

Abstract
In many solid tumors, parasympathetic input is provided by the vagus nerve, which has been shown to modulate tumor growth. However, whether cholinergic signaling directly regulates progression of pancreatic cancer (PDAC) has not been defined. Here, we found that subdiaphragmatic vagotomy in LSL-Kras+/G12D;Pdx1-Cre (KC) mice accelerated PDAC development, whereas treatment with the systemic muscarinic agonist bethanechol restored the normal KC phenotype, thereby suppressing the accelerated tumorigenesis caused by vagotomy. In LSL-Kras+/G12D;LSL-Trp53+/R172H;Pdx1-Cre (KPC) mice with established PDAC, bethanechol significantly extended survival. These effects were mediated in part through the CHRM1, which inhibited downstream MAPK/EGFR and PI3K/AKT pathways in PDAC cells. Enhanced cholinergic signaling led to a suppression of the CSC compartment, CD11b+ myeloid cells, TNF-α levels, and metastatic growth in the liver. Therefore, these data suggest that cholinergic signaling directly and indirectly suppresses growth of PDAC cells, and therapies that stimulate muscarinic receptors may be useful in the treatment of PDAC.
Keywords: Pancreatic cancer, vagotomy, cholinergic signaling, muscarinic receptor, cancer stem cells
Introduction
Extensive efforts in cancer research over recent decades have led to a steady increase in survival for most cancer types, but less so for pancreatic ductal adenocarcinoma (PDAC), which continues to have the lowest 5-year survival rate for cancers at 8% (1). With few specific symptoms and no reliable test for early detection, PDAC is typically diagnosed at a locally advanced or distant stage (2). After careful staging, only 15–20% of patients are eligible for upfront resection (3). Even after potential curative resection, most patients will eventually have recurrence, and 5-year survival of completely resected patients is only 26% (3). In the metastatic setting, FOLFIRINOX and nab-paclitaxel–gemcitabine are standard treatment options in patients with good performance status and have shown benefit over previously standard gemcitabine monotherapy, but even in those highly selected patients, overall survival remains poor (3).
The resistance of PDAC to treatment has been attributed in part to the tumor microenvironment and the complex desmoplastic stroma (4), which includes immune cells, endothelial cells, stellate cells, matrix proteins, and nerves (5,6). In this regard, the nervous system is increasingly recognized as a central regulator of both normal stem cell function and cancer growth across a range of tissues (7). In a pathogenic context, neurons are emerging as a critical microenvironmental element in a variety of cancers i.e. prostate (8,9), skin (10), gastric (11–13), and pancreatic (14–17) cancers. Furthermore, growing evidence suggests that there is increased crosstalk between tumor cells and nerves, with tumors able to induce active axonogenesis (13,17–19).
In many cases, nerves appear to promote the growth of tumors, but the result of neural input is likely site-specific, influenced largely by distinct nerve-tumor interactions. The stomach in particular is regulated predominantly by the parasympathetic nervous system, with the vagus nerve strongly promoting epithelial proliferation, stem cell activity, and tumorigenesis. Thus, in the case of gastric cancer, abrogation of cholinergic input by vagotomy or chemical denervation inhibits the growth of gastric cancer (11,12). In addition to directly regulating the epithelium, nerves have been shown to act indirectly through effects on the tumor stroma or microenvironment (8,9). In mouse models of prostate cancer, for example, cholinergic signals transduced in the tumor stroma by the muscarinic type 1 receptor (CHRM1) promote tumor invasion (9).
Similar to many other solid organs, the pancreas is innervated by both sympathetic and parasympathetic nerves (20). In the pancreas, the autonomic nervous system regulates both exocrine and endocrine function, and influences normal pancreatic development (21). In addition, the vagus nerve has been shown to stimulate proliferation of the normal exocrine pancreas, such that ventromedial hypothalamic lesions that induce vagal hyperactivity stimulate pancreatic proliferation and lead to pancreatic hypertrophy, whereas vagotomy leads to decreased pancreatic acinar growth (22). On the other hand, clinical studies have suggested that vagus nerve signaling may actually slow pancreatic tumor progression (23). In this regard, a higher vagal nerve activity as expressed by a higher heart rate variability was reported to correlate significantly with lower risk of death in metastatic PDAC. These results are in line with possible vagal nerve protection in this fatal cancer (23). Furthermore, a higher incidence of PDAC was found in patients who underwent vagotomy for gastric ulcer disease in the past (24). Similarly, a study in orthotopic and syngeneic PDAC mouse models demonstrated that vagotomy promoted tumor growth and shortened overall survival, although this was largely attributed to indirect effects on tumor-associated macrophages and elevated TNF-α levels (16). In addition, tumor-bearing animals that underwent chemical or surgical vagotomy showed enhanced metastasis of breast cancer cells (25).
Nevertheless, although these earlier studies suggested that vagal signaling could be suppressive in some cancer models, a direct role for cholinergic signaling in genetically engineered mouse models (GEMM) and metastatic models of PDAC has not been demonstrated, nor has the mechanism been elucidated. Consequently, we investigated the direct contribution of the vagus nerve, cholinergic signaling and muscarinic receptors to PDAC development and progression in GEMMs (LSL-Kras+/G12D;Pdx1-Cre (KC), LSL-Kras+/G12D;LSL-Trp53+/R172H;Pdx1-Cre (KPC)), which phenocopy human PDAC (26), and metastatic models.
Results
Subdiaphragmatic Vagotomy Promotes Pancreatic Tumorigenesis.
We first investigated the specific effects of parasympathetic denervation on PDAC development. Genetically engineered KC mice underwent a subdiaphragmatic vagotomy with a subsequent pyloroplasty (KC+VxPP) at 8 weeks of age. When analyzed at 20 weeks (Supplementary Fig. 1A), KC+VxPP mice showed a similar ratio of pancreas weight to body weight (PW/BW) compared to control KC mice, which received pyloroplasty only (KC + PP) (Supplementary Fig. 1B). Analysis of mRNA expression revealed significantly increased pancreatic expression of the muscarinic type 1 receptor (Chrm1) (p < 0.05), whereas expression levels of other muscarinic receptors did not differ when compared to the control group (KC+PP) at 20 weeks (Fig. 1A). This finding was confirmed by immunofluorescent staining (IHC-F), where CHRM1 was more highly expressed in pancreatic acinar and tumor epithelial cells in KC+VxPP mice (n = 13) compared with KC+PP mice (n = 10) (Fig. 1B and C). Morphometric quantification of PanIN lesions in pancreata from KC+VxPP mice at 20 weeks revealed a significantly larger area affected by PanINs than in KC+PP mice (p < 0.01) (Fig. 1D-F). Importantly, PDAC lesions, not observed in KC+PP mice, were present in 40% of the KC+VxPP mice (Fig. 1E and G).
Figure 1. Subdiaphragmatic Vagotomy Promotes Pancreatic Tumorigenesis.
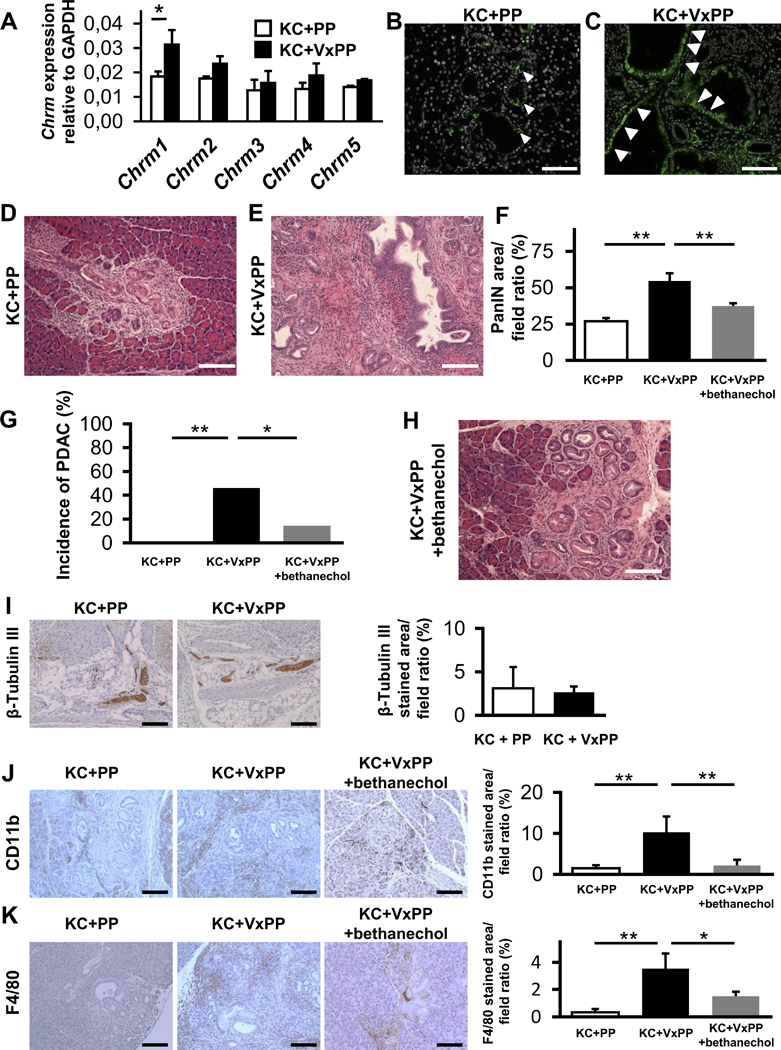
A. Relative quantification of mRNA expression of Chrm1 to Chrm5 in KC+PP mice pancreata compared to KC+VxPP mice pancreata at 20 weeks (n = 3, each group). B. Representative image of CHRM1 immunofluorescent staining (IHC-F) of pancreata from KC+PP at 20 weeks. White arrowheads indicate CHRM1 positive cells in PanIN lesions (CHRM1; green, DAPI; white). C. Representative image of CHRM1 IHC-F of pancreata from KC+VxPP at 20 weeks. White arrowheads indicate CHRM1 positive cells in PanIN lesions. D. Representative image of H&E stained pancreatic section from KC+PP mice at 20 weeks showing low grade PanIN lesions. E. Representative image of H&E stained pancreata from KC+VxPP mice at 20 weeks showing high-grade PanIN / PDAC lesions. F. Percentage of PanIN area in higher power fields in pancreata from KC+VxPP mice (n = 12), KC+PP mice (n = 10) and KC+PP+bethanechol (n = 14) at 20 weeks. G. Percentage of KC+VxPP mice (n = 13) compared to KC+PP mice (n = 10) and KC+PP+bethanechol mice (n = 14) that developed pancreatic cancer at 20 weeks. H. Representative image of H&E stained pancreata from KC+VxPP+bethanechol mice at 20 weeks showing low-grade PanIN lesions. I. Representative images of pancreatic immunohistochemical staining (IHC) for β-Tubulin III in KC+PP and KC+VxPP mice at 20 weeks (n = 4, each group). Bar graph showing quantification of β-Tubulin III stained area in pancreata from KC+PP and KC+VxPP mice. J. Representative images of pancreatic IHC for CD11b in KC+PP, KC+VxPP and KC+VxPP+bethanechol mice at 20 weeks. Bar graph showing quantification of CD11b stained area in pancreata from KC+PP, KC+VxPP and KC+VxPP+bethanechol mice (n = 3, each group). K. Representative images of pancreatic IHC for F4/80 in KC+PP, KC+VxPP and KC+VxPP+bethanechol mice at 20 weeks. Bar graph shows quantification of F4/80 stained area in pancreata from KC+PP, KC+VxPP and KC+VxPP+bethanechol mice (n = 3, each group). *p < 0.05; **p < 0.01. Means ± SD. Scale bars, 100 μm.
Given that subdiaphragmatic vagotomy appeared to accelerate PDAC development and upregulate expression of CHRM1 on epithelial cells, we sought to determine whether systemic muscarinic stimulation could rescue the regular KC phenotype and suppress PDAC development. Thus, we examined the effect of systemically administered bethanechol, a broad muscarinic agonist, on KC mice that had undergone vagotomy. Bethanechol treatment was initiated in KC mice at 8 weeks, immediately after the mice had undergone surgery (Vx+PP) and continued until 20 weeks of age (Supplementary Fig. 1C). Administration of bethanechol led to a significant reduction in PanIN area and pancreatic tumor incidence in KC+VxPP mice (n = 14) (p < 0.01 and p < 0.05, respectively) compared to untreated KC+VxPP mice (n = 13) (Fig. 1E-H).
Since CD44 expression is known to mark a subpopulation of PDAC cells that harbor a higher grade of plasticity and greater resistance to treatment (27), we investigated expression of CD44 in the pancreas of these mice. Herein, we found increased levels of CD44 in pancreata of KC+VxPP versus KC+PP mice (p < 0.05) at 20 weeks (Supplementary Fig. 1D). In contrast, bethanechol treated KC+VxPP mice showed significantly reduced pancreatic CD44 expression compared to KC+VxPP mice (Supplementary Fig. 1D).
The pancreas has multiple sources of innervation, such that subdiaphragmatic vagotomy typically would not be expected to lead to a significant reduction in overall nerve density (28). Nevertheless, we sought to confirm the efficacy of vagotomy procedure. First, we performed immunohistochemical studies of pancreatic sections with an antibody recognizing β-Tubulin III, thereby staining neuronal fibers. As expected, we found only minimal alteration in neuronal density in KC+PP versus KC+VxPP mice (Fig. 1I). However, when we analyzed the density of cholinergic fibers, which were stained with an antibody against the vesicular acetylcholine transporter (VAChT) in pancreata of Wild-type C57BL/6 (WT)+PP versus WT+VxPP mice, a significant reduction in VAChT positive structure was detected (Supplementary Fig. 1E and F).
As an effective subdiaphragmatic vagotomy should result in gastric distention through the inability of pyloric musculature relaxation (29), we measured in our study animals the gastric diameter as the distance from the mid of the lesser curvature to the greater curvature in a perpendicular way. An increase of more than 1.5-fold was used as a cutoff for indicating a successful surgical procedure, which was documented in all study animals (Supplementary Fig. 1G and H). In addition, the completeness of vagotomy was verified during postmortem inspection of vagal nerve endings using microscopic inspection.
Since the biological effects of vagotomy are complex, and thus could modulate cancer development indirectly through effects on the tumor microenvironment, we analyzed effects of vagal transection in KC+VxPP versus KC+PP mice on the stromal compartment. As demonstrated in the past (30), subdiaphragmatic vagotomy resulted in an increase in systemic and splenic levels of TNF-α in KC+VxPP mice compared the KC+PP mice (Supplementary Fig. 1I-K). Surprisingly, treatment with bethanechol resulted in suppression of TNF-α levels, both in the spleen and circulation (Supplementary Fig. 1I-K). Further analysis of the immune cell compartment showed increased levels of CD11b+ myeloid cells (Fig. 1J) and F4/80+ cells (Fig. 1K) in pancreata of KC+VxPP mice compared to KC+PP mice, consistent with greater inflammation. The increased number of immune cells was significantly suppressed when KC+VxPP mice were treated by bethanechol (Fig. 1J and K). Flow cytometric analysis of the spleen revealed differences only in F4/80+ cells (Supplementary Fig. 1L). In contrast, we found no major differences in the levels of T-(CD3) or B-(B220) lymphoid cells in the spleen (Supplementary Fig. 1M and N). Furthermore, immunohistochemical analysis of CD3+ cells, B220+ cells, myofibroblasts (α-SMA+), endocrine cells (CgA+), and endothelial cells (CD31+) showed no significant changes in these stromal lineages in the pancreas of KC+VxPP mice compared to control KC+PP mice (Supplementary Fig. 1O-S). Taken together, these data suggest that vagal denervation in the presence of an oncogenic Kras mutation promotes pancreatic tumorigenesis through an expansion of CD44+ epithelial cells and potentially through enhanced inflammation.
Muscarinic Stimulation Suppresses Pancreatic Tumorigenesis and Extends Overall Survival in KPC Mice
Next, we investigated the role of cholinergic signaling in established PDAC, using the KPC mouse model, which develops frank PDAC at a median age of 17–19 weeks (31). KPC mice with pancreatic tumors that measured 3–5 mm in diameter on ultrasound were randomized to treatment with gemcitabine (GEM) alone, or GEM+bethanechol (Fig. 2A). Treatment with GEM+bethanechol extended the overall survival of KPC mice treated with GEM alone from 29 to 48 days (p < 0.001) (Fig. 2B). Pancreatic tumor sections stained by H&E at the time of necropsy did not reveal obvious differences in histology between the two groups (Fig. 2C and D). However, treatment with GEM+bethanechol led to a significant reduction in pancreatic CD44 expression compared to GEM alone (p < 0.05) (Fig. 2E).
Figure 2. Muscarinic Stimulation Suppresses Pancreatic Tumorigenesis and Extends Overall Survival in KPC Mice.
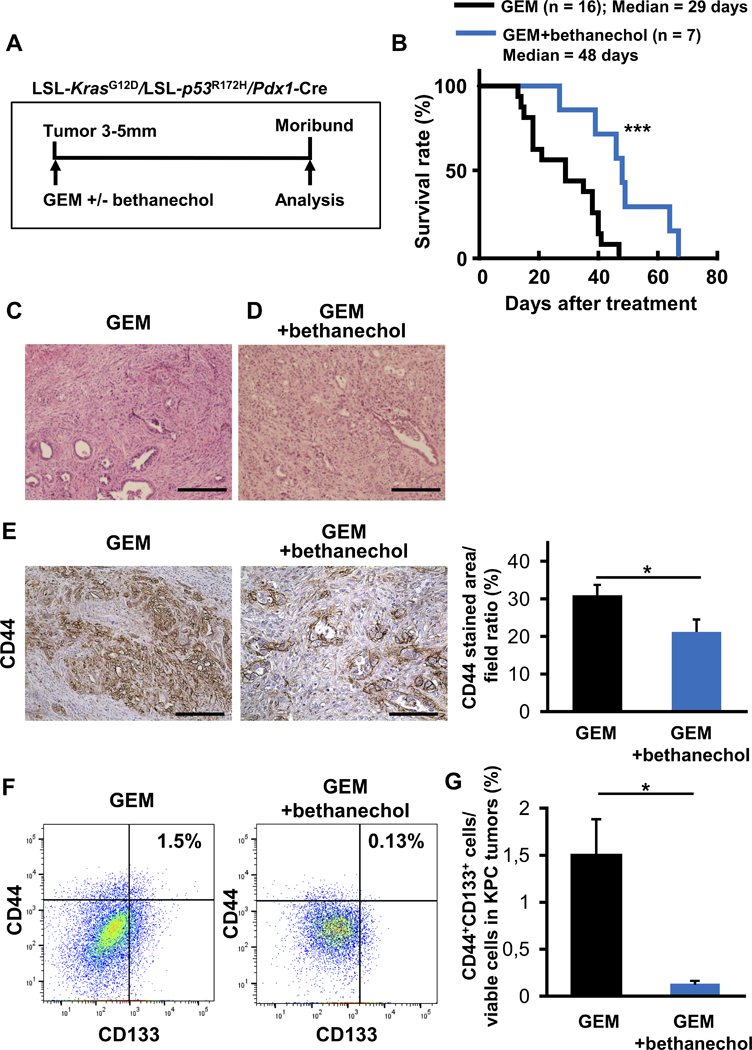
A. Experimental setup: KPC mice were enrolled with tumors of 3–5 mm, confirmed by high resolution ultrasound, and treated with gemcitabine (GEM) (100 mg/kg) biweekly (n = 15) or GEM + bethanechol in the drinking water (400 μg/ml) (n = 10) until they became moribund and needed to be sacrificed. B. Kaplan-Meier curve comparing overall survival of KPC mice treated with GEM (n = 15) or GEM+bethanechol (n=10) after initiation of the respective therapy. C. Representative images of H&E stained pancreatic ductal adenocarcinoma (PDAC) from GEM treated KPC mice. D. Representative images of H&E stained PDAC from GEM+bethanechol treated KPC mice. E. Representative images of IHC for CD44 in tumors from KPC mice treated by GEM or GEM+bethanechol. Bar graph showing quantification of the area stained positive for CD44 in PDAC from KPC mice treated with GEM (n = 15) or GEM + bethanechol (n = 10). F. Representative flow cytometric plot of CD44+CD133+ cells in PDAC from KPC mice treated with GEM or GEM + bethanechol. Numbers are showing ratio of CD44+CD133+ cells to viable cells. G. Bar graph shows quantification of CD44+CD133+ cells in PDAC from KPC mice treated with GEM (n = 3) or GEM + bethanechol (n = 4). Scale bars, 100 μm. Means ± SE in Fig. 2E and Means ± SD in Fig. 2G. *p < 0.05; ***p < 0.001.
To further characterize this CD44+ population, we examined the expression of markers used to isolate cancer stem cells (CSC) in human tumors (32), as specific CSC markers for murine PDAC have been less well validated. In this regard, the CD44+CD133+ population was described earlier as putative murine CSCs (33,34). Intriguingly, this double positive population was significantly reduced in the GEM+bethanechol group compared to control (p < 0.05) (Fig. 2F and G). In addition, analysis of the CD44+CD24+EpCAM+ population revealed also a trend toward suppression of the CD44+CD24+EpCAM+ population in tumors of KPC mice treated with GEM+bethanechol versus GEM only treatment in KPC mice (Supplementary Fig. 2A and B). Taken together, these data suggest that muscarinic agonists can suppress pancreatic tumorigenesis in part by suppression of the CSC compartment.
Cholinergic Signaling Directly Promotes Cell Proliferation in Kras Mutant Spheres via CHRM1 and Regulates Cancer Stemness.
To determine whether cholinergic agonists suppress tumor development in part through direct stimulation of muscarinic receptors on pancreatic epithelial cells, we assessed the sphere forming capacity of KrasG12D pancreatic acinar cells in an established 3D Matrigel culture system. These cultures are known to mimic acinar to ductal metaplasia (ADM) (35), the proposed first step of PDAC tumorigenesis (36). KrasG12D mutant spheres were generated from LSL-Kras+/G12D mice by delivery of adenoviral-Cre (Adeno-Cre) and then treated with the non-selective muscarinic agonist pilocarpine, the non-selective muscarinic antagonist scopolamine, the CHRM1-selective agonist McN-34A, or the CHRM1-selective antagonist pirenzepine. Pilocarpine- or McN-34A- treated Kras mutant acinar cell cultures formed significantly fewer and smaller spheres, while acinar cultures treated with scopolamine or pirenzepine formed significantly more and larger spheres (Fig. 3A-J). In addition, we studied the effects of muscarinic agonism in 3D primary human PDAC organoids (37). After 6 days of treatment with increasing doses of pilocarpine (6.25 – 400 μM), we observed fewer viable spheres in these cultures in a dose dependent manner (Fig. 3K-M).
Figure 3. Cholinergic Signaling Directly Promotes Cell Proliferation in Kras Mutant Spheres via CHRM1 and Regulates Cancer Stemness.
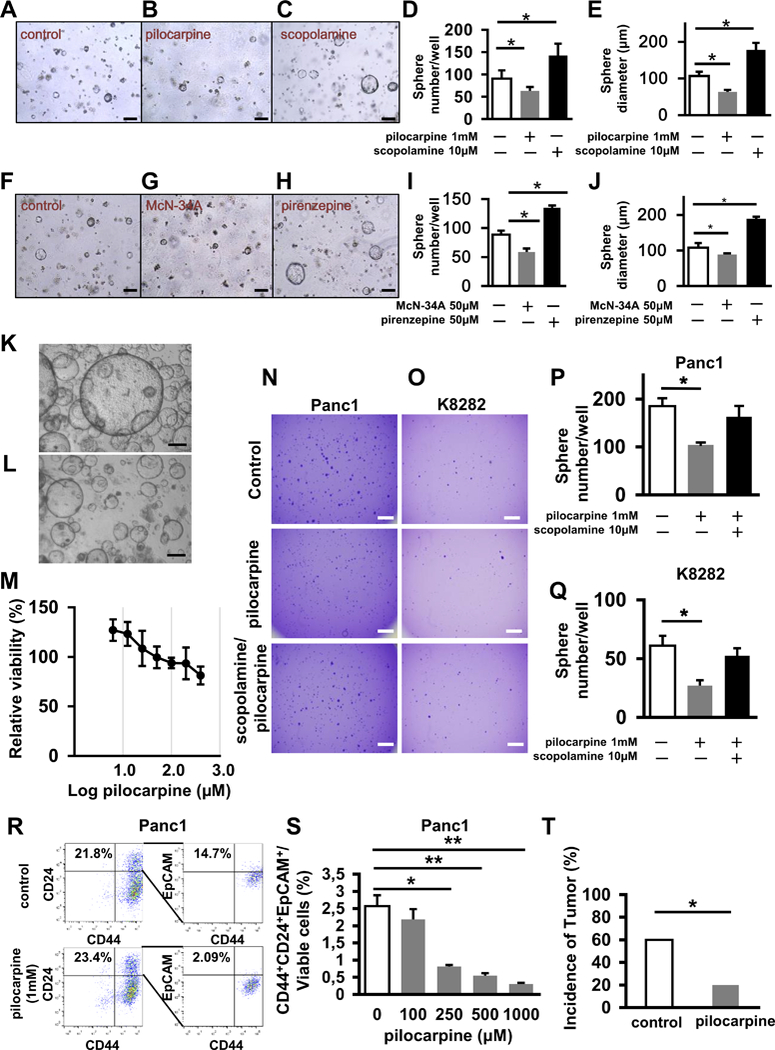
A-C. Representative images of pancreatic spheres isolated from LSL-Kras+/G12D mice and treated with an Adeno-Cre virus (A) or an Adeno-Cre virus and pilocarpine (B) or an Adeno-Cre virus and scopolamine (C). D and E. Number of spheres per well (D) and size of spheres (E) isolated from LSL-Kras+/G12D mice and cultured in the presence of an Adeno-Cre virus, which are untreated, treated with pilocarpine, or treated with scopolamine (n = 3, each group). F-H. Representative images of spheres isolated from LSL-Kras+/G12D mice and treated with an Adeno-Cre virus (F) or an Adeno-Cre virus and McN-34A (G) or Adeno-Cre virus and pirenzepine (H). I and J. Number of spheres per well (I) and size of spheres (J) isolated from LSL-Kras+/G12D mice and cultured in the presence of Adeno-Cre virus, which are untreated, treated with McN-34A, or treated with pirenzepine (n = 3, each group). K. Representative image of organoids generated from primary resected human PDAC specimen without treatment L. Representative image of organoids generated from primary resected human PDAC specimen treated with 100 μM of pilocarpine. M. Graph showing dose-dependent decrease in viability of organoids generated from primary resected human PDAC specimen on pilocarpine treatment. N. Representative images of spheres in soft agar from Panc1 cells that are untreated, treated by pilocarpine or treated by scopolamine and pilocarpine at day 14. O. Representative images of spheres in soft agar from K8282 cells that are untreated, treated by pilocarpine or treated by scopolamine and pilocarpine at day 14. P. Bar graph shows quantification of numbers of spheres from Panc1 cells plated in soft agar which are untreated, treated by pilocarpine or treated by scopolamine and pilocarpine at day 14 (n = 3, each group). Q. Bar graph showing quantification of numbers of spheres from K8282 cells plated in soft agar, which are untreated, treated by pilocarpine or treated by scopolamine and pilocarpine at day 14 (n = 3, each group). R. Flow cytometric analysis of CD44+CD24+EpCAM+ cells in Panc1 cells that are untreated, treated with pilocarpine. Numbers are showing the ratio of indicated cells to total cell number on the graphs. S. Bar graph shows quantification of CD44+CD24+EpCAM+ cells in Panc1 cells treated with different dosages of pilocarpine (n = 3). T. Percentage of NOD/SCID mice developing tumors 6 weeks after injection of 25,000 injected Panc1 cells with and without pretreatment with pilocarpine (n = 10, each group). Scale bars, 100 μm in Fig A-C and F-H, 500 μm in Fig K, L, N and O. Means ± SD in Fig D, E, I, J, M and T, Means ± SEM in Fig P, Q and S. *p < 0.05; **p < 0.01.
Analysis of muscarinic receptor expression in human (Panc1 and MiaPaca2) and murine (K8282 and Panc02) PDAC cell lines revealed higher CHRM1 and Chrm1 expression after scopolamine treatment, respectively, and lower CHRM1 and Chrm1 expression with pilocarpine treatment (Supplementary Fig. 3A-D) compared to control. In addition to Chrm1 expression, Chrm3 and Chrm4 were highly expressed in human pancreatic cancer cell lines, although they did not show significant changes with drug treatment. Moreover, MTT assays demonstrated that pilocarpine decreased cell viability of a human (Panc1) cancer cell line in a dose dependent manner (Supplementary Fig. 3E). Muscarinic agonists decreased cell viability of human (Panc1) and murine (K8282) cancer cell lines, while muscarinic antagonists (scopolamine and pirenzepine) had the opposite effect (Supplementary Fig. 3F and G). These results fueled the hypothesis that muscarinic receptors and CHRM1 in particular were largely responsible for modulating the anti-proliferative effects of muscarinic agonists. Furthermore, we also studied the effects of muscarinic agonism on non-tumorigenic, HPV-16E6E7 immortalized human pancreatic ductal epithelial (HPDE‐E6E7) cells (38). Interestingly, pilocarpine in doses up to 100 μM had no effect on the viability of these cells. In contrast the expression of mutant KrasG12D in HPDE-E6E7 cells by retroviral transduction resulted in a significant reduction in viability at doses as low as 100 μM of pilocarpine (Supplementary Fig. 3H). Similarly, we observed that pilocarpine suppresses the growth of Kras mutant pancreatic spheroids, but stimulates the growth of WT pancreatic spheroids. This study was performed using spheres that were generated from LSL-KrasG12D pancreata recombined by Adeno-Cre recombinase (Kras mutant) or without treatment of Adeno-Cre (Kras WT) (Supplementary Fig. 3I and J).
To further address the potential effects of muscarinic agonists on the CSC compartment, we studied the effects on a human pancreatic cancer cell line (Panc1) and a murine cancer cell line (K8282) in anchorage independent soft agar cultures. These experiments revealed reduced sphere forming capability after pilocarpine treatment, suggesting suppression of the CSC compartment in human and murine cancer cell lines (Fig. 3N-Q). To elaborate this observation further, we evaluated the CD44+CD133+ population or the CD44+CD24+EpCAM+ population in human (Panc1) and murine (K8282 and K2584) PDAC cell lines by flow cytometry after pilocarpine treatment. Administration of pilocarpine resulted in a dose dependent suppression of these population in the cell lines investigated (Fig. 3R and S; Supplementary Fig. 3K-N). Moreover, Panc1 cells were treated with pilocarpine for 72 hours, after which 25,000 cells were implanted subcutaneously into NOD/SCID mice. Pre-treatment with pilocarpine resulted in a significant reduction in tumor incidence (Fig. 3T) and tumor volume compared to untreated Panc1 cells (Supplementary Fig. 3O-S) (p < 0.05). Overall, these findings demonstrate that treatment with muscarinic agonists lead to an overall reduction in pancreatic cancer stem cells.
Muscarinic Signaling Inhibits Downstream EGFR/MAPK and PI3K/AKT Signaling in PDAC Cells.
To determine potential mechanisms by which muscarinic agonists are able to suppress progression of PDAC, we analyzed Panc1 cells following pilocarpine treatment using RNAseq (Supplementary Fig. 4A-C). Growth related genes that were suppressed by pilocarpine included EGFR and PI3K (Supplementary Fig. 4A). A more detailed analysis of differentially expressed genes in terms of KEGG pathways using iPathwayGuide suggested a possible mechanism of tumor suppression through the PI3K-AKT and MAPK pathways (Supplementary Fig. 4B and C). The RNAseq data are consistent with inhibition by pilocarpine of the PI3K-AKT pathway (Supplementary Fig. 4B) (39). PIK3CA (PI3K) is activated by EGFR, KDR (RTK) and ITGAV (ITGA), all of which were downregulated by pilocarpine, so that PIK3CA (PI3K) is inhibited at the signaling level as well. PIK3CA (PI3K) is an activator of AKT, so that inhibition of PIK3CA (PI3K) leads to inhibition of AKT, thus reducing proliferative signals. In the classical MAPK signaling pathway (Supplementary Fig. 4C), NTRK2 (TRKB) and BRAF (RafB), which are also pro-proliferative (40), were also downregulated according to the RNAseq results.
To test key aspects of the above model, we examined the expression of phosphorylated forms of EGFR, PI3K, and ERK1/2 (p-EGFR, p-PI3K, and p-ERK1/2) by immunohistochemical staining (IHC) in pancreatic sections from KC+PP, KC+VxPP, and KC+VxPP+bethanechol mice. Expression of p-EGFR, p-PI3K, and p-ERK1/2 was significantly higher in pancreata of KC+VxPP than in KC+PP mice (p < 0.05) and suppressed to similar levels as the control (KC+PP) when KC+VxPP mice were treated with bethanechol (KC+VxPP+bethanechol) (p < 0.05) (Fig. 4A-C). Furthermore, in pancreata of KPC mice treated with bethanechol, expression of these phosphoproteins was also significantly downregulated by IHC compared with untreated KPC mice (Supplementary Fig. 4D-F).
Figure 4. Muscarinic Signaling Inhibits Downstream EGFR/MAPK and PI3K/AKT Signaling in PDAC Cells.
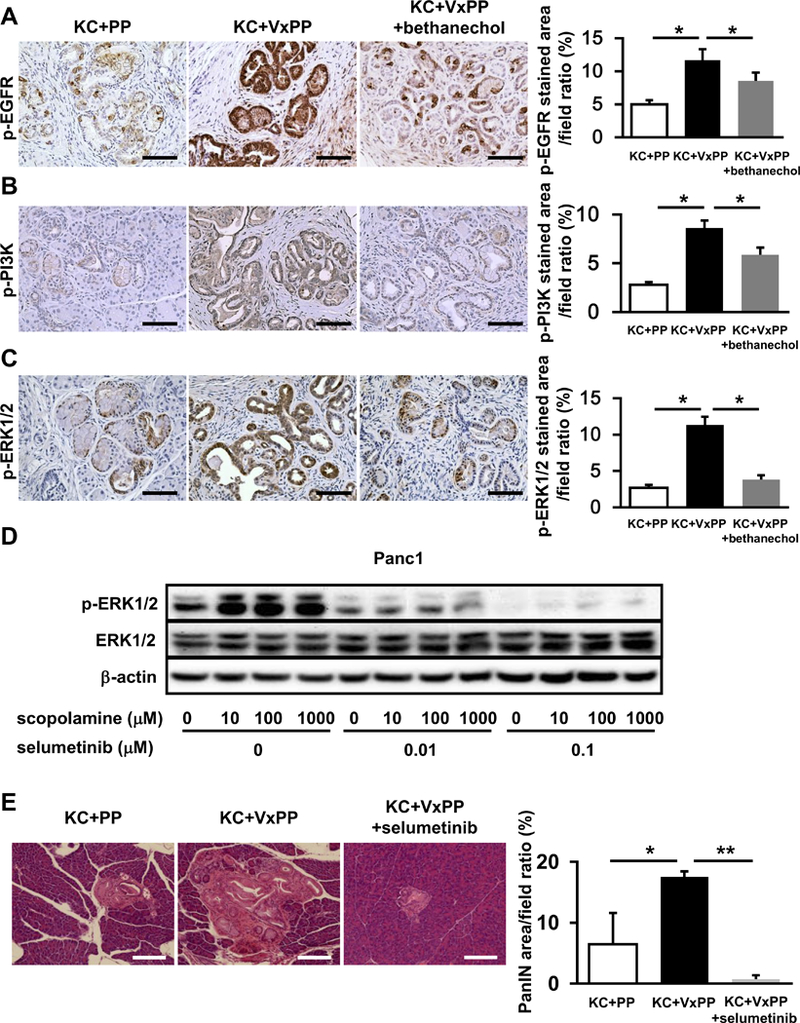
A-C. Representative images IHC for p-EGFR (A), p-PI3K (B), and p-ERK1/2 (C) in pancreatic sections from KC + PP mice, KC + VxPP mice, and KC + VxPP + bethanechol mice. Bar graphs show quantification of p-EGFR, p-PI3K or p-ERK1/2 staining in pancreata from KC + PP mice, KC + VxPP mice, and KC + VxPP + bethanechol mice (n = 3, each group). D. Representative western blot showing p-ERK1/2 and ERK1/2 relative to β-actin in Panc1 cells after treatment with indicated dosages of scopolamine and selumetinib. E. Representative H&E stained images of pancreatic sections from KC + PP mice, KC + VxPP mice, and KC + VxPP + selumetinib mice. Bar graph shows quantification of PanIN area in pancreata from KC + PP mice, KC + VxPP mice, and KC + VxPP + selumetinib mice (n = 3, each group). Scale bars, 100 μm, Means ± SD. *p < 0.05; **p < 0.01.
Next, we used a phospho-kinase array and found significant suppression of phosphorylation of AKT at S473 and T308 after treatment of Panc1 cells with 100 μM of pilocarpine (Supplementary Fig. 4G and H). To further characterize downstream signaling pathways modulated in PDAC cells in response to parasympathetic signaling in vitro, we performed western blots for key signaling proteins in human or murine PDAC cells treated with pilocarpine, scopolamine, McN-34A, or pirenzepine. First, treatment of Panc1 cells with increasing concentrations of pilocarpine resulted in a reduction of phosphorylation of EGFR and ERK1/2 in a dose dependent manner (Supplementary Fig. 4I). EGFR, BRAF, ERK1/2, PI3K, and AKT kinases were also less phosphorylated in murine (K8282) or human (Panc1) PDAC cells in response to the selective CHRM1 agonist McN-34A (Supplementary Fig. 4J-U). In contrast, antagonism with scopolamine or pirenzepine led to significantly more phosphorylation of these signaling molecules. However, following treatment with either a pan-muscarinic antagonist or a selective CHRM1 antagonist, the addition of either a pan-muscarinic agonist or a selective CHRM1 agonist completely abrogated the increases in these phosphorylated kinases (Supplementary Fig. 4 J-U).
In order to further elaborate whether cholinergic signaling directly regulates EGFR/MAPK pathway, we treated Panc1 cells with scopolamine, and then with the MEK inhibitor selumetinib (AZD6244). These studies revealed that the MEK inhibitor abolished the increase in ERK phosphorylation seen in response to scopolamine (Fig. 4D). To assess the biological significance of this observation in vivo, we treated also KC+VxPP mice with selumetinib (KC+VxPP+selumetinib). We found that administration of the MEK inhibitor reduced significantly PanIN progression that could be attributed to vagotomy (KC+VxPP) (Fig. 4E). Taken together, muscarinic receptors appear to suppress signaling through MAPK and other downstream pathways that likely contribute to pancreatic tumorigenesis.
Knockout of CHRM1 Results in Larger PanIN Area and Tumor Incidence in KC Mice and Shorter Overall Survival in KPC Mice.
Given that unselective and selective CHRM1 agonists and antagonists modulated pancreatic tumorigenesis most likely via MAPK and PI3K-AKT signaling, we sought to confirm the role of CHRM1 in pancreatic tumor development by reducing receptor expression through genetic deletion. Therefore, we crossed KC mice to Chrm1-KO mice, and generated KC/Chrm1-KO (KCM) mice (n = 11). We confirmed the absence of Chrm1 expression in KCM mice by qRT-PCR, and protein expression by IHC (Fig. 5A-C). Similar to vagotomized KC mice, KCM mice showed more advanced PanINs, including PDAC, which was absent in KC mice (Fig. 5D-G). Morphometric analysis revealed a significantly larger PanIN area in KCM mice compared with KC mice (n = 10) (p < 0.01) (Fig. 5H). Moreover, the PDAC incidence in KCM mice was significantly higher compared with control KC mice (p < 0.05, 36.7% vs. 0%) (Fig. 5I). Furthermore, IHC revealed significantly increased expression of CD44 (Fig. 5J), p-EGFR (Fig. 5K), p-PI3K (Fig. 5L), and p-ERK1/2 (Fig. 5M) in KCM mice compared with KC mice (p < 0.05, respectively) (Fig. 5N).
Figure 5. Knockout of CHRM1 Results in Larger PanIN Area and Tumor Incidence in KC Mice and Shorter Overall Survival in KPC Mice.
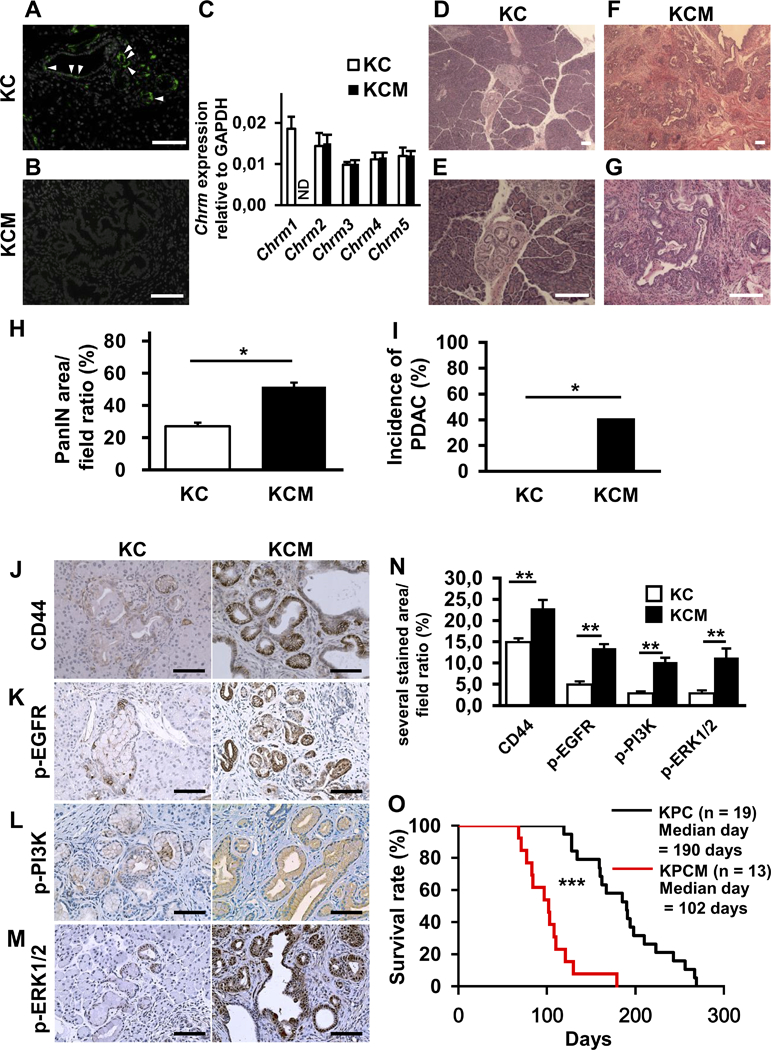
A and B. Representative images of CHRM1 immunofluorescence in pancreatic sections from KC (A) and KCM (B) mice at 20 weeks (CHRM1; green, DAPI; white). C. Relative quantification of mRNA expression of Chrm1–5 in KC compared to KCM mice at 20 weeks (n = 3, each group). D and E. Representative images of H&E stained pancreatic sections from KC mice at 20 weeks showing low-grade PanIN lesions in low (D) and high (E) power magnification. F and G. Representative images of H&E stained pancreatic sections from KCM mice at 20 weeks showing high-grade PanIN lesions/PDAC in low (F) and high (G) power magnification. H. Quantification of PanIN area in pancreatic sections from KC and KCM mice at 20 weeks (n = 5, each group) I. Percentage of KC and KCM mice with PDAC development at 20 weeks (n = 10, each group). J-M. Representative images of pancreatic IHC for CD44 (J), p-EGFR (K), p-PI3K (L), and p-ERK1/2 (M) in KC and KCM mice at 20 weeks. N. Bar graph shows quantification of CD44, p-EGFR, p-PI3K, and p-ERK staining in pancreata from KC and KCM mice at 20 weeks (n = 3, each group). O. Kaplan-Meier curve comparing overall survival of KPC (n = 19) and KPCM mice (n = 13). Scale bars, 100 μm, Means ± SD. *p < 0.05; **p < 0.01, ***p < 0.001.
Next, we assessed the sphere forming capacity of LSL-Kras+/G12D mutant spheres versus LSL-Kras+/G12D;Chrm1-KO mutant spheres in a 3D Matrigel culture system. The Kras mutation was activated in pancreatic spheres by infection with Adenoviral-Cre. As expected, the LSL-Kras+/G12D;Chrm1-KO sphere cultures generated more spheres compared with KrasG12D control spheres. Consistent with an absence of CHRM1 signaling, after pilocarpine or scopolamine treatment, LSL-Kras+/G12D;Chrm1-KO spheres showed a similar size and number compared to the untreated controls, whereas KrasG12D spheres showed a significant decrease or increase, respectively (Supplementary Fig. 5A-D).
Finally, we crossed KPC mice to Chrm1-KO mice, generating KPC/Chrm1-KO (KPCM) mice (n = 13). The median overall survival in KPCM mice was significantly decreased compared to KPC mice (n = 18) (p < 0.001) (Fig. 5O). Furthermore, IHC showed significantly increased expression of CD44, p-EGFR, p-PI3K, and p-ERK1/2 in tumors of KPCM compared to KPC mice (p < 0.05, respectively) (Supplementary Fig. 5E-H). Thus, these data suggest that an absence of CHRM1 signaling leads to enhanced EGFR/MAPK and PI3K/AKT signaling, increased CD44 expression, and accelerated PDAC progression.
Parasympathetic Signaling Influences Survival in a Model of Hepatic Metastasis.
Our studies in KC and KPC mice suggest that signaling through CHRM1 inhibits growth of primary pancreatic tumors through downregulation of growth factor pathways. However, given that many PDAC patients eventually die with liver metastases, we examined the effect of parasympathetic signaling on the growth of hepatic lesions, utilizing a well-established syngeneic model of metastatic PDAC (41). The surgical procedures used in this study are shown in Fig. 6A and Supplementary Fig. 6A-D. At 30 days after injection of murine PDAC cells (Panc02) into the spleen, cancer cells appear as large pale nodules replacing macroscopically normal liver tissue (Fig. 6B-D). To increase the detection of small metastases, Panc02 cells were stably transfected with a GFP-expressing construct, which was readily detected following splenic injection (Supplementary Fig. 6E and F). WT C57BL/6 mice received splenic injections of 2×106 GFP-labeled Panc02 cells and were then divided into 3 groups: untreated controls (n = 10), bethanechol treated (n = 10), and selective parasympathetic liver denervation by transection of the hepatic branch of the vagus (n = 11). Mice treated with bethanechol showed significantly longer survival (p < 0.001), whereas the group pretreated with selective hepatic vagotomy (SHVx) showed significantly shorter survival (p < 0.01) (Fig. 6E). As shown by quantification of number of metastases and maximum volume of metastases (Fig. 6F and G), metastatic growth was expanded by SHVx (p < 0.05), while growth was inhibited by bethanechol (p < 0.05). Moreover, to confirm that the parasympathetic pathway directly regulated tumor cells at the liver metastatic site, expression of Ki-67 (Fig. 6H), p-EGFR (Fig. 6I) and CD44 (Fig. 6J) were analyzed immunohistochemically. As expected, quantitative scoring indicated that their expression was significantly decreased in the bethanechol group (p < 0.05), and significantly increased in the selective vagotomy group (p < 0.05). Therefore, these data suggest that parasympathetic signaling suppresses the growth of not only the primary tumor, but also inhibits the growth of metastatic hepatic lesions.
Figure 6. Parasympathetic Signaling Influences Survival in a Model of Hepatic Metastasis.
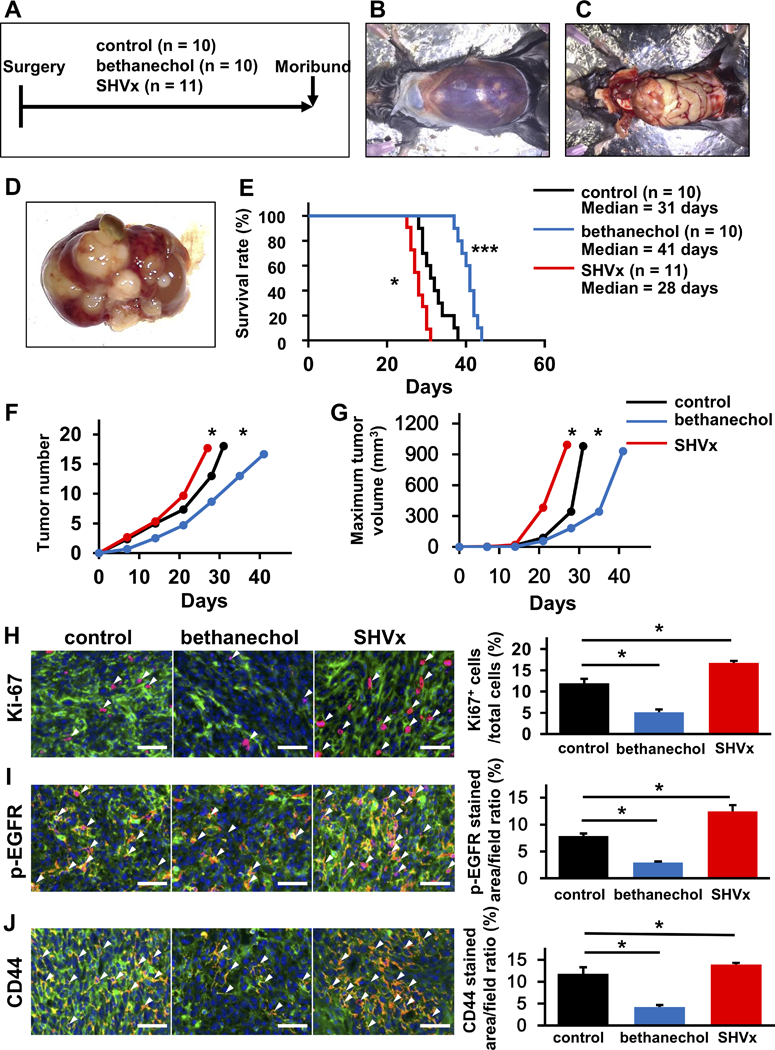
A. Experimental setup for the studies depicted in A-J. Wild-type C57BL/6 mice received splenic injections of 2×106 GFP-labelled Panc02 cells and were then divided into 3 groups: untreated controls (n = 10), bethanechol-treated (n = 10), and selective hepatic vagotomy by transection of the hepatic branch of the vagus nerve (SHVx) (n =11). Mice were observed until they became moribund and needed to be sacrificed. B. Representative images at the time of necropsy. Mouse shows massive bloody ascites. C and D. Representative images showing cancer cells replacing the normal liver tissue as large pale nodules. E. Kaplan-Meier curve comparing overall survival after splenic injection of GFP-labeled Panc02 cells in control mice (black), mice with SHVx (red) or mice treated with bethanechol (blue). F. Tumor number at necropsy in mice that received SHVx (red) and bethanechol (blue), compared to untreated control (black). G. Tumor volume in mice that received SHVx (red) and bethanechol (blue) compared to untreated control (black). H-J. Representative images of immunofluorescent staining of liver metastases from untreated control, bethanechol-treated and SHVx mice for Ki-67 (Ki-67; red) (H), p-EGFR (p-EGFR; red) (I), and CD44 (CD44; red) (J). GFP; green, DAPI; blue, white arrowheads indicate red-positive cells). Bar Graphs show quantitative analysis of positive cells for each staining in untreated control, bethanechol-treated, and SHVx mice (n = 3, each group). Scale bars, 100 μm. Means ± SD. *p < 0.05; *** p < 0.001.
Discussion
Accumulating evidence has revealed a key role for the autonomic nervous system in the development of cancer (7,42). Here, we have shown that subdiaphragmatic vagotomy accelerates PDAC progression in KC mice. Importantly, systemic administration of a broad muscarinic agonist (bethanechol) rescued the regular KC phenotype (i.e. early PanINs) in vagotomized KC mice, and significantly extended the survival of KPC mice with established PDAC. In vivo studies revealed that bethanechol suppressed the CD44+CD133+ and CD44+CD24+EpCAM+ putative CSC populations and increased levels of circulating TNF-α levels along with CD11b+ myeloid cells within the pancreas. In vitro studies confirmed that muscarinic agonists showed antiproliferative effects on Kras mutant pancreatic spheres and suppressed the CD44+CD133+ and CD44+CD24+EpCAM+ cell fraction in PDAC cell lines. We identified CHRM1 as the receptor largely responsible for cholinergic suppressive effects in mice. Further analysis of downstream signaling pathways indicated that CHRM1 signaling is able to suppress the EGFR/MAPK/PI3K/AKT pathway. Finally, we found that bethanechol treatment alone was sufficient to extend the survival of mice with hepatic metastases. Taken together, these data reveal that activation of muscarinic signaling via CHRM1 can directly and indirectly suppress the initiation and progression of PDAC (Supplementary Fig. 6G).
The nervous system in theory has the potential to influence the progression of cancer indirectly by modulating the immune system, tumor metabolism, angiogenesis, and tumor-stroma cross-talk, or through direct effects on tumor cells (9,12,17). In prostate cancer, both sympathetic and parasympathetic nerves are significantly involved in promoting all phases of murine cancer development (8,9). In gastric cancer, cholinergic signaling directly supports stem cell growth directly via the M3 muscarinic receptor (CHRM3) through Wnt signaling, and denervation of the mouse stomach markedly reduces tumor incidence and progression of gastric cancer (11).
The current study demonstrates that vagal nerve signaling can have an opposite and thus inhibitory effect on tumorigenesis in the Kras mutated pancreas. In KC, KPC and metastatic mouse models, pancreatic cancer development was accelerated by vagotomy and inhibited by cholinergic muscarinic agonists. Herein, the vagus nerve acts to directly suppress tumorigenesis in part through inhibition of CD11b+ myeloid cells, suppression of cancer stem cells and downregulation of the MAPK pathway (Supplementary Fig. 6G). These observations are consistent with clinical findings suggesting a protective role of the vagus nerve in PDAC and specifically in the metastatic stage (23).
Interestingly, a retrospective cohort study comprising 27,930 PDAC patients revealed that body/tail tumors have a much better prognosis after surgical resection than pancreatic head tumors (43). It is tempting to speculate that the transection of the vagus associated with pancreatic head (but not tail) resections may partly explains these findings.
The innervation of the pancreas is indeed quite complex, as it contains nerves derived from spinal cord as well as entero-pancreatic nerves from the myenteric plexus of the pyloric stomach and the duodenum (44,45). In addition to sympathetic and parasympathetic nerves, the pancreas contains abundant sensory nerves. Previous reports have noted that up to 80% of vagal fibers are sensory (46), suggesting that vagotomy may also reduce pancreatic input from sensory neurons. Nevertheless, given the strong evidence that sensory neurons in general strongly promote pancreatic tumorigenesis (15), this argues even more for a distinct suppressive effect by the vagal cholinergic fibers, which comprise only a minor subset of nerve fibers in the vagus.
Previous studies have established the ability of parasympathetic signaling to modulate TNF-α expression in the spleen as part of the neural inflammatory reflex (47). Recently, we extended this paradigm by showing that the neural inflammatory reflex inhibits colon cancer development in part through arresting myeloid cell expansion (48). In addition, studies using orthotopic models of pancreatic cancer have suggested that interruption of this pathway by surgical vagotomy promotes cancer growth, possibly due to upregulation of TNF-α (16). However, while cholinergic signaling mediated by the vagus nerve suppresses inflammatory responses, it is thought to do so through binding of acetylcholine to the α7 subunit of the nicotinic acetylcholine receptor (α7 nAChR). Thus, nicotinic and not muscarinic receptors were shown to mediate the vagal inhibitory effect on macrophage activation and TNF-α synthesis in cancer and sepsis (47,49).
In this study, we confirmed that subdiaphragmatic vagotomy in KC mice leads to increased TNF-α levels in the spleen and circulation, demonstrating the effectiveness of the surgical procedure. In addition, analysis of pancreatic stroma in KC+VxPP mice revealed increased levels of CD11b+ myeloid cells, along with increased levels of TNF-α in the spleen and circulation, consistent with greater inflammation. Surprisingly, treatment with a muscarinic agonist led to significant suppression of TNF-α levels in the spleen and circulation, indicating a role for muscarinic receptors in this setting. Indeed, muscarinic receptors are expressed in many cell types, including cells of the central nervous system, diverse immune (myeloid and lymphoid) cells, and other stromal cells (50). It has to be stated, that muscarinic agonists clearly had direct effects on pancreatic epithelial cells, suppressing signaling and growth in both PDAC cell lines and Kras mutant spheres. However, we cannot exclude a role for suppression of inflammation by muscarinic agonists in the inhibition of PDAC development, which will need to be elucidated in future studies.
Interestingly, pilocarpine had no effect on the viability of non-tumorigenic HPDE cells with wild type Kras, while in contrast, pilocarpine treatment of HPDE cells expressing mutant KrasG12D resulted in a significant reduction in viability. Furthermore, pilocarpine also suppressed the growth of Kras mutant pancreatic spheroids, but stimulated the growth of WT pancreatic spheroids. These data suggest that Kras mutation changes the effect of transmitted signals from muscarinic receptors from a stimulatory to an inhibitory pathway. This potential mechanism requires further study, including the potential impact of mutant Kras signaling on the allosteric modulation on receptor regulatory mechanisms such as β-arrestin recruitment, receptor internalization or endocytic trafficking. However, it is tempting to speculate that previously described distinct binding modes of β-arrestin to CHRM1 (transient or stable) (51), which can induce opposite effects on ERK activation, might explain in part the suppression of signaling by muscarinic receptors.
CD44 expression was markedly increased in vagotomized KC mice and suppressed by bethanechol to similar levels as in controls. Beyond serving as a marker of cancer stem cells in pancreatic cancer, CD44 expression level correlates with cell plasticity and invasiveness in PDAC (27). As CSC markers in murine PDAC are less validated we analyzed double (CD44+CD133+) and triple (CD44+CD24+EpCAM+) positive putative cancer stem cells in KPC tumors as well as murine and human PDAC cell lines. We found significant decreases in CD44+CD133+ CSCs in KPC tumors treated with a muscarinic agonist with a trend of suppressed CD44+CD24+EpCAM+ cells possibly owing to an insufficient number of animals. Importantly, in murine and in human PDAC cell lines the triple positive population was significantly decreased even with doses as low as 100 μM of pilocarpine. Interestingly, EGFR signaling, which was suppressed by muscarinic agonists, contributes to the acquisition of cancer stem-like properties, including the enrichment of the CD44+ population of cancer cells in breast cancer (52). CD44 also plays an important role in tumorigenesis and tumor progression by promoting cell proliferation and migration via several signaling pathways/networks, including p-AKT or p-ERK (53,54).
Multiple lines of evidence indicated that the suppression of pancreatic tumorigenesis by muscarinic signaling was mediated by CHRM1. Only Chrm1 expression was upregulated in the murine pancreas following surgical denervation, and the growth of pancreas spheres and PDAC cell lines was suppressed by CHRM1 selective agonists (McN-34A) and stimulated by CHRM1 selective antagonist (pirenzepine). Finally, genetic ablation of Chrm1 resulted in the same accelerated PDAC phenotype as seen with subdiaphragmatic vagotomy. Interestingly, CHRM1 is also expressed at high levels in the healthy prostate gland of mice, but in prostate cancer, Chrm1-KO reduced prostate cancer progression in an orthotopic mouse model (8), while Chrm1-KO accelerated PDAC. CHRM1 is a G protein coupled receptor (GPCR), associated with both Gq- and arrestin-dependent pathways, and has been shown to enhance EGFR and ERK activation and stimulate cell proliferation (55). Indeed, most GPCRs have generally been considered to be pro-tumorigenic, overexpressed or activated to drive cancer progression.
Nevertheless, it is also clear that some GPCRs can play tumor suppressive roles, including the melanocortin 1 receptor (MC1R) (56), GPRC5A (57), and the cannabinoid receptors CB1 and CB2 (58). CB2, for example, suppresses EGFR/ERK and AKT signaling in breast cancer cells, although the mechanisms have not been well defined (58). Possible mechanisms could include modulation of receptor cross-talk through heterodimerization, and altered b-arrestin interactions. Furthermore, it is known that GPCRs possess two different conformations, active and inactive, and they spontaneously alternate between the two in the absence of ligands (59), possibly explaining the increased sphere forming capacity of Chrm1-KO;LSL-Kras+/G12D sphere cultures compared to LSL-Kras+/G12D spheres. In addition, it is important to note that roles of other potential inhibitory muscarinic receptors have not been addressed in this study. Notably, we have shown that CHRM4 is the predominant receptor expressed in human pancreatic cancer cell lines (Supplementary Fig. 3A and B). Further studies are needed to elucidate the roles of the other muscarinic receptors in regulating pancreatic cancer growth. In any case, our findings that CHRM1 signaling has the potential to suppress numerous growth factor pathways broaden our view regarding the role of GPCRs in cancer.
In summary, we have demonstrated that muscarinic signaling via CHRM1 can directly suppress pancreatic tumor development through downregulation of the MAPK and PI3K/AKT signaling pathway in cancer cells, as well as potentially indirectly through suppression of myeloid cells. Although the role of muscarinic signaling in modulating crosstalk between pancreatic tumor cells and the microenvironment needs to be further investigated, the findings reveal a surprising inhibitory role for parasympathetic signaling in PDAC and point to cholinergic agonists as potentially useful adjunctive therapies in the treatment of PDAC in early and late stages.
Methods
Human Tissue Samples
Human pancreatic cancer samples for organoid cultures were obtained from the Technische Universität München (Dr. Maximilian Reichert, Dept. of Medicine II). All samples were anonymized. All protocols using human materials were approved by the ethics committee of the University of Munich or Technische Universität München. Written informed consent was obtained from all patients.
Animals
LSL-Kras+/G12D;Pdx1-Cre (KC) and LSL-Kras+/G12D;LSL-Trp53+/R172H;Pdx1-Cre (KPC) mice were described previously (26,31) and provided by Dr. Kenneth P. Olive. Chrm1-KO mice (C57BL/6-Chrm1tm1Stl/J) (M1RKO) (60) were purchased from the Jackson Laboratory. KC mice and KPC mice were crossed to M1RKO mice. In the vagotomy experiments, KC mice received pyloroplasty with or without vagotomy at 8 weeks. Bethanechol was administered at 400 μg/ml in drinking water (Sigma-Aldrich) and selumetinib (AZD6244) by subcutaneous injection (s.c.) of 250 mg/kg biweekly (Selleckchem) immediately after surgery. Animals were sacrificed at 16 or 20 weeks. KPC mice in treatment studies were palpated biweekly for pancreatic tumors. Once a tumor was suspected, mice were screened by VEVO 2100 - Ultrasound Imaging System (FUJIFILM VisualSonics) for PDAC. KPC mice were treated with gemcitabine, given by i.p. injection 100 mg/kg biweekly, with or without bethanechol, when a tumor measuring 3–5 mm in diameter was detected by ultrasound. Mice were either sacrificed when moribund or after two weeks of treatment. NOD/SCID mice were also purchased from the Jackson Laboratory and implanted with Panc1 cells (2.5 × 104 cells) subcutaneously and sacrificed at 30 days after implantation. For liver metastatic experiments, wild-type C57/B6 mice (WT) received a splenic injection of GFP-labeled Panc02 cells, followed by hemi-splenectomy. Animals were then divided into three groups. One group was untreated, another group received selective vagotomy of the hepatic vagal branch (SHVx), and a third group was treated with bethanechol after surgery. All animal studies and procedures were approved by the Institutional Animal Care and Use Committee at Columbia University (IACUC). All mice were bred under specific pathogen free conditions. Comparisons were made with age- and sex- matched control animals.
Histology, immunohistochemistry, immunofluorescence and microscopy
5 μm PFA-fixed frozen or paraffin embedded sections were prepared for immunofluorescence and immunohistochemistry, respectively. For immunofluorescence, slides were washed with 1% Triton X-100 in PBS, rinsed and blocked for 30 minutes with 2% bovine serum albumin (BSA - Sigma-Aldrich). Primary antibodies and fluorophore-conjugated secondary antibodies were diluted in 2% BSA and incubated over night at 4°C. The following primary antibodies were used; Chrm1 (1:200 Santa Cruz), VAChT (1:100 Synaptic Systems), Ki-67 (1:500 Abcam), p-EGFR (1:200 Abcam), and CD44 (1:200 BIO-RAD). For immunohistochemical staining, slides were deparaffinized in xylene. Antigen retrieval was performed by boiling the slides in citrate buffer (10 mM pH 6.0) in a water bath for 20 minutes. Endogenous peroxidase was blocked by incubation with 3% hydrogen peroxide in PBS for visualization using the peroxidase reaction. Slides were rinsed in PBS Tween 0.05% and blocked for 30 minutes with 2% BSA or 10% serum. Primary antibodies and biotinylated secondary antibodies (Jackson Immunoresearch) were diluted in 2% BSA and incubated overnight at 4°C. The following primary antibodies were used; p-EGFR (1:200 Abcam), CD44 (1:200 BIO-RAD), p-PI3K (1:100 Sigma-Aldrich), beta-Tubulin III (1:5000 Abcam), CD11b (1:5000 Abcam), F4/80 (1:200 Abcam), p-ERK1/2 (1:200 Cell Signaling), αSMA (1:500 Abcam), CgA (1:400 Abcam) and CD31 (1:500 Abcam). Subsequently, slides were incubated with peroxidase conjugated streptavidin (Vector Laboratories) and 3,3’-diaminobenzidine (Dako) as chromogens, respectively. Slides were counterstained with hematoxylin and mounted for viewing. Bright field and fluorescence images were acquired using an Eclipse TU2000-U microscope (Nikon) connected to a cooled color CCD camera (RTKE Diagnostic Instruments) using SPOT software (Spotimaging).
RNAseq Experiments
Panc1 cells were treated with or without 1 mM pilocarpine (Sigma-Aldrich) in RPMI-1640 with 0.5% FBS and 1% antibiotics for 72 hours. Panc1 cells were lysed in RNA lysis buffer supplied in ARCTURUS PicoPure RNA isolation kit (Life Technologies) (n = 4). Total RNA was isolated in accordance with the manufacturer’s protocol. cDNA was amplified and libraries were constructed by using SMARTer Ultra Low Input RNA Kit (Clontech Laboratories) and Nextera XT DNA Library Preparation Kit (Illumina) according to the respective manufacturer’s instructions. Sequencing was performed with Hiseq 2500 (Illumina) (30 M 100 bp single end reads per sample).
Accession number
The full RNA-seq data have been deposited in the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession Code GEO: GSE102880.
3D Spheroid cultures
3D spheroid were isolated from mouse pancreas and cultured as previously described (37). Tissue was digested as described in flow cytometry section with a shorter incubation (20 minutes). Cells were resuspended in Matrigel (Corning) with or without Adeno-Cre virus (provided from the University of Iowa), and 6.5 × 103 cells/well were seeded into a pre-warmed 24 well plate. Cells were cultured in RPMI-1640 containing B27 and N2 supplements (Gibco), 5% Nu-Serum IV (Corning), 100 μg/ml trypsin inhibitor, and 100 ng/ml Cholera Toxin (Sigma-Aldrich). Pilocarpine, scopolamine, McN-34A or pirenzepine (all Sigma-Aldrich) was added to the media, and the medium was replaced every day. Sphere size and number were analyzed using ImageJ software at day 5.
Human patient derived organoids (PDO) were isolated as previously described (37). Cultured PDOs were mechanically broken and furthermore enzymatically dissociated into single cells. Cell pellets were counted and 1000 cells in a mixture of Matrigel and medium were seeded in each well. After 24 hours, 10 μl of human organoid medium containing the drug was added in each well. After 72 hours the viability was measured with the CellTiter-Glo 3D Cell Viability Assay (Promega).
Morphometric analysis of murine PanIN and PDAC
Murine pancreatic intraepithelial neoplasia (PanIN) was diagnosed based on the degree of cytological atypia and epithelial proliferation. PanIN area was measured by ImageJ softeware. Microcarcinoma appeared as nodular clusters of coalescing small ducts expanded by solid neoplastic epithelial proliferation. These microcarcinomas showed foci of microinvasion and were also seen adjacent to larger poorly differentiated carcinomas. The existence of PDAC was assessed on full-face sections of the entire mouse pancreas.
Supplementary Material
Statement of significance.
Subdiaphragmatic vagotomy or Chrm1 knockout accelerates pancreatic tumorigenesis, in part via expansion of the CSC compartment. Systemic administration of a muscarinic agonist suppresses tumorigenesis through MAPK and PI3K/AKT signaling, in early stages of tumor growth and in more advanced, metastatic disease. Therefore, CHRM1 may represent a potentially attractive therapeutic target.
Acknowledgements
We thank Ms. Theresa Swayne for assistance with confocal microscopy. Images were collected in the Confocal and Specialized Microscopy Shared Resource at Columbia University. The confocal microscope was purchased with an NIH grant. These studies also used the resources of the Cancer Center Flow Core Facility at Columbia University funded in part through a Center Grant. We also thank Ms. Wendy Beth Jackelow (Medical & Scientific Illustration) for creating schematic images.
Financial support
T. C. Wang received grants from the NIH (R35 CA210088), the Pancreatic Cancer Action Network - ACCR (Innovative Grant) and the Lustgarden Foundation. B.W. Renz was supported by a grant from the German Research Foundation (Deutsche Forschungsgemeinschaft RE 3440/1–1). T. Tanaka and R. Takahashi were supported by Uehara Memorial Foundation. T. Tanaka was supported by Japanese Society of Gastroenterology (JSGE) Support for Young Gastroenterologists Studying in the United States. M. Middelhoff was supported by a grant from the Mildred-Scheel-Stiftung, Deutsche Krebshilfe, Germany (70111870). P. Oberstein was supported by a PanCAN/AACR grant. R. White was supported by the medical oncology T32 grant (T32CA203703). H. Deng was supported by the National Science Foundation of China (81770624). Y. Hayakawa was supported by a grant from the Mitsukoshi Health and Welfare Foundation and JSPS Postdoctoral Fellowships for Research Abroad. G.Valenti was partially supported by NYSTEM New York State Stem Cell Science.
Footnotes
Disclosure: The authors have no conflicts of interest or funding to disclose.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: A Cancer Journal for Clinicians. 3rd ed. 2018;97:3133–24. [DOI] [PubMed] [Google Scholar]
- 2.Werner J, Combs SE, Springfeld C, Hartwig W, Hackert T, Büchler MW. Advanced-stage pancreatic cancer: therapy options. Nat Rev Clin Oncol. 2013;10:323–33. [DOI] [PubMed] [Google Scholar]
- 3.Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15:333–48. [DOI] [PubMed] [Google Scholar]
- 4.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science [Internet]. AAAS; 2009;324:1457–61. Available from: http://stke.sciencemag.org/cgi/content/full/sci;324/5933/1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demir IE, Friess H, Ceyhan GO. Neural plasticity in pancreatitis and pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2015;12:649–59. [DOI] [PubMed] [Google Scholar]
- 6.Vennin C, Murphy KJ, Morton JP, Cox TR, Pajic M, Timpson P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology. 2018;154:820–38. [DOI] [PubMed] [Google Scholar]
- 7.Monje M Settling a Nervous Stomach: The Neural Regulation of Enteric Cancer. Cancer Cell. 2017;31:1–2. [DOI] [PubMed] [Google Scholar]
- 8.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic Nerve Development Contributes to Prostate Cancer Progression. Science. 2013;341:1236361–1. [DOI] [PubMed] [Google Scholar]
- 9.Zahalka AH, Arnal-Estapé A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science. 2017;358:321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson SC, Eberl M, Vagnozzi AN, Belkadi A, Veniaminova NA, Verhaegen ME, et al. Basal Cell Carcinoma Preferentially Arises from Stem Cells within Hair Follicle and Mechanosensory Niches. Cell Stem Cell. 2015;16:400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao C-M, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT, et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med. 2014;6:250ra115–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H, et al. Nerve Growth Factor Promotes Gastric Tumorigenesis through Aberrant Cholinergic Signaling. Cancer Cell. 2017;31:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayakawa Y, Wang TC. Nerves switch on angiogenic metabolism. Science. 2017;358:305–6. [DOI] [PubMed] [Google Scholar]
- 14.Stopczynski RE, Normolle D, Hartman DJ, Ying H, Deberry JJ, Bielefeldt K, et al. Neuroplastic changes occur early in the development of pancreatic ductaladenocarcinoma. Cancer Research. American Association for Cancer Research; 2014;74:1718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saloman JL, Albers KM, Li D, Hartman DJ, Crawford HC, Muha EA, et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. PNAS. 2016;:201512603–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Partecke LI, Käding A, Trung DN, Diedrich S, Sendler M, Weiss F, et al. Subdiaphragmatic vagotomy promotes tumor growth and reduces survival via TNFα in a murine pancreatic cancer model. Oncotarget. Impact Journals; 2017;8:22501–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell. 2018;33:75–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ayala GE, Dai H, Powell M, Li R, Ding Y, Wheeler TM, et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin Cancer Res. American Association for Cancer Research; 2008;14:7593–603. [DOI] [PubMed] [Google Scholar]
- 19.Albo D, Akay CL, Marshall CL, Wilks JA, Verstovsek G, Liu H, et al. Neurogenesis in colorectal cancer is a marker of aggressive tumor behavior and poor outcomes. Cancer. Wiley-Blackwell; 2011;117:4834–45. [DOI] [PubMed] [Google Scholar]
- 20.Borden P, Houtz J, Leach SD, Kuruvilla R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep. 2013;4:287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holmgren S, Olsson C. Autonomic control of glands and secretion: A comparative view. Autonomic Neuroscience. 2011;165:102–12. [DOI] [PubMed] [Google Scholar]
- 22.Kiba T, Tanaka K, Numata K, Hoshino M, Misugi K, Inoue S. Ventromedial hypothalamic lesion-induced vagal hyperactivity stimulates rat pancreatic cell proliferation. YGAST. 1996;110:885–93. [DOI] [PubMed] [Google Scholar]
- 23.De Couck M, Maréchal R, Moorthamers S, Van Laethem J-L, Gidron Y. Vagal nerve activity predicts overall survival in metastatic pancreatic cancer, mediated by inflammation. Cancer Epidemiol. Elsevier Ltd; 2016;40:47–51. [DOI] [PubMed] [Google Scholar]
- 24.De Couck M, Mravec B, Gidron Y. You may need the vagus nerve to understand pathophysiology and to treat diseases. Clin Sci. 2012;122:323–8. [DOI] [PubMed] [Google Scholar]
- 25.Erin N, Duymuş O, Oztürk S, Demir N. Activation of vagus nerve by semapimod alters substance P levels and decreases breast cancer metastasis. Regul Pept. 2012;179:101–8. [DOI] [PubMed] [Google Scholar]
- 26.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. [DOI] [PubMed] [Google Scholar]
- 27.Zhao S, Chen C, Chang K, Karnad A, Jagirdar J, Kumar AP, et al. CD44 Expression Level and Isoform Contributes to Pancreatic Cancer Cell Plasticity, Invasiveness, and Response to Therapy. Clin Cancer Res. 2016;22:5592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hüchtebrock HJ, Niebel W, Singer MV, Forssmann WG. Intrinsic pancreatic nerves after mechanical denervation of the extrinsic pancreatic nerves in dogs. Pancreas. 1991;6:1–8. [DOI] [PubMed] [Google Scholar]
- 29.Andrews PLR, Rothwell NJ, Stock MJ. Effects of subdiaphragmatic vagotomy on energy balance and thermogenesis in the rat. J Physiol. 1985;362:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavlov VA, Tracey KJ. Neural regulation of immunity: molecular mechanisms and clinical translation. Nat Neurosci. Nature Publishing Group; 2017;20:156–66. [DOI] [PubMed] [Google Scholar]
- 31.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 32.Abel EV, Simeone DM. Biology and clinical applications of pancreatic cancer stem cells. Gastroenterology. 2013;144:1241–8. [DOI] [PubMed] [Google Scholar]
- 33.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. Nature Publishing Group; 2014;:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins MA, Yan W, Sebolt-Leopold JS, Pasca di Magliano M. MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology. 2014;146:822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopp JL, Figura von G, Mayes E, Liu F-F, Dubois CL, Morris Iv JP, et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2012;22:737–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reichert M, Takano S, Heeg S, Bakir B, Botta GP, Rustgi AK. Isolation, culture and genetic manipulation of mouse pancreatic ductal cells. Nat Protoc. 2013;8:1354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ouyang H, Mou L, Luk C, Liu N, Karaskova J, Squire J, et al. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol. 2000;157:1623–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–88. [DOI] [PubMed] [Google Scholar]
- 40.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions Methods Mol Biol. Totowa, NJ: Humana Press; 2010;661:3–38. [DOI] [PubMed] [Google Scholar]
- 41.Soares KC, Foley K, Olino K, Leubner A, Mayo SC, Jain A, et al. A preclinical murine model of hepatic metastases. J Vis Exp. 2014;:51677–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen D, Ayala GE. Innervating Prostate Cancer. Phimister EG, editor. N Engl J Med. 2018;378:675–7. [DOI] [PubMed] [Google Scholar]
- 43.Sohal DPS, Tullio K, Khorana AA. Do patients with pancreatic body or tail cancer benefit from adjuvant therapy?A cohort study. Surg Oncol. 2018;27:245–50. [DOI] [PubMed] [Google Scholar]
- 44.Kirchgessner AL, Liu MT, Gershon MD. In situ identification and visualization of neurons that mediate enteric and enteropancreatic reflexes. J Comp Neurol. 1996;371:270–86. [DOI] [PubMed] [Google Scholar]
- 45.Kirchgessner AL, Gershon MD. Innervation of the pancreas by neurons in the gut. J Neurosci. 1990;10:1626–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chavan SS, Tracey KJ. Essential Neuroscience in Immunology. J Immunol. 2017;198:3389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. Nature Publishing Group; 2000;405:458–62. [DOI] [PubMed] [Google Scholar]
- 48.Dubeykovskaya Z, Si Y, Chen X, Worthley DL, Renz BW, Urbanska AM, et al. Neural innervation stimulates splenic TFF2 to arrest myeloid cell expansion and cancer. Nat Commun. 2016;7:10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nature Medicine. Nature Publishing Group; 2004;10:1216–21. [DOI] [PubMed] [Google Scholar]
- 50.Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, et al. Expression and Function of the Cholinergic System in Immune Cells. Front Immunol. Frontiers; 2017;8:1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung S-R, Kushmerick C, Seo JB, Koh D-S, Hille B. Muscarinic receptor regulates extracellular signal regulated kinase by two modes of arrestin binding. Proc Natl Acad Sci U S A. National Academy of Sciences; 2017;114:E5579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu H, Wu K, Tian Y, Liu Q, Han N, Yuan X, et al. CD44 correlates with clinicopathological characteristics and is upregulated by EGFR in breast cancer. Int J Oncol. Spandidos Publications; 2016;49:1343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hao J, Madigan MC, Khatri A, Power CA, Hung T-T, Beretov J, et al. In vitro and in vivo prostate cancer metastasis and chemoresistance can be modulated by expression of either CD44 or CD147. Tang DG, editor. PLoS ONE. Public Library of Science; 2012;7:e40716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiaoping L, Xiaowei Z, Leizhen Z, Weijian G. Expression and significance of CD44 and p-AKT in pancreatic head cancer. World J Surg Oncol. BioMed Central; 2015;13:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blaukat A, Barac A, Cross MJ, Offermanns S, Dikic I. G protein-coupled receptor-mediated mitogen-activated protein kinase activation through cooperation of Galpha(q) and Galpha(i) signals. Mol Cell Biol. 2000;20:6837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature. Nature Publishing Group; 2012;491:449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong S, Yin H, Liao Y, Yao F, Li Q, Zhang J, et al. Lung Tumor Suppressor GPRC5A Binds EGFR and Restrains Its Effector Signaling. Cancer Research. American Association for Cancer Research; 2015;75:1801–14. [DOI] [PubMed] [Google Scholar]
- 58.Elbaz M, Ahirwar D, Ravi J, Nasser MW, Ganju RK. Novel role of cannabinoid receptor 2 in inhibiting EGF/EGFR and IGF-I/IGF-IR pathways in breast cancer. Oncotarget. Impact Journals; 2017;8:29668–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakashima A, Takeuchi H, Imai T, Saito H, Kiyonari H, Abe T, et al. Agonist-independent GPCR activity regulates anterior-posterior targeting of olfactory sensory neurons. Cell. 2013;154:1314–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gerber DJ, Sotnikova TD, Gainetdinov RR, Huang SY, Caron MG, Tonegawa S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. PNAS. 2001;98:15312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.