

Abstract
Although smoking is a significant risk factor for pancreatic cancer (PDAC), the molecular mechanisms underlying PDAC development and progression in smokers are still unclear. Here we show the role of cAMP response element-binding protein (CREB) in the pathogenesis of smoking-induced PDAC. Smokers had significantly higher levels of activated CREB when compared to non-smokers. Cell lines derived from normal pancreas and pancreatic intraepithelial neoplasm (PanIN) exhibited low baseline pCREB levels compared to PDAC cell lines. Furthermore, elevated CREB expression correlated with reduced survival in PDAC patients. Depletion of CREB significantly reduced tumor burden after tobacco-specific nitrosamine 4-(methyl nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) treatment, suggesting a CREB-dependent contribution to PDAC growth and progression in smokers. Conversely, NNK accelerated PanIN lesion and PDAC formation via GM-CSF mediated activation of CREB in a PDAC mouse model. CREB inhibition (CREBi) in mice more effectively reduced primary tumor burden compared to control or GM-CSF blockade alone following NNK exposure. GM-CSF played a role in the recruitment of tumor associated macrophages (TAM) and Treg expansion and promotion, whereas CREBi significantly reduced TAM and Treg populations in NNK-exposed mice. Overall, these results suggest that NNK exposure leads to activation of CREB through GM-CSF, promoting inflammatory and Akt pathways. Direct inhibition of CREB, but not GM-CSF, effectively abrogates these effects and inhibits tumor progression, offering a viable therapeutic strategy for PDAC patients.
Keywords: cAMP response element binding protein (CREB), mitogen-activated protein kinase (MEK/MAPK), pancreatic intraepithelial neoplasia (PanIN), pancreatic ductal adenocarcinoma (PDAC), 4-(methyl nitrosamino)-1-(3-pyridyl)-1-butanone (NNK), granulocyte-macrophage colony-stimulating factor (GM-CSF)
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer-related death in the United States and is characterized by its poor prognosis and a dismal 5-year survival rate approaching 5% (1). It is predicted to become the second leading cause of cancer-related deaths by 2030 (2,3). Smoking is a major modifiable risk factor which accounts for almost 20–30% of PDAC cases in the United States (4). Cigarette use increases the likelihood of developing PDAC and adversely influences overall patient survival (5). Nicotine and nitrosamines are the most potent and highly abundant carcinogens present in cigarette smoke. These compounds have been shown to initiate a spectrum of oncogenic processes integral to the development of PDAC (6). In smoking patients, nicotine accumulates to high concentrations in numerous organs, including the pancreas (7,8). Similarly, nitrosamines are present at seven times higher levels in the pancreatic juice of smokers (9) compared to non-smokers. Among multiple tobacco specific nitrosamines, 4-(methyl nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is considered to be the most carcinogenic (10,11). The precise molecular pathway governing the progression from dysplasia to invasive cancer due to NNK exposure remains unclear.
Recent evidence suggests that nicotine exposure significantly increases the number of low grade PanINs in LSL-KrasG12D/+;Pdx1Cre/+ (KC) mice (11), suggesting the role of NNK in promoting tumorigenesis through multiple diverse oncogenic pathways (6). Binding of NNK to the β-adrenergic receptor activates the EGFR/MAPK pathway, leading to ERK1/2 phosphorylation and thereby promoting cellular proliferation, migration, and angiogenesis (12). Inhibition of the EGFR pathway in smokers using Erlotinib (EGFR inhibitor) monotherapy has failed to show efficacy in a phase II clinical trial (13), suggesting that alternate signaling pathways contribute to NNK-induced PDAC pathogenesis and require further investigation. NNK acts as an agonist to nAChR and α7nAChR is the most investigated subunit of nAChR, which activate PKA and PI3K/Akt signaling (14).
Cyclic AMP response element binding protein (CREB) is a transcriptional co-activator which has been shown to be activated with nicotine use (15). CREB is activated through phosphorylation at Ser133 (KID domain) by several key oncogenic pathways such as protein kinase A (PKA), Akt/protein kinase B (PKB), MAPK and p90 ribosomal S6 kinase (p90RSK) (16). Activated CREB (pCREB) then binds to its mammalian transcriptional co-activator, the CREB-binding protein (CBP) (17), enabling the recruitment of additional transcriptional machinery elements that drive malignant progression. Moreover, GM-CSF signaling can promote PDAC tumorigenesis (18) and GM-CSF production can induce activation of CREB through MEK-dependent signaling pathway (19). Recently, studies in PDAC have also been reported that GM-CSF is involved in the recruitment and polarization of tumor associated macrophages (TAMs) in the tumor microenvironment (18,20). Therefore, we hypothesized that CREB could be a possible target for smoking-induced PDAC progression through its interactions with GM-CSF.
In this study, we demonstrate that NNK promotes PanIN progression and PDAC formation through GM-CSF mediated activation of CREB. CREB inhibition (CREBi) effectively prevented this progression, suggesting CREB is a critical regulator of NNK-induced tumor progression and a novel potential target for therapy in smoking-induced PDAC.
Materials and Methods
Cell lines, reagents and primers
Human pancreatic ductal epithelial cell lines (HPNE, HPNE-Kras), and PDAC cell lines such as MiaPaCa2, PANC1, SW1990, AsPC1, CAPAN1, CEPAC, HPAC and BxPC3 were obtained from American Type Culture Collection (ATCC) and were maintained according to the ATCC guidelines. Cell authentication was performed by using STR DNA profiling (latest date: June 16, 2016 and July 21, 2017) and cell lines tested negative for Mycoplasma via Genetica cell line testing (Burlington, NC) using eMYCO plus kit (iNtRON Biotechnology). Cells with relative low passage numbers (< 20) were used in the study. Cells treated with NNK in Figure 1 and Figure 5 with relative passage numbers (< 60) were used in the study. ATCC cell lines were characterized and were free of Mycoplasma contamination, tested by Hoechst DNA stain (indirect) and agar culture (direct) methods.Figure 2,3,4,6,7
Figure 1.
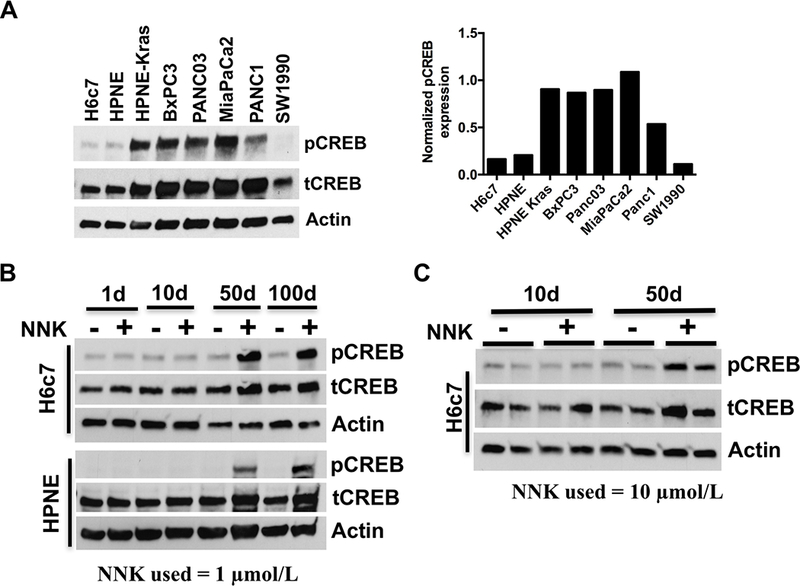
NNK exposure activates CREB in human pancreatic epithelial cells. A. Western blot demonstrating baseline expression of activated and total CREB levels in a panel of eleven human and pancreatic cell lines including PDAC cells (left panel). Actin was used as a loading control. Densitometry analyses of pCREB normalized to total CREB was shown in the right panel. B. Western blot demonstrating time-dependent pCREB expression in H6c7 and HPNE human pancreatic ductal epithelial cells (exhibiting low basal pCREB expression) upon NNK treatment (1 μmol/L) for up to 100 days. C. Western blot demonstrating time-dependent pCREB expression in H6c7 cells upon NNK exposure (10 μmol/L) for 10 and 50 days respectively.
Figure 5.

NNK administration differentially stimulates GM-CSF secretion in vitro and in vivo. A. RNA obtained from PanIN cells treated with NNK (1 μmol/L) or DMSO (control), for 5 and 50 days respectively, were subjected to RNA-seq analysis. The differentially expressed genes between the control and treatment groups were determined using hierarchical clustering and subsequent heat map generation of normalized gene expression in standardized units. Data analysis using Ingenuity Pathway Analysis (IPA) revealed the upregulated genes in the treatment group (denoted by red color). B. qPCR data of RNA collected from PanIN cells (top panel) and H6c7 cells (bottom panel) treated with NNK (1 μmol/L) or DMSO (control) for 50 days and analyzed to validate the genes upregulated in RNA-seq data (n=3). C. Human cytokine array data obtained using conditioned media collected from NNK (1 μmol/L) treated H6c7 cells and PanIN cells (left panel). Relative GM-CSF expression from the respective NNK-treated cells (H6c7 and PanIN) quantified using Image J image analysis software (right panel). D. GM-CSF levels in serum of Ptf1aCreER; LSL-KrasG12D/+ mice administered with NNK (10 mg/100g/3days) in a dose dependent manner, measured using ELISA (top panel, n=6 per group). KC (LSL-KrasG12D/+; Pdx1Cre/+), KPC (LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+) and PKT (Ptf1aCre/+; LSL-KrasG12D/+; Tgfbr2 flox/flox) mice were injected with NNK at 10 mg/100g by intraperitoneal administration, every 3 days/week for two weeks, GM-CSF levels in serum was measured using ELISA (lower panel, n=4 per group). E. Western blot demonstrating pCREB expression levels in MiaPaCa2 cells treated with NNK (1 μmol/L) with or without GM-CSF antibody blockade (αGM, 1 μg/ml) and CREBi (666–15, 1 μmol/L). F. Western blot demonstrating pAkt and pCREB expression levels in MiaPaCa2 cells treated with MK2206 (250 nMol/L) with or without recombinant GM-CSF (rGM-CSF, 100 ng/ml). G. Serum GM-CSF level was measured from PKT mice treated with CREBi (666–15) (10mg/kg/daily) for 3 weeks. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Figure 2.
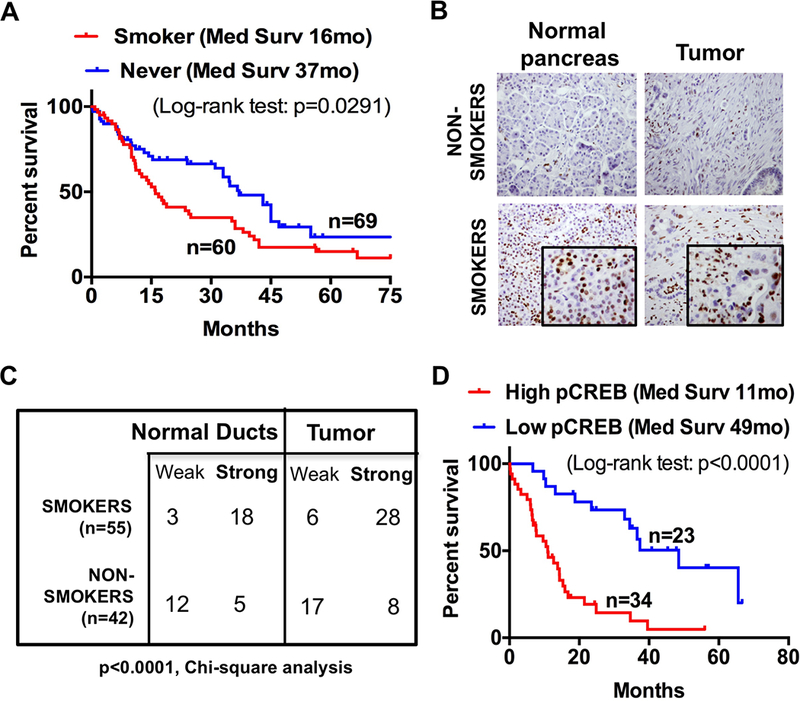
Activated CREB levels in human pancreatic tissues correlates with their smoking status. A. Kaplan-Meier survival curve comparing overall survival of PDAC patients with never smokers versus smokers (n=129; P = 0.0291). B. Representative immunohistochemical analysis of pCREB staining in tissue microarray (TMA) constructed from human pancreatic tissues obtained from smokers and non-smokers (one specimen per group shown). C. Chi-squared analysis of pCREB expression in normal pancreatic duct and tumor tissues obtained from smokers and non-smokers. (n=97, P<0.0001). D. Kaplan-Meier survival curve comparing overall survival of PDAC patients stratified by their corresponding pCREB staining levels (n=57; P< 0.0001).
Figure 3.

Smoking accelerates PanIN and PDAC formation and activates CREB in the spontaneous Ptf1aCreER; LSL-KrasG12D/+ (iPK) mouse model. A. Western blot demonstrating baseline expression of activated and total CREB levels in a panel of three mouse pancreatic cell lines including cancer cells (left panel). Actin was used as a loading control. Normalized pCREB expression levels were quantified using Image J image analysis software (right panel). B. Western blot demonstrating time-dependent pCREB expression in pre-cancerous mouse PanIN cells (exhibiting low basal pCREB expression) upon NNK treatment (1 μmol/L) for up to 100 days. C. Representative H&E, cytokeratin 19, alcian blue and claudin 18 staining of pancreatic tissues harvested from NNK (10 ppm in drinking water) treated iPK mice for up to 24 weeks. Scale bar: 50μm. D. Quantification of the number of alcian blue positive PanIN lesions (low and high grade) per high-power field (HPF) in the control and NNK-exposed iPK mice. (n=8, **, P < 0.01; ***, P < 0.001). E. Western blot demonstrating pCREB expression in pancreatic tissues harvested from NNK (10 ppm in drinking water) administered iPK mice (left panel) and densitometry analyses of pCREB normalized to total CREB protein was shown (right panel).
Figure 4.
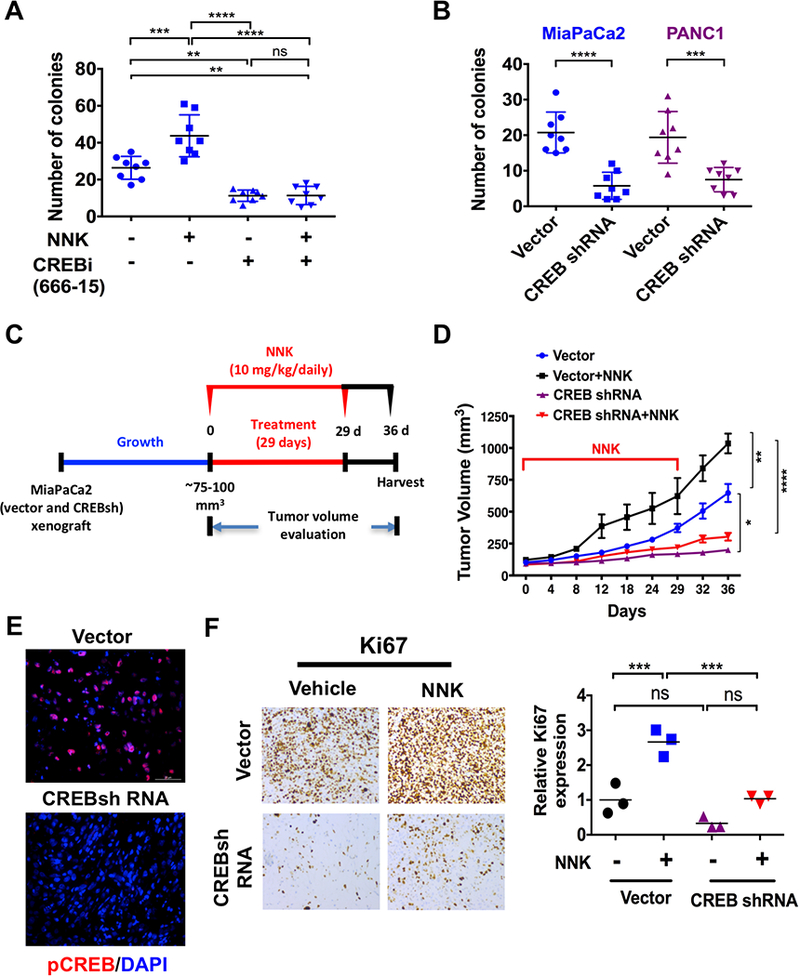
CREB inhibition (CREBi) effectively suppresses NNK-induced pancreatic tumor growth. A. MiaPaCa2 cells were treated with NNK (1 μmol/L) and/or CREB inhibitor (1 μmol/L) and analyzed for number of colonies. Colony size results were calculated from eight photographs analyzed from triplicate wells. Data are represented as mean ± S.D. B. MiaPaCa2 and PANC1 CREB shRNA cells were analyzed for number of colonies. Colony size results were calculated from eight photographs analyzed from triplicate wells. C. Experimental design for NNK treatment in CREBsh MiaPaCa2 xenograft mouse model (n=5 per experimental cohort). D. CREBsh MiaPaCa2 and empty vector cells were subcutaneously injected onto the flank of Fox1-nu/nu mice and treated with NNK at 10 mg/100g by intraperitoneal administration, three times/week for 29 days. Tumor volumes were measured up to 36 days. The tumors were later dissected and growth curves were measured and represented as mean ± S.D (n=5). E. Immunofluorescence staining of pCREB expression in vehicle or NNK-treated vector and CREBsh MiaPaCa2 flank xenografts. F. Representative Ki67 staining of vehicle or NNK-treated vector and CREBsh MiaPaCa2 flank xenograft tissues (left panel). Quantification of Ki67 staining data obtained from vehicle or NNK-treated vector and CREBsh MiaPaCa2 flank xenografts (right panel) (n=3; ns, P > 0.05; ***, P < 0.001, Student t test). ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Figure 6.

Targeting CREB decreases tumor growth and alters recruitment of tumor-associated macrophages (TAMs) and Regulatory T cells (Tregs) in NNK-exposed PKT mice. A. Experimental design for NNK treatment with or without GM-CSF blockade (αGM) and/or CREBi (666–15) in PKT mice model (n=5 per group). B. Tumor weights obtained from resected tissues obtained from PKT mice treated with NNK (10 mg/100g/3days); with or without αGM (2.5 mg/ kg/ 3days); with or without CREBi (666–15) (10 mg/kg/daily) for 3 weeks (n=5). C. Representative H&E, F4/80, FoxP3 and CD3 staining of pancreatic tissues harvested from PKT mice treated with NNK with or without 666–15 and/or αGM. Scale bar: 50μm. D. Quantification of relative expression of F4/80, FoxP3 and CD3 in the harvested PKT tumor tissues (n=5). ns, P > 0.05; *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Figure 7.
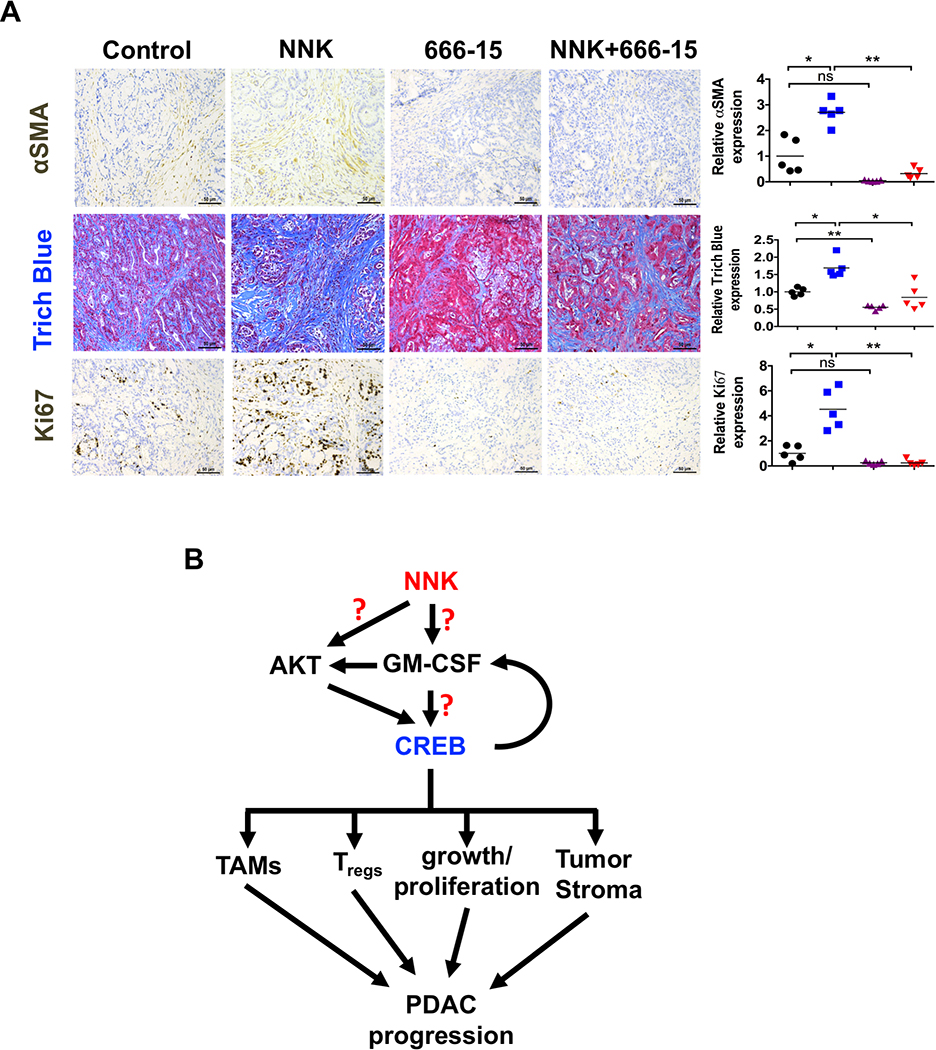
Targeting CREB decreases tumor stroma and proliferation by reducing immunosuppressive infiltrate in NNK-exposed PKT mice. A. Tissues obtained from PKT mice treated with NNK (10 mg/100g/3days); with or without CREBi (666–15) (10 mg/kg/daily) for 3 weeks (n=5) were stained for Ki67, α-SMA, trichrome blue and cleaved caspase 3 (left panel). Scale bar: 50μm. Quantification of relative expression of α-SMA, trichrome blue and Ki67 in the harvested PKT tumor tissues (right panel, n=5). ns, P > 0.05; *, P < 0.05; **, P < 0.01. B. Proposed model of the NNK-induced acceleration of the PDAC tumor growth by activating CREB. NNK induces GM-CSF that activates CREB through Akt signaling, which results in the activation of TAMs, Tregs, tumor growth/proliferation and tumor stroma, and regulates immune infiltration, which further accelerated the progression of PDAC.
Primary antibodies used for Western blot analysis, immunohistochemistry and immunofluorescence were listed in the Supplementary Table S1, and gene specific primers used for quantitative reverse transcription PCR (qRT-PCR) are listed in Supplementary Table S2.
CREB gene knockdown by short hairpin RNA
SMART vector Lentiviral Human CREB1 shRNA (#V3SH11243–00EG1385) and SMART vector Empty Vector Control (#VSC11649) were obtained from Dharmacon. Transfection was performed according to supplier protocol and as previously described (21). Lentiviral particles were prepared by co-transfecting plasmids into 293T cells. MiaPaCa2 and PANC1 cells were transduced with lentiviral vectors at an MOI (multiplicity of infection) of 40, and GFP expression was confirmed by FACS Caliber flow cytometer (BD Biosciences). The cells were then selected for 7 days with puromycin (1.5 μg/mL), and when cultures reached near confluency, cells were trypsinized and processed for FACS sorting to separate cells with highest GFP expression. To generate stable knockdown clones, these cells were plated at high dilutions in 10 cm petri dishes and colonies obtained from single cells were screened for the expression of CREB by immunoblotting technique.
RNA sequencing (RNA-seq)
Total RNA was isolated from PanIN cells treated with NNK (1 μmol/L) for 5 and 50 days and untreated cells (n = 3) using RNeasy Plus Mini Kit (Qiagen) and submitted to the Microarray Core Facility at Vanderbilt University. Ingenuity analysis was performed with help from Vanderbilt Technology for Advanced Genomics and Research Design. The upregulated and downregulated gene list for 5 days (Supplementary Tables S3 and S4) and 50 days (Supplementary Tables S5 and S6) are listed. The CREB signature genes upregulated by NNK are listed in Supplementary Table S7. The raw RNA-seq data has been deposited at Gene Expression Omnibus (GEO) under accession number (GSE119517).
Cytokine array analysis
NNK (1 μmol/L)-exposed human H6c7 and mouse PanIN cells were grown on culture flasks until 70% to 80% confluent and then cultured in serum-free medium (SFM) for 24 hours at 37°C. Conditioned medium (CM) was collected separately from these cells and stored at −80°C. The CM was collected multiple times. Human and mouse collections were separately pooled. The cells were maintained in culture with fresh media added twice weekly. CM and culture medium were concentrated using Millipore centriprep centrifugal filters (EMD Millipore, Billerica, MA) with 10k cut off and centrifuged at 2000 rpm, 4°C to concentrate. The concentrated CM and culture medium were quantified for the protein concentration and the aliquots were stored at −80°C. In the following experiments, conditioned media protein was diluted with serum-free medium (SFM) and the cytokine levels in the conditioned media were measured using human and mouse cytokine array kits (R&D systems) according to the manufacturer’s directions.
Human tissue microarray
Clinical materials were obtained from patients who provided written informed consent, according to the Vanderbilt Institutional Review Board standards and guidelines. The studies were conducted in accordance with recognized ethical guidelines (Declaration of Heisinki, CIOMS, Belmont Report, U.S. Common Rule). Tissue microarrays (TMA) were designed and constructed as previously described (22). The TMA slides were simultaneously evaluated by a pathologist (C.S). Normal pancreas, well-differentiated PDAC, moderately-differentiated PDAC, and poorly-differentiated PDAC tissue cores were used as previously detailed (22). The staining of the tissues was assessed using staining index measured as a sum of the intensity score (0, no staining; 1+, weak; 2+, moderate; 3+, strong) and distribution score (0 – no staining; 1+ – staining of <33% of cells; 2+ – between 33% and 66% of cells; and 3+ – staining of >66% of cells). Staining indices were classified as follows: 2+ to 3+ or higher – strong staining; 0 to 1+– weak staining. Activated CREB (pCREB) was scored as positive if any detectable staining was present.
Genetically engineered transgenic mice
Ptf1aCre/+;Tgfbr2flox/flox and LSL-KrasG12D/+;Tgfbr2flox/flox mice were provided by Dr. Hal Moses (Vanderbilt University Medical Center, Nashville, Tennessee, USA). These two lines were intercrossed to generate Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox mice (PKT) on a C57Bl/6 background (22). Genotyping of alleles was performed using oligonucleotide primers as described previously (23,24). Ptf1aCreER knock-in mice (provided by Dr. Christopher Wright, Vanderbilt University Medical Center, Nashville, TN) expresses CreER under endogenous Ptf1a promoter/enhancer elements (25). CreER production recapitulates endogenous Ptf1a expression, with nuclear translocation within 24 hours of tamoxifen (Tam) administration. Ptf1aCreER mice were intercrossed with LSL-KrasG12D/+ to generate Ptf1aCreER; LSL-KrasG12D/+ (iPK) mice. LSL-KrasG12D/+, Pdx1Cre/+and p53R273H/+mice were intercrossed to generate indicated LSL-KrasG12D/+; Pdx1Cre/+ (KC) and LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+ (KPC) animals. All mice were housed under a 12-h light-dark diurnal cycle with controlled temperature (21°C to 23°C) and provided with a standard rodent diet and water ad libitum throughout all experiments.
In vivo xenograft studies
Athymic nude mice – Foxn1 nu/nu (4–5 weeks old) – were purchased from Harlan Sprague Dawley, Inc. Subcutaneous tumors were established by injecting 2×106 MiaPaCa2 or PANC1 CREB knockdown (CREB shRNA) cells into the flank of a 6-week-old Fox1-nu/nu mouse (n=5 in each group) as previously detailed (22). Treatment was initiated when the subcutaneous (s.c.) tumors reached 70 – 200 mm3 size. NNK [10 mg/100 g body weight, three times/week - a dosage similar to the level of NNK found in the pancreatic juice of smokers (26)] or vehicle (saline) was administered intra-peritoneal (IP) for 29 (MiaPaCa2) and 25 (PANC1) days. The subcutaneous tumor volume and percent body weight change was recorded as previously described (22). Growth curves for tumors were plotted as the mean volume ± standard deviation (s.d.) of tumors for mice from each group. At the end of the study, animals were sacrificed and their primary tumors were removed.
Treatment of PKT mice
PKT mice were treated with vehicle, GM-CSF blockade antibody (αGM), CREB inhibitor (666–15), or a combination of αGM and 666–15. Mice in the NNK (10 mg/100 g/day), αGM (2.5 mg/kg/3days) and 666–15 (10 mg/kg/day) arm were received starting at 4 weeks of age. NNK and/or αGM were administered intraperitoneally and 666–15 or vehicle was administered by oral gavage for 3 weeks, after which mice were euthanized and dissected. Due to the irregularity of the tumor dimensions, size was determined by weighing the entire tumor. Tumor tissue was processed for further immune-histochemical examination.
Immunohistochemistry
Tissues were fixed and immunostained using antibodies against cytokeratin 19, Ki67, F4/80, FoxP3 and claudin 18 (Supplementary Table S1). These tissues were stained for alcian blue. Stained tissues were evaluated by an expert pathologist (C.S). Tissue slides were deparaffinized, antigen retrieval was carried out in citrate buffer (pH = 6.0) under pressure for 15 minutes, and endogenous peroxidase activity was blocked by incubating with 3% H2O2 for ten minutes. The sections were stained with primary antibodies at described concentrations and developed using DAB substrate (Vector, Burlingame, CA). Immunostained slides were scanned and their corresponding staining was quantified using the Ariol SL-50 platform (Leica Camera, AG). Digital slide images were adjusted to exclude areas containing obvious histologic artifacts, such as tissue folds or nonorganic material, from the digital image. Values were calculated and reported as relative expression of protein staining.
Statistical analysis
Descriptive statistics were calculated using Microsoft Excel and Prism software (Graphpad). Results are shown as values of mean ± s.d. unless otherwise indicated. Statistical analyses of immunohistochemistry data were performed using the ANOVA followed by Tukey’s multiple comparisons test to determine P-values. The two-sided Student’s t test was used for two groups comparison, with P < 0.05 considered as statistically significant, except when indicated otherwise. Survival curves were estimated with the Kaplan-Meier method and significance of the differences between groups was assessed with the log-rank test.
Ethics statement
All experiments were performed in compliance with the regulations and ethical guidelines for experimental and animal studies of the Institutional Animal Care and Use Committees at the Vanderbilt University (Nashville, TN) and the University of Miami (Miami, FL) (Protocol # 15–057, 15–099 and 18–081).
For additional experimental procedures please refer to Supplementary Materials and Methods.
Results
NNK exposure induces pCREB expression in pancreas cells
To understand the mechanistic role of smoking in pancreatic cancer progression, we first exposed human primary pancreatic ductal epithelial cells (H6c7) to NNK for up to 50 days and performed a phosphokinase array (Supplementary Fig. S1A). Array data revealed a significant upregulation of CREB and Akt phosphorylation upon NNK exposure. Akt inhibition with MK-2206 in these cells resulted in downregulation of pCREB expression, demonstrating that CREB activation is dependent, at least in part, on Akt signaling (Supplementary Fig. S1B). MK-2206 is known to inhibit the activity of Akt by decreasing the phosphorylation of serine 473 (S473) and threonine 308 (T308) on Akt (27). This provided the molecular basis to further examine the role of NNK-induced activation of CREB in pancreas cells.
A panel of normal pancreatic ductal epithelial and PDAC cell lines were screened for their respective baseline pCREB expression. This analysis showed that all PDAC cell lines except SW1990 exhibit robust expression of pCREB, whereas normal ductal epithelial cells (H6c7 and HPNE) had low basal expression (Fig. 1A). To determine if NNK exposure results in CREB activation, H6c7 and HPNE cells were treated with NNK (1 μmol/L) and immunoblotted for pCREB expression. NNK dosage of 1 μmol/L and 10 μmol/L corresponds to lowest and highest dose delivered per cigarette, respectively (6). There was a significant time-dependent increase in pCREB expression in both the cell lines upon chronic exposure to NNK (Fig. 1B). Since NNK acts as an agonist to nAChR (α7 subunit; α7nAChR), the baseline expression of this receptor and CSFR2α (Supplementary Fig. S2A) on a panel of seven pancreatic cell lines was determined. As shown in Supplementary Fig. S2B (28), robust expression of α7nAChR was seen in most pancreas cells including HPNE. Interestingly, exposure of H6c7 cells to a high concentration of NNK (10 μmol/L corresponding to equivalent dose of 5–6 cigarettes/day) for 50 days exhibited similar time-dependent CREB activation (Fig. 1C) that was observed with low concentration NNK exposure, suggesting that NNK-induced CREB activation is a phenomena dependent on duration of exposure more so than concentration.
Cigarette smokers have elevated pCREB levels and decreased survival in human PDAC
Smokers had a shorter median survival than non-smokers (median survival of 16 months vs 37 months, respectively; p=0.0291) (Fig. 2A). Tissue microarray (TMA) samples from patients with known smoking status were examined for pCREB expression (Figs. 2B and C). pCREB expression was significantly higher in both normal and malignant pancreatic tissues obtained from smokers compared to non-smokers. Furthermore, elevated pCREB expression in PDAC tumors was associated with worse overall survival (OS) compared to low pCREB levels (median survival of 11 months vs 49 months, respectively; p<0.0001, Fig. 2D).
NNK exposure accelerates PanIN lesion development in Ptf1aCreER; LSL-KrasG12D/+ mice
To determine the role of pCREB in tumor progression, we screened for pCREB levels in multiple cell lines derived from PanIN, primary PDAC (PDA) and liver metastasis (LMP) lesions which were generated from LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+ (KPC) genetically engineered mouse model (GEM) of PDAC (Fig. 3A). These cell lines serves as a predictive in vitro model of tumor progression as validated in our previous studies (29). Pre-malignant PanIN cells exhibited lowest basal expression of pCREB compared with malignant PDA and LMP cell lines. Chronic exposure of NNK (1 μmol/L) to PanIN cells for up to 50 days revealed an elevated expression of pCREB in a time-dependent fashion relative to their corresponding vehicle-treated controls (Fig. 3B), concordant with our results seen with NNK-treated human pancreatic ductal epithelial cells (Fig 1B).
Early stage PDAC development is characterized by the formation of PanINs, occurring through acinar-to-ductal metaplasia (30). In Ptf1aCreER; LSL-KrasG12D/+ (iPK) mice, KrasG12D is specifically activated in acinar cells using a tamoxifen-inducible Cre expressed by the endogenous Ptf1a locus (25,31). iPK mice can initiate PDAC by activating oncogenic KrasG12D specifically within mature acinar cells, while KrasG12D activation in adult duct cells or centroacinar cells has little or no effect on PDAC initiation (32). These mice can normally develop ADM by 9 weeks of age and PanIN lesions at 8 to 17 months of age, which arise from acinar cells (31). The endogenous Kras in iPK model rely on Ptf1aCre to obtain acinar cell-specific expression of Kras by Cre-mediated recombination of a stop cassette placed in the Kras locus. Cre-mediated recombination can excise the stop codon and permit the oncogenic protein to be expressed. The oncogenic Kras expression is for the cell’s lifetime because it is a genetic change. Thus, the cells targeted and all the cells that come from that cell will have the recombined KrasG12D allele and presumably express it. Interestingly, chronic NNK exposure led to progressive increase in cytokeratin 19 (CK19), alcian blue positive ductal precursor lesions (PanIN-1 and PanIN-2), and claudin 18 levels as early as 8 weeks in the pancreatic tissues of iPK mice, whereas these effects were not seen even at 24 weeks in the corresponding vehicle-treated mice (Figs. 3C and D). Chronic NNK exposure (upto 16 weeks and onwards) also significantly elevated pCREB expression in the pancreatic tissues of iPK mice compared to their corresponding vehicle-treated control mice (Fig. 3E). Furthermore, chronic NNK exposure upto 24 weeks also led to increase in PanIN-3 lesions. These results show that NNK exposure accelerates PDAC progression through progressive increase in number of PanIN lesions.
CREB knockdown decreases NNK-induced pancreatic tumor growth
To further delineate the role of CREB in NNK-induced pancreatic carcinogenesis, MiaPaCa2 cells were treated with a CREB inhibitor (CREBi, 666–15) with or without NNK exposure. Specificity of 666–15 has been previously investigated and shown to inhibit CREB’s transcription activity with an IC50 ~80–100 nM (33). Our results showed 666–15 in PDAC cells resulted in inhibition of cell proliferation and increase in apoptosis (Supplementary Figs. S3A and B), as well as it inhibited CREB mediated gene transcription of FOS and EGR1 (Supplementary Fig. S3C). CREBi significantly reduced NNK-induced anchorage independent growth in MiaPaCa2 cells (Fig. 4A). Next, CREB knockdown (CREBsh) cells were generated for MiaPaCa2 and PANC1 (Supplementary Figs. S4A and B), which showed a significant reduction in the number of colonies compared to a vector control (Fig. 4B).
MiaPaCa2 and PANC1 CREB shRNA xenografts were generated using cell lines via subcutaneous flank injection in athymic nude mice (Fig. 4C and Supplementary Fig. S4C). The empty vector and CREB shRNA xenografts were then treated with either NNK or vehicle (Figs. 4C and D). Administration of NNK (10 mg/100g body weight) administration by intraperitoneal injection was chosen as it recapitulates the levels of NNK found in smokers (26). CREBsh MiaPaCa2 xenografts showed absence of pCREB expression when compared to vector control xenografts (Fig. 4E). Furthermore, CREB knockdown significantly impaired NNK-induced tumor growth and proliferation, as measured by Ki67 index, in comparison to NNK-treated vector controls (Figs. 4D and F). This effect on NNK-induced tumor growth was also observed in PANC1 CREBsh xenografts (Supplementary Fig. S4C). NNK and its metabolites have a long retention time in vivo (34). Our results showed that the tumor growth rate remained unaltered in the absence of NNK. Importantly, NNK treatment did not have any impact on mouse weight throughout the treatment period (Supplementary Fig. S4D).
GM-CSF activates CREB in vitro and in vivo
To elucidate the molecular signals that drive NNK-induced pancreatic tumor growth, RNA-seq analysis was used to examine the upregulated pathways in PanIN cells exposed to NNK (for 5 and 50 days) (Fig. 5A, Supplementary Fig. S5). Ingenuity Pathway Analysis (IPA) identified top seven cellular functions, overlapping genes, upstream/downstream molecules and the top five diseases (Supplementary Fig. S5A, B, C and D). This analysis revealed that CREB1, FOS, EGR1 and GM-CSF (CSF2) were the most upregulated genes in response to chronic (50 days) NNK exposure (Fig. 5B), indicating that NNK induction activates CREB at the transcriptional level. Additionally, FOS and EGR1, known downstream target genes of CREB, were also significantly upregulated upon NNK exposure. The RNA-seq results were subsequently validated using qRT-PCR (Fig. 5B) for both mouse PanIN and human H6c7 cells treated with NNK for 50 days. Given the elevated expression of GM-CSF with NNK exposure, we then sought to delineate the role of GM-CSF in NNK-induced tumor cell growth by analyzing the conditioned media obtained from NNK treated H6c7 and PanIN cells. GM-CSF secretion was significantly increased in the conditioned media of NNK treated cells compared to their respective untreated controls, in concordance with our RNA-seq findings (Fig. 5C). This effect was also noted in vivo, where a significant increase in serum levels of GM-CSF in NNK-treated iPK (Fig. 5D, top panel), KC, KPC and PKT (Fig. 5D, lower panel) mice was observed. This indicated that NNK-induction stimulates GM-CSF release both in vitro and in vivo.
In order to determine the specific role of GM-CSF on pancreatic cancer cell function, we first examined the expression of the GM-CSF receptor protein CSF2Rα on five different primary human pancreatic cancer cell lines and two pancreatic ductal epithelial cell lines. The GM-CSF receptor CSF2Rα was expressed in all the cell lines tested (Supplementary Fig. S2). NNK induced activation of CREB was reduced by blocking GM-CSF (αGM) or CREBi or combination of αGM and CREBi in H6c7 cells (Fig. 5E). To determine if the pro-tumorigenic effects of GM-CSF were mediated through CREB, HPNE-Kras (Supplementary Fig. S6A) and MiaPaCa2 (Supplementary Fig. S6B) cells were treated with recombinant GM-CSF (rGM-CSF) with or without CREB inhibitor. CREBi significantly reduced GM-CSF mediated colony formation, suggesting that GM-CSF exerts its cellular functions, at least in part, through CREB. To understand the mechanistic link between GM-CSF and CREB, MiaPaCa2 cells were treated with rGM-CSF with or without Akt inhibitor MK-2206 (Fig. 5F). Cells treated with rGM-CSF showed activation of both Akt and CREB. We observed a significant reduction in both pAkt and pCREB levels with Akt inhibition in comparison to the controls or rGM-CSF, suggesting that GM-CSF also causes upregulation of CREB by activating Akt signaling in PDAC cells. Furthermore, there was a significant reduction in GM-CSF levels in the serum obtained from CREBi treated PKT (Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox) mice in comparison to the vehicle treated controls (Fig. 5G). These results confirm that CREBi affects GM-CSF levels.
Targeting CREB affects the immune tumor microenvironment
Tumor-derived GM-CSF plays a critical role in regulating inflammation and immune suppression within the tumor microenvironment (35). Our results demonstrating an enhanced release of GM-CSF upon NNK exposure both in vitro and in vivo compelled us to further explore the potential effects of smoking on in vivo PDAC tumor growth and its effect on the immune tumor microenvironment. NNK was administered with or without GM-CSF blockade and/or CREBi in PKT mice (Fig. 6A). CREBi (666–15) dosage has been chosen as it’s the dose that is well-tolerated in vivo (33). CREBi significantly reduced tumor burden in NNK-treated PKT mice compared to the corresponding vehicle treated controls. The arm receiving additional GM-CSF blockade did not display any additional anti-tumor effects compared with CREBi monotherapy in NNK-treated PKT mice (Fig. 6B), suggesting these effects are mediated primarily through CREB. The effect on the different immune compartments of an autochthonous and aggressive PKT mouse model of PDAC was evaluated. CREBi monotherapy significantly reduced F4/80 positive tumor-associated macrophages (TAMs) and FoxP3 positive regulatory T cells (Tregs cell population) in NNK-treated PKT mice when compared to their corresponding vehicle, αGM or combination of CREBi and αGM treated mice (Figs. 6C and D). Additionally, CREBi increased the CD3+ T cell population in the NNK treated tumors. None of the treatment modalities employed had any significant impact on mouse weights assessed during treatment as shown in Supplementary Fig. S6C. Analysis of the effect of CREB on inflammation has been performed using flow cytometry (Supplementary Table S8). Results show that CREBi increased the infiltration of CD3+ T cells while at the same time increasing the proportion of CD8+ T cells. This resulted in increased CD8/CD4 T cell ratio.
Targeting CREB affects tumor stroma and immune infiltration
To explore the relationship between CREB signaling, stroma and immune infiltration, we utilized PKT mice. Tissues from PKT mice treated with NNK with or without CREBi were stained for α-SMA and trichrome blue, which showed abundant tumor stroma (Fig. 7A). Coincident with tumor formation, the pancreatic tissue of PKT mice were highly fibrotic, as reported previously (36,37). CREBi significantly reduced tumor stroma in NNK-treated PKT mice compared to the corresponding controls. Furthermore, CREBi significantly reduced proliferation in NNK-treated PKT mice compared to the corresponding controls (Fig. 7A). These results suggest that the underlying reason of reduced tumor growth is due to reduced proliferation of the tumor cells and invoked anti-tumor immunity due to reduced infiltration of immunosuppressive cells.
Discussion
Oncogenic Kras is the most frequent mutation and a critical driver of PDAC growth and development (38). Targeting Kras has been largely ineffective, leading investigators to pursue other downstream targets for therapeutic effect. CREB is a downstream mediator in the Kras pathway and is constitutively active in most PDAC tumors. In our current study, survival outcomes are significantly worse in smokers compared with never smokers (Fig. 2A) and we confirmed that activated CREB is overexpressed in pancreatic tissues of smokers (Figs. 2B and C). Additionally, activated CREB levels were strongly associated with worse overall survival in PDAC patients, suggesting the potential role of cigarette smoking in worsening overall disease prognosis through upregulation of pCREB.
Our results demonstrate a predominant increase in pCREB expression which correlates with an accelerated progression of ADM, and low-grade and high-grade PanIN lesions during the transformation from normal epithelium to dysplastic tissue with chronic exposure to NNK (Figs. 3B, C and D). Activation of CREB in iPK mice exposed to NNK were consistent with our in vitro observations in human normal ductal epithelial and mouse pre-neoplastic cells exposed to NNK. Additionally, we noted that pCREB levels were elevated to a greater degree with chronic NNK exposure when compared to acute NNK exposure (Figs. 1 and 3), indicating that the duration of NNK exposure is an important risk factor for PDAC development (39).
Our results provide compelling evidence that CREB activation induced by NNK exposure plays a critical role in the onset (Fig. 3D) and development of PDAC as CREB knockdown significantly reduces NNK-induced PDAC tumor growth and proliferation in vivo (Figs. 4C and E). NNK is metabolized into 4-(methylnitrosamino) −1-(3-pyridyl)-1-butanol (NNAL) and [4-(methylnitrosamino)-1-(3-pyridyl) but-1-yl]-beta-O-D-glucosiduronic acid (NNAL-Gluc) in vivo (40). The slow elimination of carcinogenic metabolites found in cigarette smoke and their presence within multiple organs suggests a high retention of NNK within host tissues including the pancreas. This may indicate an increased risk for PDAC development and progression amongst former smokers even after smoking cessation. Further investigation into this phenomenon is warranted.
After delineating the strong correlation between NNK and pCREB overexpression, we successfully showed the regulatory mechanism which leads to pCREB induction after NNK exposure. The RNA seq and subsequent qPCR data obtained from NNK treated H6c7 and PanIN cells showed a significant upregulation of CREB, FOS, EGR1 and GM-CSF in comparison to their corresponding vehicle treated controls. GM-CSF blockade attenuated pancreas tumor weight gain in NNK exposed PKT mice, suggesting a pro-tumorigenic role of GM-CSF in PDAC. This is consistent with the recent report about the role of CREB in GM-CSF secretion in lung fibroblast cells (41) and smoking-induced lung cancer model. Our study revealed a strong correlation between pCREB levels and serum levels of GM-CSF in both in vitro cell models and in vivo PDAC GEMMs when were treated with NNK (Figs. 5C and D). To the best of our knowledge, these data are the first report to link GM-CSF to smoking-induced PDAC formation and progression. Furthermore, we showed that CREBi completely abrogated the adverse oncogenic effects of GM-CSF, indicating that NNK-induced activation of CREB occurs through GM-CSF (Figs. 5E and G). Recent reports have defined the role of GM-CSF in malignant progression (42), worse prognosis (43) and modulation of immune tumor microenvironment through recruitment and differentiation of myeloid progenitor cells into immunosuppressive myeloid-derived suppressor cells (MDSCs) in the surrounding stroma in PDAC (44). More importantly, we show here that CREB also regulates GM-CSF (Fig. 5G), which may have an indirect effect on these suppressive cells.
We also observed that NNK treated PKT mice exhibited elevated GM-CSF secretion, which corresponded to an increased tumoral macrophage population. Other studies have suggested the role of TAMs in ADM formation in PDAC (45). Our data suggests that NNK exposure may mediate TAM infiltration into pancreatic tissue and promote ADM through the CREB/GM-CSF pathway. It has also been shown that CREB activation promotes the induction and maintenance of Tregs (46) and generation of immunosuppressive macrophages (47). We show here that CREBi alone can significantly reduce tumor burden and decrease infiltration of these immunosuppressive TAMs and Tregs in NNK-exposed PKT mice, suggesting that CREB is a critical regulator of the immune microenvironment. Specifically, in terms of TAMs, it has been shown that CREB can promote the M2 macrophage phenotype (48). Hence, specific contribution of CREBi in modulating M1 vs. M2 phenotype of TAMs in the TME should be analyzed in the future studies. Furthermore, our results show that CREBi also increases the infiltration of CD3+ T cells into the PDAC tumors; suggesting a overall switch from immunosuppressive to tumor inhibitory TME. Further studies aimed at evaluating the direct effects of CREBi on various immune cells should inform the extent of direct effects of CREBi on immune microenvironment in the TME.
Previous reports have identified GM-CSF as a central signaling molecule in promotion of an immunosuppressive microenvironment (49). Although GM-CSF may not directly affect Tregs, it has been shown that increased GM-CSF levels may promote generation of Tregs secondary to the induction of CD11c+CD8a- DCs (38). Therefore, targeting the negative effects of GM-CSF pathway, which is an important component of multiple normal homeostatic processes, may be achieved through downstream inhibition of CREB. Therefore, CREBi further presents a novel therapeutic pathway in regulating the PDAC immune microenvironment. It’s known that GSK3 and NF-κB pathways regulate inflammatory cells through CREB (46,50). It is plausible that CREBi may regulates inflammation indirectly through GSK3 and NF-κB signaling pathways. Our results indicate that these effects proceed through CREB, thus presenting a potential pathway that can be therapeutically targeted to improve overall immune responses. A more detailed understanding of the communication between tumor and stromal cells would further aid in knowing the potential direct and indirect effects of CREBi on immune cells and role of inflammatory cells within this process.
Our work demonstrates for the first time that CREB plays a significant role in smoking-induced PDAC tumor growth through modulation of the GM-CSF pathway, mediating Akt, affecting downstream pro-inflammatory factors, tumor stroma and reducing TAMs and Treg infiltration (Fig. 7B). Furthermore, CREB depletion delays tumor growth in PDAC mouse models and appears to downregulate the Akt pathways induced by cigarette smoking. Overall, these data provide mounting evidence of the functional role of CREB in smoking-induced PDAC progression and provides with a unique therapeutic target in smoking-induced cancer models.
Supplementary Material
Significance:
Findings identify GM-CSF-induced CREB as a driver of pancreatic cancer in smokers and demonstrate the therapeutic potential of targeting CREB to reduce PDAC tumor growth.
Acknowledgements
The authors thank Mr. Frank Revetta, Dr. Xizi Dai, Ms. Jennifer Barretta and Ms. Yanhua Xiong for their technical assistance.
Grant Support: This work was supported by the NIH NCI R21 CA209536, American Cancer Society IRG 98–277-13 and Stanley Glaser Foundation Research Award (UM SJG 2017–24) to N.S. Nagathihalli and R01 CA161976 and NIH T32 CA211034 to N. B. Merchant. Histopathology Core Service was performed through the Sylvester Comprehensive Cancer Center (SCCC) support grant (N. Nagathihalli).
Footnotes
Conflict of Interest:
The authors declare no potential conflicts of interest.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: A Cancer Journal for Clinicians 2013;63:11–30 [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–21 [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013;63:11–30 [DOI] [PubMed] [Google Scholar]
- 4.Fryzek JP GD, Greenson JK, Schottenfeld D,. A review of the epidemiology and pathology of pancreas cancer. Gastrointest Cancer 1997;2 99–110 [Google Scholar]
- 5.Yuan C, Morales-Oyarvide V, Babic A, Clish CB, Kraft P, Bao Y, et al. Cigarette Smoking and Pancreatic Cancer Survival . Journal of Clinical Oncology 2017;0:JCO.2016.71.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Momi N, Kaur S, Ponnusamy MP, Kumar S, Wittel UA, Batra SK. Interplay between Smoking-induced Genotoxicity and Altered Signaling in Pancreatic Carcinogenesis. Carcinogenesis 2012;33:1617–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chowdhury P, Doi R, Chang LW, Rayford PL. Tissue distribution of [3H]-nicotine in rats. Biomed Environ Sci 1993;6:59–64 [PubMed] [Google Scholar]
- 8.Le Houezec J Role of nicotine pharmacokinetics in nicotine addiction and nicotine replacement therapy: a review. Int J Tuberc Lung Dis 2003;7:811–9 [PubMed] [Google Scholar]
- 9.Prokopczyk B, Hoffmann D, Bologna M, Cunningham AJ, Trushin N, Akerkar S, et al. Identification of tobacco-derived compounds in human pancreatic juice. Chem Res Toxicol 2002;15:677–85 [DOI] [PubMed] [Google Scholar]
- 10.Secretan B, Straif K, Baan R, Grosse Y, El Ghissassi F, Bouvard V, et al. A review of human carcinogens--Part E: tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol 2009;10:1033–4 [DOI] [PubMed] [Google Scholar]
- 11.Kumar S, Torres MP, Kaur S, Rachagani S, Joshi S, Johansson SL, et al. Smoking accelerates pancreatic cancer progression by promoting differentiation of MDSCs and inducing HB-EGF expression in macrophages. Oncogene 2015;34:2052–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaal C, Chellappan SP. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol Cancer Res 2014;12:14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fountzilas C, Chhatrala R, Khushalani N, Tan W, LeVea C, Hutson A, et al. A phase II trial of erlotinib monotherapy in advanced pancreatic cancer as a first- or second-line agent. Cancer Chemother Pharmacol 2017;80:497–505 [DOI] [PubMed] [Google Scholar]
- 14.Wu CH, Lee CH, Ho YS. Nicotinic acetylcholine receptor-based blockade: applications of molecular targets for cancer therapy. Clin Cancer Res 2011;17:3533–41 [DOI] [PubMed] [Google Scholar]
- 15.Walters CL, Cleck JN, Kuo Y-c, Blendy JA. μ-Opioid Receptor and CREB Activation Are Required for Nicotine Reward. Neuron 2005;46:933–43 [DOI] [PubMed] [Google Scholar]
- 16.Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal 2004;16:1211–27 [DOI] [PubMed] [Google Scholar]
- 17.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 1999;68:821–61 [DOI] [PubMed] [Google Scholar]
- 18.Waghray M, Yalamanchili M, Dziubinski M, Zeinali M, Erkkinen M, Yang H, et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov 2016;6:886–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellegrini M, Cheng J, Voutila J, Judelson D, Taylor J, Nelson S, et al. Expression profile of CREB knockdown in myeloid leukemia cells. BMC Cancer 2008;8:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon EM, Raines MA, Blenis J, Sakamoto KM. Granulocyte-macrophage colony-stimulating factor stimulation results in phosphorylation of cAMP response element-binding protein through activation of pp90RSK. Blood 2000;95:2552–8 [PubMed] [Google Scholar]
- 21.Nagathihalli NS, Beesetty Y, Lee W, Washington MK, Chen X, Lockhart AC, et al. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-alpha: impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res 2014;74:2062–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology 2015;149:1932–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016;531:110–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 25.Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, et al. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 2013;140:751–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexandre M, Uduman AK, Minervini S, Raoof A, Shugrue CA, Akinbiyi EO, et al. Tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone initiates and enhances pancreatitis responses. Am J Physiol Gastrointest Liver Physiol 2012;303:G696–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sangai T, Akcakanat A, Chen H, Tarco E, Wu Y, Do KA, et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res 2012;18:5816–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Wadei MH, Al-Wadei HAN, Schuller HM. Effects of chronic nicotine on the autocrine regulation of pancreatic cancer cells and pancreatic duct epithelial cells by stimulatory and inhibitory neurotransmitters. Carcinogenesis 2012;33:1745–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagathihalli NS, Castellanos JA, VanSaun MN, Dai X, Ambrose M, Guo Q, et al. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget 2016;7:65982–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50 [DOI] [PubMed] [Google Scholar]
- 31.Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JPt, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee AYL, Dubois CL, Sarai K, Zarei S, Schaeffer DF, Sander M, et al. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li BX, Gardner R, Xue C, Qian DZ, Xie F, Thomas G, et al. Systemic Inhibition of CREB is Well-tolerated in vivo. Sci Rep 2016;6:34513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nature reviews Cancer 2003;3:733–44 [DOI] [PubMed] [Google Scholar]
- 35.Bayne Lauren J, Beatty Gregory L, Jhala N, Clark Carolyn E, Rhim Andrew D, Stanger Ben Z, et al. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012;21:822–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nature medicine 2016;22:497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iacobuzio-Donahue CA. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut 2012;61:1085–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schulte A, Pandeya N, Tran B, Fawcett J, Fritschi L, Risch HA, et al. Cigarette smoking and pancreatic cancer risk: more to the story than just pack-years. European journal of cancer (Oxford, England : 1990) 2014;50:997–1003 [DOI] [PubMed] [Google Scholar]
- 40.Hecht SS, Carmella SG, Chen M, Dor Koch JF, Miller AT, Murphy SE, et al. Quantitation of urinary metabolites of a tobacco-specific lung carcinogen after smoking cessation. Cancer Res 1999;59:590–6 [PubMed] [Google Scholar]
- 41.Koga Y, Hisada T, Ishizuka T, Utsugi M, Ono A, Yatomi M, et al. CREB regulates TNF-alpha-induced GM-CSF secretion via p38 MAPK in human lung fibroblasts. Allergol Int 2016;65:406–13 [DOI] [PubMed] [Google Scholar]
- 42.Pylayeva-Gupta Y, Lee Kyoung E, Hajdu Cristina H, Miller G, Bar-Sagi D Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012;21:836–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeuchi S, Baghdadi M, Tsuchikawa T, Wada H, Nakamura T, Abe H, et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res 2015;75:2629–40 [DOI] [PubMed] [Google Scholar]
- 44.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63:1769–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Habtezion A, Edderkaoui M, Pandol SJ. Macrophages and pancreatic ductal adenocarcinoma. Cancer Lett 2016;381:211–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. Journal of immunology 2010;185:6413–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalkman HO, Feuerbach D. Modulatory effects of alpha7 nAChRs on the immune system and its relevance for CNS disorders. Cell Mol Life Sci 2016;73:2511–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A 2009;106:17475–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohanbash G, McKaveney K, Sakaki M, Ueda R, Mintz AH, Amankulor N, et al. GM-CSF Promotes the Immunosuppressive Activity of Glioma-Infiltrating Myeloid Cells through Interleukin-4 Receptor-α. Cancer Research 2013;73:6413–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 2005;6:777–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.