

Summary:
Copy-number changes generate phenotypic variability in health and disease. Whether organisms protect against copy-number changes is largely unknown. Here, we show that Saccharomyces cerevisiae monitors the copy number of its ribosomal DNA (rDNA) and rapidly responds to copy-number loss with the clonal amplification of extrachromosomal rDNA circles (ERCs) from chromosomal repeats. ERC formation is replicative, separable from repeat loss, and reaches a dynamic steady state that responds to the addition of exogenous rDNA copies. ERC levels are also modulated by RNAPI activity and diet, suggesting that rDNA copy number is calibrated against the cellular demand for rRNA. Lastly, we show that ERCs reinsert into the genome in a dosage-dependent manner, indicating that they provide a reservoir for ultimately increasing rDNA array length. Our results reveal a DNA-based mechanism for rapidly restoring copy number in response to catastrophic gene loss that shares fundamental features with unscheduled copy-number amplifications in cancer cells.
Keywords: rRNA genes, genome instability, eccDNA, copy-number variations, Fob1, Hmo1
eTOC Blurb
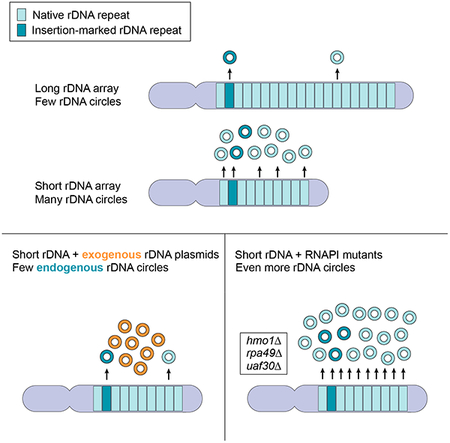
Copy number variation is associated with both disease and chromosomal evolution. Mansisidor et al. show that healthy yeast cells continuously monitor the copy number of ribosomal DNA repeats and that one strategy for balancing copy number loss is through the amplification of repeats as DNA circles.
Graphical Abstract
Introduction:
Copy-number changes are a potent source of genetic disorders and arise by a variety of mechanisms, including chromosome missegregation and errors in DNA repair. Copy-number changes are particularly common in repetitive genomic regions because of the inherent propensity of DNA repeats to undergo non-allelic homologous recombination (NAHR). One of the best-studied examples is the ribosomal RNA gene cluster (rDNA), which codes for the RNA components of the ribosome and is arranged in one or multiple gene arrays in most eukaryotes (Eickbush and Eickbush, 2007). rDNA array sizes are highly variable between individuals and large-scale changes can occur from one generation to the next (Averbeck and Eickbush, 2005; Gibbons et al., 2015; Michel et al., 2005; Stults et al., 2008; West et al., 2014). rDNA copy number alterations are common in cancers (Stults et al., 2009; Udugama et al., 2018; Wang and Lemos, 2017; Xu et al., 2017) and aging cells (Gaubatz et al., 1976; Hallgren et al., 2014; Lindstrom et al., 2011). Moreover, complete loss of individual rDNA arrays is a frequent consequence of Robertsonian fusions, the most common chromosome rearrangements in humans (Hurley and Pathak, 1977; Wolff and Schwartz, 1992).
In part, the phenotypic robustness to rDNA copy number changes may be explained by the fact that only a fraction of all rDNA repeats are actively expressed (Dammann et al., 1993; French et al., 2003; Hamperl et al., 2013; McStay and Grummt, 2008). However, differences in rDNA copy number are also associated with variation in global gene expression and mitochondrial copy number (Gibbons et al., 2014; Michel et al., 2005), and low rDNA copy numbers are linked to increased DNA damage sensitivity and cancers (Ide et al., 2010; Wang and Lemos, 2017; Xu et al., 2017).
Perhaps as a result of these selective pressures, several forms of rDNA copy number regulation have been observed. The single rDNA array of the yeast Saccharomyces cerevisae gradually shrinks and grows to restore array size after large-scale copy-number changes (Kobayashi, 2011; Kobayashi et al., 1998). Relative copy number regulation is also implied for the human 5S and 45S rRNA genes, which co-vary in copy number in some tissues despite mapping to unlinked repetitive arrays (Gibbons et al., 2015). In addition, rDNA copy number in yeast and flies is coupled with metabolic demands through trans-generational array contraction and repeat amplification (Aldrich and Maggert, 2015; Ha and Huh, 2011; Jack et al., 2015). Most of these changes, however, occur over the course of several dozens to hundreds of cell divisions. Thus, it is unclear if copy number control is purely arising from selective pressures that differentially expand cell populations with advantageous copy number, or if cells also directly assess the number of rDNA copies in the short term.
The S. cerevisiae rDNA is encoded in a single ~1Mb tandem array that accounts for nearly 10% of the yeast genome and is comparable in size to a typical human 45S rDNA array (Stults et al., 2008). Each of the 100–200, 9.1-kb repeats encodes 35S and 5S rRNA genes, separated by intergenic spacers that encode a unidirectional replication fork barrier (RFB) and a replication origin, respectively. The protein Fob1 mediates fork barrier activity at RFBs but also causes the frequent formation of replication-dependent DNA double-strand breaks (DSBs) at these sites (Johzuka and Horiuchi, 2002; Kobayashi et al., 1998; Tsang and Carr, 2008; Weitao et al., 2003). Repair of these lesions can result in unequal sister chromatid exchange (USCE), which occurs if a lesion on one chromatid interacts with a non-allelic repeat on the sister chromatid during homologous repair. USCE manifests itself as changes in array length and the apparent loss of reporter inserts (Gangloff et al., 1996; Szostak and Wu, 1980). Because USCE can result in both larger and smaller array sizes, it is assumed to be the major driver of rDNA copy-number homeostasis (Kobayashi, 2011). In fob1 mutants, USCE is reduced and aberrant rDNA arrays sizes remain stable (Kobayashi et al., 1998).
FOB1 activity also leads to the production of extrachromosomal rDNA circles (ERCs) (Defossez et al., 1999). ERCs arise when a lesion is repaired with a different repeat on the same chromatid (intrachromatid recombination). Whether ERCs also contribute to copy-number homeostasis is unclear, as the mechanism of ERC production remains controversial. ERC formation is thought to occur primarily by RAD52-dependent circularization of one or multiple rDNA repeats at the collapsed replication fork (Ganley et al., 2009; Park et al., 1999; Takeuchi et al., 2003). It remains unclear, however, what consequence this loop-out of repeats has for the integrity of the rDNA array. As ERCs are frequently observed in mutants with elevated rDNA instability, one widely cited model is that the looped-out repeats are lost from the genome, leading to a net shortening of the rDNA array (Blake et al., 2006; Ganley et al., 2009; Kobayashi, 2011; Lindstrom et al., 2011; Sinclair et al., 1998). However, because ERC formation occurs during replication, a neighboring replication fork could restore the looped-out copies, leaving array length unchanged (Ganley et al., 2009; Takeuchi et al., 2003). ERC numbers are typically low in wild-type yeast cells (Clark-Walker and Azad, 1980) but accumulate to high levels in old cells and many mutants with rDNA instability (Blake et al., 2006; Kobayashi, 2011; Lindstrom et al., 2011; Sinclair and Guarente, 1997; Sinclair et al., 1998). Intriguingly, ERC levels are high in rDNA instability mutants with short rDNA arrays but low in mutants that accumulate extra-long rDNA arrays (Ivessa et al., 2000), suggesting a potential link between rDNA copy number and ERC production.
Here, we show that large-scale deletions in the rDNA of S. cerevisiae trigger the rapid and proportional production of ERCs without increasing rDNA instability. ERC production is self-limiting and responds to diet and to rRNA transcriptional output, implying that ERC production is linked to the demand for rRNA. Strains with higher ERC levels exhibit more frequent ERC re-integrations into the chromosomal rDNA array, providing a potential additional mechanism for array expansion.
Results:
ERC production responds to chromosomal rDNA copy number
To probe the cellular response to copy-number changes, we created a collection of S. cerevisiae strains with different rDNA array lengths. Partial deletions of the rDNA array were induced by ends-apart insertion of a URA3-marked targeting plasmid (Figure 1A). Based on pulsed-field gel electrophoresis (PFGE) analysis, we selected 12 strains with rDNA arrays comprising between ~90 and ~180 repeats (Figures 1B, S1A, and Table S1). These array lengths roughly span the copy number considered wild-type for this organism (100–200 repeats) (Salim et al., 2017; Schweizer et al., 1969). Array lengths were stable at the population level over at least 150 divisions as well as through meiosis (Figure S1B), even though the rDNA fork barrier protein Fob1, which promotes expansion of critically short arrays (Kobayashi et al., 1998), was left intact. The relative stability of arrays >80 repeats is consistent with previous findings (Kwan et al., 2013; Michel et al., 2005). Minor expansion of short arrays was only observed after repeated isolation of single colonies, a process that allows for the detection of rare events (Figure S1C) (Kobayashi et al., 1998).
Figure 1: ERC levels anti-correlate with chromosomal rDNA array size.

(A) Random deletion of chromosomal rDNA repeats using a URA3-targeting plasmid containing homologies to the rDNA intergenic sequence (orange and pink). (B) PFGE analysis of deletion strains. Southern probe (URA3) detects the ura3 locus (Chr.5) and the URA3 insertion in rDNA (Chr. 12). (C) Southern analysis of ERCs probing against rDNA sequence and GAT1 (loading control) after digestion by AfeI. ERC signals labeled “a-f” based on electrophoretic mobility. (D) Total ERC signal (bands “a-e”) relative to GAT1 as function of array size; P < 4.5×10‒5 (Spearman’s rank correlation) and r2 fit to a power law function. Band f was not quantified due to proximity to wells, which is subjected to non-reproducible smearing and cracking during transfer. (E) Southern analysis of total ERCs with and without digestion by exonuclease RecBCD.
Intriguingly, Southern assays revealed increased levels of ERCs in strains with short rDNA arrays (Figures 1C and 1D). ERCs are generated by FOB1-stimulated intrachromatid NAHR (Defossez et al., 1999) and typically contain 1 to 5 rDNA repeats (Burkhalter and Sogo, 2004). Because of their circular nature, ERCs are detected above and below the sheared linear rDNA signal when undigested rDNA is separated by standard gel electrophoresis (Figure 1C) (Sinclair and Guarente, 1997). In addition, ERC signals were resistant to RecBCD exonuclease, which digests linear DNA fragments (Figure 1E), demonstrating that the signals represent bona fide circular DNA molecules. Quantification against a single-copy loading control revealed a strong dependence on array size, as strains with ~90 chromosomal rDNA repeats exhibited 5–10 times more ERCs than strains with ~180 chromosomal repeats (Figures 1C and 1D). This effect requires FOB1 (Figure 1C) and is observed in multiple genetic backgrounds (Figures 1C–D and S1D–F). It is also observed when specifically probing for ERCs originating from the chromosomal rDNA repeat containing the URA3 construct (Figures 2A, 2B, and S2A). Furthermore, ERC signals exhibit the expected dependence on the repair factors RAD52 and RAD50, but not RAD51 (Figure S2B) (Park et al., 1999). We conclude that shortening of the rDNA array is linked to a proportional FOB1-dependent increase in ERC levels. This increase is not connected to the well-established accumulation of ERCs in replicatively old yeast cells (Sinclair and Guarente, 1997), because replicatively old cells (>15 divisions) comprise 0.003% of the logarithmically growing cultures used in this study, and lifespan is unaffected by a reduction of array size to 75–80 repeats (Michel et al., 2005; Saka et al., 2013).
Figure 2: URA3-marked ERCs anti-correlate with array size, but not with marker loss.

(A-B) Southern analysis of URA3-marked ERCs as function of array size after digestion with NdeI. (C) 3.5-fold dilutions of rDNA deletion strains spotted onto synthetic complete (SC) media with and without 5-FOA. (D-E) Southern analysis of multi-copy, URA3-marked ERCs and quantification relative to single-copy ERCs. Data are represented as mean of n=3 clonal replicates ± standard deviation. Exonuclease RecBCD was used to digest linear DNA fragments, then heat inactivated before subsequent digestion with NdeI.
ERC formation is functionally separable from rDNA repeat instability
To test whether high ERC levels reflect increased rDNA instability, we monitored the loss of the URA3 construct. Cells were grown for 150–200 generations without selection before plating on 5-fluoroorotic acid (5-FOA), which kills cells expressing URA3. Irrespective of chromosomal rDNA copy number, approximately 1 in 4.0×104 cells were able to grow on 5-FOA (Figure 2C). 5-FOA resistance (FOAR) reflected repeat loss and was not the result of gene silencing, because no recovery was observed after transfer to media lacking uracil (Figure S2C). Loss of the URA3 marker was also evident by Southern analysis (Figure S2D). The fraction of FOAR cells aligns with previous measurements of marker loss in wild-type cells (Defossez et al., 1999; Gangloff et al., 1996; Ganley and Kobayashi, 2011) and indicates that shortening the rDNA array down to ~90 repeats does not detectably increase rDNA instability.
The accumulation of URA3-marked ERCs without substantial marker loss from the array suggests that ERCs are copied from a chromosomal repeat. This notion is supported by the relative abundance of URA3-marked ERCs. Southern analysis after linearizing repeats showed that single-copy ERCs containing the URA3 construct accumulate to ~15% of a genomic ura3 loading control in ~90-repeat strains (Figures 2A and 2B). Isolation of RecBCD exonuclease-resistant species further indicated that multi-copy ERCs derived from the same repeat are present at 2.5–3 times the level of single-copy ERCs (Figures 2D and 2E). Together, these data indicate that ERCs containing the URA3 construct accumulate to ~40% of a genomic loading control in ~90-repeat strains. Thus, ERCs derived from single chromosomal repeats are orders of magnitude higher than the loss rates of the same repeats, strongly implying that ERCs are copied from array-encoded repeats.
ERC production is rapid and self-limiting
To monitor the kinetics of ERC accumulation, we rendered ERC formation conditional by placing FOB1 under the control of an estrogen-inducible expression system in a ~90-repeat strain (Figures 3A and 3B) (Benjamin et al., 2003). This system induces FOB1 at near endogenous levels (Figure S3A). Cells maintained in log phase prior to FOB1 induction recapitulated the low ERC levels of fob1Δ mutants (Figure 3B). Upon estradiol addition, all major ERC species became detectable within 5 divisions, with levels reaching a steady state after about 11 divisions. Analysis of single-copy ERCs derived from the URA3-marked repeats yielded similar kinetics and showed that ERC levels stabilized at 15–20% relative to a genomic loading control (Figures S3B and S3C). This level is similar to the ERC levels observed in strains that have undergone 150–200 divisions (Figure 2B), suggesting that ERC levels reach a steady state that can be maintained for more than 150 divisions.
Figure 3: ERC accumulation responds to copy number.

(A-D) Southern analysis of ERCs in ~90-repeat strains. (A-B) Kinetics of ERC accumulation (bands a-e) relative to GAT1 after FOB1 induction with 100nM beta-estradiol. (C-D) ERC levels (bands a, c, d, e) relative to GAT1 after introduction of high-copy (2) rDNA plasmids or after further increasing plasmid copy number (leu2); ERC b (#) overlapped with plasmid signal and was not quantified. Data are represented as mean of n=4 biological replicates ± standard deviation. **P < 0.01, ***P < 0.001 (one-tailed t-test with Holm correction).
The rapid implementation of a steady-state ERC level that depends on array size implies that cells assess the total copy number of the rDNA and reduce ERC formation once sufficient rDNA copies have accumulated. To test this model, we transformed cells with an rDNA-bearing 2μ plasmid that exists at 40–50 copies in the cell and whose copy number can be amplified to ~100 copies by selection in the absence of leucine (Chernoff et al., 1994). Transformation of this plasmid into a ~90-repeat strain led to a strong reduction in endogenous ERC levels and a further reduction when plasmid copy number was boosted in the absence of leucine (Figures 3C, 3D, and S3D). The new steady-state levels strongly indicate that cells assess total rDNA copy number. No obvious nucleolar defects were observed upon copy number amplification of the 2μ plasmid, nor among strains with different chromosomal rDNA copy numbers (Figure S3E), arguing against secondary effects of altered nucleolar integrity. The responsiveness to plasmid-borne rDNA copies also indicates that copy-number is determined regardless of whether rDNA copies are chromosomal or episomal. These data indicate that ERC production is under negative feedback regulation.
Diet and rRNA transcription modulate ERC levels
We sought to investigate the mechanisms by which cells monitor total rDNA copy number. Metabolic demand has long-term effects on rDNA copy number in yeast and flies, suggesting that cells respond to the need for rRNA (Aldrich and Maggert, 2015; Jack et al., 2015). To test whether metabolic demand impacts ERC levels, we analyzed the effect of slower logarithmic growth, which is expected to reduce the demand for ribosomes and rRNA (Waldron and Lacroute, 1975). Cells were shifted from rich medium containing dextrose (doubling time of ~90 min) to rich medium containing glycerol (doubling time of ~180 min) and maintained in log phase for 30 doublings. Upon shift to glycerol, ERC levels diminished within 7 cell divisions and gradually approached a new steady-state level 2–3 times lower than in the presence of dextrose (Figures 4A and 4B). Quantification of URA3-marked single-copy ERCs revealed identical kinetics (Figures 4C and 4D). The decrease in URA3-marked ERC levels was not the result of repeat loss because the amount of FOAR colonies was indistinguishable before and after growth in glycerol (Figure S4). The new steady state suggests that ERC levels respond to metabolic demand.
Figure 4: Glycerol metabolism decreases ERC levels to a new steady state.

(A-D) Southern analysis of ERCs in ~90-repeat strains after shift from dextrose (YPD, t=0) to glycerol media. (A) Representative Southern blot of total ERCs. (B) Average signal of biological replicates, n=4 (*P < 0.05, **P < 0.01, ***P < 0.001). (C) Representative Southern blot of URA3-marked ERCs. (D) Average signal of biological replicates, n=4, performed in duplicate (*P < 0.05, **P < 0.01, ***P < 0.001).
To further test whether ERC formation is linked to the demand for rRNA, we reduced rRNA transcription using hmo1Δ, rpa49Δ, and uaf30Δ mutants, which lack important activators of rRNA transcription (Albert et al., 2013; Gadal et al., 2002; Siddiqi et al., 2001). Under glucose growth conditions, each mutant exhibited a significant increase in steady-state ERC levels (Figures 5A and S5A) despite having array sizes similar to wild type (Figures S5B and S5C). Specific analysis of URA3-marked repeats similarly revealed a 3-fold increase in steady-state ERC levels in the hmo1Δ mutant strains (Figures 5B and 5C). Intriguingly, hmo1Δ mutants still respond to differences in chromosomal rDNA copy number, but with a consistent 3-fold overproduction of ERCs. These data suggest that reduced rRNA output leads to miscalibration of the copy-number response. We note that marker loss rate is also increased in hmo1Δ mutants (Figure S5D), but this effect is genetically separable. Increased ERC formation in hmo1Δ mutants requires FOB1 (and RAD52; Figure S5E), whereas increased marker loss is FOB1-independent (Figure S5D). We conclude that ERC production is increased in response to low rRNA levels.
Figure 5: ERC formation is linked to rRNA levels.

(A) Quantification of ERC levels in ~120-repeat strains (bands a-e; URA3 probe) relative to GAT1 upon deletion of HMO1 (n=4), RPA49 (n=6), or UAF30 (n=4); data are represented as mean ± standard deviation *P < 0.05, **P < 0.01, ***P < 0.001 (two-tailed t-test with Holm correction; all experiments represent biological replicates). (B-C) URA3-marked ERCs in WT and hmo1Δ mutants by arrays size after digestion by NdeI; data are represented as mean ± standard deviation, **P < 0.01 (two-tailed t-test; clonal replicates n=3). WT data in (C) is same as in Figure 2B.
For additional mechanistic insight into the regulation of ERC production, we analyzed binding of Fob1 to the rDNA as a key initial step for ERC production (Figure 6A) (Defossez et al., 1999). Quantitative PCR (qPCR) after chromatin immunoprecipitation (ChIP) revealed that Fob1 binding to the replication-fork barrier (RFB) is significantly increased in short-array strains (Figure 6B), in line with their increased ERC levels. A similar effect is also observed when comparing Fob1 binding to the RFB in short-array strains growing in glycerol versus dextrose (Figure 6C). These data indicate that the observed changes in ERC production are linked to altered Fob1 interaction with the RFB.
Figure 6: Copy number alters Fob1 binding and ERC reinsertion.

(A-C) ChIP-qPCR analysis of Fob1 levels in the rDNA (A) and at the RFB (B, C) in indicated strains and conditions; data are represented as mean ± standard deviation, *P < 0.05, **P < 0.01 (two-tailed t-test; biological replicates, n=3). (D) Strategy for detecting cells with multiple URA3 insertions (green) by targeting URA3 with a LEU2 construct (blue). (E) leu+ ura+ transformants relative to the total leu+ transformants in the indicated strains; data are represented as mean ± standard deviation, *P < 0.05, **P < 0.01 (pair-wise, two-tailed t-tests with Holm correction; clonal replicates, n=3).
Consequences of increased ERC production
Our findings imply a cellular need for compensatory ERC production, even though cells can survive with as few as 20 chromosomal rDNA repeats (Ide et al., 2010). Strains with very low chromosomal copy number (20–60) are sensitive to the DNA damaging agent MMS (Ide et al., 2010). However, wild-type and fob1Δ strains harboring either 90 or 180 chromosomal rDNA repeats were indistinguishable over a wide range of MMS concentrations (Figure S6A). The same result was also obtained in a sensitized rad9Δ background (data not shown), indicating that ERC formation is dispensable in these situations.
We asked whether ERCs might insert into the genome to help re-expand the rDNA array after repeat loss. Such events are difficult to isolate directly because all repeats can serve as potential ERC sources and integration sites, creating substantial heterogeneity. However, reinsertion of a URA3-marked ERC should create a duplication of the URA3 marker elsewhere in the rDNA array that can be captured by targeted URA3 disruption with LEU2 (Figure S6B). If multiple URA3 markers are present, leu+ ura+ transformants will be observed when at least one but not all of the URA3 markers is disrupted (Figure 6D). Applying this assay to six URA3 insertion strains with rDNA chromosomal copy number from 86–123 repeats revealed a FOB1-dependent, positive correlation between the fraction of leu+ ura+ transformants and ERC levels (Figures 6E and 2B), consistent with dosage-dependent ERC insertion into the rDNA. As marker loss rates do not show a copy-number dependence among these strains (Figure 2C), the strain-dependent differences in marker duplication are unlikely to have arisen from USCE.
To directly show that ERCs can insert into the rDNA, we amplified URA3-marked ERCs in bacteria and used them to transform unmarked cells. ERC preparations were treated with exonuclease RecBCD to eliminate sheared DNA circles prior to transformation. In most transformants, the marked ERC was propagated episomally, giving rise to highly sectored colonies (Figure S6C). Micromanipulation revealed that these colonies consisted mostly of cells that were unable to grow on -ura plates, consistent with the asymmetric retention of ERCs in mother cells (Sinclair and Guarente, 1997), although about 2% of cells (n=1614) were able to again form sectored colonies (Figure S6D). Importantly, approximately 1 in 5×10−4 cells from these colonies gave rise to nonsectored colonies (Figure S6C), indicative of stable marker inheritance. Southern analysis of these colonies confirmed that the marked ERC had inserted into the rDNA (Figure S6E). These data demonstrate that ERCs spontaneously reinsert into the rDNA and suggest that ERCs provide a reservoir for regrowth of shortened rDNA arrays.
Discussion:
Catastrophic loss of repeats by NAHR is an inherent risk to tandem repetitive DNA arrays. Here, we show that S. cerevisiae cells respond to large-scale rDNA deletions by proportionally increasing the production of circular rDNA minichromosomes. This process is self-limiting and likely provides substrates for the regeneration of the rDNA array.
Our results suggest a replicative mode of ERCs production. As ERC production relies on one-ended DSB formation at the RFB (Burkhalter and Sogo, 2004; Weitao et al., 2003), a conceivable model is that the looped-out sequences are re-replicated by the oncoming replication fork (Figure 7). A similar mechanism has been suggested for the formation of cancer-associated circular EGFR amplicons, which also form without loss of the chromosomal EGFR copies (Vogt et al., 2004). The dependence of ERC production on the strand-annealing factor Rad52 but not the recombinase Rad51 (Park et al., 1999) further suggests that ERC-associated NAHR initiates without strand invasion. It is possible that ERCs arise from combined fork breakage at two neighboring replication forks, followed by Rad52-dependent end annealing and circularization (Figure 7). This mode of ERC formation is supported by the fact that fork breakage only occurs on the leading strand and involves nicks that could yield complementary overhangs (Burkhalter and Sogo, 2004). Moreover, the estimated size of rDNA replicons correlates well with the distribution of ERC sizes (Brewer and Fangman, 1988; Burkhalter and Sogo, 2004). Alternatively, recombinase-independent ERC formation may also occur if a resected DSB anneals with transcription-associated single-stranded DNA near the RFB (Houseley and Tollervey, 2011). We note that re-replication by the oncoming fork will inherently limit the number of ERCs formed per S phase at a given repeat, which may explain why it takes ~10 population doublings for ERC levels to reach a steady state.
Figure 7: Model for Fob1-dependent ERC formation.

(A-E) Model for ERC formation that was adapted from (Vogt et al., 2004) and modified to include the unipolar directionality of rDNA replication (Brewer and Fangman, 1988). (A) Asterisks represent origin firing, which typically occurs in a clustered fashion (Pasero et al., 2002). (B) Rightward moving forks are stalled at the Fob1 binding site (RFB), which is vulnerable to collapse and DSB formation. (C) When neighboring RFBs collapse into DSBs, newly synthesized DNA may be excised and (D) circularize through the stand-annealing activity of Rad52. Passive replication from the rightmost fork could then re-replicate repeats that have formed ERCs, (E) ultimately amplifying rDNA copy number.
We interpret the appearance of a steady state as evidence that cells actively control ERC levels in response to total rDNA copy number. However, a steady state could also result if selective pressures caused accumulation of cells with an optimal number of ERCs. We do not favor this possibility for two reasons. First, ERCs are almost entirely retained in the mother cell during cell division (Sinclair and Guarente, 1997), which in itself precludes inheritance of an optimal ERC number, although we note that ~2% of cells temporarily escape this regulation (Figure S6D). Second, back-of-the envelope calculations (see Supplementary Methods) show that even under the most conservative estimates, cells with an optimal (i.e. steady-state) number of ERCs would need to grow about 20% faster than cells with no ERCs to sweep the population within ~10 population doublings. Analysis of individual growth rates, however, showed no difference (Figure S7), in line with previous analyses (French et al., 2003; Ide et al., 2010; Kobayashi et al., 1998). These considerations do not support an adaptive model of ERC accumulation.
There are additional indications that yeast cells respond to copy number loss in the rDNA. Two known examples of rDNA copy-number monitoring include regulating the density of transcriptionally active rDNA repeats (French et al., 2003; Ide et al., 2010), and regulating the cellular levels of histone deacetylase Sir2 (Michel et al., 2005). Either of these effects could be connected to the observed increase in ERC levels. A link to the density of active repeats is appealing, because origin firing in the rDNA, and thus RFB-associated DSB formation, is connected to transcription of the neighboring rRNA gene (Muller et al., 2000). A coupling of ERC production to the relative density of active rDNA repeats could explain why ERCs are high in short array strains but lower when growth rate is reduced (Fahy et al., 2005; Sandmeier et al., 2002). It could also explain why ERC production is affected by altering rRNA output, which is known to impact the number of active rDNA repeats (Catala et al., 2008). Sir2 reduction also provides an appealing mechanistic link, as Sir2 is a well-known negative regulator of ERC production (Kaeberlein et al., 1999). Sir2 affects DSB repair partner choice (Kobayashi et al., 2004), which may be sufficient to alter ERC production. Moreover, regulation by Sir2 may explain the observed changes in Fob1 ChIP signal, as Sir2 counteracts Fob1-dependent gene looping in the rDNA (Zaman et al., 2016). One argument against a role of lower Sir2 levels is that reporter gene silencing is unaffected when rDNA copy number is reduced (Michel et al., 2005), although gene silencing and ERC formation may be differentially sensitive to Sir2 dosage (Kaeberlein et al., 1999).
Our data indicate that ERC levels can be modulated in both directions. Down-regulation of ERCs in a culture likely occurs as a combined consequence of reduced ERC production and passive dilution by cell division. At the cellular level, this dilution is highly asymmetric, because ERCs are almost entirely retained in the mother cell during cell division (Sinclair and Guarente, 1997). Importantly, given that newborn daughters are essentially free of ERCs, this setup allows every newborn daughter to specifically assess the number of rDNA copies in the chromosomal rDNA array before initiating appropriate cell-autonomous ERC production over the subsequent cell divisions. We note that compensatory ERC production may also help explain the elevated ERC levels observed in many mutants experiencing rDNA instability (Ivessa et al., 2000; Torres et al., 2004). rDNA instability will lead to a higher incidence of catastrophic array shortening, and thus more frequent activation of compensatory ERC production.
Although ERCs have primarily been studied in the context of yeast aging (Sinclair et al., 1998), our findings suggest that ERCs can provide a physiological benefit by serving as a reservoir for regrowing short rDNA arrays. Importantly, unlike USCE, where an array grows at the expense of another, replicative amplification of ERCs followed by reinsertion is purely additive. However, ERC insertion events are quite infrequent (Figure S6C) (Sinclair and Guarente, 1997). This low frequency may partly explain why it takes nearly 100 generations for an array to grow from 40 to 150 repeats (Kobayashi et al., 1998; Kobayashi and Ganley, 2005).
Extrachromosomal circular DNAs (eccDNAs) are observed in a variety of cell types although their function remains largely unknown (Paulsen et al., 2018). In cancers, eccDNAs are major contributors to tumor heterogeneity (Turner et al., 2017) and are generally seen as markers of genome instability. However, eccDNAs are also detected in healthy cells and can occur in a tissue-specific or developmentally regulated pattern (Cohen et al., 2003, 1999, Møller et al., 2018, 2015; Shoura et al., 2017), implying a possible physiological role. In human cells, ERCs are prime candidates for functional regulation, as they are the most abundant eccDNA molecule in healthy tissue and they are highly overrepresented (8.1% of all eccDNA) compared their proportion of chromosomal rDNA arrays (~0.25% of all chromosomal DNA) (Gibbons et al., 2015; Møller et al., 2018). The regulated formation of ERCs in yeast indicates that the formation of eccDNAs does not have to be the result of genome instability and that the study of ERC regulation may serve as a powerful model for understanding eccDNA production in healthy cells.
STAR METHODS:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andreas Hochwagen (andi@nyu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The majority of yeast strains used in this study are derivatives of the laboratory strain SK1 and are listed in Table S1. Yeasts strains derived from the laboratory strain W303 are specified when applicable.
METHOD DETAILS
Yeast strains, plasmids, transformation, and growth conditions
All strains used in this study are of the SK1 background, unless specified otherwise (Table S1). To make the collection of strains with different chromosomal rDNA copy number, we transformed cells with SphI-digested plasmid pRS306 that had the rDNA intergenic sequence cloned into it between BamHI and EcoRI restriction sites (Fig. 1A–B) (Vader et al., 2011). Lithium acetate transformation was used for insertion of pRS306-rDNA (Fig. 1A–B) and for URA3-to-LEU2 insertion swap (pRS306-to-pRS305) (Fig. 6D–E). Cells were incubated at 30°C in 0.1M l ithium acetate, 33% PEG-3350, 100 g salmon sperm single-stranded DNA, and 200–700 ng of insertion-targeting DNA for 30 minutes prior to a 15-minute heat-shock at 42°C. Due to changes in rDNA size upon lithium acetate transformation (Kwan et al., 2016), electroporation was used for transforming the high-copy rDNA plasmid (pRDN-hyg1 leu2-D; (Chernoff et al., 1994)) into a ~90-repeat strain (Fig. 3C–D) and for transforming RecBCD-treated pRS306-rDNA into a ~140 repeat strain (Fig. S6C–E). Cells were resuspended in chilled 1M sorbitol and subjected to an electro-pulse (1.5kV, 25uF, and 200 Ohms) using a Bio-Rad GenePulser Xcell. When analyzing the effect of plasmid overexpression or mutations, experiments were performed using strains with the same rDNA array size to exclude rDNA size as a variable. To make the collection of W303 strains with different chromosomal rDNA copy number, a strain with 20 chromosomal rDNA copies (Sasaki and Kobayashi, 2017) was mated with a FOB1 W303 strain. All strains were grown at 30°C in YEP + 2% dextrose or 3% glycerol when indic ated.
Plug preparation and pulsed-field gel electrophoresis conditions
Cultures were grown to saturation and 8.5 × 105 cells were embedded in agarose using Bio-rad plug molds followed by incubations in zymolyase T100 (200 g/mL) overnight at 37°C, proteinase K (4 mg/mL) and 1% N-lauroyl sarko sine overnight at 50°C, and PMSF (1 mM) for one hour at 4°C. PMSF and remaining detergent were removed by washing plugs with 1 mL of CHEF TE (10 mM Tris.HCl pH 7.5, 50 mM EDTA) three times for thirty minutes. Plugs were subsequently run in a 1 × TAE, 0.8% agarose gel using a Bio-rad CHEF-DR II at settings: 3V/cm, 50 hrs, 250 seconds start switch time, and 900 seconds end switch time (Figs. 1B, S1A-D, S3B-C, S6E). H. wingei ladder (Biorad) was used as size marker.
Genomic DNA extraction and gel electrophoresis conditions
8.5 × 107 cells undergoing mid-logarithmic growth (OD600 0.5–1.0) were collected and treated with 250 g/mL zymolyase T100 in 1M sorbitol, 42 mM K2HPO4, 8mM KH2PO4, 5mM EDTA at 37°C for 30 minutes, followed by treatm ent with proteinase K (285 g) and SDS (0.5%) at 65°C for 1.5 hours. SDS was then precipitated out using potassium acetate. Nucleic acids were ethanol precipitated and treated overnight at 4°C with RNase A (60 g). DNA was subsequently purified using phenol/chloroform (Roche) and precipitated using isopropanol. Genomic DNA was digested for 4 hours at 37°C using 2 L AfeI (NEB) for Figs. 1C, 3A, 4A, S1C, S3B, S5A; 4 hours at 37°C with 2 L XhoI (NEB) for Figs. 3C, S1E; 4 hours at 37°C with 2 L NdeI + 2 L AvrII (NEB) for Figs. 2A, 2D, 4C, 5B, S3B, S5D; and 24-hours at 37°C with 2 L AfeI + 2 L AvrII + 2 L RecBCD (NEB) for Figs. 1C, 2D, S2A. Digested DNA was subsequently run in a 1× TBE, 0.7% agarose gel for 20 hours at 1.5 V/cm.
Southern blotting, phosphoimaging, and quantification
Southern blotting was performed by alkaline transfer using Hybond-XL membranes (GE). Blots were subsequently probed with 32P-labeled DNA complementary to [GAT1 and NTS1] or URA3. Signal from blots was detected using FujiFilm imaging plates and imaged using Typhoon FLA9000, at non-saturating conditions for ERC bands and single-copy loading controls. Signals were quantified using ImageJ. Background signal above and below each band was averaged and subtracted from signal of interest to account for well-to-well variation in the amount of DNA shearing. ERC signals were then summed and divided by intensity measurement of single-copy loading controls, GAT1 or ura3.
Spot tests for growth on 5-FOA and DNA damage sensitivity
Cells were serially diluted 3.5-fold and spotted onto 2% dextrose synthetic complete (SC) media with and without 11.5mM 5-FOA (Toronto Research Chemicals) (Fig. 1C and Fig. S4). For DNA damage sensitivity assay, cells were similarly spotted onto 2% YPD with and without methyl methanesulfonate (MMS; Sigma-Aldrich) (Fig. S6A).
Immunofluorescence
Immunofluorescence analysis was performed as described previously (Wang et al., 2016). Briefly, 2 OD cells were fixed by addition of formaldehyde to a final concentration of 3.7% (v/v). Cells were pelleted, resuspended in 500 l of 3.7% formaldehyde in 0.1M potassium phosphate (pH 6.4) and fixed for 15 minutes at room temperature. Cells were washed 3× in in 0.1M potassium phosphate (pH 6.4) and once in sorbitol-citrate (128mM KH2P04, 36mM citrate, 1.2M sorbitol), followed by spheroplasting with zymolyase and glusulase in sorbitol-citrate. Spheroplasts were fixed on polylysine-coated slides, incubated with primary antibody overnight at 4°C, and secondary antibody for 4 hours at room temperature. Antibodies were diluted in PBS/BSA (50mM potassium phosphate, pH 7.4, 150mM NaCl, 1% bovine serum albumin, 0.1% sodium azide). Nop1 was detected using a mouse monoclonal Nop1 antibody (EnCor Biotechnology) at a 1:500 dilution and an anti-mouse fluorescein (FITC) antibody (Jackson ImmunoResearch) at 1:200. Nuclear DNA was detected using DAPI staining (Vectashield). Images were captured as z-stacks using a Delta Vision Elite System with EMCCD camera (Applied Precision, Issaquah, WA). Stacks were deconvolved, and compressed into a 2D image using SoftWoRX2.50 software (Applied Precision).
Chromatin Immunoprecipitation
8.5 × 107 cells undergoing mid-logarithmic growth (OD600 0.5–1.0) were collected and fixed with 1% formaldehyde for 30 minutes and quenched with glycine. Cells were resuspended in lysis buffer (50mM HEPES/KOH pH 7.5, 140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na-Deoxycholate, 1mM PMSF, 1mM Benzamidine, 1mg/mL Bacitracin, Roche Protease Inhibitor Tablet) and lysed by glass-bead biopulverizeration (MP Biomedicals). Chromatin was then subjected to shearing using Branson Digital Sonifier Model 250 at 15% amplitude for 15 seconds, repeated five times, and centrifugally separated from cell debris, followed by incubation with Anti-Fob1 (kindly provided by Stephen P. Bell) for 1 hour and a subsequent incubation with magnetic protein G beads (NEB) for 1 hour. Immunoprecipitated chromatin was then washed with lysis buffer (50mM HEPES/KOH pH 7.5, 140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na-Deoxycholate), high salt buffer (50mM HEPES/KOH pH 7.5, 500mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na-Deoxycholate), lithium chloride buffer (10mM Tris, 0.25M LiCl, 0.5% NP-40, 0.5% Dexoycholate, 1mM EDTA), and TE, followed by elution at 65°C and DNA purification using Qiagen P CR purification columns. Quantitative PCR was performed on both immunoprecipitated and nonimmunoprecipitated input samples, and then quantified using percent input method: 100*2^(Adjusted input – IP).
Assay for detection of multiple rDNA insertions
Cells containing an integrated URA3 marker were transformed with pRS305 that had been digested at BamHI (NEB) and HindIII (NEB) sites. Positive transformants were grown on leucine dropout media, and after three days of growth at 30°C, selection plates were replica-plated onto uracil dropout media and 11.5mM 5-FOA. The proportion of leu+ ura+ transformations was quantified relative to the total number of leu+ transformants. A total of 450 or more leu+ transformants were scored for each strain from three transformations. Statistical comparisons were performed using two-tailed, pairwise t-tests with a Holm correction for multiple testing.
rDNA expansion experiment
Strains were repeatedly bottlenecked by streaking for single colonies on 2% YPD and incubated for 2 days. The number of vegetative cell divisions were estimated by colony size (2mm was estimated to be equivalent to approximately 20 cell divisions) (Kobayashi et al., 1998).
RNA preparation and qPCR analysis
10 g of total RNA was phenol/chloroform extracted and treated with TURBO DNase (Invitrogen, 1 L per sample) at 37°C for 1 hour. DNase inactivatio n reagent (Invitrogen, 5 L per sample) was added and incubated with samples for 5 minutes at room temperature. 1 g of this RNA was used for reverse transcription (RT). Samples for RT were incubated with Revert Aid (Fermentas, 1 L per sample), Revert Aid buffer (Fisher), Oligo-dT (100 M, Fisher, 1 L per sample) and RNaseOut recombinant ribonuclease inhibitor (Invitrogen, 1 L per sample) at 42°C for 1 hour in a 20 L reaction. Samples without RT were run to control for the DNase step. Reactions were terminated by incubation at 70°C for 10 minutes. Samples were diluted to 400 L for analysis by qPCR. For qPCR analysis, 8 L samples (diluted ChIP samples or diluted RT samples) and 1 L of each primer (20nM) were mixed with Quantifast SYBR green master mix (Qiagen, 10 L per reaction) and placed in a BioRad CFX96 Real-Time System. Program: 95°C, 5 minutes; 39 cycles: 95°C, 10 secon ds; 60°C, 30 seconds.
QUANTIFICATION AND STATISTICAL ANALYSIS
For analyses of both total ERC levels and URA3-marked ERC levels as a function of rDNA array size, P values were determined by Spearman’s rank correlation tests. These data also showed high correlations to power law functions as determined by coefficient of determination (r2) values. Error bars represent standard deviations of measurements in all main and supplemental figures. Number of replicates are specified as “biological” (distinct meiotic segregants of the same genotype) or “clonal” (independent experiments using same clonal population).
Estimating the benefits of ERC Production under an adaptive model
We used the kinetics of reaching steady-state ERC levels (Fig. 3B) to obtain a back-ofthe-envelope estimate for the minimal growth benefits of ERC production under an adaptive model. From that data, the signal at 11 doublings represents the steady state, which, under an adaptive model, indicates that cells with an optimal ERC number would have taken over the population. For simplicity, we assumed the presence of two cell types: cells with no ERCs and cells with the optimal number of ERCs. Based on the signal-to-noise ratio of our assay, we expect to confidently detect any change that is 10% or larger in the number of ERCs in the population. Therefore, the most extreme assumption is that Fob1 induction creates about 10% of cells with the correct ERC number within the first cell doubling ([N]0 = 0.1; i.e. just below our level of detection). These cells then take 11 doublings (t = 11) to sweep the populations to within our levels of detection ([N]11 = 0.9) by competing against cells that did not induce ERCs in that time frame. Solving a simple exponential equation
yields α = 0.2, indicating that cells with the optimal number of ERCs would have to grow 20% faster than cells with no ERCs to overtake the population in the given time frame This simplistic back-of-the-envelope estimate obviously ignores the possibility of cells with optimal ERC levels arising at later time points as well as the potential existence of cells with suboptimal (or deleterious) ERC levels.
DATA AND SOFTWARE AVAILABILITY
Data have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/9z25hjzjdc.1.
Supplementary Material
Table S1. Yeast strains used in this research, Related to all main and supplemental figures.
Highlights.
rDNA circles form through a replicative mechanism independent of array instability.
Production of rDNA circles anti-correlates with total rDNA copy number.
Steady-state level of rDNA circles is regulated by diet and rRNA production.
rDNA circles re-insert into the chromosomal rDNA array.
Acknowledgements:
We thank S.P. Bell for the Fob1 antibody, L. Steinmetz and T. Kobayashi for strains, and D. Gresham, J. Hubbard, R. Rothstein, M. Siegal, and D. Smith for helpful discussions. This work was supported by NIH grants R01GM088248 and R01GM111715 to A.H. and NIH grant R01CA146940 to H.K. A.R.M. was supported by NIH fellowship T32HD7520–17 and a Dean’s Dissertation Fellowship (NYU).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
The authors declare no competing interests.
References:
- Albert B, Colleran C, Léger-Silvestre I, Berger AB, Dez C, Normand C, Perez-Fernandez J, McStay B, Gadal O (2013). Structure-function analysis of Hmo1 unveils an ancestral organization of HMG-Box factors involved in ribosomal DNA transcription from yeast to human. Nucleic Acids Res. 41, 10135–10149. doi: 10.1093/nar/gkt770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich JC, Maggert KA (2015). Transgenerational Inheritance of Diet-Induced Genome Rearrangements in Drosophila. PLOS Genet. 11, e1005148. doi: 10.1371/journal.pgen.1005148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averbeck KT, Eickbush TH (2005). Monitoring the mode and tempo of concerted evolution in the Drosophila melanogaster rDNA locus. Genetics 171, 1837–1846. doi: 10.1534/genetics.105.047670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin KR, Zhang C, Shokat KM, Herskowitz I (2003). Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes Dev. 17, 1524–1539. doi: 10.1101/gad.1101503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake D, Luke B, Kanellis P, Jorgensen P, Goh T, Penfold S, Breitkreutz BJ, Durocher D, Peter M, Tyers M (2006). The F-box protein Dia2 overcomes replication impedance to promote genome stability in Saccharomyces cerevisiae. Genetics 174, 1709–1727. doi: 10.1534/genetics.106.057836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL (1988). A replication fork barrier at the 3’ end of yeast ribosomal RNA genes. Cell 55, 637–643. doi: 10.1016/0092-8674(88)90222-X [DOI] [PubMed] [Google Scholar]
- Burkhalter MD, Sogo JM (2004). rDNA enhancer affects replication initiation and mitotic recombination: Fob1 mediates nucleolytic processing independently of replication. Mol. Cell 15, 409–421. doi: 10.1016/j.molcel.2004.06.024 [DOI] [PubMed] [Google Scholar]
- Catala M, Tremblay M, Samson E, Conconi A, Abou Elela S (2008). Deletion of Rnt1p Alters the Proportion of Open versus Closed rRNA Gene Repeats in Yeast. Mol. Cell. Biol 28, 619–629. doi: 10.1128/MCB.01805-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff YO, Vincent A, Liebman SW (1994). Mutations in eukaryotic 18S ribosomal RNA affect translational fidelity and resistance to aminoglycoside antibiotics. EMBO J. 13, 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Walker GD, Azad AA (1980). Hybridizable sequences between cytoplasmic ribosomal RNAs and 3 micron circular DNAs of Saccharomyces cerevisiae and Torulopsis glabrata. Nucleic Acids Res. 8, 1009–1022. doi: 10.1093/nar/8.5.1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Menut S, Méchali M (1999). Regulated formation of extrachromosomal circular DNA molecules during development in Xenopus laevis. Mol. Cell. Biol 19, 6682–6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Yacobi K, Segal D (2003). Extrachromosomal Circular DNA of Tandemly Repeated Genomic Sequences in Drosophila Extrachromosomal Circular DNA of Tandemly Repeated Genomic Sequences in Drosophila 1133–1145. doi: 10.1101/gr.907603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammann R, Lucchini R, Koller T, Sogo JM (1993). Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 21, 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defossez PA, Prusty R, Kaeberlein M, Lin SJ, Ferrigno P, Silver PA, Keil RL, Guarente L (1999). Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol. Cell 3, 447–455. doi: 10.1016/S1097-2765(00)80472-4 [DOI] [PubMed] [Google Scholar]
- Eickbush TH, Eickbush DG (2007). Finely orchestrated movements: Evolution of the ribosomal RNA genes. Genetics 175, 477–485. doi: 10.1534/genetics.107.071399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy D, Conconi A, Smerdon MJ (2005). Rapid changes in transcription and chromatin structure of ribosomal genes in yeast during growth phase transitions. Exp. Cell Res. 305, 365–373. doi: 10.1016/j.yexcr.2005.01.016 [DOI] [PubMed] [Google Scholar]
- French SL, Osheim YN, Cioci F, Nomura M, Beyer AL, French SL, Osheim YN, Cioci F, Nomura M, Beyer AL (2003). In Exponentially Growing Saccharomyces cerevisiae Cells, rRNA Synthesis Is Determined by the Summed RNA Polymerase I Loading Rate Rather than by the Number of Active Genes. Mol. Cell. Biol 23, 1558–1568. doi: 10.1128/MCB.23.5.1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadal O, Labarre S, Boschiero C, Thuriaux P (2002). Hmo1, an HMG-box protein, belongs to the yeast ribosomal DNA transcription system. EMBO J. 21, 5498–5507. doi: 10.1093/emboj/cdf539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, Zou H, Rothstein R (1996). Gene conversion plays the major role in controlling the stability of large tandem repeats in yeast. EMBO J. 15, 1715–1725. [PMC free article] [PubMed] [Google Scholar]
- Ganley ARD, Ide S, Saka K, Kobayashi T (2009). The Effect of Replication Initiation on Gene Amplification in the rDNA and Its Relationship to Aging. Mol. Cell 35, 683–693. doi: 10.1016/j.molcel.2009.07.012 [DOI] [PubMed] [Google Scholar]
- Ganley ARD, Kobayashi T (2011). Monitoring the rate and dynamics of concerted evolution in the ribosomal DNA repeats of Saccharomyces cerevisiae using experimental evolution. Mol. Biol. Evol 28, 2883–2891. doi: 10.1093/molbev/msr117 [DOI] [PubMed] [Google Scholar]
- Gaubatz J, Prashad N, Cutler RG (1976). Ribosomal RNA gene dosage as a function of tissue and age for mouse and human. Biochim. Biophys. Acta - Nucleic Acids Protein Synth. 418, 358–375. doi: 10.1016/0005-2787(76)90297-5 [DOI] [PubMed] [Google Scholar]
- Gibbons JG, Branco AT, Godinho SA, Yu S, Lemos B (2015). Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc. Natl. Acad. Sci 112, 201416878. doi: 10.1073/pnas.1416878112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons JG, Branco AT, Yu S, Lemos B (2014). Ribosomal DNA copy number is coupled with gene expression variation and mitochondrial abundance in humans. Nat Commun 5, 4850. doi: 10.1038/ncomms5850 [DOI] [PubMed] [Google Scholar]
- Ha CW, Huh WK (2011). Rapamycin increases rDNA stability by enhancing association of Sir2 with rDNA in Saccharomyces cerevisiae. Nucleic Acids Res. 39, 1336–1350. doi: 10.1093/nar/gkq895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallgren J, Pietrzak M, Rempala G, Nelson PT, Hetman M (2014). Neurodegeneration-associated instability of ribosomal DNA. Biochim. Biophys. Acta - Mol. Basis Dis 1842, 860–868. doi: 10.1016/j.bbadis.2013.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Wittner M, Babl V, Perez-Fernandez J, Tschochner H, Griesenbeck J (2013). Chromatin states at ribosomal DNA loci. Biochim. Biophys. Acta - Gene Regul. Mech 1829, 405–417. doi: 10.1016/j.bbagrm.2012.12.007 [DOI] [PubMed] [Google Scholar]
- Houseley J, Tollervey D (2011). Repeat expansion in the budding yeast ribosomal DNA can occur independently of the canonical homologous recombination machinery. Nucleic Acids Res. 39, 8778–8791. doi: 10.1093/nar/gkr589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JE, Pathak S (1977). Elimination of nucleolus organizers in a case of 13/14 Robertsonian translocation. Hum. Genet 35, 169–73. [DOI] [PubMed] [Google Scholar]
- Ide S, Miyazaki T, Maki H, Kobayashi T (2010). Abundance of ribosomal RNA gene copies maintains genome integrity. Science 327, 693–696. doi: 10.1126/science.1179044 [DOI] [PubMed] [Google Scholar]
- Ivessa a S., Zhou JQ, Zakian VA (2000). The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell 100, 479–489. doi: 10.1016/S0092-8674(00)80683-2 [DOI] [PubMed] [Google Scholar]
- Jack CV, Cruz C, Hull RM, Keller MA, Ralser M, Houseley J (2015). Regulation of ribosomal DNA amplification by the TOR pathway. Proc. Natl. Acad. Sci 112, 201505015. doi: 10.1073/pnas.1505015112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johzuka K, Horiuchi T (2002). Replication fork block protein, Fob1, acts as an rDNA region specific recombinator in S. cerevisiae. Genes to Cells 7, 99–113. doi: 10.1046/j.1356-9597.2001.00508.x [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L (1999). The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580. doi: 10.1101/gad.13.19.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T (2011). Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell. Mol. Life Sci 68, 1395–1403. doi: 10.1007/s00018-010-0613-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ganley ARD (2005). Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 309, 1581–1584. doi: 10.1126/science.1116102 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Heck DJ, Nomura M, Horiuchi T (1998). Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: Requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev. 12, 3821–3830. doi: 10.1101/gad.12.24.3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Horiuchi T, Tongaonkar P, Vu L, Nomura M (2004). SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 117, 441–453. doi: 10.1016/S0092-8674(04)00414-3 [DOI] [PubMed] [Google Scholar]
- Kwan EX, Foss EJ, Tsuchiyama S, Alvino GM, Kruglyak L, Kaeberlein M, Raghuraman MK, Brewer BJ, Kennedy BK, Bedalov A (2013). A natural polymorphism in rDNA replication origins links origin activation with calorie restriction and lifespan. PLOS Genet. 9, e1003329. doi: 10.1371/journal.pgen.1003329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan EX, Wang XS, Amemiya HM, Brewer BJ, Raghuraman MK (2016). rDNA Copy Number Variants Are Frequent Passenger Mutations in S. cerevisiae Deletion Collections and De Novo Transformants. G3: Genes|Genomes|Genetics 6, 2829–2838. doi: 10.1534/g3.116.030296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom DL, Leverich CK, Henderson KA, Gottschling DE (2011). Replicative age induces mitotic recombination in the ribosomal RNA gene cluster of Saccharomyces cerevisiae. PLOS Genet. 7. doi: 10.1371/journal.pgen.1002015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay B, Grummt I (2008). The epigenetics of rRNA genes: from molecular to chromosome biology. Annu. Rev. Cell Dev. Biol 24, 131–157. doi: 10.1146/annurev.cellbio.24.110707.175259 [DOI] [PubMed] [Google Scholar]
- Michel AH, Kornmann B, Dubrana K, Shore D (2005). Spontaneous rDNA copy number variation modulates Sir2 levels and epigenetic gene silencing. Genes Dev. 19, 1199–1210. doi: 10.1101/gad.340205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller HD, Mohiyuddin M, Prada-Luengo I, Sailani MR, Halling JF, Plomgaard P, Maretty L, Hansen AJ, Snyder MP, Pilegaard H, Lam HYK, Regenberg B (2018). Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat. Commun 9, 1–12. doi: 10.1038/s41467-018-03369-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller HD, Parsons L, Jørgensen TS, Botstein D, Regenberg B (2015). Extrachromosomal circular DNA is common in yeast. Proc. Natl. Acad. Sci 201508825. doi: 10.1073/pnas.1508825112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Lucchini R, Sogo JM (2000). Replication of yeast rDNA initiates downstream of transcriptionally active genes. Mol. Cell 5, 767–777. doi: 10.1016/S1097-2765(00)80317-2 [DOI] [PubMed] [Google Scholar]
- Park PU, Defossez PA, Guarente L (1999). Effects of mutations in DNA repair genes on formation of ribosomal DNA circles and life span in Saccharomyces cerevisiae. Mol. Cell. Biol 19, 3848–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, Bensimon A, Schwob E (2002). Single-molecule analysis reveals clustering and epigenetic regulation of replication origins at the yeast rDNA locus. Genes Dev. 2479–2484. doi: 10.1101/gad.232902.GENES [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen T, Kumar P, Koseoglu MM, Dutta A (2018). Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends Genet. 34, 270–278. doi: 10.1016/j.tig.2017.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka K, Ide S, Ganley ARD, Kobayashi T (2013). Cellular senescence in yeast is regulated by rDNA noncoding transcription. Curr. Biol 23, 1794–1798. doi: 10.1016/j.cub.2013.07.048 [DOI] [PubMed] [Google Scholar]
- Salim D, Bradford WD, Freeland A, Cady G, Wang J, Pruitt SC, Gerton JL (2017). DNA replication stress restricts ribosomal DNA copy number. PLOS Genet. doi: 10.1371/journal.pgen.1007006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmeier JJ, French S, Osheim Y, Cheung WL, Gallo CM, Beyer AL, Smith JS (2002). RPD3 is required for the inactivation of yeast ribosomal DNA genes in stationary phase. EMBO J. 21, 4959–4968. doi: 10.1093/emboj/cdf498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Kobayashi T (2017). Ctf4 Prevents Genome Rearrangements by Suppressing DNA Double-Strand Break Formation and Its End Resection at Arrested Replication Forks. Mol. Cell 66, 533–545.e5. doi: 10.1016/j.molcel.2017.04.020 [DOI] [PubMed] [Google Scholar]
- Schweizer E, MacKechnie C, Halvorson HO (1969). The redundancy of ribosomal and transfer RNA genes in Saccharomyces cerevisiae. J. Mol. Biol 40, 261–277. doi: 10.1016/0022-2836(69)90474-4 [DOI] [PubMed] [Google Scholar]
- Shoura MJ, Gabdank I, Hansen L, Merker J, Gotlib J, Levene SD, Fire AZ (2017). Intricate and Cell Type-Specific Populations of Endogenous Circular DNA (eccDNA) in Caenorhabditis elegans and Homo sapiens. G3 Genes, Genomes, Genet 7, 3295–3303. doi: 10.1534/g3.117.300141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi IN, Dodd JA, Vu L, Eliason K, Oakes ML, Keener J, Moore R, Young MK, Nomura M (2001). Transcription of chromosomal rRNA genes by both RNA polymerase I and II in yeast uaf30 mutants lacking the 30 kDa subunit of transcription factor UAF. EMBO J. 20, 4512–4521. doi: 10.1093/emboj/20.16.4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L (1997). Extrachromosomal rDNA circles - A cause of aging in yeast. Cell 91, 1033–1042. doi: 10.1016/S0092-8674(00)80493-6 [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Mills K, Guarente L (1998). Molecular mechanisms of yeast aging. Trends Biochem. Sci 23, 131–134. doi: 10.1016/S0968-0004(98)01188-8 [DOI] [PubMed] [Google Scholar]
- Stults DM, Killen MW, Pierce HH, Pierce AJ (2008). Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18, 13–18. doi: 10.1101/gr.6858507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stults DM, Killen MW, Williamson EP, Hourigan JS, Vargas HD, Arnold SM, Moscow J. a., Pierce AJ (2009). Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104. doi: 10.1158/0008-5472.CAN-09-2680 [DOI] [PubMed] [Google Scholar]
- Szostak JW, Wu R (1980). Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature 284, 426–430. doi: 10.1038/284426a0 [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Horiuchi T, Kobayashi T (2003). Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev. 17, 1497–1506. doi: 10.1101/gad.1085403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres JZ, Bessler JB, Zakian VA (2004). Local chromatin structure at the ribosomal DNA causes replication fork pausing and genome instability in the absence of the S. cerevisiae DNA helicase Rrm3p. Genes Dev. 18, 498–503. doi: 10.1101/gad.1154704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang E, Carr AM (2008). Replication fork arrest, recombination and the maintenance of ribosomal DNA stability. DNA Repair (Amst). 7, 1613–1623. doi: 10.1016/j.dnarep.2008.06.010 [DOI] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, Kornblum HI, Taylor MD, Kaushal S, Cavenee WK, Wechsler-Reya R, Furnari FB, Vandenberg SR, Rao PN, Wahl GM, Bafna V, Mischel PS (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125. doi: 10.1038/nature21356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugama M, Sanij E, Voon HPJ, Son J, Hii L, Henson JD, Chan FL, Chang FTM, Liu Y, Pearson RB, Kalitsis P, Mann JR, Collas P, Hannan RD, Wong LH (2018). Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. Proc. Natl. Acad. Sci. U. S. A 201720391. doi: 10.1073/pnas.1720391115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G, Blitzblau HG, Tame MA, Falk JE, Curtin L, Hochwagen A (2011). Protection of repetitive DNA borders from self-induced meiotic instability. Nature 477, 115–119. doi: 10.1038/nature10331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt N, Lefèvre S-H, Apiou F, Dutrillaux A-M, Cör A, Leuraud P, Poupon M-F, Dutrillaux B, Debatisse M, Malfoy B (2004). Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc. Natl. Acad. Sci. U. S. A 101, 11368–11373. doi: 10.1073/pnas.0402979101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron C, Lacroute F (1975). amounts of ribosomal and Effect of Growth Rate on the Amounts of Ribosomal and Transfer Ribonucleic Acids in Yeast. J. Bacteriol 122, 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Lemos B (2017). Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic context, nucleolus activity, and proliferation. PLOS Genet. 13, e1006994. doi: 10.1371/journal.pgen.1006994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitao T, Budd M, Hoopes LLM, Campbell JL (2003). Dna2 helicase/nuclease causes replicative fork stalling and double-strand breaks in the ribosomal DNA of Saccharomyces cerevisiae. J. Biol. Chem 278, 22513–22522. doi: 10.1074/jbc.M301610200 [DOI] [PubMed] [Google Scholar]
- West C, James SA, Davey RP, Dicks J, Roberts IN (2014). Ribosomal DNA sequence heterogeneity reflects intraspecies phylogenies and predicts genome structure in two contrasting yeast species. Syst. Biol 63, 543–554. doi: 10.1093/sysbio/syu019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff DJ, Schwartz S (1992). Characterization of Robertsonian translocations by using fluorescence in situ hybridization. Am. J. Hum. Genet 50, 174–81. [PMC free article] [PubMed] [Google Scholar]
- Xu B, Li H, Perry JM, Singh VP, Unruh J, Yu Z, Zakari M, McDowell W, Li L, Gerton JL (2017). Ribosomal DNA copy number loss and sequence variation in cancer. PLOS Genet. 13, 1–25. doi: 10.1371/journal.pgen.1006771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman S, Choudhury M, Jiang JC, Srivastava P, Mohanty BK, Danielson C, Humphrey SJ, Jazwinski SM, Bastia D (2016). Mechanism of Regulation of Intrachromatid Recombination And Long Range Chromosome Interactions In Saccharomyces cerevisiae. Mol. Cell. Biol 36, MCB.01100–15. doi: 10.1128/MCB.01100-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Yeast strains used in this research, Related to all main and supplemental figures.