

Abstract
Oncogenic tyrosine kinase fusions involving the anaplastic lymphoma kinase (ALK) are detected in numerous tumor types. Although more than 30 distinct 5’ fusion partner genes have been reported, treatment of ALK-rearranged cancers is decided without regard to which 5’ partner is present. There is little data addressing how the 5’ partner affects the biology of the fusion or responsiveness to ALK tyrosine kinase inhibitors (TKIs). Based on the hypothesis that the 5’ partner influences the intrinsic properties of the fusion protein, cellular functions that impact oncogenic potential, and sensitivity to ALK TKIs, clonal 3T3 cell lines stably expressing seven different ALK fusion variants were generated Biochemical and cellular assays were used to assess the efficacy of various ALK TKIs in clinical use, transformative phenotypes, and biochemical properties of each fusion. All seven ALK fusions induced focus formation and colonies in soft agar, albeit to varying degrees. IC50s were calculated for different ALK TKIs (crizotinib, ensartinib, alectinib, lorlatinib) and consistent differences (5–10 fold) in drug sensitivity were noted across the seven ALK fusions tested. Finally, biochemical analyses revealed negative correlations between kinase activity and protein stability. These results demonstrate that the 5’ fusion partner plays an important biological role that affects sensitivity to ALK TKIs.
Implications
This study shows that the 5’ ALK fusion partner influences ALK TKI drug sensitivity. As many other kinase fusions are found in numerous cancers, often with overlapping fusion partners, these studies have ramifications for other kinase-driven malignancies.
Keywords: Anaplastic Lymphoma Kinase, tyrosine kinase fusion, tyrosine kinase inhibitor, chromosomal rearrangement, EML4-ALK
Introduction
Genomic rearrangements involving the gene which encodes anaplastic lymphoma kinase (ALK) have been described in a broad spectrum of malignancies including anaplastic large cell lymphoma (ALCL), diffuse large B cell lymphoma, inflammatory myofibroblastic tumor (IMT), glioma, non-small cell lung cancer (NSCLC), colorectal, breast, ovarian, and esophageal cancer (1). ALK rearrangements have been identified in up to 8% of NSCLC cases (2) and in up to 50% of IMTs, a soft tissue tumor predominantly diagnosed in children (3). The resulting ALK fusion proteins all retain the entire kinase domain of ALK at the C-terminus, and the N-terminus consists of an entirely different protein. These fusion proteins are validated therapeutic targets. Several large international trials have now validated that patients with ALK positive (ALK+) lung cancer derive improved clinical outcomes from treatment with ALK TKIs (Supplementary Table S1), leading to FDA approval of agents such as crizotinib, ceritinib, alectinib, brigatinib, and lorlatinib. Similar trials have also been completed in ALK+ IMT and ALCL (4), and there have been case reports of response to ALK TKI therapy in patients with renal cell carcinoma and colon carcinoma harboring ALK fusions (5).
Although ALK fusions are validated targets for cancer therapy, precision regarding how to best target these fusions clinically is lacking in comparison to other oncogenic drivers like mutant Epidermal Growth Factor Receptor (EGFR) or retinoic acid receptor alpha (RARα) fusions, where it is known that different variants dictate drug sensitivity (6,7). For example, in EGFR-mutant NSCLC, it is known that different EGFR kinase domain mutations confer varying degrees of sensitivity or resistance to EGFR directed therapies (6). Likewise, for RARα fusions found in subtypes of leukemia, it is known that the particular gene fused to RARα not only affects response to therapy but also can be a therapeutic target itself (7). Greater than 30 distinct ALK fusion partners have been identified, including TPM-3/−4, CLTC, LMNA, PRKAR1A, EML4, RANBP2, TFG, FN1, KIF5B, and many others (3,8). Although many different 5’ partner genes have been reported for ALK, even within the same cancer type, there is currently little data to address the question of how different fusion partners may affect pretreatment clinical characteristics, disease responsiveness to targeted therapies, or acquired resistance. Therefore, treatment of ALK+ cancers is currently decided without considering which fusion partner is present. However, recent retrospective clinical studies in ALK+ NSCLC suggest a difference in progression-free survival based on the specific ALK fusion variant present (9–11). As next-generation sequencing technologies continue to be approved by regulatory agencies, clinicians will know both the 5’ partner and the 3’ kinase involved in the fusion. Therefore, it is imperative that the nuances of the various ALK fusions are better understood, including determining the therapeutic implications of the various ALK fusions to bring more precision to patient care. Herein, we sought to test the hypothesis that the 5’ ALK fusion partner influences the intrinsic properties of the fusion protein as well as the cellular functions that impact overall oncogenic potential and sensitivity to ALK targeted therapy.
Materials and Methods
Cell Culture:
NIH 3T3 cells were a kind gift from Dr. William Pao (12). NIH3T3 cells stably expressing ALK F1174L were a kind gift of Dr. Marc Ladanyi (13). Cell line authentication was not performed after receipt of the cells. NIH3T3 cells were maintained in DMEM (Mediatech, Corning, NY, USA), supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA, USA), and penicillin (100U/mL) / streptomycin (100μg/mL) (Mediatech) at 37°C, 5% CO2 for maintenance culture and all experiments unless noted otherwise. Plat GP cells were obtained from Cell Biolabs and cultured in RPMI 1640 (Mediatech), supplemented with 10% FBS (Atlanta Biologicals), and 2μg/mL blasticidin (Invitrogen, Grand Island, NY, USA). All cell lines were routinely evaluated for mycoplasma contamination. The latest date these cell lines were tested was September 2017.
Expression Constructs:
cDNAs for EML4-ALK E13:A20 (variant 1, V1), EML4-ALK E6a/b:A20 (variant 3, V3), KIF5B-ALK, TFG-ALK, PRKAR1A-ALK, FN1-ALK, RANBP2-ALK, and wild-type (WT) ALK receptor were synthesized by GeneArt, (ThermoFisher, Grand Island, NY, USA) and subcloned into the pMXs-Puro retroviral vector (Cell Biolabs, San Diego, CA, USA) using EcoRI and NotI or PacI. Genomic breakpoints we previously reported for TFG-ALK, PRKAR1A-ALK, and RANBP2-ALK were used to generate these cDNAs (3). The EML4-ALK V1, EML4-ALK V3, KIF5B-ALK, and FN1-ALK cDNA sequences were generated using previously described breakpoints (14–17).
Viral Transduction and Clonal Selection:
Retroviral vectors were individually transfected into the Plat GP packaging cell line (HEK293 cells stably expressing a gag-pol internal ribosome entry site) (Cell Biolabs). Viral media was harvested 48 hours post transfection and pelleted. The viral pellet was re-suspended in DMEM and applied to NIH 3T3 cells. Transduced NIH 3T3 cells were treated with 2μg/mL puromycin (Invitrogen) beginning at 48 hours post transduction for a minimum of two weeks. Single cell clones were grown until confluent in a 6-well plate at which time half of the cells were harvested for lysate, and half were frozen and stored for later culture. Lysate for each clone was run on the same gel and probed for total ALK expression (Cell Signaling, #3333). Clones exhibiting relatively equal expression for all ALK fusions were selected for expansion, except EML4-ALK V3, for which the lowest expressing clone of 20 tested (data not shown) was selected. Cells were maintained in DMEM, supplemented with 10% FBS, penicillin (100U/mL) / streptomycin (100μg/mL), and 2μg/mL puromycin at 37°C, 5% CO2 for maintenance culture and experiments unless noted otherwise. Cells were cultured and used for experiments for up to 12 weeks after thawing. All cell lines were routinely evaluated for mycoplasma contamination. The latest date these cell lines were tested was September 2017.
Immunoblot and Antibodies:
The following primary antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA): ALK mAb Rabbit (#3333), ALK mAb Mouse (3791S), ALK (D5F3) mAb Rabbit (#3633) and pALK Y1604 (#3341S). The actin antibody (A2066) was purchased from Sigma-Aldrich (St. Louis, MO, USA). The following secondary antibodies were obtained from LiCor (Lincoln, NE, USA): IRDye 680RD Goat anti-Mouse (#92668070), and IRDye 800 Goat anti-Rabbit (#92632211). For infrared (IR) immunoblot, cells were harvested, washed in PBS, and lysed in KRAS buffer [50 mM Tris-HCl, 150 mM NaCl, 5 mM MgCl2, 1% triton X-100, 0.5% Na Deoxy Cholate, 0.1% SDS, 40 nmol/L sodium fluoride, 1mM sodium orthovanidate, and complete protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA)]. Signal detection was obtained using the LiCor Odyssey scanner system and quantified using iMageStudio Lite software (LiCor).
Reverse Phase Protein Lysate Microarrays (RPPA):
NIH 3T3 cell lines stably expressing ALK fusion variants, WT ALK, ALK F1174L, and empty vector control cells were grown in normal culture conditions. Lysates were harvested using the lysis buffer recommended by the MD Anderson RPPA core [1% Triton X‐100, 50 mM HEPES, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM Na pyrophosphate, 1mM sodium orthovanidate, 10% glycerol, containing freshly added protease and phosphatase inhibitors]. Protein concentrations were adjusted to 1.5μg/μL. Samples were prepared as instructed by the MD Anderson RPPA Core using 4xSDS sample buffer [40% Glycerol, 8% SDS, 0.25M Tris‐HCL, pH 6.8. β-mercaptoethanol added at 1/10 of the volume immediately before use)], boiled for 5 minutes, and stored at −80°C until sample submission. Three separate biological replicates for each cell line type were tested. All of the samples were assayed for the expression of 304 phospho-protein or total protein analytes using the reverse phase protein lysate microarrays (RPPA), as described (https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core/rppa-process.html).
RPPA Analysis: The loading control normalized and log2 transformed RPPA data were imported and analyzed using R packages. Unsupervised hierarchical clustering analysis was performed to check the data quality and the association between cell lines. The heat map shows that the empty vector transferred and wild type cell line are well separated with other cell lines.
RPPA Statistical analysis: One-Way analysis of variance (ANOVA) was used to assess the differences in protein expressions between cell lines on a feature-by-feature basis. First, for one protein at a time, we carried out an over-all F test to detect any significant difference among the means of all the groups. Next, for the proteins identified in this process, we then compared between desired cell line groups to identify the sources of difference. The R library “multcomp” was used for this purpose. Note that the fold change (FC) values were calculated as the estimated ratio between the 2 groups in comparison, with the following conventional modification: For the ratios > 1 (up-regulation), FCs were noted as the same as the ratio. For the ratios ≤ 1 (down-regulation), FCs were noted as the negative inverse of the ratio. Furthermore, to account for multiple testing, we estimated the false discovery rates (FDR) of the overall test of the model using the Benjamini-Hochberg method. The criteria of protein selection for each pairwise comparison were: 1. Significant in overall F-test (FDR adjusted p-value<0.05); 2. Significant in pairwise comparison. (Raw p-value<0.05). 3. The FC between groups is at least 1.5.
In order to calculate pathway activity scores, RPPA data were median-centered and normalized by standard deviation across all samples for each component to obtain the relative protein level. The pathway score is then the sum of the relative protein level multiplied by its weight of all components in a particular pathway. The similar comparisons as protein level were applied to the pathway scores.
Focus Formation:
NIH 3T3 cell lines stably expressing ALK fusion variants were trypsinized to disrupt cell adhesions, seeded at 4000 cells/well in 24 well plates, and allowed to grow for 8 days in a cell culture incubator containing an IncuCyte ZOOM (Essen BioSciences, Ann Arbor, MI, USA). Foci were quantified manually from daily images acquired by the IncuCyte ZOOM.
Soft Agar Assays:
NIH 3T3 cell lines stably expressing ALK fusion variants were trypsinized to disrupt cell adhesions. 1.5mL of 0.5% agar/complete media were added to each well in a 6 well plate and allowed to solidify. Cells were seeded on top of the 0.5% agar/complete media layer at a density of 5000 cells/well in complete media containing 0.3% agar. After the agar solidified, 200μL of media was added on top and changed every week. Cultures were grown at 37°C in 5% CO2 for 6 weeks. Colony formation was quantified using the GelCount system (Oxford Optronix, United Kingdom).
Inhibitors:
Crizotinib, alectinib, lorlatinib, and ganetespib were purchased from Selleck Chemicals (Houston, TX, USA). Ensartinib was obtained from Xcovery (Palm Beach Gardens, FL). Cycloheximide was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Drug Response Curves:
NIH 3T3 cell lines stably expressing ALK fusion variants were seeded at 2000 cells/well in 96 well plates using the Multidrop Combi dispenser (ThermoFisher). Cells were treated with increasing doses of each inhibitor. 72 hours post drug treatment propidium Iodide (PI) (Sigma-Aldrich) and Hoechst 33342 (Invitrogen) were added to a concentration of 0.4μg/mL and 4μg/mL respectively using the Multidrop Combi dispenser and allowed to incubate for 20 minutes at 37°C. For automated high-content imaging, the ImageXpress Micro XL (Molecular Devices, San Jose, CA) is integrated with a Thermo F3 robotic arm. Whole-well imaging was captured with a 4x objective using DAPI and Texas Red filters. Total nuclei and PI+ cells were counted using the multi-wavelength cell scoring application module in the MetaXpress software. Dose response curves, using live cell counts (total nuclei - PI+), and IC50s were generated using GraphPad Prism™ version 7 (GraphPad software, La Jolla, CA, USA).
Kinase Assays:
ALK variants were immunoprecipitated from NIH 3T3 cells lines stably expressing ALK fusion variants and controls using a monoclonal anti-ALK antibody (Cell Signaling #3633) Lysates were pre-cleared with Protein A Sepharose beads (Invitrogen). After 1 hour in primary antibody, Protein A Sepharose beads (Invitrogen) were added for 1 hour. Beads were washed 4x in PBS as directed in the manufacturer’s protocol. Kinase assays were performed using the Universal Kinase Assay Kit (Takara, Clontech, Japan) according to the manufacturer’s instructions. An aliquot of the immunoprecipitate, equal to the amount used in each in vitro kinase assay, was assessed by immunoblot for total ALK (Cell Signaling #3333). After completion of the in vitro kinase assay, the data was analyzed with a two-step normalization process: (1) Total ALK expression for each variant was calculated using iMageStudio Lite (LiCor) and normalized to (divided by) the WT ALK expression. (2) Kinase activity of each fusion was then normalized to (divided by) expression and represented as relative to WT ALK. Kinase assays were performed in the absence of known ALK ligand.
Protein Stability Assays:
NIH 3T3 cell lines stably expressing ALK fusion variants were seeded in 6 well plates. 24 hours after seeding, cells were treated with 50μg/mL cycloheximide (Sigma-Aldrich) for 2, 8, 16, or 24 hours, harvested for lysate, and probed for total ALK protein (Cell Signaling, #3191S) by IR Western blot. Total ALK variant expression was normalized to the actin loading control.
Data Analysis and Statistical Considerations:
Each experiment was performed with three biological replicates and repeated at least three times. IR Western blots were visualized using the LiCor Odyssey scanner system and quantified using ImageStudio Lite (LiCor), and ALK phosphorylation and total ALK protein expression were normalized to the actin loading control. Kinase assays were measured using the Synergy MX microplate reader (BioTek). All graphs and statistical analyses were generated using GraphPad Prism™ software and a p value<0.05 as the threshold for statistical significance. For comparing more than two conditions, the Kruskal-Wallis or Tukey tests were used.
Results
Expression of ALK fusion variants in NIH3T3 cells
At present, there are limited cell lines that harbor endogenous ALK fusions, and these models are limited to EML4-ALK and NPM-ALK. To study the effects of multiple 5’ partners, we selected seven different ALK fusions for exogenous expression: FN1-ALK, KIF5B-ALK, RANBP2-ALK, TFG-ALK, PRKAR1A-ALK, EML4-ALK E13:A20 (variant 1, V1; exon 13 of EML4 fused to exon 20 of ALK), and EML4-ALK E6b:A20 (variant 3b, V3b, exon 6b of EML4 fused to exon 20 of ALK) (Table 1, Fig. 1). These fusion variants have all been detected in IMT and/or NSCLC (3,15,17), the two tumor types with the most clinical data regarding the efficacy of ALK inhibition. Of the greater than 30 ALK fusion partners which have been described, we selected these particular variants because they produce fusion proteins of different molecular weights which differ in protein subdomain structure, particularly with respect to the putative oligomerization domains (Fig. 1), and subcellular localizations (Table 1). Full-length ALK receptor, either WT or F1174L mutated, were used as controls. The F1174L mutation in the ALK kinase domain confers ligand-independent, constitutive activity of the receptor, representing a positive comparator for oncogenic activity while the full length WT ALK receptor is expected to be inactive unless ligand is added (18,19).
Table 1: Properties of the ALK fusion variants described in this study.
This table details the ALK fusions chosen for this study, published functions of the 5’ partner protein, protein length, expected molecular weight, cancers in which the fusion variant has been reported, and published subcellular localization of the ALK fusion protein. Genomic breakpoints used to synthesize the cDNA’s that generate these proteins came from literature reports and are cited in the ALK Variant column. #Protein length represented as the number of amino acids. *Data from findings in this study.
| ALK Fusion Variant | Protein Length# (predicted MW) | Tumor Types | Reported Subcellular Localization | Reported function of the 5’ fusion partner |
|---|---|---|---|---|
| FN1-ALK (14) | 1799 (198kD) |
IMT (3) Ovarian (14) | Plasma membrane, Cytoplasmic (14) |
Fibronectin, FN1: glycoprotein found in a dimer or multimer at the cell surface and in extracellular matrix (43). |
| KIF5B-ALK (15) | 1481 (163kD) |
NSCLC(15) IMT* |
Cytoplasmic (15) |
Kinesin Heavy Chain, KIF5B: microtubule-dependent motor involved in distribution of mitochondria and lysosomes and regulation of centrosome and nuclei (44). |
| RANBP2-ALK (3) | 1427 (157kD) |
IMT (3) DLBCL (45) | Nuclear periphery, Cytoplasmic (34) |
Ran Binding Protein, RANBP2: large scaffold and mosaic cyclophilin-related nucleoporin involved in the Ran-GTPase cycle (46). |
| TFG-ALK (3) | 800 (88kD) |
NSCLC (47) IMT (3) ALCL (48) PTC (49) |
Cytoplasmic (50) |
Tyrosine Kinase Fused Gene, TFG: localizes to the endoplasmic reticulum (ER) exit sites and modulates ER export.(51) May function at the ER/ERGIC interface to concentrate COPII-coated vesicles and link exit sites on the ER to ERGIC membranes (52). Inhibitory regulator of the ubiquitin-proteasome system (53). |
| PRKAR1A-ALK (3) | 727 (80kD) |
IMT (3) | Cytoplasmic* | Protein Kinase A Type 1 Regulatory subunit, PRKAR1A: dissociated from the inactive holoenzyme upon cAMP binding (54,55). |
| EML4-ALK V1 (17) | 1057 (116kD) |
NSCLC (17) | Cytoplasmic (37) |
Echinoderm Microtubule Associated Protein-Like 4, EML4: WD-repeat protein possibly affecting microtubule formation and dynamics (31). |
| EML4-ALK V3b (16) | 781 (86kD) |
NSCLC (16) | Cytoplasmic (37) |
Echinoderm Microtubule Associated Protein-Like 4, EML4: WD-repeat protein possibly affecting microtubule formation and dynamics (31). |
Figure 1. Schematic representation of ALK fusion variants utilized in this study.
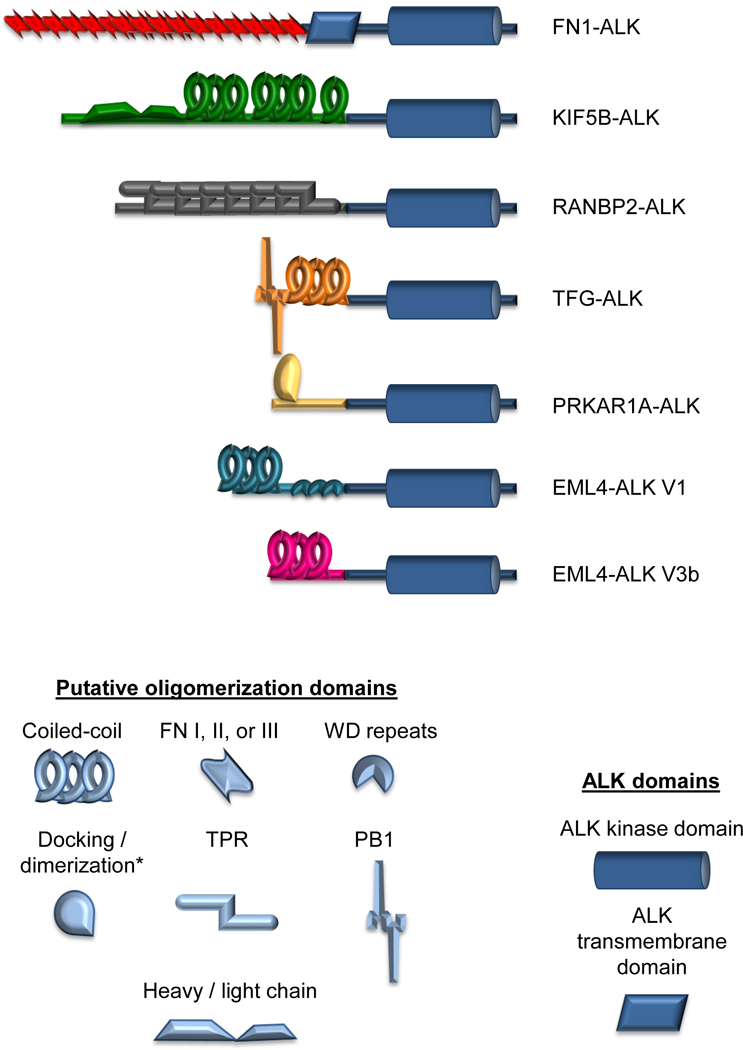
Depicted are the structures of each ALK fusion variant in this study (scaled relative to each other). The ALK portion of the protein is represented in dark blue. The 5’ partner proteins are represented in red, green, gray, orange, yellow, teal, or pink. Putative oligomerization domains within the 5’ partner protein are also represented. * The “dimerization/docking” domain of PRKAR1A(56). Note that the shape of the oligomerization domain in this figure does not necessarily represent the true shape of the protein domain.
A challenge to consider in studying ALK fusion variants comparatively is that each variant is under the control of the 5’ fusion partner’s regulatory elements, which vary from fusion partner to fusion partner and cell type to cell type. To address this issue, all variants were placed under the control of the LTR in the PMXs-Puro retroviral vector. We stably transduced the seven distinct ALK variants (Table 1, Fig. 1) into NIH 3T3 cells to generate isogenic cell line models(20) and selected clones in which the specific ALK fusion proteins were expressed at relatively equivalent levels (Supplementary Fig. S1). We confirmed the expression of each fusion at the expected molecular weight (Table 1, Supplementary Fig. S1). For FN1-ALK, we noted an additional lower molecular weight band at approximately 75kD, as previously described for this fusion variant (14).
In further support of our cell line models, we performed Reverse Phase Protein Array (RPPA) in order to quantitatively evaluate changes in protein expression induced by each active ALK variant (Fig. 2, Supplementary Fig. S2). This extensively validated assay evaluates the relative expression of 304 cancer related proteins and phospho-proteins from each sample simultaneously (Supplementary Table S1), allowing for confident, quantitative comparisons. The most significantly changed proteins between the negative control cell lines (WT ALK, empty vector) and those expressing an ALK fusion or the constitutively active mutant, ALK F1174L are shown in Fig. 2 (Supplementary Table S2). We observed a clear shift in the protein expression profile between the negative control cell lines (WT ALK, empty vector) and those expressing an ALK fusion or the constitutively active mutant, ALK F1174L (Fig. 2).
Figure 2. Active ALK variants change the protein expression profile of NIH 3T3 cells.
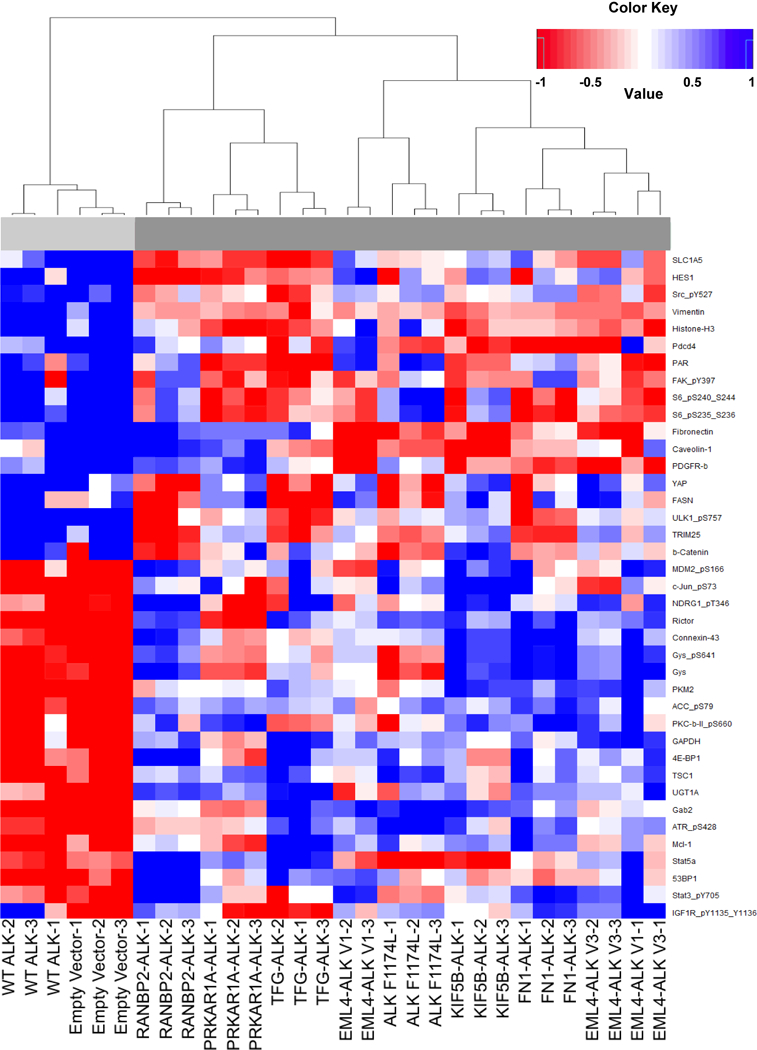
This heatmap depicts those proteins that are the most significantly differentially expressed between the WT ALK and Empty Vector cell lines (group1-light gray) and all other cell lines which expression active ALK variant (group2-dark gray). This data was generated using a Reverse Phase Protein Array (RPPA). The experiment was performed using biological replicates run in triplicate. Note that blue is representative of increased expression (from the mean), and red is decreased expression (from the mean). A hierarchical tree is depicted on the right to further distinguish delineation of the cell lines. The protein names and phosphorylation sites are represented at the bottom.
ALK fusion partners confer differential oncogenic properties
Next, we sought to determine if the 5’ partner affects transforming and proliferative properties. Using the NIH 3T3 cell line models, we first assessed focus formation, an established assay used to evaluate the ability of oncogenes to overcome contact inhibition. As expected, cells expressing empty vector and WT ALK did not support focus formation, while the ALK F1174L cells (the positive control) lost contact inhibition and were able to form foci (Fig. 3A). We observed that all seven distinct ALK fusion variants also supported focus formation, albeit to different degrees. Notably, FN1-ALK exhibited the most robust ability to form foci, more than double the amount of the positive control (ALK F1174L). Similarly, when assessed for anchorage independent growth (Fig. 3B), all fusions produced colonies in soft agar. However, only FN1-ALK produced significantly more colonies than the other fusions (Fig. 3B). Like ALK F1174L, FN1-ALK retains the trans-membrane domain of ALK (Fig. 1) – due to an alternative breakpoint within the ALK locus - and exhibits similar doubling time (data not shown), yet FN1-ALK forms significantly more foci and more colonies in soft agar than ALK F1174L, suggesting that properties belonging to FN1 specifically are likely responsible for this hyperactive phenotype. These findings reaffirm the hypothesis that the 5’ fusion partner plays a role in oncogenic properties.
Figure 3. ALK fusion variants confer differential transformation ability.
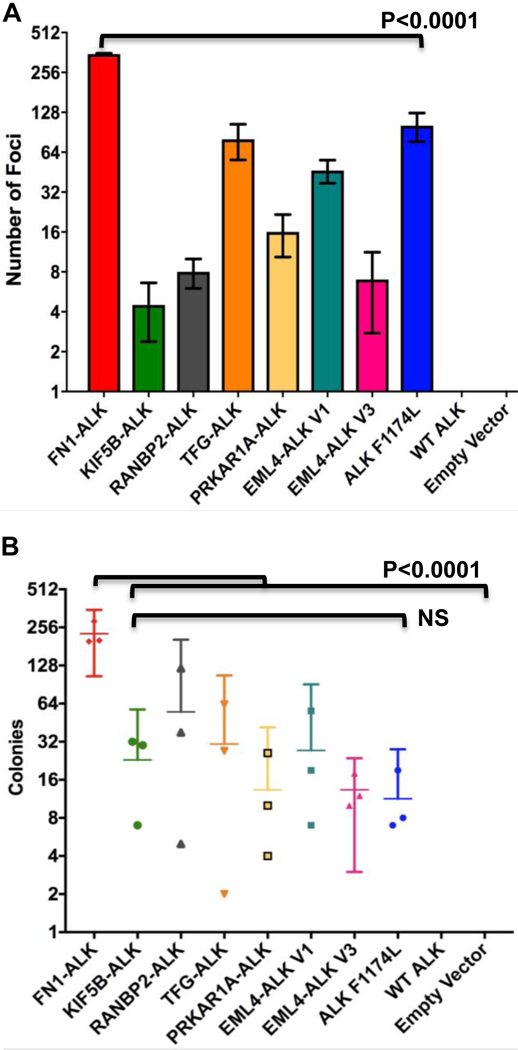
(A) ALK variant 3T3 cell line models were seeded in triplicate at 4000 cells per well in 24 well dishes and allowed to grow for 8 days. Using the Incucyte ZOOM, 16 images were obtained daily for each well. The number of foci present at day 8 is represented. Experiments were repeated three times. (B) ALK variant 3T3 cell line models were seeded in triplicate atop a 0.5% solid agar layer in complete media containing 0.3% agar at 5000 cells/well in 6 well plates and allowed to grow for 6 weeks. Colonies were counted using the GelCount machine. Data represented are the number of colonies at week 6. Experiments were repeated three times. Note: all 3 replicate experiments are represented in both A and B.
ALK fusion variants exhibit differential response to ALK tyrosine kinase inhibition
Previous retrospective clinical studies have suggested that tumors harboring different ALK fusion variants may display different responses to ALK TKI therapy (9–11). It is unclear from a biochemical perspective why this is the case since each ALK fusion protein contains the entire ALK tyrosine kinase domain, and all of the ALK TKIs in clinical use function as ATP mimetics. The efficacy of ALK TKIs against different ALK variants, differing only by the 5’ fusion partner, has not been directly compared in vitro. We generated eight-point dose response curves to evaluate the efficacy of crizotinib, the first FDA-approved ALK TKI, against each ALK fusion variant. 3T3 cells stably transduced with empty vector, full length WT ALK receptor, or full length ALK receptor encoding known activating mutation, F1174L, were used as controls. We observed a dose dependent decrease in cell viability across all cell lines (Fig. 4A, Supplementary Fig. S3). However, we noted that the efficacy of crizotinib was not uniform across all fusions tested. To verify on-target effects of crizotinib, we assessed ALK auto-phosphorylation by immunoblot (Supplementary Fig. S3). We calculated IC50 values for crizotinib against each ALK variant cell line and found a > 5 fold difference in responsiveness (Fig. 4C). Cells expressing TFG-ALK exhibited the lowest IC50 (87.36 nM), while cells expressing PRKAR1A-ALK exhibited the highest IC50 (461.8 nM) (Fig. 4A, C).
Figure 4. ALK fusion variants exhibit differential response to ALK TKIs.
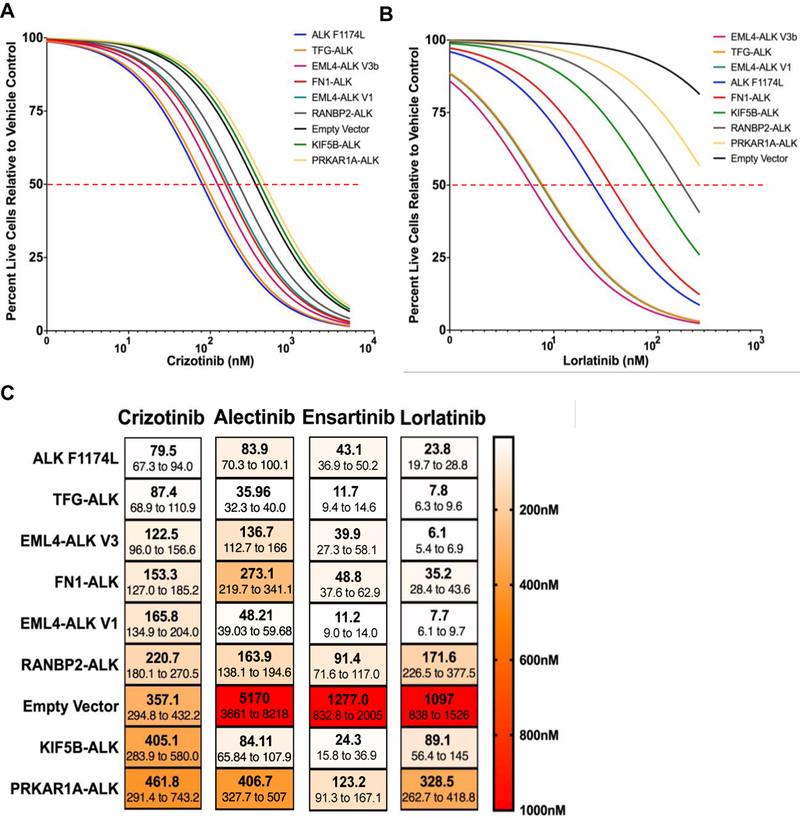
NIH 3T3 cells expressing the indicated ALK variants were treated with increasing concentrations of crizotinib (A) or lorlatinib (B) for 72 hours. Propidium iodide (PI) and Hoechst 33342 were added and allowed to incubate for 20 minutes before imaging on the ImageXpress. Dose response curves were generated by determining total live cells (# of Hoechst positive cells minus # of PI positive cells) at each dose of ALK TKI. A non-linear regression of the data is represented here. Experiments were performed with 6 replicates per drug concentration and repeated three times. One representative experiment is shown here. (C) IC50s with 95% confidence intervals were generated with the data from the dose response curves (in 3A,B, S3A, B) using GraphPad Prism.
To determine if this was a drug class effect, we also investigated the efficacy of second generation ALK TKIs, alectinib and ensartinib, as well as the third generation ALK TKI, lorlatinib (Fig. 4B, Supplementary Fig. S3, Supplementary Table S3). These inhibitors are known to be more potent against the target (ALK) compared to the first generation ALK TKI, crizotinib (21–23). Notably, there was a >10 fold difference in IC50s across the fusions for second and third generation ALK TKIs and less efficacy against the empty vector control cells. When comparing across inhibitors, PRKAR1A-ALK was consistently the least sensitive fusion (highest IC50 values) to all 4 distinct ALK TKIs evaluated. However, we noted that the order of fusion sensitivity did not remain consistent for the other 6 ALK fusion variants (Fig. 4C). For instance, KIF5B-ALK was highly sensitive to ensartinib (IC50 24.3 nM) but was one of the least sensitive fusions to crizotinib (IC50 405.1 nM) and lorlatinib (IC50 89.1 nM). These data support the hypothesis that drug efficacy is influenced by the specific fusion partner present and also highlight the importance of selecting the “right” ALK inhibitor for each fusion.
Tyrosine kinase activity of ALK fusion variants
Many structural and biochemical properties of the fusion proteins have the potential to contribute to differences in response to ALK TKIs that we observed (Fig. 4), including dimerization/oligomerization, intrinsic kinase activity, and protein stability. Previous studies have shown that the fusion partner contributes a dimerization domain to assist in auto-activation of the kinase. For example, EML4 contains a coiled-coiled domain in the N-terminus, and when this coiled-coil domain is abrogated through mutation, the resultant EML4-ALK fusion loses transforming properties (17). Many fusion partners contain coiled-coil domains or other known dimerization domains; however, not all fusion partners have an obvious dimerization motif. Similar to other tyrosine kinases, ALK must dimerize to auto-activate and signal downstream (17,24,25). Pathway analysis of the RPPA data, comparing WT ALK to each ALK fusion variant or ALK F1174L individually, revealed significant up-regulation of the RAS/MAPK pathway in FN1-ALK, KIF5B-ALK, RANBP2-ALK, and ALK F1174L (Supplementary Table S4). When assessing the auto-phosphorylation site of ALK, a surrogate for enzymatic activity of the ALK tyrosine kinase domain, we observed differences in the ratio of phospho-ALK to total ALK from one fusion to the next (Fig. 5A). Interestingly, those variants, which exhibited a pALK/ALK ratio of 0.5 or greater, also had significant RAS/MAPK pathway activation (Supplementary Table S4).
Figure 5. ALK fusion variants exhibit differences in kinase activity and protein turnover.
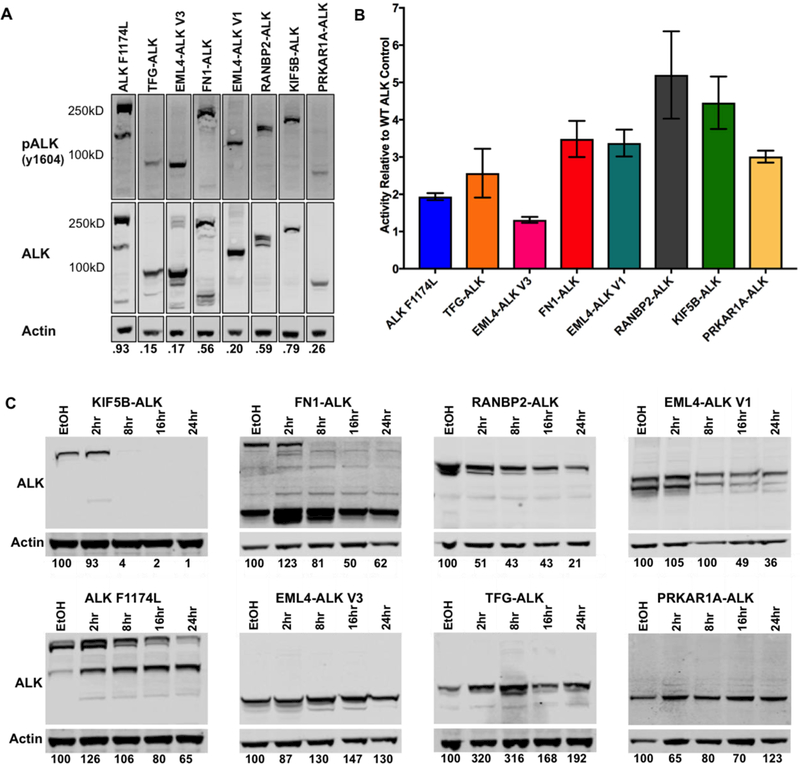
(A) Lysates from the ALK Variant 3T3 cell line models were run out by SDS-PAGE and blotted for pALK (Y1604), total ALK, and actin. Membranes were visualized using the LiCor Odyssey and quantified with iMageStudio Lite. The pALK and ALK signals were normalized to the actin in the corresponding lane. The respective pALK/ALK ratios are shown below each lane. (B) Kinase assays were performed in triplicate for each ALK variant. ALK variants were immunoprecipitated from the 3T3 cell line model lysates using a polyclonal ALK antibody (Cell Signaling) and Sepharose A beads. An additional aliquot of the immunoprecipitated product was run on SDS-PAGE for quantification of total ALK per sample. Kinase activity was determined using a standard curve provided with the assay. Activity was normalized to the total ALK immunoprecipitated. Represented is the expression-normalized activity relative to the WT ALK control. Experiments were performed three times. One representative experiment is shown here. (C) ALK variant expressing 3T3 cell line models were treated with 50ug/mL of cycloheximide for 2, 8, 16, or 24 hours. Lysates were run on SDS-PAGE and probed for total ALK (Cell Signaling). Percent ALK remaining in each sample compared to the vehicle control treated sample is depicted below each respective lane. NOTE: Quantifications for FN1-ALK and ALK F1174L include both the high and low molecular weight bands. Experiments were repeated three times. Once representative experiment is shown here.
It is currently unknown if different fusions vary with respect to kinase activity, but structural differences between the various 5’ partners, including the oligomerization domains (Fig. 1), could alter the dimerization ability and therefore activation dynamics of the fusion and/or accessibility of the catalytic domain in ALK. To further evaluate relative kinase activity, we immunoprecipitated the ALK fusions and measured the enzymatic activity against an exogenous peptide substrate, using previously validated assays (10,13). Controlling for the amount of protein immunoprecipitated, we calculated the kinase activity for each variant relative to WT ALK (Fig. 5B, Supplementary Fig. S4). As expected, the known activating mutation, ALK F1174L, exhibited two-fold greater kinase activity compared to WT ALK. We also observed that all 7 ALK fusion proteins possessed in vitro kinase activity, albeit to different extents. RANBP2-ALK and KIF5B-ALK exhibited the highest relative activity; approximately 5 fold greater than the activity of WT ALK. These results are notable because the portion of ALK in each of these fusion proteins is the same, with the exception of FN1, which contains the ALK trans-membrane domain in addition to the ALK kinase domain and C terminus. Interestingly, kinase activity towards an exogenous substrate (Fig. 5B) did not directly correlate with ALK auto-phosphorylation ratios (Fig. 5A).
Stability of ALK fusion proteins
Next, we sought to evaluate the stability or turnover rates of the seven distinct ALK fusions (26). We performed a time course study to assess fusion protein stability by treating cells with cycloheximide to inhibit new protein synthesis. We observed marked differences in the turnover rates of the various ALK fusion proteins (Fig. 5C). Levels of EML4-ALK V3, PRKAR1A-ALK, and TFG-ALK did not decrease over the cycloheximide time course. In contrast, KIF5B-ALK, FN1-ALK, RANBP2-ALK, and EML4-ALK V1 all showed a time dependent decrease in ALK protein levels, with KIF5B-ALK having the highest turnover rate. To ensure that the rapid degradation of KIF5B-ALK and FN1-ALK was not a result of cytotoxicity, we performed the experiment with shorter time intervals. As expected, we saw a gradual decrease in total ALK over time (Supplementary Fig. S5A). Overall, we were able to confirm a negative correlation between kinase activity and protein stability at 16 hours post cycloheximide treatment (Supplementary Fig. S5B). While we observed a negatively correlative pattern between protein stability and crizotinib sensitivity, it did not reach statistical significance (Supplementary Fig. S5C).
Previous studies have shown EML4-ALK to be a client of Heat Shock Protein 90 (HSP90), and HSP90 inhibitors have been in clinical trials for ALK fusion NSCLC (27). In light of our cycloheximide results, we thought the FN1-ALK, KIF5B-ALK, and RANBP2-ALK may show significant sensitivity to HSP90 inhibition. Therefore, we challenged our cell lines with ganetespib, an HSP90 inhibitor in clinical use. Surprisingly, only EML4-ALK variant 1 and 3 were sensitive to HSP90 inhibition (Supplementary Fig. S5D). This suggests that the instability or turnover of FN1-ALK, KIF5B-ALK, and RANBP2-ALK may not be due to difficulties in protein folding.
Discussion
While clinical results with ALK TKIs are promising, more granular data is needed to increase therapeutic precision. Studies involving point mutations in the ALK kinase domain that confer resistance to ALK TKI therapy have revealed that different point mutations confer varying degrees of sensitivity and resistance to the numerous ALK TKIs in clinical use (28). However, our understanding of how the 5’ partner protein plays a role in therapeutic response is lacking. As next-generation sequencing technologies continue to come to the forefront of clinical diagnostics, clinicians will not only know the mutational status of the ALK kinase domain but also the 5’ partner involved in the fusion. Therefore, it is vital that we lay the foundation for determining the therapeutic implications of the 5’ protein.
Our studies demonstrate that the 5’ partner affects the biochemical and cellular properties of the ALK fusion protein, including kinase activity, protein stability, transformative potential, and – importantly – response to ALK TKIs. For example, FN1-ALK exhibited the greatest ability to form foci and colonies in soft agar. Of note, FN1-ALK is the only ALK fusion studied which retains the ALK trans-membrane domain (Fig. 1). However, it is unlikely that the retention of this domain alone results in increased colony formation because the full length ALK receptor harboring the constitutively active F1174L mutation retains the ALK trans-membrane domain but did not display the same level of transformative ability. Two cases of EML4-ALK fusions, which retain the trans-membrane domain, have been reported (29,30). Given that EML4 is not a secreted protein with known extracellular interactions (31), it is unknown how this non-canonical breakpoint may affect transformation in this case.
In our biochemical analyses, we identified fusions which exhibited more rapid protein turnover than others, yet these data did not correlate with sensitivity to HSP90 inhibition. We noted that the fusions, which exhibited the shortest half-life (KIF5B-ALK, FN1-ALK, RANBP2-ALK, and EML4-ALK V1), also had the highest kinase activity. While ALK trafficking biology is not completely understood, especially in the context of ALK fusions, it is known that many RTKs will turnover upon activation. One study of the full length ALK receptor has shown that ALK undergoes lysosomal degradation upon activation rather than receptor recycling (32). This could be a potential explanation for the differential degradation times of the fusions.
Furthermore, we evaluated the efficacy of four distinct and clinically relevant ALK TKIs across the seven fusion variants. At present, the assumption in the field remains that since all ALK fusion proteins retain the entire ALK kinase domain, that the efficacy of ALK TKI therapy will be analogous across all the fusions. However, our data point to a different conclusion. We consistently noted a 5–10 fold difference in the measured IC50 value for the four ALK TKIs – crizotinib, alectinib, ensartinib, and lorlatinib – against the seven distinct fusions tested, supporting our hypothesis that the 5’ partner protein can affect response to ALK inhibition (Fig. 4A, Supplementary Fig. S3). Indeed, IMT patients harboring the RANBP2-ALK fusion, which is less sensitive to ALK TKI in our study, also seem to exhibit more aggressive disease and less sensitivity to ALK TKI therapy (33,34). There are many structural and biochemical properties of the fusion proteins which have the potential to contribute to differences in response to ALK TKIs, including: 1) the type of oligomerization domains present; 2) the stoichiometry of oligomerization: dimers (35), trimers (36,37), and multimers (38,39); 3) the intrinsic kinase activity of the fusion protein (13); 4) protein-protein interactions, which may differ based on the domain structure of the fusion partner; 5) length of the 5’ partner; 6) protein folding, tertiary structure, and degree of disorder; and 7) protein stability. Additional in silico modeling coupled with rigorous model systems will be necessary to dissect these critical issues.
Kinase fusions are known to localize differently based on the 5’ partner (Table 1), even for those which share the same 5’ partner (and therefore promoter) but vary in the amount of that protein present in the fusion, such as the EML4-ALK variants (37,40). This could allow for different protein interactions. Differential pathway activation based on ALK localization has been shown in Medves et al. 2011. Additionally, in Armstrong et al. 2004 where investigators explored phenotypes of 5 different ALK fusions in NIH 3T3 cell lines, differences in pathway activation were also seen. Our RPPA data clearly shows significant changes in total and phospho-protein expression profiles when an active ALK variant is expressed (Figure 2) and also shows expression differences between the ALK variants (Figure 2, Supplementary Figure S2, and Supplementary Tables 2–3). The RPPA data was revisited throughout the study in search for insight into different phenotypes assessed. The only correlation that we found was that those variants, which exhibited a pALK/ALK ratio of 0.5 or greater, also had significant RAS/MAPK pathway activation. While the RPPA panel at the MD Anderson core includes many of the most highly implicated cancer associated proteins, the panel is, by definition, limited in scope. Therefore, if there are other proteins (or additional phosphorylation sites) that are playing important roles in the phenotypes reported in this study, they may be missed. There is also a similar bias in the pathway analysis pipeline in that there are pathways defined that are mostly confined or well understood in other adult solid tumors such as breast cancer. This pathway analysis pipeline also does not include pathway definitions for other important cancer phenotypes such as migration/metastasis and metabolism.
Finally, we noted that the relative sensitivity (in terms of ranked IC50 value) of a given fusion was not consistent across all inhibitors tested. For example, while KIF5B-ALK was one of the least sensitive fusions towards crizotinib, this same fusion was one of the most sensitive towards ensartinib (Fig. 1C), suggesting that drug selection may necessitate knowledge of the specific fusion partner in order to select the most efficacious inhibitor amongst the several FDA-approved or investigational ALK TKIs in clinical use (Supplementary Table S3). In support of these data, two recent retrospective clinical analyses have shown that NSCLC patients whose tumors harbor different EML4-ALK fusion variants – defined by the amount of EML4 contained within the fusion (the ALK portion is the same) – experience different responses upon treatment with distinct ALK TKIs. In one study, NSCLC patients with tumors harboring EML4-ALK variant 3a/b exhibited longer progression free survival (PFS) when treated with lorlatinib compared to patients whose tumors harbored EML4-ALK variant 1. However, the reciprocal responses where observed with crizotinib (11). Overall, our data, coupled with this retrospective clinical data, support the need for further prospective investigation regarding the role of the 5’ fusion partner in dictating therapeutic response.
Adding to the complexity, greater than 30 different 5’ fusion partners have been reported for ALK fusions alone (Fig. 6). Furthermore, several oncogenic tyrosine kinase fusions have been reported in solid tumor and hematologic malignancies, including ROS1, RET, and NTRK1, amongst others(8), and the 5’ fusion partners can overlap between these different kinases (Fig. 6). For example, TFG has been a reported 5’ partner for ALK, ROS1, and NTRK fusions (3,41). It is not feasible to extensively study each kinase fusion in the laboratory. To approach this complex problem, a high-throughput, computational approach to assess the effect of the 5’ protein on the kinase and how it may alter drug binding could be greatly beneficial. Interestingly, a recent study in RET-rearranged NSCLC reported that patients harboring the KIF5B-RET fusion exhibited significantly less sensitivity to RET inhibition than patients harboring other RET fusions (42). In our study KIF5B-ALK was also one of the least sensitive ALK fusions to crizotinib and lorlatinib. Given that KIF5B reduced the response for both the ALK and RET kinase fusion, we may be able to make conclusions about the effects of a 5’ partner that can be applied across multiple kinase fusions such as ALK, RET, ROS1, and NTRK1/2/3.
Figure 6. The heterogeneity of tyrosine kinase fusion partners.
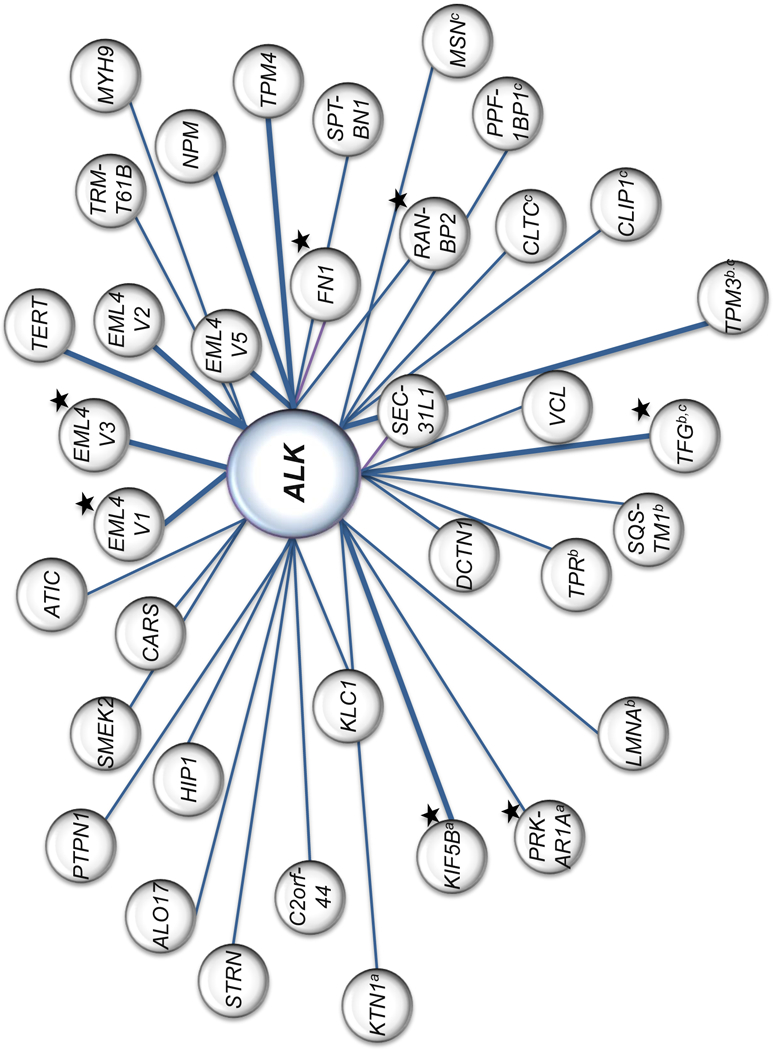
The ALK tyrosine kinase is represented in larger center circle. 5’ fusion partners reported for ALK are depicted in gray circles. The weight of the connecting line between the kinase and 5’ fusion partner reflects frequency of occurrence relative to other 5’ partners reported. These fusions are not restricted to NSCLC or IMT. This figure is not a comprehensive representation of literature report(8,57–60). ALK fusions represented in our study are marked with a black star. 5’ fusion partners which are also reported for RET, NTRK, and ROS1 fusions are marked witha,b, or c respectively.
Current limitations in this study include the lack of clinical data to corroborate our findings. This is partly due to the lack of reporting of the specific fusion variant including the 5’ partner in clinical studies. Additionally, studies that may report the specific variants, such as Drilon et al. 2018, may not be statistically powered to compare outcomes based on the variant. In order to fill this gap in knowledge, the design of future clinical trials will need to include prospective analysis of the specific fusion variants.
In summary, our findings contribute to the preliminary studies in the emerging field of fusion biology, adding further evidence that the 5’ partner of kinase fusion variants plays a significant role in the biological functions of the fusion that can affect cellular phenotypes and response to targeted therapies. Future clinical trials will need to prospectively consider ALK TKI efficacy in terms of the fusion partner present upon enrollment into the trial. Furthermore, given the large number of possible fusion variants that may be detected in the clinic, high-throughput computational and modeling studies will be necessary in order to support therapeutic decision-making.
Supplementary Material
Acknowledgements
The authors would like to thank Yingjun Yan for her technical support and Josh Bauer and the Vanderbilt High Throughput Sequencing CORE for their assistance with the drug response assays. The authors also thank the members of the Lovly laboratory for helpful discussions and critical review of the manuscript, particularly Yunkai Zhang for this help with editing. This study was supported in part by the For the Linds Foundation, the TJ Martell Foundation, the American Cancer Society (Institutional Research Grant #IRG-58–009-54), and by the National Institutes of Health (NIH) and National Cancer Institute (NCI) R01CA121210 (CML) and U10CA180864. CML was also supported by a Damon Runyon Clinical Investigator Award, a LUNGevity Career Development Award, a V Foundation Scholar-in-Training Award, an AACR-Genentech Career Development Award (15–20-18-LOVL), and by an IASLC/LCFA Lori Monroe Scholarship. MAC was supported by the NCI (F31 CA213744–02) and (T32 CA009592). The MD Anderson RPPA Core Facility is supported by NCI #CA16672.
Abbreviations:
- ALK
anaplastic lymphoma kinase
- NSCLC
non-small cell lung cancer
- IMT
inflammatory myofibroblastic tumor
- ALCL
anaplastic large cell lymphoma
- DLBCL
diffuse large B-cell lymphoma
- FDA
Food and Drug Administration
- USA
United States of America
- TKI
tyrosine kinase inhibitor
- EGFR
epidermal growth factor receptor
- TPM3/4
non-muscle tropomyosin
- CLTC
clatherin heavy chain
- LMNA
lamin A
- PRKAR1A
protein kinase A type 1 regulatory subunit
- EML4
echinoderm microtubule associated protein-like 4
- RANBP2
ran binding protein 2
- TFG
tyrosine fused gene
- FN1
fibronectin
- KIF5B
kinesin family 5 B
- kD
kilodalton
- MW
molecular weight
- RPPA
reverse phase protein array
- HSP90
heat shock protein 90
- NTRK
neurotropic receptor tyrosine kinase
- PI
propidium iodide
- RARα
retinoic acid receptor alpha
- PFS
progression free survival
- LTK
leukocyte receptor tyrosine kinase
- IGF-1R
insulin growth factor receptor 1
- WT
wild-type
- FC
fold change
- FDR
false discovery rate
- IR
infrared
Footnotes
Conflicts of interest: CML has served as a consultant for Pfizer, Novartis, Astra Zeneca, Genoptix, Sequenom, ARIAD, Takeda, and Foundation Medicine, serves on an Advisory Board for Cepheid Oncology, and has been an invited speaker for Abbott and Qiagen. MAC and SMH report no conflicts of interest. MAD has served as a consultant for GSK, Novartis, Genentech/Roche, BMS, Sanofi-Aventis, Syndax, Veridex, and Nanostring, and has been the PI of research funding to his institution from GSK, Genentech/Roche, BMS, Sanofi-Aventis, Oncothyreon, AstraZeneca, and Myriad.
References
- 1.Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 2013;13(10):685–700 doi 10.1038/nrc3580. [DOI] [PubMed] [Google Scholar]
- 2.Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II , et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. Jama 2014;311(19):1998–2006 doi 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT , et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 2014;4(8):889–95 doi 10.1158/2159-8290.CD-14-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J , et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol 2013;14(6):472–80 doi 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J , et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372–001 and STARTRK-1). Cancer Discov 2017;7(4):400–9 doi 10.1158/2159-8290.CD-16-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayeni D, Politi K, Goldberg SB. Emerging Agents and New Mutations in EGFR-Mutant Lung Cancer. Clin Cancer Res 2015;21(17):3818–20 doi 10.1158/1078-0432.CCR-15-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 1999;93(10):3167–215. [PubMed] [Google Scholar]
- 8.Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer 2013;13(11):772–87 doi 10.1038/nrc3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshida T, Oya Y, Tanaka K, Shimizu J, Horio Y, Kuroda H , et al. Differential Crizotinib Response Duration Among ALK Fusion Variants in ALK-Positive Non-Small-Cell Lung Cancer. J Clin Oncol 2016;34(28):3383–9 doi 10.1200/JCO.2015.65.8732. [DOI] [PubMed] [Google Scholar]
- 10.Woo CG, Seo S, Kim SW, Jang SJ, Park KS, Song JY , et al. Differential protein stability and clinical responses of EML4-ALK fusion variants to various ALK inhibitors in advanced ALK-rearranged non-small cell lung cancer. Annals of oncology : official journal of the European Society for Medical Oncology 2017;28(4):791–7 doi 10.1093/annonc/mdw693. [DOI] [PubMed] [Google Scholar]
- 11.Lin JJ, Zhu VW, Yoda S, Yeap BY, Schrock AB, Dagogo-Jack I , et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J Clin Oncol 2018:JCO2017762294 doi 10.1200/JCO.2017.76.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Red Brewer M, Yun CH, Lai D, Lemmon MA, Eck MJ, Pao W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc Natl Acad Sci U S A 2013;110(38):E3595–604 doi 10.1073/pnas.1220050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiesner T, Lee W, Obenauf AC, Ran L, Murali R, Zhang QF , et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015;526(7573):453–7 doi 10.1038/nature15258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren H, Tan ZP, Zhu X, Crosby K, Haack H, Ren JM , et al. Identification of anaplastic lymphoma kinase as a potential therapeutic target in ovarian cancer. Cancer Res 2012;72(13):3312–23 doi 10.1158/0008-5472.CAN-11-3931. [DOI] [PubMed] [Google Scholar]
- 15.Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K , et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res 2009;15(9):3143–9 doi 10.1158/1078-0432.CCR-08-3248. [DOI] [PubMed] [Google Scholar]
- 16.Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S , et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res 2008;68(13):4971–6 doi 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 17.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S , et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448(7153):561–6 doi 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 18.Lee CC, Jia Y, Li N, Sun X, Ng K, Ambing E , et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J 2010;430(3):425–37 doi 10.1042/BJ20100609. [DOI] [PubMed] [Google Scholar]
- 19.Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V , et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008;455(7215):967–70 doi 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong F, Duplantier MM, Trempat P, Hieblot C, Lamant L, Espinos E , et al. Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene 2004;23(36):6071–82 doi 10.1038/sj.onc.1207813. [DOI] [PubMed] [Google Scholar]
- 21.Lovly CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C , et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res 2011;71(14):4920–31 doi 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M , et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer cell 2015;28(1):70–81 doi 10.1016/j.ccell.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinoshita K, Asoh K, Furuichi N, Ito T, Kawada H, Hara S , et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorganic & medicinal chemistry 2012;20(3):1271–80 doi 10.1016/j.bmc.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 24.Amano Y, Ishikawa R, Sakatani T, Ichinose J, Sunohara M, Watanabe K , et al. Oncogenic TPM3-ALK activation requires dimerization through the coiled-coil structure of TPM3. Biochemical and biophysical research communications 2015;457(3):457–60 doi 10.1016/j.bbrc.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 25.Roskoski R Jr. Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacological research 2013;68(1):68–94 doi 10.1016/j.phrs.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Heuckmann JM, Balke-Want H, Malchers F, Peifer M, Sos ML, Koker M , et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res 2012;18(17):4682–90 doi 10.1158/1078-0432.CCR-11-3260. [DOI] [PubMed] [Google Scholar]
- 27.Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C , et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov 2013;3(4):430–43 doi 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R , et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov 2016;6(10):1118–33 doi 10.1158/2159-8290.CD-16-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ , et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 2012;18(5):1472–82 doi 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Penzel R, Schirmacher P, Warth A. A novel EML4-ALK variant: exon 6 of EML4 fused to exon 19 of ALK. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2012;7(7):1198–9 doi 10.1097/JTO.0b013e3182598af3. [DOI] [PubMed] [Google Scholar]
- 31.Eichenmuller B, Everley P, Palange J, Lepley D, Suprenant KA. The human EMAP-like protein-70 (ELP70) is a microtubule destabilizer that localizes to the mitotic apparatus. J Biol Chem 2002;277(2):1301–9 doi 10.1074/jbc.M106628200. [DOI] [PubMed] [Google Scholar]
- 32.Mazot P, Cazes A, Dingli F, Degoutin J, Irinopoulou T, Boutterin MC , et al. Internalization and down-regulation of the ALK receptor in neuroblastoma cell lines upon monoclonal antibodies treatment. PLoS One 2012;7(3):e33581 doi 10.1371/journal.pone.0033581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen ST, Lee JC. An inflammatory myofibroblastic tumor in liver with ALK and RANBP2 gene rearrangement: combination of distinct morphologic, immunohistochemical, and genetic features. Hum Pathol 2008;39(12):1854–8 doi 10.1016/j.humpath.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 34.Marino-Enriquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD , et al. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol 2011;35(1):135–44 doi 10.1097/PAS.0b013e318200cfd5. [DOI] [PubMed] [Google Scholar]
- 35.Baumann H, Kunapuli P, Tracy E, Cowell JK. The oncogenic fusion protein-tyrosine kinase ZNF198/fibroblast growth factor receptor-1 has signaling function comparable with interleukin-6 cytokine receptors. J Biol Chem 2003;278(18):16198–208 doi 10.1074/jbc.M300018200. [DOI] [PubMed] [Google Scholar]
- 36.Medves S, Noel LA, Montano-Almendras CP, Albu RI, Schoemans H, Constantinescu SN , et al. Multiple oligomerization domains of KANK1-PDGFRbeta are required for JAK2-independent hematopoietic cell proliferation and signaling via STAT5 and ERK. Haematologica 2011;96(10):1406–14 doi 10.3324/haematol.2011.040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richards MW, O’Regan L, Roth D, Montgomery JM, Straube A, Fry AM , et al. Microtubule association of EML proteins and the EML4-ALK variant 3 oncoprotein require an N-terminal trimerization domain. Biochem J 2015;467(3):529–36 doi 10.1042/BJ20150039. [DOI] [PubMed] [Google Scholar]
- 38.Tognon CE, Mackereth CD, Somasiri AM, McIntosh LP, Sorensen PH. Mutations in the SAM domain of the ETV6-NTRK3 chimeric tyrosine kinase block polymerization and transformation activity. Mol Cell Biol 2004;24(11):4636–50 doi 10.1128/MCB.24.11.4636-4650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao X, Ghaffari S, Lodish H, Malashkevich VN, Kim PS. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat Struct Biol 2002;9(2):117–20 doi 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]
- 40.Richards MW, Law EW, Rennalls LP, Busacca S, O’Regan L, Fry AM , et al. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical beta-propeller domain. Proc Natl Acad Sci U S A 2014;111(14):5195–200 doi 10.1073/pnas.1322892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greco A, Mariani C, Miranda C, Lupas A, Pagliardini S, Pomati M , et al. The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol Cell Biol 1995;15(11):6118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M , et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17(12):1653–60 doi 10.1016/S1470-2045(16)30562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci 2002;115(Pt 20):3861–3. [DOI] [PubMed] [Google Scholar]
- 44.Splinter D, Tanenbaum ME, Lindqvist A, Jaarsma D, Flotho A, Yu KL , et al. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS biology 2010;8(4):e1000350 doi 10.1371/journal.pbio.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SE, Kang SY, Takeuchi K, Ko YH. Identification of RANBP2-ALK fusion in ALK positive diffuse large B-cell lymphoma. Hematol Oncol 2014;32(4):221–4 doi 10.1002/hon.2125. [DOI] [PubMed] [Google Scholar]
- 46.Lin DH, Zimmermann S, Stuwe T, Stuwe E, Hoelz A. Structural and functional analysis of the C-terminal domain of Nup358/RanBP2. J Mol Biol 2013;425(8):1318–29 doi 10.1016/j.jmb.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H , et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007;131(6):1190–203 doi 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 48.Hernandez L, Bea S, Bellosillo B, Pinyol M, Falini B, Carbone A , et al. Diversity of genomic breakpoints in TFG-ALK translocations in anaplastic large cell lymphomas: identification of a new TFG-ALK(XL) chimeric gene with transforming activity. Am J Pathol 2002;160(4):1487–94 doi 10.1016/S0002-9440(10)62574-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McFadden DG, Dias-Santagata D, Sadow PM, Lynch KD, Lubitz C, Donovan SE , et al. Identification of oncogenic mutations and gene fusions in the follicular variant of papillary thyroid carcinoma. J Clin Endocrinol Metab 2014;99(11):E2457–62 doi 10.1210/jc.2014-2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hernandez L, Pinyol M, Hernandez S, Bea S, Pulford K, Rosenwald A , et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood 1999;94(9):3265–8. [PubMed] [Google Scholar]
- 51.Beetz C, Johnson A, Schuh AL, Thakur S, Varga RE, Fothergill T , et al. Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure. Proc Natl Acad Sci U S A 2013;110(13):5091–6 doi 10.1073/pnas.1217197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson A, Bhattacharya N, Hanna M, Pennington JG, Schuh AL, Wang L , et al. TFG clusters COPII-coated transport carriers and promotes early secretory pathway organization. The EMBO journal 2015;34(6):811–27 doi 10.15252/embj.201489032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yagi T, Ito D, Suzuki N. Evidence of TRK-Fused Gene (TFG1) function in the ubiquitin-proteasome system. Neurobiology of disease 2014;66:83–91 doi 10.1016/j.nbd.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 54.Prinz A, Diskar M, Erlbruch A, Herberg FW. Novel, isotype-specific sensors for protein kinase A subunit interaction based on bioluminescence resonance energy transfer (BRET). Cell Signal 2006;18(10):1616–25 doi 10.1016/j.cellsig.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 55.Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annual review of biochemistry 1990;59:971–1005 doi 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 56.Horvath A, Bertherat J, Groussin L, Guillaud-Bataille M, Tsang K, Cazabat L , et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Human mutation 2010;31(4):369–79 doi 10.1002/humu.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davare MA, Tognon CE. Detecting and targetting oncogenic fusion proteins in the genomic era. Biology of the cell 2015;107(5):111–29 doi 10.1111/boc.201400096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin JJ, Shaw AT. Recent Advances in Targeting ROS1 in Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2017;12(11):1611–25 doi 10.1016/j.jtho.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD , et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 2018;378(8):731–9 doi 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pavlick D, Schrock AB, Malicki D, Stephens PJ, Kuo DJ, Ahn H , et al. Identification of NTRK fusions in pediatric mesenchymal tumors. Pediatric blood & cancer 2017;64(8) doi 10.1002/pbc.26433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.