

Abstract
Background
The ventral tegmental area (VTA) is important for alcohol‐related reward and reinforcement. Mouse VTA neurons are hyposensitive to γ‐aminobutyric acid (GABA) during ethanol (EtOH) withdrawal, and GABA responsiveness is normalized by in vitro treatment with histone deacetylase inhibitors (HDACi). The present study examined the effect of a systemically administered HDACi, suberanilohydroxamic acid (SAHA) on GABA sensitivity, and related molecular changes in VTA neurons during withdrawal after chronic EtOH intake in rats.
Methods
Sprague Dawley male adult rats were fed with Lieber‐DeCarli diet (9% EtOH or control diet) for 16 days. Experimental groups included control diet‐fed and EtOH diet‐fed (0‐ or 24‐hour withdrawal) rats treated with either SAHA or vehicle injection. Single‐unit recordings were used to measure the response of VTA neurons to GABA. Immunohistochemistry was performed to examine levels of HDAC2, acetylated histone H3 lysine 9 (acH3K9), and GABAA receptor α1 and α5 subunits in the VTA; quantitative polymerase chain reaction was performed to examine the mRNA levels of HDAC2 and GABAA receptor subunits.
Results
VTA neurons from the withdrawal group exhibited GABA hyposensitivity. In vivo SAHA treatment 2 hours before sacrifice normalized the sensitivity of VTA neurons to GABA. EtOH withdrawal was associated with increased HDAC2 and decreased acH3K9 protein levels; SAHA treatment normalized acH3K9 levels. Interestingly, no significant change was observed in the mRNA levels of HDAC2. The mRNA levels, but not protein levels, of GABAA receptor α1 and α5 subunits were increased during withdrawal.
Conclusions
Withdrawal from chronic EtOH exposure results in a decrease in GABA‐mediated inhibition, and this GABA hyposensitivity is normalized by in vivo SAHA treatment. Disruption of signaling in the VTA produced by alteration of GABA neurotransmission could be 1 neuroadaptive physiological process leading to craving and relapse. These results suggest that HDACi pharmacotherapy with agents like SAHA might be an effective treatment for alcoholism.
Keywords: SAHA, Epigenetic, Dopamine, Ethanol, VTA
The ventral tegmental area (VTA) and its associated circuitry are highly involved in alcohol reward and reinforcement (Koob and Volkow, 2010). Neurons of the VTA provide dopamine (DA) to many brain areas implicated in drug and alcohol addiction, including the nucleus accumbens (NAc), prefrontal cortex, and basolateral amygdala (Albanese and Minciacchi, 1983; Koob and Volkow, 2016; Oades and Halliday, 1987). VTA DA neurons are under inhibitory control by γ‐aminobutyric acid (GABA), which is released from interneurons (Margolis et al., 2012) as well as from afferent neurons from the rostromedial tegmental nucleus (Jhou et al., 2009), NAc (Rahman and McBride, 2000), lateral hypothalamus (Kallo et al., 2015), and amygdala (Janak and Tye, 2015). Increased DA release in the NAc is a common feature of drugs of abuse (Arora et al., 2010; di Chiara and Imperato, 1988; Luscher and Ungless, 2006), which is achieved either by increasing the firing rate of DA VTA neurons or by blocking re‐uptake of DA. Ethanol (EtOH) administration in vitro or in vivo increases DA VTA neuronal activity (Brodie et al., 1990, 1999; Gessa et al., 1985). Ultimately, the DA output of the VTA is controlled by a number of factors, including the release of GABA. Therefore, studying the effects of GABA in the context of the control of DA neuronal activity may yield insight into mechanisms of alcohol actions related to addiction.
Chronic alcohol administration alters brain neurocircuitry during the development of addiction and alcohol use disorder (Koob and Volkow, 2016). In response to repeated alcohol exposure and withdrawal, altered epigenetic factors regulate withdrawal‐related physiological changes (Pandey et al., 2008a). Assessment of the effects of chronic and repeated alcohol on the brain should take epigenetic modifications into account as a potential mechanism for the persistence of neuroadaptive changes (Pandey et al., 2008a, 2017). Specific epigenetic alterations such as DNA methylation and histone modifications are consistently induced by chronic alcohol exposure (Berkel and Pandey, 2017; Kalsi et al., 2009; Moonat et al., 2010). Changes in histone acetylation associated with chronic alcohol treatment have been demonstrated in the VTA (Arora et al., 2013; Shibasaki et al., 2011), the amygdala (Pandey et al., 2008a), and in numerous other brain areas, including cerebral cortex, prefrontal cortex, dorsal striatum, hippocampus, and NAc (Bohnsack et al., 2017; Botia et al., 2012; D'Addario et al., 2013; Dominguez et al., 2016; Finegersh et al., 2015; Hashimoto et al., 2017; Qiang et al., 2011; Simon‐O'Brien et al., 2015). Notably, reversal of alcohol‐induced epigenetic modifications by histone deacetylase inhibitor (HDACi) treatment has been observed in the VTA (Arora et al., 2013) and amygdala (Sakharkar et al., 2012; You et al., 2014). The histone deacetylase (HDAC) isoform HDAC2 has been shown to be up‐regulated after alcohol and opiate treatment (Arora et al., 2013; Authement et al., 2016; Pandey et al., 2017; Sakharkar et al., 2014), and this increase results in decreased histone acetylation, including at the histone H3 lysine 9 (H3K9) site, that decreases gene transcription (Krishnan et al., 2014; Moser et al., 2014). The reduction in both GABAA receptor α1 subunit (Gabra1) mRNA expression and responsiveness to GABA in the prefrontal cortex after chronic alcohol exposure and withdrawal is associated with increased Hdac2 and Hdac3 mRNA levels and decreased histone H3K9 and H3K14 acetylation at the Gabra1 promoter; HDACi administration normalized Gabra1 expression and response to GABA (Bohnsack et al., 2018).
We previously demonstrated that repeated EtOH treatment of C57BL/6J mice decreased the sensitivity of DA VTA neurons to GABA inhibition (Brodie, 2002). Additional studies from our group demonstrated that the changes in GABA sensitivity in the mouse VTA are correlated with decreased acetylated histone and increased HDAC2 immunoreactivity (Arora et al., 2013). In those studies, brain slices were incubated in vitro with HDACi for 2 hours prior to recording, and this treatment normalized the responses of VTA neurons to GABA (Arora et al., 2013). While an epigenetic mechanism for reversing the GABA hyposensitivity localized to the VTA region was indicated, the question remained whether in vivo HDACi could also reverse VTA neuronal hyposensitivity, as this mode of drug delivery is more relevant to pharmacotherapeutic approaches for alcoholism. For the present study, we chose to use the Lieber‐DeCarli liquid diet that is a standard method for inducing withdrawal symptoms after chronic drinking and that has been used by us and numerous other investigators in the field (Baldwin et al., 1991; Lieber et al., 1989; Pandey et al., 2008a; Sharda et al., 2012). We investigated VTA neuronal sensitivity to GABA inhibition, and explored changes in HDAC2 and acetylated histone H3 lysine 9 (acH3K9) protein levels, as well as expression of mRNA for HDAC2 and GABAA receptor α1 and α5 subunits, after withdrawal from chronic EtOH exposure. In addition, we determined whether in vivo treatment with an HDACi, suberanilohydroxamic acid (SAHA; vorinostat), could restore GABA sensitivity to VTA neurons and normalize molecular changes induced by withdrawal.
Materials and Methods
Animals
Male Sprague Dawley adult rats were purchased from Envigo (Indianapolis, IN). Animals weighing between 270 and 290 g were used in all experiments and randomly assigned to experimental groups. All rats were group housed and acclimatized for a week and then single‐cage housed in a temperature‐ and humidity‐controlled facility with a 12‐hour light/dark cycle (6 am to 6 pm) during EtOH treatment. All experiments were performed in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the University of Illinois at Chicago Institutional Animal Care and Use Committee.
Lieber‐DeCarli Control/EtOH Diet Treatment
EtOH administration was accomplished by oral Lieber‐DeCarli EtOH‐containing diet feeding as described previously (You et al., 2014). Male adult Sprague Dawley rats were individually housed and offered 80 ml of the Lieber‐DeCarli control diet (Bio‐Serv, Inc., Frenchtown, NJ) as their source of food and fluid for 3 days. After random assignment to experimental groups, rats in the pair‐fed control groups continued to receive the control liquid diet for the entire length of treatment. Rats in the EtOH‐fed groups were gradually introduced to EtOH over a 7‐day period (concentrations increased daily) and then maintained on 9% (v/v) EtOH‐containing Lieber‐DeCarli liquid diet for 15 (withdrawal groups) or 16 (EtOH‐vehicle group) days. The withdrawal groups were withdrawn from EtOH and given control diet for 24 hours before sacrifice, while the other groups remained on their diet for the final 24 hours. Fresh diet was provided daily between 5:00 and 6:00 pm, right before the beginning of the dark cycle of the housing facility. Control and EtOH diet rats were pair‐fed as reported earlier (You et al., 2014). In this model, the amount of alcohol diet consumed by rats reliably produces blood levels of 172 to 198 mg%, but after 24‐hour withdrawal, the alcohol level in blood is undetectable (Pandey et al., 1992, 2008a; You et al., 2014). At 24‐hour withdrawal, rats given the same chronic alcohol treatment exhibited anxiety‐like behavior in the elevated plus maze and light/dark box (Pandey et al., 2008a; You et al., 2014).
When injections were made, SAHA (6.25 mg/ml, 50 mg/kg, and see below for preparation method) or vehicle was administered intraperitoneally (i.p.) 2 hours before sacrifice. Rat brains were sectioned for electrophysiological recording to assess the response to GABA or perfused for immunohistochemistry (IHC) to measure protein levels of GABAA receptor subunit α1 or α5, acetylated H3K9 and HDAC2, or collected for quantitative polymerase chain reaction (qPCR) measurement of HDAC2 or GABAA receptor subunits.
The groups for electrophysiology recordings were as follows: control diet fed and vehicle injected (CV, n = 8), control diet fed and SAHA injected (CS, n = 9), withdrawn from EtOH diet and vehicle injected (WV, n = 14), and withdrawn from EtOH diet and SAHA injected (WS, n = 10). The groups for protein levels of acetylated H3K9, HDAC2, and GABA subunits, measured by gold immunolabeling, were the same as those indicated for the electrophysiology recordings, with the addition of an EtOH diet‐fed, vehicle‐injected group (EV); 5 rats in each of these 5 groups were used. For qPCR measurement of mRNA expression of GABAA receptor subunits, the numbers of animals used in each group were as follows: CV, 5; CS, 4; EV, 4; WV, 4; WS, 5. Finally, for qPCR measurement of mRNA expression of HDAC2 isoform, no injections were made prior to sacrifice, and 3 treatment groups were used, control diet fed (C), EtOH diet fed with no withdrawal (E), and 24‐hour withdrawal from EtOH diet (W) (n = 10 for each group).
SAHA Administration to Rats
SAHA (Vorinostat, Selleck Chemicals, Houston, TX) solution was prepared by dissolving 62.5 mg SAHA in 0.2 ml Dimethyl sulfoxide (DMSO) and vortexing until completely dissolved. Then, 4 ml of PEG 300, 0.5 ml propylene glycol, 0.1 ml Tween‐80, 5.2 ml normal saline were added to the solution sequentially and vortexed after adding each compound. The final concentration of SAHA in the solution was 6.25 mg/ml, 2% DMSO, 40% PEG300, 5% propylene glycol, and 1% Tween‐80 in saline. Control vehicle solution was prepared with the same solvents and in the same concentrations, again in saline. SAHA was injected at a dose of 50 mg/kg body weight and was given i.p. 2 hours before sacrifice.
In additional supplementary experiments, to correlate with our earlier in vitro mouse study, brain slices were made from rats withdrawn from chronic 9% EtOH Lieber‐DeCarli diet for 24 hours (W) and these brain slices were incubated in artificial cerebrospinal fluid (aCSF) with either 3 μM SAHA in DMSO (n = 8) or 0.1% DMSO vehicle (n = 9) for 2 hours before recordings were made.
Extracellular Recording
The brain slice preparation technique has been described previously (Arora et al., 2013; Nimitvilai and Brodie, 2010). Briefly, after deep isoflurane anesthesia, each rat was sacrificed and the brain was rapidly removed from the cranium. Coronal sections (400 μm) containing the VTA were cut and immediately placed in the recording chamber in which aCSF flowed at 2 ml/min at 35°C. The composition of the aCSF in these experiments was as follows (in mM): NaCl 126, KCl 2.5, NaH2PO4 1.24, CaCl2 2.4, MgSO4 1.3, NaHCO3 26, glucose 11. The composition of the cutting solution was as follows (in mM): KCl 2.5, CaCl2 2.4, MgSO4 1.3, NaHCO3 26, glucose 11, and sucrose 220. All solutions were saturated with 95% O2/5% CO2 (pH = 7.4).
Extracellular recording electrodes were made from 1.5‐mm‐diameter glass tubing with filament and were filled with 0.9% NaCl; tip resistance of all microelectrodes ranged from 2 to 4 MΩ. A high‐gain extracellular amplifier (x‐Cell; FHC, Inc., Bowdoin, ME) was used in conjunction with a PC‐based data acquisition system (ADInstruments, Inc., Colorado Springs, CO). Firing rate was determined before and during drug application and was calculated over 1‐minute intervals throughout the entire recording period. The change in firing rate was expressed as a percentage of the baseline firing rate prior to administration of each concentration of GABA, to control for changes in firing rate that may occur over time.
GABA was added to the aCSF using a calibrated infusion pump from stock solutions 100 to 1,000 times the desired final concentrations. Electrophysiological recording was performed on spontaneously firing neurons while single concentrations of GABA (50, 100, 200, or 500 μM) were added to the superfusate in the recording chamber for 4 minutes, and followed by 8 minutes of washout for each concentration until baseline firing rate recovered, before the next concentration was tested. Neurons that did not return to at least 85% of the baseline firing rate during washout after GABA administration were excluded from data analysis. Final concentrations were calculated from aCSF flow rate, pump infusion rate, and concentration of drug stock solution. The small volume chamber (about 300 μl) used in these studies permitted the rapid application and washout of drug solutions. Salts used to prepare the extracellular media were purchased from Sigma (St. Louis, MO). The method for in vitro SAHA administration shown in Fig. S1 is described in the legend to that figure.
Cell Identification
During electrophysiological recording, the recording electrodes were placed in the VTA under visual control. Only those neurons which were located within the lateral VTA and which conformed to the criteria for DA neurons established in the literature and in this laboratory (Brodie et al., 1990; Lacey et al., 1989; Mueller and Brodie, 1989) were studied. As extracellular recordings were performed, it is not technically feasible to inject dye or use single‐cell RT‐PCR to determine whether we were monitoring neuronal activity specifically of dopaminergic neurons. Although not all units have been tested, neurons conforming to these electrophysiological criteria that were tested with baclofen in our laboratory have all been inhibited. Sensitivity to baclofen inhibition has been reported to be a more rigorous identification of DA neurons than testing them with other agents like DA (Margolis et al., 2012).
Gold Immunolabeling of Acetylated H3K9, HDAC2, and GABAA Receptor α1 and α5 Subunits
The IHC method used in our laboratory has been described previously (Moonat et al., 2011; Pandey et al., 2004, 2008b; You et al., 2014). Rats were anesthetized with pentobarbital and perfused with 200 ml of normal saline, followed by 300 ml of 4% ice‐cold paraformaldehyde (PFA) fixative prepared in 0.1M phosphate buffer (PB; pH 7.4). Following perfusion, brains were removed, postfixed overnight in PFA at 4°C, and cryoprotected using sequential placement in sucrose solutions (10, 20, and 30%) prepared in 0.1M PB. Brains were then frozen, and 20 μm brain sections containing the VTA were cut using a cryostat. Brain sections were washed in phosphate‐buffered saline (PBS) twice for 10 minutes, then incubated in RPMI 1640 for 30 minutes, followed by two 30‐minute blocking steps: in 10% normal goat serum (NGS) in phosphate‐buffered saline with Tween‐20 (PBST), then in 1% bovine serum albumin (BSA) in PBST. The sections were then incubated for 18 hours or overnight with primary antibody [HDAC2: 1:100 dilution (JM‐3602‐100; MBL International, Woburn, MA); acH3K9: 1:500 dilution (06‐942; Millipore, Billerica, MA); GABAA α1 subunit: 1:200 dilution (ab33299; Abcam, Cambridge, MA); GABAA α5 subunit: 1:200 dilution (NB300‐195; Novus Biologicals, LLC, Littleton, CO)] dissolved in BSA‐PBST. Sections were subsequently washed with PBS (twice for 10 minutes) and 1% BSA‐PBS (twice for 10 minutes) before incubation with the gold‐conjugated secondary antibody (IgG [H + I]: 1:200 dilution; Ted Pella Inc., Redding, CA) for 1 hour. After washing with 1% BSA‐PBS (3 times, 3 minutes each), then with distilled water (3 times, 3 minutes each), Silver Enhance Solution (Ted Pella, Inc.) was applied to develop the staining. Sections were washed, mounted on glass slides, and then dehydrated and coverslipped.
Gold‐immunolabeled HDAC2, acetylated H3K9, GABAA α1, and α5 in the VTA were quantified using an image analysis system (Loats Associates, Westminster, MD) at 100× magnification as described previously (Arora et al., 2013). The threshold of each image was set so that an area without staining yielded zero counts. Under these conditions, gold particles in 3 randomly selected object fields per section within the VTA in each of 3 adjacent brain sections (bregma: −5.2 to −6 mm; Paxinos and Watson, 1998) from each rat (9 total object fields per rat) were counted and the values averaged for each rat. The results are reported below as mean (±SEM) of the number of immunogold particles/100 μm2 area for 5 rats per group.
Quantitative PCR
For qPCR, rats were euthanized with isoflurane; the brains were rapidly harvested, and the VTA was dissected on ice using RNase‐free conditions. RNA was isolated from VTA tissue using RNeasy mini kit (Qiagen, Valencia, CA) and subjected to first‐strand cDNA synthesis using reverse transcriptase (Thermo Fisher, Waltham, MA). Quantitative real‐time PCR was used to determine the mRNA levels for HDAC2 (Hdac2) and GABAA receptor α1 (Gabra1) and α5 (Gabra5) subunits using specific primers and SYBR green PCR master mix (Bio‐Rad, Hercules, CA). Specific primers are listed in Table S1 and SYBR green PCR master mix (Bio‐Rad). PCR conditions were 95°C for 3 minutes, followed by 40 cycles of 95°C for 30 seconds, 58°C for 30 seconds, and 72°C for 30 seconds. Relative mRNA levels were determined by normalization to Gapdh using the ΔΔCt method (Livak and Schmittgen, 2001).
Statistical Analysis
Statistical analyses were performed with Origin (OriginLab, Northampton, MA) or GraphPad Prism version 6.05 (GraphPad Software, Inc., La Jolla, CA). Comparison of differences in firing rate in response to GABA (Fig. 1) was analyzed by 3‐way analysis of variance (ANOVA) (EtOH diet * SAHA treatment * GABA concentration). Protein levels of HDAC2 and acH3K9 (Figs 2 and 3) and GABAA receptor subunits α 1 and α 5 (Fig. 4) were compared using 1‐way ANOVA. qPCR results of mRNA levels of HDAC2 (Fig. 3) and GABAA receptor subunits α 1 and α 5 (Fig. 4) were compared with a 1‐way ANOVA. Tukey's post hoc tests were used for multiple comparisons testing as appropriate.
Figure 1.
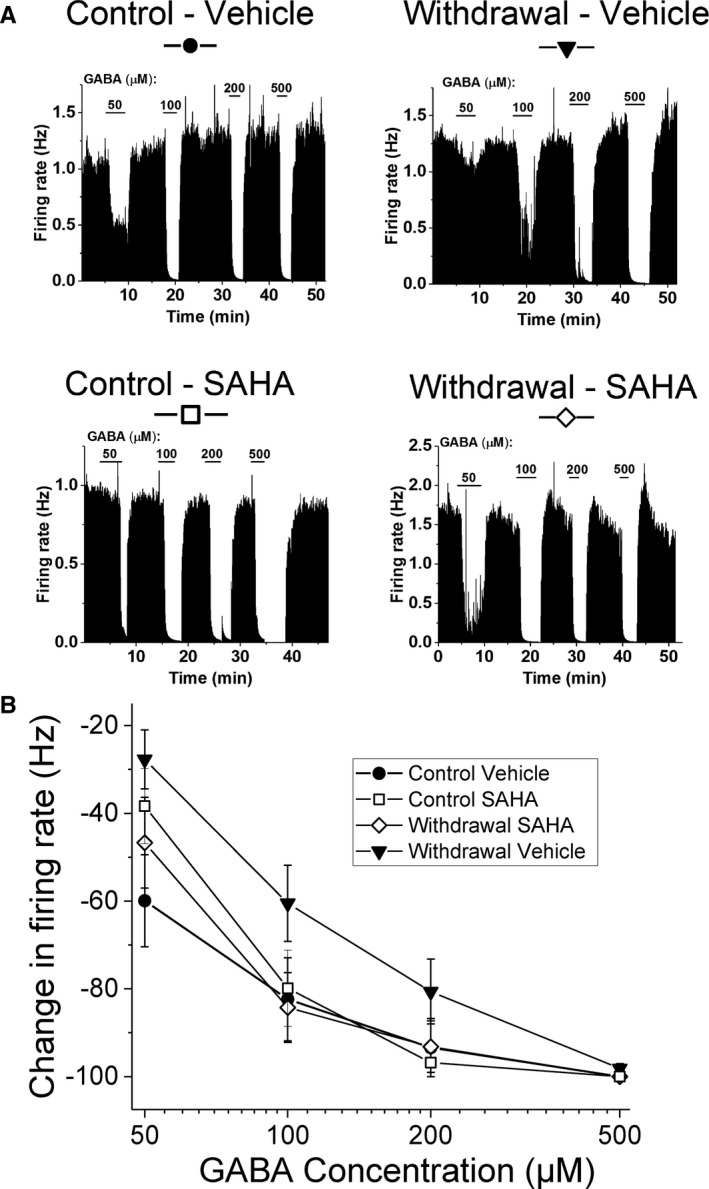
Effects of ethanol (EtOH) withdrawal and SAHA on GABA inhibition of ventral tegmental area (VTA) neurons. (A) Ratemeter graphs of single‐neuron firing rates: Each graph represents the firing rate of a single VTA neuron over time; vertical bars are proportional to the firing rate over 5‐second intervals. The duration of application of each GABA concentration (in μM) is indicated by horizontal bars. Neurons were recorded in brain slices obtained from EtOH‐withdrawn rats after chronic EtOH exposure. Slices were made 2 hours after i.p. injection of SAHA or vehicle. Addition of GABA to the extracellular medium resulted in a dose‐dependent reduction in firing in all of these neurons, although the responsiveness of the VTA neuron from the Withdrawal+Vehicle (WV)–treated rat was less than the responsiveness of VTA neurons from rats in the other treatment groups. (B) Mean concentration–response curves: Rats were randomly assigned into 4 groups as described in the Methods: GABA (50 to 500 μM) inhibited VTA neurons in a concentration‐dependent manner, 3‐way ANOVA, F(2, 111) = 35.0, main GABA effect, p < 0.001. VTA neurons of rats from the WV group (filled triangle) showed significant reduction in sensitivity to GABA, compared to Control+Vehicle (CV) and Withdrawal+Vehicle (WS) groups, 3‐way ANOVA, F(1, 111) = 6.78 for (EtOH diet * SAHA) effect, p < 0.01; Tukey post hoc WV different from CV and WS p < 0.05.
Figure 2.
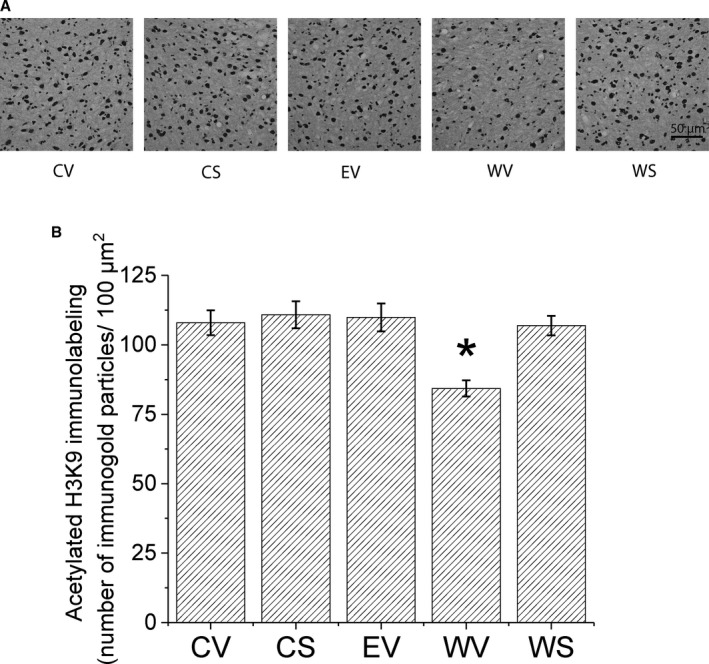
Effects of SAHA treatment on acetylated histone H3 lysine 9 (acH3K9) protein expression in ventral tegmental area (VTA) during ethanol (EtOH) withdrawal using gold immunolabeling. (A) Representative photomicrographs (scale bar = 50 μm) showing acH3K9 gold immunolabeling in the VTA. Sprague Dawley rats were randomly assigned to 5 groups: control Lieber‐DeCarli diet with SAHA (CS) or vehicle (CV) injections, or 9% EtOH Lieber‐DeCarli diet for 16 days with vehicle injection (EV), or EtOH diet for 15 days followed by 24‐hour withdrawal and either SAHA (WS) or vehicle (WV) injections. Brains were collected 2 hours after i.p. injection of SAHA or vehicle. (B) Bar diagram showing mean (± SEM) acH3K9 immunolabeling in the VTA. There was a significant decrease, 1‐way ANOVA, F(4, 20) = 6.85, p < 0.002, Tukey post hoc comparison *p < 0.05, n = 5, of acH3K9 protein in the VTA of rats withdrawn from chronic EtOH (WV), compared with all the other groups (CV, CS, EV, and WS).
Figure 3.
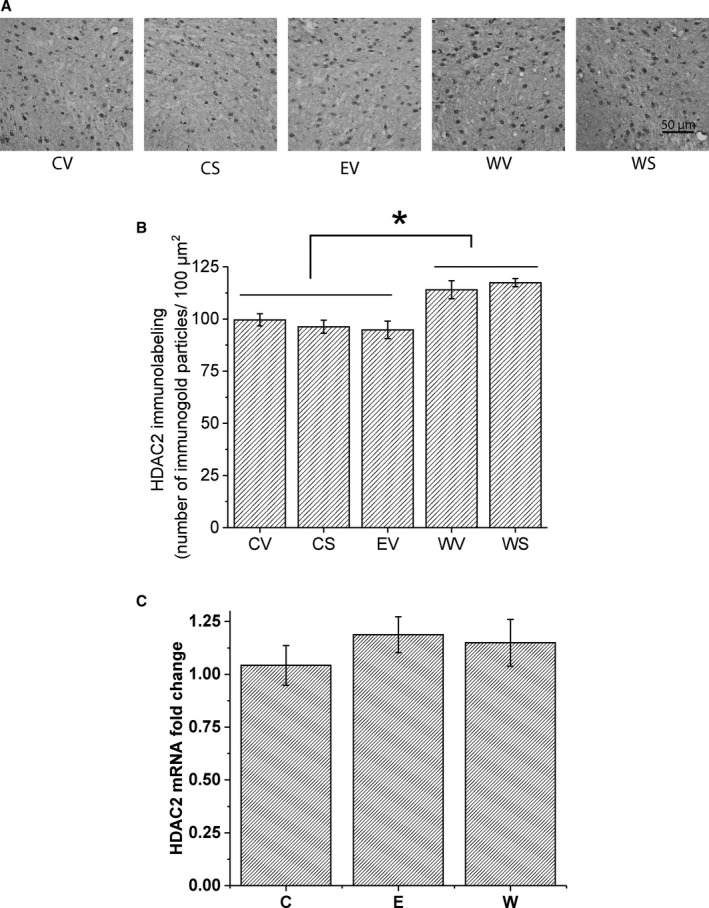
Changes in HDAC2 expression in ventral tegmental area (VTA) during ethanol (EtOH) withdrawal. (A) Representative photomicrographs (scale bar = 50 μm) showing HDAC2 gold immunolabeling in the VTA. The same treatment groups were used for HDAC2 protein measurement as those for the acetylated acH3K9. Sprague Dawley rats were randomly grouped to CV, CS, EV, WV, and WS. Brains were collected 2 hours after i.p. injection of SAHA (6.25 mg/ml, 50 mg/kg) or vehicle. (B) Bar diagram showing mean (± SEM) HDAC2 immunolabeling in the VTA. There was a significant increase in HDAC2, 1‐way ANOVA, F(4, 20) = 7.891, p < 0.002, Tukey post hoc comparison *p < 0.05, n = 5, in the VTA of rats withdrawn from chronic EtOH (WV and WS) compared to controls (CV and CS) or to EtOH diet‐fed (EV) rats. (C) Bar diagram showing mean HDAC2 mRNA expression in the VTA. Three treatment groups were used: control diet fed (C), EtOH diet fed for 16 days (E), and 24 hours withdrawal from 15 days of EtOH diet (W). No significant difference between treatment groups was observed (1‐way ANOVA; p > 0.05, n = 10).
Figure 4.
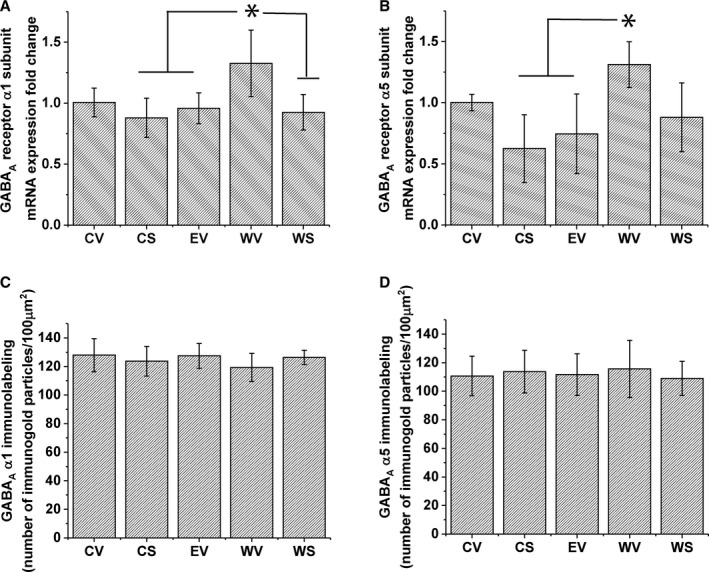
GABAA receptor subunits α1 and α5 mRNA and protein level expression in the ventral tegmental area (VTA). Five treatment groups were used: control fed with vehicle injection (CV), control fed with SAHA injection (CS), ethanol (EtOH) fed with vehicle injection (EV), EtOH fed with 24‐hour withdrawal and vehicle injection (WV), EtOH fed with 24‐hour withdrawal and SAHA injection (WS), as described in the Methods section. (A) Bar diagram showing mean GABAA receptor α1 subunit (Gabra1) mRNA expression in the VTA. There was a significant increase, 1‐way ANOVA, F(4, 17) = 4.478, p < 0.05; Tukey's post hoc test *p < 0.05, n = 4 to 5, of Gabra1 mRNA in the VTA of rats withdrawn from chronic EtOH (WV), compared to CS, EV, and WS groups. (B) Bar diagram showing mean GABAA receptor α5 subunit (Gabra5) mRNA expression in the VTA. There was a significant increase, 1‐way ANOVA, F(4, 17) = 4.823, p < 0.01; Tukey's post hoc test *p < 0.05, n = 4 to 5, of Gabra5 mRNA in the VTA of rats withdrawn from chronic EtOH (WV), compared to CS and EV treatment groups. (C) Bar diagram showing mean GABAA receptor α1 subunit protein levels in the VTA. No significant difference between treatment groups was observed (1‐way ANOVA, p > 0.05, n = 5). (D) Mean GABAA receptor α5 subunit protein levels in the VTA. No significant difference between treatment groups was observed (1‐way ANOVA p > 0.05, n = 5).
Results
Effect of SAHA Treatment on VTA GABA Hyposensitivity Induced by Withdrawal from Chronic EtOH
A total of 41 VTA neurons from 19 rats were used for electrophysiological recordings in this study. These neurons had firing rates that ranged from 0.65 to 2.4 Hz, with an overall mean firing rate of 1.46 ± 0.07 Hz. All of the neurons were sensitive to inhibition by GABA. Up to 4 neurons were recorded per slice, with an average number of neurons per slice of 2.1 ± 0.25. Representative single‐cell firing ratemeter graphs are shown in Fig. 1 A.
The summary of GABA‐induced inhibition of cells from all 4 treatment groups (CV, CS, WV, and WS) is shown in Fig. 1 B. Consistent with our previous report in mice (Arora et al., 2013), concentration‐dependent inhibition of firing by GABA was observed in VTA neurons from Sprague Dawley rats in all cells recorded, 3‐way ANOVA, F(2, 111) = 35.0, for main GABA effect, p < 0.001. There was a significant main effect of EtOH diet, 3‐way ANOVA, F(1, 111) = 4.04, for effect of EtOH diet, p < 0.05, but not a main effect of SAHA, and a significant interaction of EtOH diet * SAHA, F(1, 111) = 6.98, p < 0.001. There was no difference in the response of VTA neurons to GABA in control diet‐fed rats injected with either SAHA (CS) or vehicle (CV). When rats were withdrawn from chronic EtOH diet and given vehicle injection (WV), the VTA neurons of these animals showed significant reduction in sensitivity to GABA, compared to the CV and WS groups, 3‐way ANOVA, F(1, 111) = 6.78, for EtOH diet * SAHA effect, p < 0.01 (Tukey post hoc WV different from CV and WS, p < 0.05). These data are consistent with our previous finding (Arora et al., 2013) that showed that withdrawal from repeated treatment with EtOH in mice produced a decrease in the response to GABA in VTA neurons. Interestingly, in the VTA of withdrawn rats that were injected with SAHA (WS), the response to GABA was not different from the control groups (CV and CS). These results indicate that rat VTA neurons are hyposensitive to GABA after withdrawal from chronic EtOH diet treatment and that in vivo administration of SAHA normalizes the attenuated GABA responses.
Our earlier study in mice demonstrated a decreased GABA sensitivity of VTA neurons during withdrawal from repeated EtOH treatment that was reversed by incubation with 3 μM SAHA in vitro (Arora et al., 2013). To correlate with this previous study, we also incubated brain slices from rats after 24‐hour withdrawal (W) with either 3 μM SAHA in DMSO (n = 8) or 0.1% DMSO vehicle (n = 9) for 2 hours before recordings were made. There was no significant difference in the baseline firing rate between the 2 groups (2.18 ± 0.35 Hz for slices incubated with DMSO; 2.33 ± 0.35 Hz for slices incubated with SAHA; t‐test, t = 0.31, p > 0.05), and incubation with SAHA significantly increased GABA sensitivity of VTA neurons from rats in withdrawal from EtOH diet, 2‐way ANOVA, F(1, 7) = 5.65, p < 0.05 (Fig. S1). This result confirms our initial finding in mice and further establishes that a 2‐hour incubation, in vivo or in vitro, can counteract the GABA hyposensitivity of VTA neurons during withdrawal after chronic EtOH exposure.
Effects of Chronic EtOH Exposure and Withdrawal on acH3K9 Levels in the VTA
Our electrophysiological results showed that the HDACi SAHA reversed the GABA hyposensitivity of VTA neurons of rats withdrawn from chronic EtOH diet, suggesting that epigenetic modifications, especially histone acetylation, might have played a role in this response. Previous studies have shown a close association between alcohol withdrawal and acH3K9 (Pandey et al., 2017; Renthal and Nestler, 2008; Shibasaki et al., 2011). In our previous study in mice, we found decreased acH3K9 during withdrawal (Arora et al., 2013); therefore, we measured the levels of acH3K9 in rat VTA using gold immunolabeling. For these experiments, all 5 treatment groups were used (CV, CS, EV, WV, WS).
Figure 2 A shows representative photographs of acH3K9 immunostaining, and the bar graph in Fig. 2 B shows the mean acH3K9 protein levels in the VTA. Withdrawal from chronic EtOH exposure (WV) produced a significant decrease, 1‐way ANOVA, F(4, 20) = 6.85, p < 0.002, n = 5, in acH3K9 in the VTA, compared with all other groups (Tukey's post hoc test p < 0.05); note that the WS group was similar to the CV, CS, and EV groups. This indicates that SAHA normalized acH3K9 levels to control values. SAHA had no effect on the control fed group (CS vs. CV) (Tukey's post hoc test p > 0.05), indicating that SAHA only increased acH3K9 when the withdrawal‐induced deficit was present.
Effects of Chronic EtOH Exposure and Withdrawal on mRNA and Protein Levels of HDAC2 in VTA
The electrophysiology results above indicate that in vivo administration of the HDAC inhibitor SAHA reversed the GABA hyposensitivity induced by withdrawal from chronic EtOH diet. Although there are 11 HDAC isoforms, a limited number of HDAC isoforms have been implicated in alcohol and substance abuse. Among them, the HDAC2 isoform is associated with synaptic plasticity (Guan et al. 2009) and alcohol drinking behaviors (Moonat et al., 2013; Sakharkar et al., 2014). We have also demonstrated an increase in HDAC2 immunoreactivity during EtOH withdrawal in mice (Arora et al., 2013). HDAC2 removal of acetyl groups on histone H3 (H3K9) has been associated with alcohol and drug‐induced changes in gene expression (Pandey et al., 2017; Renthal and Nestler, 2008; Shibasaki et al., 2011). Because acH3K9 was significantly decreased during chronic EtOH withdrawal (Fig. 2), we measured the protein levels of HDAC2 in the VTA using gold immunolabeling. The same animals used to assess acH3K9 levels (CV, CS, EV, WV, WS) were used for the measurement of HDAC2 protein.
Figure 3 A shows photomicrographs of HDAC2 gold immunolabeling in the VTA, and Fig. 3 B shows the quantitative assessment of gold particle density (mean ± SEM of the number of immunogold particles/100 μm2 area) in the VTA. The results indicate a significant increase in HDAC2 protein in the VTA after withdrawal from chronic EtOH exposure compared with control groups, 1‐way ANOVA, F(4, 20) = 7.891, p < 0.002, n = 5. The WV and WS groups were significantly different from the CV, CS, and EV groups (Tukey's post hoc test p < 0.05). The WV and WS groups were not different from each other, nor were the CV, CS, and EV groups different from each other. SAHA treatment also did not induce changes in the HDAC2 in either control (CS vs. CV) or withdrawal groups (WS vs. WV) (Tukey's post hoc test p > 0.05). Note that the EV group was not different from the CV and CS groups (Tukey's post hoc test p > 0.05). These results indicate that GABA hyposensitivity and decreased acH3K9 might be associated with an overexpression of HDAC2 in the VTA after withdrawal from chronic EtOH diet treatment.
We also performed qPCR to determine whether a change in mRNA expression paralleled the change in protein expression. Interestingly, HDAC2 mRNA expression was not changed after chronic EtOH diet (E) and withdrawal (W) compared to control diet (C) (Fig. 3 C). This indicates that the regulation of HDAC2 expression is at the protein but not the transcript level.
Effects of Chronic EtOH Exposure and Withdrawal on mRNA and Protein Levels of GABAA Receptor Subunits in the VTA
SAHA reversed withdrawal‐induced GABA hyposensitivity (Fig. 1), and this phenotype after alcohol treatment and withdrawal is associated with decreased histone acetylation (Fig. 2), which in turn could regulate transcription of genes associated with GABA sensitivity. One group of obvious candidates are genes encoding GABA receptors (Arora et al., 2013; Bohnsack et al., 2017). We have previously shown that blockade of GABAB receptors does not change the response to bath‐applied GABA in VTA neurons (Arora et al., 2013); therefore, the response to these concentrations of exogenously administered GABA is primarily mediated by GABAA receptors. As it is known that changes in GABAA receptor subunit composition can alter the efficacy of the receptor (Whittemore et al., 1996), we measured the mRNA expression levels of GABAA receptor subunits. Results shown in Fig. 4 indicate that the mRNA for GABAA α1, Fig. 4 A, 1‐way ANOVA, F(4, 17) = 4.478, *p < 0.05, and α5, Fig. 4 B, 1‐way ANOVA, F(4, 17) = 4.823, *p < 0.01, subunits is increased after withdrawal from chronic EtOH diet. The induction of Gabra1, but not Gabra5, mRNA was reversible by in vivo SAHA administration.
Since we observed a significant increase in mRNA levels of GABAA receptor α1 and α5 subunits, we measured the protein levels of these 2 subunits in the VTA using the immunogold labeling technique. Figure 4 shows that despite the changes in the mRNA expression, the protein levels of GABAA receptor subunits α1 (Fig. 4 C) and α5 (Fig. 4 D) are not different among the treatment groups (1‐way ANOVA, p > 0.05), indicating that the functional reduction in responsiveness to GABA during withdrawal is not caused by altered protein expression of GABAA receptor subunits α1 and α5.
Discussion
The present study demonstrates that in vivo SAHA treatment reverses GABA hyposensitivity of VTA neurons in rats withdrawn from chronic EtOH exposure. Furthermore, withdrawal was associated with an increase in HDAC2 protein and a decrease in acH3K9 protein levels. The GABA hyposensitivity observed in the present study is consistent with our previous report (Arora et al., 2013) that described the effect of in vitro HDACi treatment on GABA hyposensitivity of VTA neurons in mice repeatedly injected with EtOH. GABA hyposensitivity during withdrawal may be 1 physiological effect of chronic alcohol exposure that is common among different forms of EtOH treatment. Furthermore, in vivo treatment with SAHA 2 hours prior to sacrifice reversed the GABA hyposensitivity. This extends our previous observations and identifies withdrawal‐induced alteration of histone acetylation as a factor in neuroadaptive changes in the VTA. The involvement of histone acetylation was also confirmed with immunohistochemical examination of acH3K9 and HDAC2, which indicates that withdrawal after chronic EtOH exposure increases HDAC2 protein levels and decreases histone acetylation (acH3K9) in the VTA, and that the HDACi SAHA reverses this deficit in histone acetylation. The reversal of the GABA hyposensitivity and acH3K9 by in vivo SAHA further supports the role of HDACi for treatment of alcohol‐induced brain neuropathology.
The results of the present study are further supported by a number of recent findings examining the link between HDACs and brain changes induced by EtOH exposure and withdrawal. Condensation of chromatin as a result of H3K9 deacetylation (e.g., by up‐regulation of HDAC2) should result in decreased gene expression, and HDACi could restore these deficits (Krishnan et al., 2014). Treatment with an HDACi during withdrawal from chronic EtOH exposure restored H3K9 acetylation deficits, prevented the development of anxiety‐like behaviors in rats (Pandey et al., 2008a; You et al., 2014), diminished bingelike drinking in mice (Warnault et al., 2013), and reduced operant alcohol self‐administration in rats (Jeanblanc et al., 2015; Legastelois et al., 2013; Simon‐O'Brien et al., 2015) as well as drinking in alcohol‐preferring rats (Sakharkar et al., 2014). Our own work in the VTA of mice that received repeated EtOH injections indicated a relationship between GABA responsiveness in the VTA and HDAC2 (Arora et al., 2013); the current study reinforces this observation. A variety of epigenetic changes that underlie the GABA hyposensitivity in the VTA during EtOH withdrawal may be correlated with an increase in HDAC2 expression. The immunoreactivity for the HDAC2 isoform in the VTA is increased by withdrawal after chronic EtOH exposure, but the mRNA of HDAC2 was not altered. The lack of change in mRNA levels of HDAC2 suggests that the increase in HDAC2 is mediated post‐translationally; additional studies will be needed to examine any alteration in the protein levels of other HDAC isoforms. We also found that SAHA treatment did not normalize the increase in HDAC2 observed during withdrawal from chronic EtOH exposure. These results suggest that SAHA normalized deficits in acH3K9 most likely via inhibition of HDAC activity but not by reducing HDAC2 protein levels in VTA.
Despite our earlier studies indicating that EtOH directly excites VTA DA neurons (Brodie et al., 1999), numerous other factors clearly alter EtOH‐induced excitation of DA VTA neurons, and, in turn, could affect the rewarding value of EtOH. EtOH hyperpolarizes GABA‐containing neurons of the VTA (Gallegos et al., 1999; Xiao and Ye, 2008) and can affect glutamate action on VTA DA neurons (Stobbs et al., 2004). Although the neurons recorded in this study had electrophysiological characteristics of DA VTA neurons, we do not strictly know whether the neuronal activity recorded in the present study is that of DA VTA neurons or non‐DA neurons. However, a decrease in sensitivity to GABA inhibition for any neuron in the VTA could lead to a disruption of reward signal processing. In addition to affecting the sensitivity to GABA inhibition, withdrawal from chronic EtOH treatment may alter the responsiveness of VTA neurons to other neurotransmitters. Although we have previously observed GABA hyposensitivity with no change in the response to NMDA (Arora et al., 2013; Brodie, 2002), it is possible that the increase in membrane resistance induced by decreased activation of GABA receptors could affect responsiveness to other neurotransmitters and agents, like DA. Increased inhibition by DA has been shown following in vivo EtOH experience (Perra et al., 2011), and we have shown disruption of D2 receptor desensitization on VTA neurons by EtOH (Nimitvilai et al., 2012), which together could shift the balance of the synaptic inputs to these neurons toward less activity. This combination of events could underlie the decrease in VTA dopaminergic neuronal activity during EtOH withdrawal (Bailey et al., 2001; Diana et al., 1992; Ludlow et al., 2009; Shen and Chiodo, 1993). Dynamic neuroadaptation in regulation of VTA neuronal activity could result in additional changes in DA VTA neuronal activity. For example, loss of GABAergic inhibition in the VTA, in combination with alteration in autoreceptor function, could result in a hyperdopaminergic state during protracted abstinence (Hirth et al., 2016). Extensive time‐course studies will be necessary to establish how withdrawal alters neurotransmission in the VTA over time, and how these alterations might shift the balance from reward to aversion during abstinence to promote relapse (You et al., 2018).
Although activation of GABAB receptors can cause inhibition of DAergic VTA neurons (Lacey et al., 1988; Mueller and Brodie, 1989), we have shown that the inhibition of VTA neuronal firing by bath‐applied GABA is mediated primarily by GABAA receptors (Arora et al., 2013). GABAA sensitivity can be altered in several ways: by a shift in GABAA subunit composition (Liang et al., 2007; Papadeas et al., 2001), by other changes in the number or type of subunit expressed via alterations in gene expression, posttranslational modifications, synaptic localization, and intracellular signaling (Kumar et al., 2004). It is also possible that GABA responsiveness is achieved by changing receptor trafficking, by altering subunit composition such as a decrease in GABAA β3 subunit (Parker et al., 2011), or by reduction in endogenous GABA modulators like neurosteroids (Cook et al., 2014a,b; Olsen, 2018). We found that GABAA α1 and α5 mRNA increased significantly after withdrawal from chronic EtOH exposure. We did not examine other GABAA receptor subunit mRNA, so there may be reduction in 1 or more of the other 17 subunits (Sigel and Steinmann, 2012). Despite the mRNA changes for GABAA α1 and α5 subunits, we did not observe differences in protein expression of α1 or α5 subunit. These results indicate that changes in GABAA receptor subunit expression levels (α1 or α5) during EtOH withdrawal were not likely to have been responsible for the decreased sensitivity to GABA. It is possible that the increase in GABAA α1 and α5 transcripts is due to a homeostatic response to decreased GABA sensitivity induced during EtOH withdrawal. Interestingly, during EtOH withdrawal in mice, we noted alterations of GABAA α1 subunit composition (Arora et al., 2013). Therefore, physiological changes in rat VTA in response to withdrawal from Lieber‐DeCarli diet differ from withdrawal‐induced changes in mouse VTA after repeated EtOH injections. Although there are numerous differences in the 2 methods, the most prominent differences are that the injection schedule that we used in our previous studies in mice was more intermittent, the administration in the mice was i.p. not oral, the bolus EtOH administration in the mice differed from the more regular intake of the Lieber‐DeCarli diet in rats, and, of course, the species difference. It is known that different alcohol administration paradigms or durations can produce different withdrawal‐induced alterations (Knapp and Breese, 2012; Lopez and Becker, 2005), including different shifts in GABAA receptor subunit gene expression (Matthews et al., 1998). Alteration of sensitivity to GABA could occur through other mechanisms, including decreased production of allopregnanolone (Beattie et al., 2017; Hasirci et al., 2017; Maldonado‐Devincci et al., 2014), redox modulation (Calvo and Beltran Gonzalez, 2016), or regulation of synthesis, transport, or degradation of GABA (Roth and Draguhn, 2012). Withdrawal‐induced alteration of processes (e.g., allopregnanolone synthesis) in other types of cells (e.g., astrocytes or microglia) within the VTA could result in a decrease in GABAergic neurotransmission. It should be noted that increases in allopregnanolone levels were observed in the VTA of human alcoholics (Hasirci et al., 2017), but no difference in allopregnanolone was observed in the VTA between controls and alcohol‐exposed mice (Maldonado‐Devincci et al., 2014). The reversal of GABA hyposensitivity by SAHA and changes in HDAC2 and acH3K9 associated with withdrawal would be consistent with a more condensed chromatin during withdrawal and a decrease in mRNA expression of some of the factors increasing GABA sensitivity. Extensive additional studies will be needed to identify the factors and cell types under epigenetic control that are altered during withdrawal. Most interesting and perhaps most importantly, GABA hyposensitivity during withdrawal is reversed by HDACi in both mice and rats, despite differences between these species and the alcohol treatment methods that were used. This suggests that alcohol withdrawal‐induced hyposensitivity to GABA inhibition in the VTA is a robust phenomenon and may be an important and fundamental response to alcohol withdrawal.
Electrophysiological responsiveness assessed the functional inhibitory action of GABA. We had shown previously that incubating brain slices with HDACi restored GABA sensitivity. Importantly, here we report that in vivo administration of the HDACi SAHA resulted in a normalization of GABA sensitivity as well. As with our previous study, chronic EtOH treatment and withdrawal produced a decrease in histone acetylation and up‐regulation of HDAC2. In our current study, SAHA was given in vivo, and no additional SAHA was administered to the brain slices during the superfusion. Therefore, once in vivo SAHA reversed the withdrawal‐induced GABA hyposensitivity, normal GABA responsiveness was sustained throughout the duration of the experimental day (from 1 to 7 hours after sacrifice). Furthermore, in vivo treatment with SAHA restored GABA sensitivity of VTA neurons during withdrawal but did not alter GABA responsiveness of control VTA neurons, suggesting SAHA produced a normalization rather than simply a general increase in GABA sensitivity. Interestingly, our current study shows that 2 hours between systemic SAHA injection and sacrifice was sufficient time for the phenotypic reversal of GABA sensitivity in VTA neurons. This was the same amount of time allowed for in vitro HDACi treatment both in our previous study (Arora et al., 2013) and in our experiment in rat VTA slices (Fig. S1), indicating that whether the HDACi is administered in vivo or in vitro, 2 hours is a physiologically relevant time frame for changes in histone acetylation and subsequent transcription of those elements related to the GABA sensitivity of VTA neurons.
Alteration of responsiveness of VTA neurons to GABA may be a physiologically significant factor during withdrawal. The association of these changes in the effects of GABA and epigenetic factors is consistent across the 2 models we have examined thus far and indicates that HDACi can reverse this withdrawal‐induced alteration in physiology of VTA neurons. Whether other withdrawal‐induced changes in the VTA, like increased sensitivity to EtOH excitation (Brodie, 2002), are also regulated by HDACs or other epigenetic factors is another interesting question to be answered. The change in HDAC activity during withdrawal might be linked to withdrawal‐induced craving, with reversal by an HDACi like SAHA. The fact that in vivo SAHA reverses changes in GABA responsiveness in the VTA during withdrawal after chronic EtOH exposure increases the potential for the use of systemic HDACi administration as therapeutic treatment for alcohol use disorder.
Conflicts of Interest
CY, BJV, HZ, AWL, and MSB reported no potential conflict of interests. SCP reports that a U.S. patent application entitled “Histone acetyltransferase activators and histone deacetylase inhibitors in the treatment of alcoholism” (serial number 60/848237 filed on September 29, 2006) is currently pending.
Supporting information
Fig. S1. In vitro SAHA increases GABA sensitivity of VTA neurons during withdrawal.
Table S1. Primers used for qPCR in VTA.
Acknowledgments
The authors thank Ying Chen for help with RNA isolation. The authors gratefully acknowledge that this work was supported by grants P50AA022538 (SCP, AWL, MSB) and R01AA05846 (MSB) from National Institute on Alcohol Abuse and Alcoholism and Senior Research Career Scientist award to SCP from the Department of Veterans Affairs.
References
- Albanese A, Minciacchi D (1983) Organization of the ascending projections from the ventral tegmental area: a multiple fluorescent retrograde tracer study in the rat. J Comp Neurol 216:406–420. [DOI] [PubMed] [Google Scholar]
- Arora D, Haluk DM, Kourrich S, Pravetoni M, Fernandez‐Alacid L, Nicolau JC, Lujan R, Wickman K (2010) Altered neurotransmission in the mesolimbic reward system of Girk mice. J Neurochem 114:1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora DS, Nimitvilai S, Teppen TL, McElvain MA, Sakharkar AJ, You C, Pandey SC, Brodie MS (2013) Hyposensitivity to gamma‐aminobutyric acid in the ventral tegmental area during alcohol withdrawal: reversal by histone deacetylase inhibitors. Neuropsychopharmacology 38:1674–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Authement ME, Langlois LD, Kassis H, Gouty S, Dacher M, Shepard RD, Cox BM, Nugent FS (2016) Morphine‐induced synaptic plasticity in the VTA is reversed by HDAC inhibition. J Neurophysiol 116:1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, O'Callaghan MJ, Croft AP, Manley SJ, Little HJ (2001) Alterations in mesolimbic dopamine function during the abstinence period following chronic ethanol consumption. Neuropharmacology 41:989–999. [DOI] [PubMed] [Google Scholar]
- Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT (1991) CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology 103:227–232. [DOI] [PubMed] [Google Scholar]
- Beattie MC, Maldonado‐Devincci AM, Porcu P, O'Buckley TK, Daunais JB, Grant KA, Morrow AL (2017) Voluntary ethanol consumption reduces GABAergic neuroactive steroid (3alpha,5alpha)3‐hydroxypregnan‐20‐one (3alpha,5alpha‐THP) in the amygdala of the cynomolgus monkey. Addict Biol 22:318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkel TD, Pandey SC (2017) Emerging role of epigenetic mechanisms in alcohol addiction. Alcohol Clin Exp Res 41:666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack JP, Hughes BA, O'Buckley TK, Edokpolor K, Besheer J, Morrow AL (2018) Histone deacetylases mediate GABAA receptor expression, physiology, and behavioral maladaptations in rat models of alcohol dependence. Neuropsychopharmacology 43:1518–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack JP, Patel VK, Morrow AL (2017) Ethanol exposure regulates Gabra1 expression via histone deacetylation at the promoter in cultured cortical neurons. J Pharmacol Exp Ther 363:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botia B, Legastelois R, Alaux‐Cantin S, Naassila M (2012) Expression of ethanol‐induced behavioral sensitization is associated with alteration of chromatin remodeling in mice. PLoS One 7:e47527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie MS (2002) Increased ethanol excitation of dopaminergic neurons of the ventral tegmental area after chronic ethanol treatment. Alcohol Clin Exp Res 26:1024–1030. [DOI] [PubMed] [Google Scholar]
- Brodie MS, Pesold C, Appel SB (1999) Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin Exp Res 23:1848–1852. [PubMed] [Google Scholar]
- Brodie MS, Shefner SA, Dunwiddie TV (1990) Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain Res 508:65–69. [DOI] [PubMed] [Google Scholar]
- Calvo DJ, Beltran Gonzalez AN (2016) Dynamic regulation of the GABAA receptor function by redox mechanisms. Mol Pharmacol 90:326–333. [DOI] [PubMed] [Google Scholar]
- di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic dopamine system of freely moving rats. Proc Natl Acad Sci USA 85:5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JB, Dumitru AM, O'Buckley TK, Morrow AL (2014a) Ethanol administration produces divergent changes in GABAergic neuroactive steroid immunohistochemistry in the rat brain. Alcohol Clin Exp Res 38:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JB, Werner DF, Maldonado‐Devincci AM, Leonard MN, Fisher KR, O'Buckley TK, Porcu P, McCown TJ, Besheer J, Hodge CW, Morrow AL (2014b) Overexpression of the steroidogenic enzyme cytochrome P450 side chain cleavage in the ventral tegmental area increases 3alpha,5alpha‐THP and reduces long‐term operant ethanol self‐administration. J Neurosci 34:5824–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Addario C, Caputi FF, Ekstrom TJ, di Benedetto M, Maccarrone M, Romualdi P, Candeletti S (2013) Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex. J Mol Neurosci 49:312–319. [DOI] [PubMed] [Google Scholar]
- Diana M, Pistis M, Muntoni A, Rossetti ZL, Gessa G (1992) Marked decrease of A10 dopamine neuronal firing during ethanol withdrawal syndrome in rats. Eur J Pharmacol 221:403–404. [DOI] [PubMed] [Google Scholar]
- Dominguez G, Dagnas M, Decorte L, Vandesquille M, Belzung C, Beracochea D, Mons N (2016) Rescuing prefrontal cAMP‐CREB pathway reverses working memory deficits during withdrawal from prolonged alcohol exposure. Brain Struct Funct 221:865–877. [DOI] [PubMed] [Google Scholar]
- Finegersh A, Ferguson C, Maxwell S, Mazariegos D, Farrell D, Homanics GE (2015) Repeated vapor ethanol exposure induces transient histone modifications in the brain that are modified by genotype and brain region. Front Mol Neurosci 8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos RA, Lee RS, Criado JR, Henriksen SJ, Steffensen SC (1999) Adaptive responses of gamma‐aminobutyric acid neurons in the ventral tegmental area to chronic ethanol. J Pharmacol Exp Ther 291:1045–1053. [PubMed] [Google Scholar]
- Gessa GL, Muntoni F, Collu M, Vargiu L, Mereu G (1985) Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain Res 348:201–203. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459:55‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto JG, Gavin DP, Wiren KM, Crabbe JC, Guizzetti M (2017) Prefrontal cortex expression of chromatin modifier genes in male WSP and WSR mice changes across ethanol dependence, withdrawal, and abstinence. Alcohol 60:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasirci AS, Maldonado‐Devincci AM, Beattie MC, O'Buckley TK, Morrow AL (2017) Cellular GABAergic neuroactive steroid (3alpha,5alpha)‐3‐hydroxy‐pregnan‐20‐one (3alpha,5alpha‐THP) immunostaining levels are increased in the ventral tegmental area of human alcohol use disorder patients: a postmortem study. Alcohol Clin Exp Res 41:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirth N, Meinhardt MW, Noori HR, Salgado H, Torres‐Ramirez O, Uhrig S, Broccoli L, Vengeliene V, Rossmanith M, Perreau‐Lenz S, Kohr G, Sommer WH, Spanagel R, Hansson AC (2016) Convergent evidence from alcohol‐dependent humans and rats for a hyperdopaminergic state in protracted abstinence. Proc Natl Acad Sci USA 113:3024–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janak PH, Tye KM (2015) From circuits to behaviour in the amygdala. Nature 517:284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanblanc J, Lemoine S, Jeanblanc V, Alaux‐Cantin S, Naassila M (2015) The class I‐specific HDAC inhibitor MS‐275 decreases motivation to consume alcohol and relapse in heavy drinking rats. Int J Neuropsychopharmacol 18:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Fields HL, Baxter MG, Saper CB, Holland PC (2009) The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron 61:786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallo I, Molnar CS, Szoke S, Fekete C, Hrabovszky E, Liposits Z (2015) Area‐specific analysis of the distribution of hypothalamic neurons projecting to the rat ventral tegmental area, with special reference to the GABAergic and glutamatergic efferents. Front Neuroanat 9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsi G, Prescott CA, Kendler KS, Riley BP (2009) Unraveling the molecular mechanisms of alcohol dependence. Trends Genet 25:49–55. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Breese GR (2012) Models of chronic alcohol exposure and dependence. Methods Mol Biol 829:205–230. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35:217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2016) Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3:760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan HR, Sakharkar AJ, Teppen TL, Berkel TD, Pandey SC (2014) The epigenetic landscape of alcoholism. Int Rev Neurobiol 115:75–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Fleming RL, Morrow AL (2004) Ethanol regulation of gamma‐aminobutyric acid A receptors: genomic and nongenomic mechanisms. Pharmacol Ther 101:211–226. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA (1988) On the potassium conductance increase activated by GABAB and dopamine D2 receptors in rat substantia nigra neurones. J Physiol (Lond) 401:437–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA (1989) Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. J Neurosci 9:1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legastelois R, Botia B, Naassila M (2013) Blockade of ethanol‐induced behavioral sensitization by sodium butyrate: descriptive analysis of gene regulations in the striatum. Alcohol Clin Exp Res 37:1143–1153. [DOI] [PubMed] [Google Scholar]
- Liang J, Suryanarayanan A, Abriam A, Snyder B, Olsen RW, Spigelman I (2007) Mechanisms of reversible GABAA receptor plasticity after ethanol intoxication. J Neurosci 27:12367–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM, Sorrell MF (1989) Experimental methods of ethanol administration. Hepatology 10:501–510. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Becker HC (2005) Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology 181:688–696. [DOI] [PubMed] [Google Scholar]
- Ludlow KH, Bradley KD, Allison DW, Taylor SR, Yorgason JT, Hansen DM, Walton CH, Sudweeks SN, Steffensen SC (2009) Acute and chronic ethanol modulate dopamine D2‐subtype receptor responses in ventral tegmental area GABA neurons. Alcohol Clin Exp Res 33:804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Ungless MA (2006) The mechanistic classification of addictive drugs. PLoS Med 3:e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado‐Devincci AM, Cook JB, O'Buckley TK, Morrow DH, McKinley RE, Lopez MF, Becker HC, Morrow AL (2014) Chronic intermittent ethanol exposure and withdrawal alters (3alpha,5alpha)‐3‐hydroxy‐pregnan‐20‐one immunostaining in cortical and limbic brain regions of C57BL/6J mice. Alcohol Clin Exp Res 38:2561–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Toy B, Himmels P, Morales M, Fields HL (2012) Identification of rat ventral tegmental area GABAergic neurons. PLoS One 7:e42365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DB, Devaud LL, Fritschy JM, Sieghart W, Morrow AL (1998) Differential regulation of GABA(A) receptor gene expression by ethanol in the rat hippocampus versus cerebral cortex. J Neurochem 70:1160–1166. [DOI] [PubMed] [Google Scholar]
- Moonat S, Sakharkar AJ, Zhang H, Pandey SC (2011) The role of amygdaloid brain‐derived neurotrophic factor, activity‐regulated cytoskeleton‐associated protein and dendritic spines in anxiety and alcoholism. Addict Biol 16:238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC (2013) Aberrant HDAC2‐mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biol Psychiatry 73:763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Starkman BG, Sakharkar A, Pandey SC (2010) Neuroscience of alcoholism: molecular and cellular mechanisms. Cell Mol Life Sci 67:73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MA, Hagelkruys A, Seiser C (2014) Transcription and beyond: the role of mammalian class I lysine deacetylases. Chromosoma 123:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AL, Brodie MS (1989) Intracellular recording from putative dopamine‐containing neurons in the ventral tegmental area of Tsai in a brain slice preparation. J Neurosci Methods 28:15–22. [DOI] [PubMed] [Google Scholar]
- Nimitvilai S, Arora DS, McElvain MA, Brodie MS (2012) Ethanol blocks the reversal of prolonged dopamine inhibition of dopaminergic neurons of the ventral tegmental area. Alcohol Clin Exp Res 36:1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimitvilai S, Brodie MS (2010) Reversal of prolonged dopamine inhibition of dopaminergic neurons of the ventral tegmental area. J Pharmacol Exp Ther 333:555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oades RD, Halliday GM (1987) Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res 434:117–165. [DOI] [PubMed] [Google Scholar]
- Olsen RW (2018) GABAA receptor: positive and negative allosteric modulators. Neuropharmacology 136:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Kyzar EJ, Zhang H (2017) Epigenetic basis of the dark side of alcohol addiction. Neuropharmacology 122:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Piano MR, Schwertz DW, Davis JM, Pandey GN (1992) Effect of ethanol administration and withdrawal on serotonin receptor subtypes and receptor‐mediated phosphoinositide hydrolysis in rat brain. Alcohol Clin Exp Res 16:1110–1116. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H, Xu T (2004) Partial deletion of the cAMP response element‐binding protein gene promotes alcohol‐drinking behaviors. J Neurosci 24:5022–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A (2008a) Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci 28:3729–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Ugale R, Prakash A, Xu T, Misra K (2008b) Effector immediate‐early gene arc in the amygdala plays a critical role in alcoholism. J Neurosci 28:2589–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadeas S, Grobin AC, Morrow AL (2001) Chronic ethanol consumption differentially alters GABA(A) receptor alpha1 and alpha4 subunit peptide expression and GABA(A) receptor‐mediated 36 Cl(‐) uptake in mesocorticolimbic regions of rat brain. Alcohol Clin Exp Res 25:1270–1275. [PubMed] [Google Scholar]
- Parker JG, Wanat MJ, Soden ME, Ahmad K, Zweifel LS, Bamford NS, Palmiter RD (2011) Attenuating GABAA receptor signaling in dopamine neurons selectively enhances reward learning and alters risk preference in mice. J Neurosci 31:17103–17112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The Rat Brain in Stereotaxic Coordinates Academic Press, San Diego. [Google Scholar]
- Perra S, Clements MA, Bernier BE, Morikawa H (2011) In vivo ethanol experience increases D(2) autoinhibition in the ventral tegmental area. Neuropsychopharmacology 36:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang M, Denny A, Lieu M, Carreon S, Li J (2011) Histone H3K9 modifications are a local chromatin event involved in ethanol‐induced neuroadaptation of the NR2B gene. Epigenetics 6:1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, McBride WJ (2000) Feedback control of mesolimbic somatodendritic dopamine release in rat brain. J Neurochem 74:684–692. [DOI] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ (2008) Epigenetic mechanisms in drug addiction. Trends Mol Med 14:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth FC, Draguhn A (2012) GABA metabolism and transport: effects on synaptic efficacy. Neural Plast 2012:805830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Zhang H, Tang L, Baxstrom K, Shi G, Moonat S, Pandey SC (2014) Effects of histone deacetylase inhibitors on amygdaloid histone acetylation and neuropeptide Y expression: a role in anxiety‐like and alcohol‐drinking behaviours. Int J Neuropsychopharmacol 17:1207–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC (2012) Histone deacetylases (HDAC)‐induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol Clin Exp Res 36:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharda DR, Miller‐Lee JL, Kanski GM, Hunter JC, Lang CH, Kennett MJ, Korzick DH (2012) Comparison of the agar block and Lieber‐DeCarli diets to study chronic alcohol consumption in an aging model of Fischer 344 female rats. J Pharmacol Toxicol Methods 66:257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R‐Y, Chiodo LA (1993) Acute withdrawal after repeated ethanol treatment reduces the number of spontaneously active dopaminergic neurons in the ventral tegmental area. Brain Res 622:289–293. [DOI] [PubMed] [Google Scholar]
- Shibasaki M, Mizuno K, Kurokawa K, Ohkuma S (2011) Enhancement of histone acetylation in midbrain of mice with ethanol physical dependence and its withdrawal. Synapse 65:1244–1250. [DOI] [PubMed] [Google Scholar]
- Sigel E, Steinmann ME (2012) Structure, function, and modulation of GABA(A) receptors. J Biol Chem 287:40224–40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon‐O'Brien E, Alaux‐Cantin S, Warnault V, Buttolo R, Naassila M, Vilpoux C (2015) The histone deacetylase inhibitor sodium butyrate decreases excessive ethanol intake in dependent animals. Addict Biol 20:676–689. [DOI] [PubMed] [Google Scholar]
- Stobbs SH, Ohran AJ, Lassen MB, Allison DW, Brown JE, Steffensen SC (2004) Ethanol suppression of ventral tegmental area GABA neuron electrical transmission involves N‐methyl‐D‐aspartate receptors. J Pharmacol Exp Ther 311:282–289. [DOI] [PubMed] [Google Scholar]
- Warnault V, Darcq E, Levine A, Barak S, Ron D (2013) Chromatin remodeling–a novel strategy to control excessive alcohol drinking. Transl Psychiatry 3:e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittemore ER, Yang W, Drewe JA, Woodward RM (1996) Pharmacology of the human gamma‐aminobutyric acidA receptor alpha 4 subunit expressed in Xenopus laevis oocytes. Mol Pharmacol 50:1364–1375. [PubMed] [Google Scholar]
- Xiao C, Ye JH (2008) Ethanol dually modulates GABAergic synaptic transmission onto dopaminergic neurons in ventral tegmental area: role of mu‐opioid receptors. Neuroscience 153:240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You C, Vandegrift B, Brodie MS (2018) Ethanol actions on the ventral tegmental area: novel potential targets on reward pathway neurons. Psychopharmacology 235:1711–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You C, Zhang H, Sakharkar AJ, Teppen T, Pandey SC (2014) Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment. Int J Neuropsychopharmacol 17:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. In vitro SAHA increases GABA sensitivity of VTA neurons during withdrawal.
Table S1. Primers used for qPCR in VTA.