of ER, PgR, and HER2, as previously described (35).
Immunohistochemical analysis of GR and p-GR
Immunohistochemical staining was performed for total and phospho-Ser134 GR as previously described (36). Briefly, 5 μm sections were deparaffinized, rehydrated and endogenous peroxidases were blocked with 2% hydrogen peroxide in methanol. Sections were then boiled in citrate for antigen retrieval and blocked in 2.5% horse serum for 1 hour. Primary antibodies were incubated at 4°C overnight: GR antibody clone D8H2 (1:50, Cell Signaling, MA) and human specific phospho-Ser314 GR custom antibody clone 8391 (1:100) were used to visualize total and p-GR respectively. Biotinylated horse anti-rabbit secondary antibodies (1:500, DAKO Carpentaria, CA) were incubated for 1 hour at room temperature. Slides where incubated in ABC reagent conjugated with horseradish peroxidase (Vector Laboratories Burlingame, CA) for 1 hour. Finally, staining was visualized by 3,3′ diaminobenzidine tetrahydrochloride (DAB substrate) and counterstained with hematoxylin QS (Vector Laboratories Burlingame, CA).
Stained slides were scored manually per tissue core by a pathologist who was blinded to the clinical data. Immunostaining data were registered semi-quantitatively in two ways. Staining intensity (0, no staining; 1, weak staining; 2, moderate staining; and 3, intense staining, staining examples ranging from 0–3 are provided in Supp. Figure 6A) and the proportion of stained cells (0, no staining; 1, 1–25% staining; 2, 26–50%; 3, 51–75%; and 4, if more than 75% of the tumor cells were positive (37)). These two values were added into a single histology score (0–7 total) that was used in subsequent analyses (Supp. Figure 6B).
Immunoblotting
For experiments without hormone treatment, cells were plated and cultured for 24h prior to preparation of whole-cell lysates as previously described (4). Lysates (50 μg per lane) were resolved on SDS-PAGE gels, transferred to PVDF membranes and the membranes probed with the following primary antibodies: Brk (Santa Cruz, sc-1188), GR (Santa Cruz, sc-1003), ERK1/2 (Cell Signaling, 9102L), HIF-1α (Novus Biologicals, NB100-479), HIF-2α (Novus Biologicals, NB100-122), p38 MAPK (Cell Signaling, 9212), phospho-p38 MAPK (Cell Signaling, 4511p), phospho-Ser134 GR (custom made, Pierce Biotechnology), AhR (Cell Signaling, 83200S) or PELP1 (Bethyl Labs, A300-180A). Representative images of triplicate experiments are shown. Densitometry was performed via ImageJ analysis and signals normalized to the loading control as noted in Supplementary Figure 1A.
qRT-PCR
Quantitative real-time PCR (qRT-PCR) assays were conducted as previously described (4) with cDNA prepared from total RNA extracted from MDA-MB-231 cells. Briefly, cells were plated at 5 × 104 cells per well in 6-well plates. Following specific treatments (noted in figure legends), cells were washed twice with 1X PBS and RNA was isolated via trizol, as per manufacturer protocol. Relative gene expression was normalized to the expression of internal control genes, either TATA-binding protein (TBP), Actin (beta-actin), or 18S rRNA.
ChIP assays
Chromatin immunoprecipitation (ChIP) assays were conducted and data analyzed as previously described (4), with chromatin prepared from MDA-MB-231 cells or U2OS cells expressing WT-GR (34). Briefly, wells were plated at 3 × 106 cells per 150mm dish. Following specific treatments (noted in the figure legends), chromatin was isolated via Active Motif ChIP-IT Express protocol with 5 minute formaldehyde fixation and 30 minute shearing. Immunoprecipitation was performed with 2μg of the specified antibody for 4 hours (anti-GR) and 18 hours (all other immunoprecipitations).
Co-immunoprecipitation assays
Co-immunoprecipitation (Co-IP) assays were performed as previously described (38). Briefly, MDA-MB-231 were plated for 24 hours before treatment with Taxol (10nM) or DMSO vehicle for 24h. Cells were lysed with ELB buffer (50 mM HEPES, 1% NP40, 250mM NaCl, 5mM EDTA) and immunoprecipitation was performed with the indicated antibody for 18 hours at 4 degrees Celsius. Lysates were analyzed by western blotting, as previously described.
Soft agar assays
Soft agar experiments were performed as previously described (39) and the results presented are representative of 3 independent experiments; treatment conditions are noted in the figure legend. Briefly, MDA-MB-231 cells were plated in 1X low-melt point agar with DMSO vehicle or 10nM Taxol. Cells were maintained at 37°C, 5% CO2 and colonies were counted (3 fields per well) 2 weeks after plating.
Scratch wound migration assay
MDA-MB-231 cells were plated in 6-well plates at a density of 3×105 cells/well to achieve monolayer growth at 100% confluency. Cells were allowed to attach to plates for 18hrs. A 200 μL sterile pipette tip was used to create the scratch in the central portion of the plate in a horizontal fashion. Cells were maintained in 37°C, 5% CO2 with DMEM media supplemented with 10% FBS. Independent wells were imaged at 0, 6 and 24h after scratching using a confocal light microscope. ImageJ (v1.51) was used for quantification of width of wound. Three biological replicates were used per cell line. A representative experiment is shown.
Gene expression analysis
Results are represented as means +/− SEM. Statistical significance for qRT-PCR and ChIP-qPCR assays was determined via the unpaired Student’s t test. ANOVA followed by Tukey’s multiple comparison test was performed when appropriate.
RESULTS
GR Ser134 phosphorylation is elevated in TNBC
GR expression in luminal breast cancers predicts a good prognosis. In sharp contrast, high GR expression in TNBC is associated with poor outcome (25). The reasons for these underlying biological differences are unknown. We speculated that TNBCs may exist in a molecular context in which GR is persistently phosphorylated. To investigate the phosphorylation state of GR at Ser134 in human primary luminal and TNBC tumor samples, we created and validated a custom antibody recognizing human phospho-Ser134 GR (Fig. 1A; (32)). Specificity for phospho-Ser134 GR was demonstrated in vehicle or H2O2-treated U2OS osteosarcoma cells (naturally GR-null/low) stably expressing either an empty vector control, wildtype (WT) GR, or a serine 134 to alanine mutant (S134A) GR, which cannot be phosphorylated at Ser134 (Fig. 1A). Notably, no basal or inducible GR phosphorylation was observed in the vector control U2OS cells lacking appreciable endogenous GR expression or in cells expressing S134A GR. In addition, only a single band representing phospho-Ser134 GR was visible following Western blotting of whole cell extracts harvested from multiple cell line models and using full-length SDS PAGE (8-10%) gels (data not shown).
Figure 1.
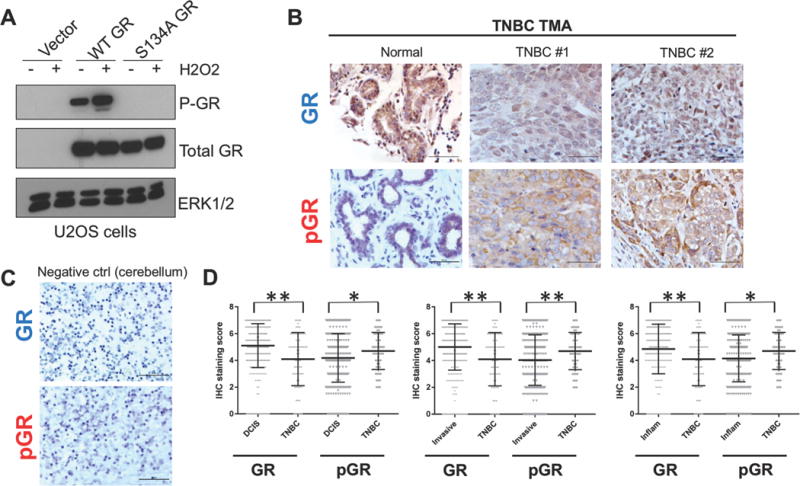
Total GR and phospho-Ser134 GR IHC staining of a custom breast cancer tissue microarray (TMA). (A) U2OS cells stably expressing either empty vector, wild type (WT) GR, or a serine 134 to alanine (S134A) GR mutant were treated with either vehicle or 100 μM H2O2 for 1 h and then subjected to Western blot analysis to detect P-GR, total GR, or ERK1/2 (loading control). (B) Immunostaining for total GR and phospho-Ser134 GR was also performed on a separate TNBC TMA containing containing 72 patient samples with matched ‘cancerized’ adjacent normal-like tissue (see methods) and representative images of staining patterns are shown. (C) As a negative control for total GR and phospho-Ser134 GR staining, GR-null cerebellum tissue was used (see methods) (D) IHC staining scores were quantified using the approach outlined in the methods, and the significance between histologically identified subgroups was assessed by unpaired Student’s t-test (*P>0.05, **P<0.01).
We then performed immunohistochemistry (IHC) staining of both luminal and TNBC tissue microarrays (TMA) for total GR and phospho-Ser134 GR. Two independent TMAs were analyzed. One TMA was generated at the University of Minnesota and was comprised of 209 primarily luminal patient breast tumors represented as 2754 individual tumor spots divided into separate pathological regions by a clinical pathologist and designated as either adjacent-normal-like tissue within tumor-containing tissue (normal), ductal carcinoma in situ (DCIS), invasive, or inflammatory (Supplementary Figure 2). An additional TMA was generated at the University of Texas Southwestern Medical Center at Dallas and contained cancer-adjacent normal tissue and TNBC samples from 72 patients with high-grade primary invasive TNBC, as represented in duplicate spots (Fig. 1B). IHC staining of cerebellum tissue for total and phospho-GR was used as a negative control and to further confirm specificity of the antibodies in our IHC protocols (Fig. 1C). Numerous studies have reported total GR protein to be present at varying levels in normal breast tissue, as well as in ER+/PR+ and ER-/PR- breast tumors (24,25). In concordance with these reports, we observed varying intensity of total GR staining in all categories of breast tissue, including normal-like tissues located adjacent to tumor tissues. GR was present in both luminal and TNBC specimens and was localized to both the cytoplasm and nucleus of cancer cells (Supp. Fig. 2B and Fig. 1B). Similarly, phospho-Ser134 GR staining was observed in both normal and breast tumor samples, again present in both the cytoplasm and the nucleus of cancer cells represented on both TMAs (Supp. Fig 2B and Fig. 1B–D). This result was expected, as phosphorylation of GR Ser134 is a basal or constitutive event that is relatively insensitive to GR ligands, but robustly induced upon exposure to physiologic stress stimuli (32,34). Additionally, the ‘normal’ tissue used in both of the analyzed TMAs was derived from tissue adjacent to the primary tumor but within the tumor-containing tissue, and therefore likely contains ‘cancerized’ stromal cells, unlike true normal mammary tissue derived from women with no prior history or presence of breast cancer. A comparison of phospho-Ser134 GR expression levels between lower grade, luminal breast carcinoma samples (i.e. DCIS) and the higher grade TNBC tumor samples revealed significantly higher levels of phospho-Ser134 GR staining in the TNBC samples relative to either the DCIS tumors, invasive tumors or inflammatory tumors (Fig. 1D). In contrast, total GR levels were significantly decreased in TNBC samples relative to samples from luminal breast cancer subtypes (Fig. 1D). Notably, ligand-bound and transcriptionally activated steroid hormone receptors undergo rapid protein turnover relative to inactive receptors (40). Thus, a decrease in total GR protein levels in TNBC may indirectly indicate the increased presence of transcriptionally active phospho-GR species. This interpretation requires further study, but is consistent with our finding of elevated (i.e. active) phospho-GR in TNBC samples represented in this TMA. Together, these results demonstrate that while phospho-Ser134 GR is readily detectable in both luminal and TNBC samples, its expression is elevated in primary human TNBC relative to GR+ luminal breast cancers.
Brk expression is induced by clinically relevant chemotherapies
GR is an important sensor for both host and cellular stress (32,34). We hypothesized that chemotherapy-associated oxidative stress may inadvertently induce Brk expression via p-GR-dependent signaling. MDA-MB-231 cells, which lack expression of ER and PR but express high levels of GR, were treated for up to 3 days with DMSO vehicle control or 10nm Taxol, a chemotherapy commonly used to treat stage IV breast cancer. Brk protein was significantly induced after 1 day of treatment and elevated levels were maintained up to 3 days of Taxol treatment relative to vehicle controls (Fig. 2A). Notably, Taxol treatment increased activation of p38 MAPK and phosphorylation of GR Ser134; GR Ser134 is a ligand-independent site known to be phosphorylated by p38 MAPK (32,34). Moreover, we observed strong induction of HIF-1α, a modest rise in HIF-2α expression, and surprisingly, increased expression of PELP1 protein following Taxol treatment; all of these factors are critical for Brk induction in response to physiologic stress (32). Brk mRNA expression was also significantly induced by Taxol at multiple time points compared to DMSO vehicle (Fig. 2B). These data suggest a feed-forward mechanism of phosphorylation of p38 and GR associated with prolonged induction of Brk expression. While Brk protein and mRNA were robustly induced early, following 1 day of Taxol treatment, and phosphorylation of p38 and GR were readily observed upon day 2 of Taxol treatment relative to day 0, multiple complex pathways regulate Brk expression via both HIF-dependent and HIF independent mechanisms (41). These data suggest that phospho-p38 and phospho-GR induce sustained Brk expression. To determine if Brk induction by chemotherapy was Taxol-specific, we also treated MDA-MB-231 cells with 5-Fluorouracil (5FU) for up to 3 days. Again, Brk mRNA and protein were significantly induced following 1-2 days of 5FU treatment. However, in contrast to Taxol treatment, Brk induction was not sustained (Supp. Figure 3A). Although HIF1/2 were clearly elevated in response to 5FU, phospho-p38 MAPK (i.e. activated), phospho-GR, and PELP1 were not appreciably induced relative to vehicle controls (Supp. Figure 3A). Notably, Brk and HIF-2α (i.e. both are GR-target genes) were elevated in Taxol-resistant MCF-7 TaxR cells relative to parental MCF-7 cells (Fig. 2C), suggesting that stress signaling is relevant to the maintenance of Taxol-resistance.
Figure 2.

Exposure to conventional chemotherapy induces Brk mRNA and protein expression. (A) MDA-MB-231 cells were treated with either vehicle or 10 nM Taxol for 0-3 days and then subjected to western blot analysis with antibodies specific to Brk, P-GR, total GR, HIF-1α, HIF-2α, PELP1, P-p38, or total p38 (loading control). Arrow indicates band specific to HIF-1α. (B) MDA-MB-231 cells were treated with either DMSO vehicle (day 0) or 10nM Taxol for 1-4 days and Brk mRNA levels were assessed by qRT-PCR after normalization to TBP; significance was assessed by the unpaired Student’s t-test (*, P < 0.05, **, P < 0.01, ***, P < 0.001). (C) To generate MCF-7 TaxR cells, MCF-7 cells purchased from ATCC were cultured in increasing concentrations of Taxol (10nM-10μM) continuously over a period of 6 months. Cells were then maintained in media containing 10μM Taxol. Lysates from MCF-7 parental and MCF-7 TaxR cells was subjected to Western blotting to assess protein expression of Brk, HIF-2α, and Actin (loading control).
Both HIF-1α and HIF-2α are recruited to numerous hypoxia response elements (HREs) throughout the Brk promoter (4). As we specifically identified HIF-2α as a novel GR target gene (4), we sought to determine if GR, HIF-2α and PELP1 were directly recruited to the Brk promoter in response to Taxol. Thus, chromatin immunoprecipitation (ChIP) assays were performed in MDA-MB-231 cells following dex, Taxol, or combined treatment. Increased recruitment of GR was detected on the Brk promoter following either dex (positive control) or Taxol treatment relative to vehicle control, while the combination of both dex and Taxol, a condition in which GR is highly phosphorylated at Ser134 and ligand activated, led to enhanced recruitment to the same GRE-containing region (Fig. 3A). PELP1 and HIF-2α were similarly recruited to the Brk promoter following treatment with either dex or Taxol alone, and in response to the combination of both agents (Fig. 3A). Importantly, recruitment of GR to the classic GR target gene, DUSP1, was unaltered by Taxol or combined treatment, suggesting specific promoter selectivity of the phospho-Ser134 GR species (Fig. 3A). These studies were repeated in U2OS osteosarcoma cells expressing either WT GR or serine 134 to alanine (S134A) mutant GR (34). As above, following 1 hour of dex treatment, GR was recruited to the Brk promoter relative to vehicle control. However, S134A GR recruitment was significantly impaired in the presence of dex relative to WT GR (Fig. 3B). Again, similar recruitment of WT GR and S134A GR to the DUSP1 promoter was observed following dex treatment, suggesting a high degree of phospho-GR specificity for the Brk promoter. Notably, when ChIP assays were performed using a phospho-Ser134 GR antibody in MDA-MB-231 cells treated with either vehicle or Taxol plus dex, we again observed specific phospho-GR recruitment to the Brk promoter (Fig. 3C). Collectively, these results support our model of a shared mechanism for Taxol-induced Brk expression via activation of p38 MAPK and formation of p-GR/PELP1/HIF-2α transcriptional complexes as previously observed for host (cortisol) and cellular (H2O2, hypoxia, nutrient starvation) stressors in TNBC models (32).
Figure 3.
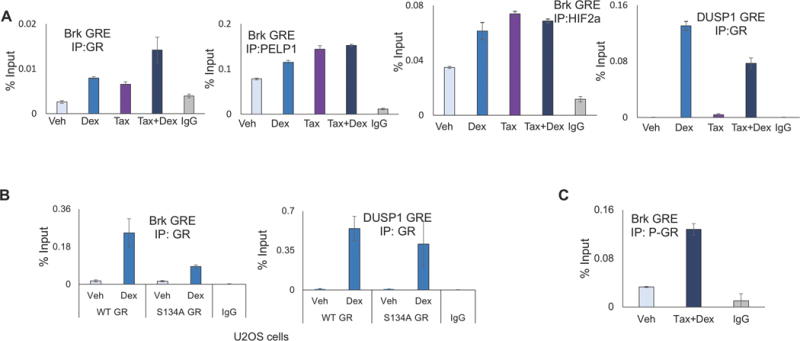
A phospho-GR, PELP1, HIF-2α complex is recruited to the Brk promoter in response to Taxol and dex treatment. (A) MDA-MB-231 cells were pre-treated with vehicle or 10 nM Taxol for 30 minutes, followed by 1 h treatment with either vehicle or 1μM dex and then ChIP assays were performed using antisera specific to GR, PELP1, HIF-2α, or isotype-matched (negative control) antibodies and qPCR was performed and quantitated as described in the methods. (B) U2OS cells stably expressing either WT GR or S134A GR were treated for 1 h with either vehicle or 1μM dex and then ChIP assays were performed with GR antisera or isotype-matched antibodies and qPCR was performed. (C) MDA-MB-231 cells were pre-treated with vehicle or 10 nM Taxol followed by 1 h treatment with either vehicle or 1μM dex and then ChIP assays were conducted using P-GR antisera or isotype-matched control antibodies and qPCR was performed.
Growth in suspension/low attachment induces p-GR/AhR complexes in TNBC
Cancer cell survival in the absence of attachment to a basement membrane is a crucial step in the metastatic cascade that allows tumor cells of epithelial origin to survive in circulation and successfully metastasize (42,43). However, growth in suspension represents a stressful condition for epithelial cells. As such, we next sought to determine if Brk expression was induced in non-adherent breast cancer cells, to ultimately aid their survival in the stressful condition of suspension. To test this, MDA-MB-231 cells were cultured on adherent dishes (attached) or in forced-suspension (ultra-low attachment; ULA) for 24 hours. Western blot analysis demonstrated higher levels of Brk protein expression in ULA conditions relative to attached cells (Fig. 4A). Remarkably, expression all pathway members was induced in response to ULA, including HIF-1α, HIF-2α, activated phospho-p38 MAPK, and phospho-Ser134 GR (i.e. a p38 substrate). Expression of Brk and HIF-2A mRNAs were also significantly induced in response to ULA (Fig. 4B). The aryl hydrocarbon receptor (AhR) and downstream AhR-regulated genes are critical mediators of survival implicated in TNBC cells (44). In addition to induction of Brk and HIF-2A mRNAs, mRNA levels for AhR and the classic AhR target gene, CYP1A1, were also significantly induced in MDA-MB-231 cells cultured in ULA conditions relative to attached conditions (Fig. 4B). ChIP assays performed using chromatin isolated from MDA-MB-231 cells cultured either in attached or ULA culture conditions for 24 h revealed that both GR and AhR were recruited to the same region of the Brk promoter under each condition, with greater recruitment observed in ULA conditions (Fig. 4C), suggesting that Brk is an AhR target gene and that upregulation of Brk mRNA occurs via p-GR/AhR collaboration.
Figure 4.

Brk expression is induced in suspension culture and is a directed GR/AhR target gene. (A) MDA-MB-231 cells were grown in either attached (Att.) or as single cells in forced-suspension culture (using ultra-low attachment dishes; ULA) for 24 hours and analyzed via western blot with antibodies for Brk, HIF-1α, HIF-2α, PELP1, P-GR, total GR, P-p38, or total p38 (loading control). (B) Brk, HIF-2α, AhR, and CYP1A1 mRNA levels were assessed using biologic triplicates by qRT-PCR following normalization to TBP expression. (C) MDA-MB-231 cells were grown in attached or ULA conditions for 24 h and ChIP assays were performed with antisera for GR, AhR or isotype-matched (negative control) antibodies and qPCR was performed. (D) MDA-MB-231 cells were treated with vehicle or 10 nM Taxol for 0-4 days and mRNA expression levels were determined by qRT-PCR following normalization to 18S rRNA. (E) MDA-MB-231 cells were pre-treated for 30 minutes with CH223191 or with vehicle, followed by 48 hours of vehicle or 10 nM Taxol treatment and then mRNA levels were analyzed by qRT-PCR after normalization to TBP expression. (F) MDA-MB-231 cells were treated with vehicle or 10nM Taxol for 24 h and ChIP assays were conducted with AhR antisera or isotype-matched control antisera and qRT-PCR was performed. (G) MDA-MB-231 cells were treated with vehicle or 10 nM Taxol for 24 h. Cell lysates were subjected to immunoprecipitation (IP) with AhR antisera or rabbit IgG (control) and assessed by western blotting using GR or AhR antibodies; input lysates are also shown. (H) MDA-MB-231 cells were treated with either vehicle or 10nM Taxol for 24 h and cell lysates were subjected to IP with PELP1 antibody or rabbit IgG (control) input lysates are also shown. Immunoprecipitated lysates and input control were analyzed via western blot with AhR or PELP1 antibodies. (I) MDA-MB-231 cells were pre-treated with either vehicle or 10 μM of a PELP1 inhibitor (D2), followed by 72 hours of treatment with either vehicle or 10 nM Taxol and mRNA prepared to measure CYP1B1 levels relative to TBP expression via qRT-PCR. Statistical significance was assessed by the unpaired Student’s t-test (*, P < 0.05, **, P < 0.01, ***, P < 0.001), or ANOVA followed by Tukey’s multiple comparison test (†, P < 0.001).
AhR has diverse ligands, including many environmental toxins, such as polycyclic aromatic hydrocarbons (PAHs) (3,45). We hypothesized that Taxol treatment may activate AhR in TNBC cells. To address this question, MDA-MB-231 cells were cultured in Taxol for 0 to 4 days and assessed mRNA expression of AhR target genes. Interestingly, mRNA expression of AhR and two canonical AhR target genes, CYP1A1 and CYP1B1, were significantly induced in response to Taxol treatment relative to vehicle at multiple time points (Fig. 4D). To specifically implicate AhR in the increased expression of CYP1A1 and CYP1B1 mRNA in response to Taxol treatment, MDA-MB-231 cells were pretreated with the AhR inhibitor, CH223191, followed by vehicle or Taxol treatment and observed a significant reduction in CYP1A1 and CYP1B1 mRNA expression following treatment with CH223191, demonstrating the AhR dependence of these events (Fig. 4E). Moreover, ChIP assays demonstrated that in response to Taxol treatment, AhR was directly recruited to AhR binding motifs in the CYP1A1 and CYP1B1 promoters relative to vehicle treatment (Fig. 4F). Similar to results observed with p-GR, PELP1, and HIF-2a, ChIP assays also showed recruitment of AhR to the same region of the Brk promoter following 24 hours of Taxol treatment relative to vehicle, suggesting direct transcriptional regulation of Brk by p-GR, PELP1, HIF-2α and AhR in response to Taxol (Fig. 4F). Together, these data indicate that AhR is transcriptionally activated in response to Taxol treatment, resulting in the induction of classic AhR target genes CYP1A1 and CYP1B1, as well as Brk, a newly defined AhR target gene and mediator of stress-induced signaling.
No studies have assessed GR/AhR interaction in breast cancer models. To investigate this question, co-immunoprecipitation (co-IP) experiments were performed in MDA-MB-231 cells treated with vehicle or Taxol for 24 h. Notably, high levels of GR were observed in AhR immunoprecipitates in both vehicle and Taxol treated samples (Fig. 4G), indicating strong basal interaction between GR and AhR in MDA-MB-231 cells. As PELP1 is an important co-activator for a diverse group of nuclear receptors, including GR, and has been previously shown to interact directly with GR (32), we next sought to determine if PELP1 interacts with AhR as a co-factor in TNBC cells. Interestingly, as found with AhR and GR, PELP1 and AhR co-IP experiments revealed basal interaction between PELP1 and AhR, but increased levels of AhR protein in PELP1 immunoprecipitates treated with Taxol relative to vehicle (Fig. 4H). To determine if PELP1 is acting as a co-activator of AhR transcriptional activity, MDA-MB-231 cells were treated with Taxol, the PELP1 peptidomimetic inhibitor D2, or both agents and assessed AhR target gene expression. As expected, expression of the AhR target gene CYP1B1 was significantly induced in response to Taxol treatment, but this induction was greatly reduced when combined with D2 treatment (Fig. 4I). Consistent with these results, D2 blocked dex-induced Brk mRNA expression in TNBC cells (32). These data collectively suggest that AhR interacts with GR and PELP1 and identify PELP1 as a novel co-activator for AhR transcriptional activity in response to Taxol treatment.
Brk is required for TNBC cell survival and growth in Taxol or ULA conditions
To determine if the induction of Brk expression in response to Taxol is HIF-dependent, we generated a HIF1A/HIF2A double knockout (DKO) line of pooled clonal lines using CRISPR/Cas9 “nickase” technology to increase the efficiency of the gene knockout relative to shRNA approaches. In these experiments, control cells expressed the empty vector (pX462). Control or CRISPR HIF DKO cells were treated with either vehicle or 10nM Taxol for 24 hours and Brk expression was assessed by Western blot analysis. We found that Brk protein and mRNA were induced in the pX462 cells in response to Taxol relative to vehicle treatment, as previously observed for the shRNA control cells (pLKO.1, Fig. 5A). Notably, the regulation of Brk protein expression by Taxol was completely lost in HIF DKO cells compared to pX462-empty vector cells (Fig. 5A). Regulation of Brk mRNA followed a similar pattern, in which total levels of Brk mRNA in HIF DKO cells following Taxol treatment were significantly reduced relative to control cells, although the ratio of Brk mRNA in parental cells compared to HIF DKO cells remained similar (Fig. 5B). Taxol also failed to induce PELP1 protein in HIF DKO cells (Supp. Figure 4). To assess the requirement of HIFs for the induction of Brk expression in response to ULA, MDA-MB-231 cells were cultured on adherent dishes or in ULA conditions for 24hrs. Again, we observed the induction of Brk protein (Fig. 5C) and mRNA (Fig. 5D) in ULA culture relative to attached culture conditions, and this induction in ULA culture was substantially reduced in the HIF DKO cells. Collectively, these data indicate that HIFs are critical for the induction of Brk expression in response to either stress induced by Taxol therapy or during anoikis.
Figure 5.
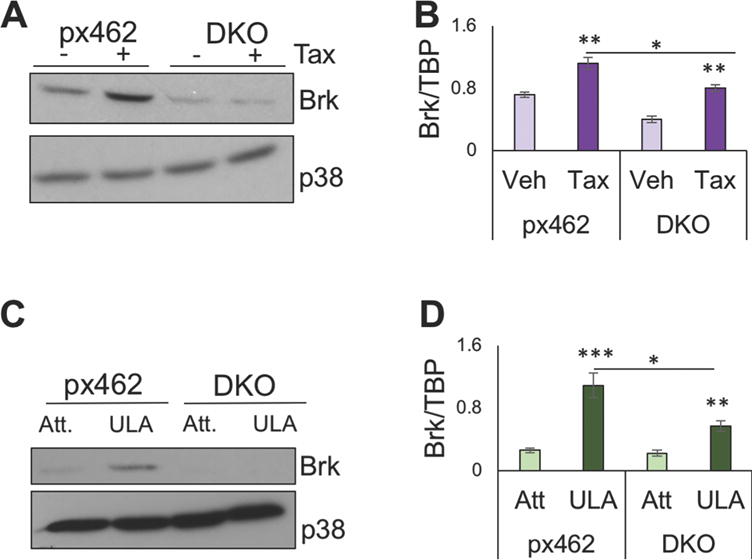
Brk expression induction in response to Taxol treatment or suspension culture stress ULA is HIF dependent. MDA-MB-231 cells expressing the control empty vector, pX462-puromcyin (px462) or in which HIF-1α and HIF-2α were deleted via CRISPR-Cas9n-mediated gene editing (DKO) cells were treated with either vehicle or 10 nM Taxol for 24 h. (A) Brk protein levels were compared by western blotting with antibodies for Brk and p38 MAPK (loading control) and (B) mRNA expression was measured by following normalization to TBP expression. (C) MDA-MB-231 pX462 control cells or HIF-DKO cells were cultured on either adherent dishes (Att) or as single cells in ULA dishes for 24 h and assessed by western blotting with Brk and p38 MAPK (loading control) antibodies or (D) by qRT-PCR analysis to determine Brk mRNA expression levels relative to TBP expression. Statistical significance was assessed by the unpaired Student’s t-test (*, P < 0.05, **, P < 0.01, ***, P < 0.001).
To test if Brk is critical for breast cancer cell survival in Taxol and ULA, we created Brk knock-out (Brk KO) cells in the MDA-MB-231 cells (pLKO.1 shRNA empty vector) via CRISPR/Cas9 technology. Brk knock-out was achieved in two independent clones, clone 27 and clone 70, as shown by Western blot analysis (Fig. 6A). While no significant differences were observed in basal growth rates (i.e. in untreated cells; Fig. 6B) between the parental (pLKO.1) cells and the Brk KO clones, Brk KO clone 27 and Brk KO clone 70 both exhibited significantly decreased growth as measured by the number of viable cells relative to the parental cells in the presence of Taxol treatment at days 3 and 6 (Fig. 6B). These data suggest that Brk is not required for basal proliferation of MDA-MB-231 cells, but that upon exposure to Taxol treatment, Brk expression is vital for maintenance of viability during this stressful condition, presumably due to enhanced cell survival of Brk+ relative to Brk-deficient clones. To determine if Brk expression is also critical for anchorage-independent survival of breast cancer cells when combined with Taxol treatment, MDA-MB-231 parental cells or MDA-MB-231 Brk KO cells were grown in soft agar in the presence of Taxol for 2 weeks and assessed colony forming ability. Notably, Brk KO clone 27 and 70 both formed significantly fewer colonies relative to the parental cells (Fig. 6C), suggesting that Brk expression is an important mediator of Taxol-resistant cell survival in TNBC cells. In similar experiments, equal numbers of untreated MDA-MB-231 parental cells or Brk KO clones 27 and 70 were plated in ULA conditions for 48 h, at which point the total number of total viable cells was quantified. Brk KO clone 27 and Brk KO clone 70 both exhibited significantly decreased cell density, numbers of cells per mL, relative to parental MDA-MB-231 cells in ULA (Fig. 6D). These data indicate that Brk expression enhances survival of MDA-MB-231 cells growing in ULA culture conditions.
Figure 6.
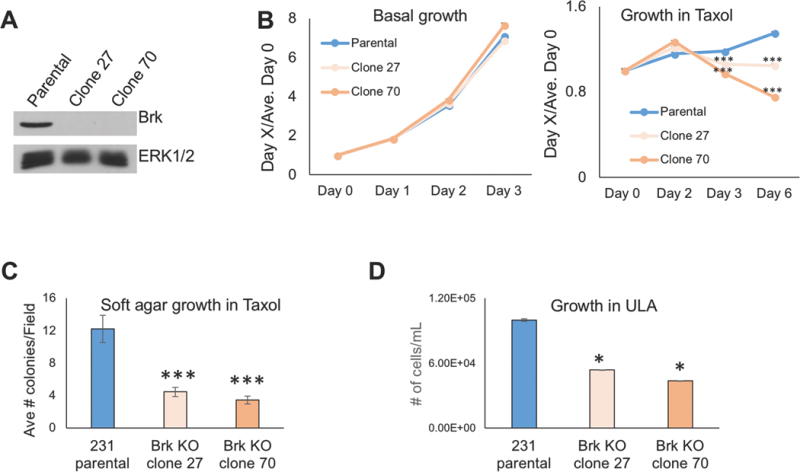
Brk enhances TNBC tumor cell growth in the presence of Taxol or during soft agar or ULA culture conditions. (A) MDA-MB-231 parental cells or MDA-MB-231 cells in two independent clonal lines which Brk was deleted via CRISPR/Cas9 gene editing (clones 27 and 70) were analyzed by western blotting with antibodies for Brk and ERK1/2 (loading control). (B) MTT cellular proliferation assays were conducted with MDA-MB-231 parental cells and Brk KO clones 27 and 70 in basal growth media with DMSO vehicle control or in media supplemented with 10 nM Taxol for the indicated amounts of time. Data are for each genotype per day relative to Day 0 levels and statistical significance is presented for each genotype relative to parental cells at the indicated time point. (C) Soft agar colony formation assays were used to compare the average number of colonies per field among MDA-MB-231 parental cells, Brk KO clone 27 or Brk KO clone 70 cells grown in the presence of 10 nM Taxol for 2 weeks. (D) Total cell density (# cells/mL) of MDA-MB-231 parental cells, Brk KO clone 27 or Brk KO clone 70 cells grown in suspension conditions for 48 h (0.5% methyl-cellulose in ULA dishes). Statistical significance was assessed by the unpaired Student’s t-test (*, P < 0.05, ***, P < 0.001).
In addition to enabling loss of contact inhibition, pro-survival pathways typically aid or accompany enhanced cancer cell migration. To assess the requirement of Brk for cell migration, scratch wound migration assays were conducted in the parental MDA-MB-231 cells or the Brk KO clones 27 and 70. Cells grown to confluent monolayers were scratched and images were taken immediately after scratching (t=0) and then again after 6 and 24 h (Fig. 7A). The rate of wound/scratch closure in Brk KO clones 27 and 70 was significantly impaired relative to parental MDA-MB-231 cells at all time points assessed (Fig. 7B), suggesting that Brk is a key mediator of basal cell migration in MDA-MB-231 cells. These results collectively demonstrate that Brk expression is critical for numerous aggressive biological phenotypes involved in the metastatic cascade in TNBC cells, including enhanced pro-survival that enables anchorage independent growth and enhanced cell migration.
Figure 7.
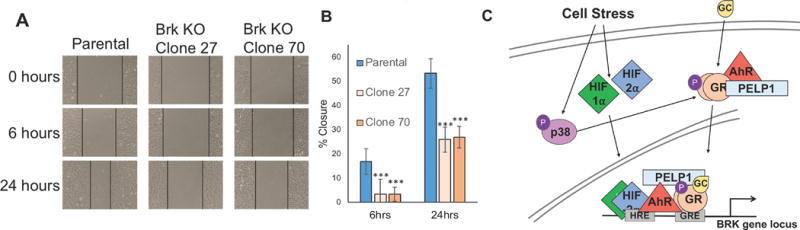
Loss of Brk impairs cell migration. A scratch wound assay was used to compare migratory phenotypes between MDA-MB-231 parental cells, and two Brk KO clones (27 and 70). (A) Images of wound closure over time (0 to 24 h) are shown; the black lines indicate the wound margins. (B) The percent of the original wound area that had closed (% closure) was determined by quantitating images using ImageJ. The grand mean of three independent biological replicate experiments is shown +/− S.E.M. Statistical significance was assessed by the unpaired Student’s t-test (*, P < 0.05, ***, P < 0.001). (C) Cartoon depiction of GR ligand independent phosphorylation at Ser134 by p38 MAPK, HIF stabilization in response to physiologic stress stimuli, and novel AhR/PELP1/GR interaction basally and in response to Taxol treatment. Upon ligand binding, phospho-GR, HIFs, AhR and PELP1 are recruited to the Brk promoter.
DISCUSSION
Herein, we report that Brk protein and mRNA levels are induced in a HIF-dependent manner following Taxol-induced stress and in conditions that mimic survival of single cells, or small clumps of tumor cells, in the circulation, demonstrating that Brk expressed is increased in response to a variety of stress inputs, including hypoxia and oxidative stress. Importantly, we have also discovered that chemotherapy and suspension culture result in direct recruitment of p-GR complexes that contain AhR to the Brk promoter, demonstrating that Brk is an AhR direct target gene (Fig. 7C).
We found that p-GR is readily detectable in human primary breast cancer tissue samples (Figure 1), and that p-GR levels are significantly enriched in TNBC samples relative to luminal samples. This observation is clinically relevant since high GR expression in TNBC tumors is associated with decreased overall survival and increased metastasis (25). Our results suggest that TNBC tumors that have high p-GR, which is ligand-activated following glucocorticoid treatment and potentially further phosphorylated in response to chemotherapy, may induce persistent Brk expression that contributes to aggressive and therapy-resistant tumor biology phenotypes. Notably, HIF-2α is a dex-induced GR target gene, while PELP1 is a HIF-2α target gene. Thus, dex-induced expression of HIF-2α (via GR) and PELP1 (via HIF-2α) induces a cascade of events in a feed-forward signaling pathway that enables formation of p-GR/HIF-2α/PELP1 complexes required for sustained stress-induced Brk expression. Indeed, high levels of Brk protein in primary human TNBC explants cultured ex vivo track with higher levels of GR and p-GR protein (32). Notably, we observed rapid induction of Sik expression, the mouse homolog of Brk, in the mammary glands of adult adrenalectomized C57Bl/6 mice just 6 hrs after a single dose of dex (Supp. Figure 1A–C), which demonstrates the remarkable inducibility of Sik/Brk in vivo. Taken together, our results suggest that, Brk may be inadvertently induced in the cancerous mammary tissue of patients following co-administration of glucocorticoids and chemotherapy. Further studies are needed to determine if administration of GCs during chemotherapy may be contraindicated in women diagnosed with TNBC.
A growing body of evidence has shown that Brk is a critical mediator of diverse phenotypes in breast cancer cells permissive for cancer cells to successfully metastasize. Notably, suspension conditions, which breast cancer cells must tolerate during their many transitions towards colonization to distant organs, result in AhR activation, recruitment of AhR to the Brk promoter, and elevated Brk expression. Our studies identify Taxol as a novel, and clinically relevant, activator of AhR transcriptional activity in TNBC cells (Figure 4). In addition to p-GR, we found that AhR interacts directly with the steroid receptor co-factor PELP1; the PELP1 peptido-mimetic inhibitor, D2, blocks AhR mediated induction of AhR target gene expression (including Brk) in response to Taxol treatment. Finally, Brk expression is critical for cancer cell viability in the presence of Taxol, survival in suspension conditions, and for associated cell migration. Overall, our data indicate that inhibiting Brk expression in TNBC cells likely diminishes the ability of cancer cells to effectively metastasize to distant organs.
While a wide range of progesterone (PR) and estrogen (ER) receptors are expressed in luminal breast cancers, their absence along with Her2 absence defines TNBC. However, SR-regulated gene programs remain highly active in selected TNBC subtypes (46,47). Closely related GR and PR exhibit a high degree of functional overlap. Namely, these SRs bind to the same hormone response element (HRE) consensus sequences in promoters and enhancers of target genes, and also interact with a shared subset of co-factors (48). Like PR, GR expression predicts good prognosis in ER+ luminal breast cancers (25). However, PR is emerging as a key mediator of ER-negative breast cancer stem or progenitor cell expansion (i.e. via paracrine signaling;(49)) and in particular, Ser294-phosphorylated PRs regulate gene sets associated with endocrine resistance (38) and breast cancer stem cell outgrowth (50). Given that high GR expression predicts poor outcome in TNBC, it is tempting to speculate that p-GR in TNBC shares functional redundancy with p-PR in luminal breast cancer, including regulation of EMT and stemness gene programs. Further study of these phospho-receptors in both luminal and TNBC tumors is warranted and may reveal novel targets for breast cancer patients with high expression of either p-PR or p-GR.
Collectively, our studies suggest that targeting the p-GR/HIF/PELP1/AhR complex and/or directly inhibiting Brk signaling is predicted to improve responses to Taxol-containing therapies and repress the metastatic cascade in TNBC patients. In future, combination therapies targeting p-GR (as opposed to GR) and/or associated factors (HIFs, AhR, PELP1) could be employed to avoid the unintended induction of stress-associated oncogenic gene expression programs (Brk, HIFs, AhR), thus allowing patients to continue beneficial glucocorticoid therapy while maximizing their tumor responses to chemotherapy.
Supplementary Material
Implication.
Breast cancer cells enlist intracellular stress response pathways that evade chemotherapy by increasing cancer cell survival and promoting migratory phenotypes.
Acknowledgments
This work was supported in part by NIH P30 CA77598 utilizing the following Masonic Cancer Center, University of Minnesota shared resource(s): Genomic Engineering Shared Resource (GESR). This work was supported by NIH/NCI R01 CA192178 (to C.A. Lange) and the Tickle Family Land Grant Endowed Chair in Breast Cancer Research (held by C.A. Lange) and by an NIH/NCI F31 predoctoral fellowship (CA195877-01 to T.M. Regan Anderson), an NIH/NCI T32 training grant fellowship (CA009138 to T.M. Regan Anderson) and NIH/NCI R01 CA138488 (to T.N. Seagroves).
List of Abbreviations
- Brk
Breast tumor kinase
- AhR
Aryl hydrocarbon receptor
- GR
glucocorticoid receptor
- HIF
hypoxia inducible factor
- TNBC
triple-negative breast cancer
- p-GR
phospho-Ser134 glucocorticoid receptor
- IHC
immunohistochemistry
- ULA
ultra-low attachment
- ER
estrogen receptor
- PR
progesterone receptor
- HER2
human epidermal growth factor receptor 2
- bHLH
basic helix-loop-helix
- PAS
Per-ARNT-Sim
- PAH
polycyclic aromatic hydrocarbon
- GC
glucocorticoid
- TMA
tissue microarray
- qRT-PCR
quantitative real-time polymerase chain reaction
- ChIP
chromatin immunoprecipitation
- Co-IP
co-immunoprecipitation
- Dex
dexamethasone
- DCIS
ductal carcinoma in situ
- PELP1
proline, glutamate and leucine rich protein 1
- 5FU
5-fluorouracil
- HRE
hypoxia response element
- DKO
double knockout
- KO
knockout
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–64. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 2.Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35(4):645–54. doi: 10.1007/s10555-016-9648-7. [DOI] [PubMed] [Google Scholar]
- 3.Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A. 1992;89(17):8185–9. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regan Anderson TM, Peacock DL, Daniel AR, Hubbard GK, Lofgren KA, Girard BJ, et al. Breast tumor kinase (Brk/PTK6) is a mediator of hypoxia-associated breast cancer progression. Cancer research. 2013;73(18):5810–20. doi: 10.1158/0008-5472.CAN-13-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lew BJ, Collins LL, O’Reilly MA, Lawrence BP. Activation of the aryl hydrocarbon receptor during different critical windows in pregnancy alters mammary epithelial cell proliferation and differentiation. Toxicol Sci. 2009;111(1):151–62. doi: 10.1093/toxsci/kfp125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hushka LJ, Williams JS, Greenlee WF. Characterization of 2,3,7,8-tetrachlorodibenzofuran-dependent suppression and AH receptor pathway gene expression in the developing mouse mammary gland. Toxicol Appl Pharmacol. 1998;152(1):200–10. doi: 10.1006/taap.1998.8508. [DOI] [PubMed] [Google Scholar]
- 7.Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov. 2002;1(8):609–20. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- 8.Brooks J, Eltom SE. Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr Cancer Drug Targets. 2011;11(5):654–69. doi: 10.2174/156800911795655967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ostrander JH, Daniel AR, Lofgren K, Kleer CG, Lange CA. Breast tumor kinase (protein tyrosine kinase 6) regulates heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells. Cancer research. 2007;67(9):4199–209. doi: 10.1158/0008-5472.CAN-06-3409. [DOI] [PubMed] [Google Scholar]
- 10.Peng M, Emmadi R, Wang Z, Wiley EL, Gann PH, Khan SA, et al. PTK6/BRK is expressed in the normal mammary gland and activated at the plasma membrane in breast tumors. Oncotarget. 2014;5(15):6038–48. doi: 10.18632/oncotarget.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Lu Y, Liang K, Hsu JM, Albarracin C, Mills GB, et al. Brk/PTK6 sustains activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR. Oncogene. 2012;31(40):4372–83. doi: 10.1038/onc.2011.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen HY, Shen CH, Tsai YT, Lin FC, Huang YP, Chen RH. Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Molecular and cellular biology. 2004;24(24):10558–72. doi: 10.1128/MCB.24.24.10558-10572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostrander JH, Daniel AR, Lange CA. Brk/PTK6 signaling in normal and cancer cell models. Current opinion in pharmacology. 2010;10(6):662–9. doi: 10.1016/j.coph.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harvey AJ, Pennington CJ, Porter S, Burmi RS, Edwards DR, Court W, et al. Brk protects breast cancer cells from autophagic cell death induced by loss of anchorage. Am J Pathol. 2009;175(3):1226–34. doi: 10.2353/ajpath.2009.080811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irie HY, Shrestha Y, Selfors LM, Frye F, Iida N, Wang Z, et al. PTK6 regulates IGF-1-induced anchorage-independent survival. PloS one. 2010;5(7):e11729. doi: 10.1371/journal.pone.0011729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ai M, Liang K, Lu Y, Qiu S, Fan Z. Brk/PTK6 cooperates with HER2 and Src in regulating breast cancer cell survival and epithelial-to-mesenchymal transition. Cancer biology & therapy. 2013;14(3) doi: 10.4161/cbt.23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castro NE, Lange CA. Breast tumor kinase and extracellular signal-regulated kinase 5 mediate Met receptor signaling to cell migration in breast cancer cells. Breast cancer research : BCR. 2010;12(4):R60. doi: 10.1186/bcr2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gatza ML, Kung HN, Blackwell KL, Dewhirst MW, Marks JR, Chi JT. Analysis of tumor environmental response and oncogenic pathway activation identifies distinct basal and luminal features in HER2-related breast tumor subtypes. Breast cancer research : BCR. 2011;13(3):R62. doi: 10.1186/bcr2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gatza ML, Silva GO, Parker JS, Fan C, Perou CM. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat Genet. 2014;46(10):1051–9. doi: 10.1038/ng.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto Y, Ibusuki M, Okumura Y, Kawasoe T, Kai K, Iyama K, et al. Hypoxia-inducible factor 1alpha is closely linked to an aggressive phenotype in breast cancer. Breast cancer research and treatment. 2008;110(3):465–75. doi: 10.1007/s10549-007-9742-1. [DOI] [PubMed] [Google Scholar]
- 22.Dales JP, Garcia S, Meunier-Carpentier S, Andrac-Meyer L, Haddad O, Lavaut MN, et al. Overexpression of hypoxia-inducible factor HIF-1alpha predicts early relapse in breast cancer: retrospective study in a series of 745 patients. International journal of cancer Journal international du cancer. 2005;116(5):734–9. doi: 10.1002/ijc.20984. [DOI] [PubMed] [Google Scholar]
- 23.Liu L, Liu W, Wang L, Zhu T, Zhong J, Xie N. Hypoxia-inducible factor 1 mediates intermittent hypoxia-induced migration of human breast cancer MDA-MB-231 cells. Oncol Lett. 2017;14(6):7715–22. doi: 10.3892/ol.2017.7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buxant F, Engohan-Aloghe C, Noel JC. Estrogen receptor, progesterone receptor, and glucocorticoid receptor expression in normal breast tissue, breast in situ carcinoma, and invasive breast cancer. Appl Immunohistochem Mol Morphol. 2010;18(3):254–7. doi: 10.1097/PAI.0b013e3181c10180. [DOI] [PubMed] [Google Scholar]
- 25.Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer research. 2011;71(20):6360–70. doi: 10.1158/0008-5472.CAN-11-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C, Wenger T, Mattern J, Ilea S, Frey C, Gutwein P, et al. Clinical and mechanistic aspects of glucocorticoid-induced chemotherapy resistance in the majority of solid tumors. Cancer biology & therapy. 2007;6(2):278–87. doi: 10.4161/cbt.6.2.3652. [DOI] [PubMed] [Google Scholar]
- 27.Pang D, Kocherginsky M, Krausz T, Kim SY, Conzen SD. Dexamethasone decreases xenograft response to Paclitaxel through inhibition of tumor cell apoptosis. Cancer biology & therapy. 2006;5(8):933–40. doi: 10.4161/cbt.5.8.2875. [DOI] [PubMed] [Google Scholar]
- 28.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer research. 2004;64(5):1757–64. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Dong J, Zou T, Du C, Li S, Chen C, et al. Dexamethasone induces docetaxel and cisplatin resistance partially through up-regulating Kruppel-like factor 5 in triple-negative breast cancer. Oncotarget. 2017;8(7):11555–65. doi: 10.18632/oncotarget.14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nanda R, Stringer-Reasor EM, Saha P, Kocherginsky M, Gibson J, Libao B, et al. A randomized phase I trial of nanoparticle albumin-bound paclitaxel with or without mifepristone for advanced breast cancer. Springerplus. 2016;5(1):947. doi: 10.1186/s40064-016-2457-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agyeman AS, Jun WJ, Proia DA, Kim CR, Skor MN, Kocherginsky M, et al. Hsp90 Inhibition Results in Glucocorticoid Receptor Degradation in Association with Increased Sensitivity to Paclitaxel in Triple-Negative Breast Cancer. Horm Cancer. 2016;7(2):114–26. doi: 10.1007/s12672-016-0251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Regan Anderson TM, Ma SH, Raj GV, Cidlowski JA, Helle TM, Knutson TP, et al. Breast Tumor Kinase (Brk/PTK6) Is Induced by HIF, Glucocorticoid Receptor, and PELP1-Mediated Stress Signaling in Triple-Negative Breast Cancer. Cancer research. 2016;76(6):1653–63. doi: 10.1158/0008-5472.CAN-15-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gradishar WJ, Anderson BO, Balassanian R, Blair SL, Burstein HJ, Cyr A, et al. NCCN Guidelines Insights: Breast Cancer, Version 1.2017. J Natl Compr Canc Netw. 2017;15(4):433–51. doi: 10.6004/jnccn.2017.0044. [DOI] [PubMed] [Google Scholar]
- 34.Galliher-Beckley AJ, Williams JG, Cidlowski JA. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Molecular and cellular biology. 2011;31(23):4663–75. doi: 10.1128/MCB.05866-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sutton LM, Cao D, Sarode V, Molberg KH, Torgbe K, Haley B, et al. Decreased androgen receptor expression is associated with distant metastases in patients with androgen receptor-expressing triple-negative breast carcinoma. Am J Clin Pathol. 2012;138(4):511–6. doi: 10.1309/AJCP8AVF8FDPTZLH. [DOI] [PubMed] [Google Scholar]
- 36.Strand DW, Jiang M, Murphy TA, Yi Y, Konvinse KC, Franco OE, et al. PPARgamma isoforms differentially regulate metabolic networks to mediate mouse prostatic epithelial differentiation. Cell Death Dis. 2012;3:e361. doi: 10.1038/cddis.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11(2):155–68. [PubMed] [Google Scholar]
- 38.Knutson TP, Daniel AR, Fan D, Silverstein KA, Covington KR, Fuqua SA, et al. Phosphorylated and sumoylation-deficient progesterone receptors drive proliferative gene signatures during breast cancer progression. Breast cancer research : BCR. 2012;14(3):R95. doi: 10.1186/bcr3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Molecular and cellular biology. 2007;27(2):466–80. doi: 10.1128/MCB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ismail A, Nawaz Z. Nuclear hormone receptor degradation and gene transcription: an update. IUBMB Life. 2005;57(7):483–90. doi: 10.1080/15216540500147163. [DOI] [PubMed] [Google Scholar]
- 41.Pires IM, Blokland NJ, Broos AW, Poujade FA, Senra JM, Eccles SA, et al. HIF-1alpha-independent hypoxia-induced rapid PTK6 stabilization is associated with increased motility and invasion. Cancer biology & therapy. 2014;15(10):1350–7. doi: 10.4161/cbt.29822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer Lett. 2008;272(2):177–85. doi: 10.1016/j.canlet.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 43.Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. 2013;1833(12):3481–98. doi: 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 44.D’Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer research. 2015;75(21):4651–64. doi: 10.1158/0008-5472.CAN-15-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol. 1982;22:517–54. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- 46.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PloS one. 2016;11(6):e0157368. doi: 10.1371/journal.pone.0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leehy KA, Regan Anderson TM, Daniel AR, Lange CA, Ostrander JH. Modifications to glucocorticoid and progesterone receptors alter cell fate in breast cancer. J Mol Endocrinol. 2016;56(3):R99–R114. doi: 10.1530/JME-15-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465(7299):803–7. doi: 10.1038/nature09091. [DOI] [PubMed] [Google Scholar]
- 50.Knutson TP, Truong TH, Ma S, Brady NJ, Sullivan ME, Raj G, et al. Posttranslationally modified progesterone receptors direct ligand-specific expression of breast cancer stem cell-associated gene programs. J Hematol Oncol. 2017;10(1):89. doi: 10.1186/s13045-017-0462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.