

Abstract
The sodium iodide symporter (NIS) is a classical iodide pump typically localized within the cell plasma membrane in thyroid cells, where NIS expression is believed to ensure success of mainstay radioiodide therapy in thyroid cancers. Although radioiodide uptake is generally reduced in thyroid cancer tissue, intracellular non-membranous NIS has been reported to increase, suggesting that NIS serves a pump-independent function. Thyroid cancer is one of the major component cancers of Cowden syndrome, a subset of which is caused by germline mutations in PTEN. In this study, we explored the non-canonical tumorigenic role of NIS in thyroid cancer cells in relation to PTEN signaling. PTEN knockdown in thyroid cancer cell lines stabilized intracellular NIS protein by promoting an interaction with NIS-LARG (leukemia associated RhoA guanine exchange factor). Increased protein levels of cytoplasmic NIS enhanced RhoA activation and resulted in a pro-migration tumorigenic phenotype. Inhibition of NIS glycosylation through activation of the PI3K/AKT/mTOR signaling pathway contributed to mislocalization of NIS in the cytoplasm, facilitating its non-pump tumorigenic function through an interaction with LARG, which predominantly localized in the cytoplasm. Moreover, PTEN or PI3K/AKT/mTOR signaling could affect DPAGT1, a glycosylating enzyme involved in the initial step of N-linked glycosylation, to inhibit glycosylation of NIS. In summary, our results elucidate a pump-independent, pro-tumorigenic role for NIS in thyroid cancer via its crosstalk with PTEN signaling.
Introduction
The sodium iodide symporter (NIS), encoded by the SLC5A5 gene (solute carrier family 5, member 5), is a plasma membrane protein expressed at high levels in the thyroid gland and the lactating breast (1–3). NIS mediates iodide uptake from the bloodstream into thyroid follicular cells for thyroid hormone biosynthesis, and iodide secretion into breastfeeding milk (4). NIS-mediated iodide uptake is the basis for diagnostic nuclear imaging and radioiodine therapy in thyroid-related diseases. In differentiated thyroid cancer (DTC), radioiodine-131 (I-131) is routinely utilized for remnant ablation and post-surgical adjuvant/targeted therapy (5). Therefore, while NIS is frequently studied in thyroid cancers, focus has been on its classical iodide-pump function.
Radioiodide uptake is generally reduced in thyroid cancer compared with normal thyroid tissue, and decreased NIS expression is widely believed to cause resistance (6). However, studies of NIS expression levels in DTC have yielded divergent data (2,7–13). Studies reporting increased NIS levels show mostly intracellular localization, and thus associated with reduced radioiodide uptake in these cancers. Similarly, NIS has been reported to be over-expressed, but largely retained intracellularly in 70–80% of breast cancers (13,14) and a number of other primary non-thyroidal cancers (15–17). We therefore hypothesized that in addition to the canonical iodide-pump function, NIS could have iodide pump-independent function when localized intracellularly in thyroid cancer cells. This hypothesis is important because the mainstay of treatment of advanced thyroid cancers remains radioiodine.
Interestingly, the two main cancers with reportedly elevated NIS, namely thyroid and breast cancers, are major phenotypic components of Cowden syndrome (CS). CS is an autosomal dominant, difficult-to-recognize and under-diagnosed disorder, characterized by high lifetime risks of thyroid, breast and other cancers (18,19). A subset of CS is caused by germline mutations in the tumor suppressor gene phosphatase and tensin homolog (PTEN) (18,20,21). PTEN antagonizes the PI3K/AKT/mTOR signaling pathway and is involved in many different sporadic cancer-related signaling pathways (22). Though the direct association between PTEN alterations and NIS is unknown, PI3K signaling upregulation has been reported to be associated with reduced iodide uptake in thyroid cancer cells (23). We therefore hypothesized that PTEN alterations in thyroid cancer can affect NIS protein levels or subcellular localization, which can, in turn, promote tumorigenesis independent of its iodide-pump function. Hence, we investigated the non-pump function of NIS in human thyroid cancer, downstream cellular phenotypes, and how PTEN and downstream signaling regulate these functions.
Materials and Methods
Cell lines and culture conditions
We utilized BCPAP, 8505C and FTC-133 thyroid cancer cell lines (Supplementary Table S1) stably expressing full-length human NIS (FL hNIS) (24). BCPAP cells were grown in RPMI-1640 medium, and 8505C, FTC-133 cells were cultured in Modified MEM medium (Sigma M0325, St. Louis, MO), supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cell lines were maintained at 37°C and 5% CO2 culture conditions and tested negative upon routine mycoplasma testing using the MycoAlert Mycoplasma Detection Kit (Lonza, Allendale, NJ). All experiments were conducted with cells at passage numbers between 3 and 15. All cell lines were authenticated through the American Type Culture Collection (ATCC) human cell authentication service (ATCC® 135-XV™) and were 100% matched to the reported STR profiles in the DSMZ database (test date 19/04/2018).
Reagents
Tunicamycin, Brefeldin A and rapamycin were purchased from Sigma. LY294002 and MK-2206 were obtained from Selleckchem (Houston, TX). Dimethyl sulfoxide (DMSO, Sigma) served as vehicle control for experiments involving de-glycosylation drug or PI3K/AKT/mTOR inhibitor treatments. Rabbit anti-NIS (Pr 2890, Rb 4430) is an in-house generated and validated antibody (25).
RNA extraction and qRT-PCR
RNA was extracted from the cell lines using the RNeasy Mini kit (Qiagen, Germantown, MD), purified using Turbo DNase treatment (Life Technologies, Grand Island, NY), and reverse transcribed using Superscript III reverse transcriptase (Life Technologies). Primers were designed for gene transcripts of interest and cDNA quantified using SYBR Green (Life Technologies). We utilized the Applied Biosystems 7500 Real-Time PCR System. Results were analyzed using the standard ΔΔCT method.
Immunoblotting
Protein was extracted from whole cell lysates using the Mammalian Protein Extraction Reagent M-PER (Thermo Scientific Pierce, Rockford, IL) supplemented with a cocktail of protease and phosphatase inhibitors (Sigma) and quantified through the BCA protein assay (Thermo Scientific Pierce). Lysates were separated by SDS-PAGE and transferred onto nitrocellulose membranes. We probed for rabbit anti-NIS at 1:4000, anti-PTEN (6H2.1) mouse monoclonal (Cascade Bioscience, Winchester, MA) at 1:1000, anti-LARG mouse monoclonal (EMD Millipore, Temecula, CA) at 1:5000, and anti-GAPDH rabbit monoclonal (Cell Signaling #2118) at 1:20000 dilution. Blots were scanned digitally using the GE Amersham Imager 600 (GE Healthcare Life Science, Chicago, IL). Densitometry was performed using ImageJ software. Uncropped versions of cropped immunoblots are included in Supplementary Fig. S1A-G.
PTEN transient transfection
Cells in 6-well plates were transfected with 0.2 μg GFP-tagged wild type PTEN plasmid (pcDNA3.1 vector) or empty vector pcDNA3.1 with Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cells were grown for at least 48 hours before harvesting.
RhoA pull-down activation assay
Rho activity was measured using the RhoA pull-down activation assay kit (Cytoskeleton, Denver, CO) as previously reported [18]. Relative variations in normalized RhoA-GTP level is defined as the ratio of the level of activated GTP-bound forms of RhoA and the level of total RhoA in NIS stable cells, divided by the same ratio in control cells.
Immunoprecipitation
Whole cell lysates were prepared by using M-PER as mentioned above, and were pre-cleared by incubation with Thermo Protein A/G Dynabeads (Thermo Scientific Pierce) for 3 hours at 4°C on a rotator. Pre-cleared protein lysates were quantified with the BCA Protein Assay Kit, and 2 mg/ml lysates were prepared. We used anti-NIS, anti-LARG and anti-RhoA antibodies for pull-down and immunoblotting at the recommended dilutions. Cell lysates were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Blots were scanned digitally using the GE Amersham Imager 600 (GE Healthcare Life Science).
Knockdown of NIS, PTEN, LARG, or DPATG1 through siRNA
Cells in 6-well plates were transfected with NIS, PTEN, LARG or DPAGT1 siRNA smartpool (Dharmacon, Lafayette, CO) using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. We used ON-TARGET non-targeting siRNA pool (Dharmacon) as a control (siNT). Cells were collected for downstream analysis 12 to 96 hours after transfection. We used Western blot and qRT-PCR analysis to confirm knockdown.
Immunofluorescence
Cells grown on coverslips were fixed with 4% paraformaldehyde for 2 minutes and then incubated with 1% Triton X-100 for 2 minutes at room temperature (RT). Cells were blocked with 10% goat serum (Vector Laboratories Inc, Burlingame, CA) for 1 hour at RT, and incubated with rabbit anti-NIS and/or mouse anti-LARG (both at 1:500 dilution) overnight at 4°C. Secondary antibodies (Alexa 488/568) were incubated at 1:2000 dilution for 1 hour in the dark at RT. Coverslips were mounted using ProLong Gold Antifade mountant with DAPI (Invitrogen). Slides were visualized and images obtained using the Leica TCS SP5 II confocal microscope (Leica Microsystems, Heidelberg, Germany).
Quantification of cell surface and cytoplasmic NIS
Cell surface protein was isolated using Pierce™ Cell Surface Protein Isolation Kit (Thermo Fisher Scientific), as previously reported (26). The cytoplasmic fraction was isolated using Mem-PER™ Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific). Briefly, cell pellets were washed with wash buffer and incubated in 500 μl of permeabilization buffer for 10 minutes at 4°C with constant mixing. We centrifuged the permeabilized cells for 15 minutes at 16,000×g. The resultant supernatant contains cytoplasmic proteins. NIS in cell surface and cytoplasmic fractions was analyzed by immunoblotting.
Cycloheximide (CHX) chase assay
To test NIS protein stability (27), BCPAP-FL hNIS and 8505C-FL hNIS cell lines were seeded in 6-well plates and allowed to grow overnight. Cells were transfected with PTEN siRNA or non-targeting siRNA for 48 hours. At 48 hours, 100 μg/ml CHX was added to cell media. Following 0, 1, 2, 4, 8, 12, 24, 48 and 72 hours, lysates were prepared with MPER and Western blot analysis performed with anti-NIS and anti-GAPDH antibodies.
In Vitro migration assay
For wound-healing assays, confluent-transfected cells monolayers were scraped with a fine sterile pipette tip, placed in the cell incubator (5% CO2, 37°C). Image were acquired at 0 and 20 hours after scraping using a Leica DMI3000B microscope (Leica Microsystems). For migration inhibitor tests, Rho-associated coiled-coil kinase (ROCK) inhibitor Y-27632 (Sigma) at 30 μM was added at 1 hour before scraping. The wound margin area was determined by image processing using ImageJ software. The average wound-healing rates were calculated from 3 independent experiments.
Iodide uptake assay
Iodine uptake of BCPAP-FL hNIS and 8505C-hNIS cells transiently transfected with siPTEN or siNT was performed with a non-radioiodine uptake assay, as previously reported (28). The iodide uptake of experimental wells was determined after linear regression of the calibration curve and normalized to DNA amount determined in parallel wells. Non-NIS-mediated iodide uptake was examined by conducting parallel experiments in the presence of perchlorate (Sigma), a selective inhibitor for NIS-mediated iodide uptake.
Statistical analyses
Experimental data between control and experimental cells are given as means ± SEM with n corresponding to the number of experiments performed. The Student’s t test was used for significance testing as indicated in figure legends. All statistical tests were two-sided, and p-values <0.05 deemed significant.
Results
Overexpression of NIS increases cell migration in thyroid cancer cells
Since NIS is not expressed in vitro in most of the established human thyroid or thyroid cancer cell lines (29) (Supplementary Fig. S2), we utilized a set of thyroid cancer cell lines stably transfected with FL hNIS for our experiments, including BCPAP and 8505C which have wildtype PTEN, and FTC-133 which is PTEN null (Supplementary Table S1). NIS is a membrane glycoprotein whose function is regulated by posttranslational glycosylation (30). On immunoblot analysis, NIS displays a hyper-glycosylated form of molecular weight of approximately 70 to 90 kDa and a hypo-glycosylated form of 60 kDa (Supplementary Fig. S2). The hyper-glycosylated NIS represents the mature and main form of iodide-pumping NIS on the plasma membrane (31).
To investigate whether NIS affects cell migration in a thyroid cancer context, we performed the wound-healing assay on BCPAP, 8505C, and FTC-133 cells. We found that overexpression of NIS increased cell migration rates, 1.3-fold in BCPAP cells, 1.8-fold in 8505C cells, and 2.3-fold in FTC-133 cells, compared with corresponding vector control cells (P=0.006, 0.004 and 0.003, respectively; Fig. 1A). To further validate that this phenotype is NIS-dependent, we performed transient NIS knockdown through siRNA in 8505C-FL hNIS cells and compared the migration rate (change of wound-healing capacity). The migration rate was reduced following NIS knockdown at 48 hours and 72 hours (P=0.017 and 0.0006, respectively; Fig. 1B). Taken together, these data provide evidence that NIS increases cell migration in thyroid cancer cells.
Figure 1. Exogenous NIS increases cell migration and RhoA activation through interaction with LARG in thyroid cancer cell lines.
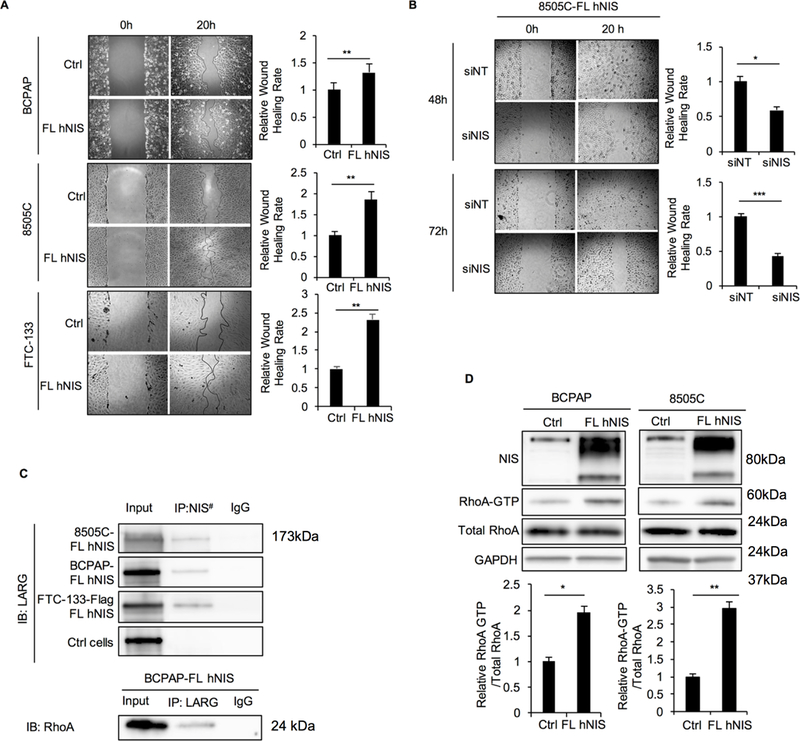
A. Wound-healing assay for BCPAP, 8505C, and FTC-133 cells stably transfected with FL hNIS or vector control. Pictures were taken at 0 and 20 hours after wound scratch. The bar graphs represent relative wound healing rates of BCPAP, 8505C and FTC-133 with FL hNIS expression normalized to controls. Data represent means ± SEM of 3 independent experiments. B. Wound-healing assay for 8505C-FL hNIS transiently transfected with non-targeting siRNA control (siNT) or NIS siRNA (siNIS) for 48 hours and 72 hours. Data represent means ± SEM of 3 independent experiments. C. Co-immunoprecipitation (IP) of NIS-LARG and LARG-RhoA using lysates prepared from thyroid cancer cell lines. BCPAP cells transfected with empty vector were set as control. Input, 20 μg of total proteins from each cell line. The exposure time was 2 minutes. Up panel, LARG immunoblots from lysates immunoprecipitated with anti-NIS (#anti-Flag in FTC-133-Flag FL hNIS cells) or control IgG in BCPAP, 8505C and FTC-133 cell with FL hNIS expression and BCPAP with vector control. Down panel, RhoA immunoblots from lysates immunoprecipitated with anti-LARG or control IgG in BCPAP-FL hNIS cells. Data are representative of 3 sets of independent experiments.D. Immunoblots from total cell lysates of NIS, RhoA and RhoA after GST-Rhotekin pull-down (RhoA-GTP form) in BCPAP-, 8505C-FL hNIS and vector control cells. Bar charts, normalized ratios of active RhoA relative to total RhoA levels. Data represent means ± SEM of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.001.
NIS activates RhoA to enhance cell migration through interaction with LARG
RhoA activation, which is controlled by Rho guanine-nucleotide-exchange factors (Rho GEFs), is known to play a key role in cancer cell motility (32). One Rho GEF, LARG, has been reported to bind with NIS directly through a PDZ-PDZ domain interaction in breast and liver cancer cell lines (33). Here, we show that NIS-LARG interaction is also valid in thyroid cancer (Fig. 1C). By co-immunofluorescence analysis, we observed NIS expressed at the plasma membrane as well as the cytoplasm, while LARG predominantly localized to the cytoplasm (Supplementary Fig. S3A), with some at the leading (migrating) edges of the cells, suggesting that NIS and LARG localize in similar subcellular regions, thus facilitating their physical interaction.
Next, we investigated downstream activation of RhoA in thyroid cancer cells. We observed that the relative level of GTP-bound (activated) form of RhoA was significantly higher in BCPAP- and 8505C-FL hNIS cells than in control cells (1.95 and 2.32 fold, P=0.018 and 0.001, respectively; Fig. 1D). Moreover, Rho-associated kinase (ROCK) inhibitor Y-27632 suppressed the migration rate in cells with exogenous FL hNIS expression (in BCPAP-FL hNIS, fold change=0.59, P=0.031; Supplementary Fig. S3B), which further support that the RhoA-ROCK pathway is involved in the enhancement of cell migration by NIS.
To confirm that LARG is involved in the NIS-mediated RhoA activation and enhancement of cell migration, we measured relative RhoA-GTP levels in BCPAP-vector control and -FL hNIS cells after siLARG knockdown. In all cells, LARG knockdown decreased activated RhoA-GTP level, which was more pronounced in BCPAP-FL hNIS than in BCPAP-vector control cells (46% versus 58%, P=0.023; Supplementary Fig. S3C). Relatedly, the wound-healing rate also decreased significantly after LARG knockdown (Supplementary Fig. S3D). Taken together, these data show that NIS enhances RhoA activation and downstream cellular migration through interaction with LARG.
PTEN siRNA knockdown increases NIS protein levels
Following the observation that NIS increases cell migration in a thyroid cancer context, to test our hypothesis that PTEN might affect NIS levels or subcellular localization, we knocked down PTEN by siRNA in BCPAP- and 8505C-FL hNIS cells (both PTEN wildtype) over different time points. PTEN protein levels decreased starting at 12 hours after siPTEN transfection. We observed that NIS protein first decreased after PTEN knockdown at/before 24 hours compared with controls, then increased significantly after 24 hours (Fig. 2A and Supplementary Fig. S4A). Moreover, the increase of hypo-glycosylated NIS was more prominent than that of hyper-glycosylated NIS. For example, in BCPAP-FL hNIS cells, at the 72-hour time point, the fold increase of hypo- and hyper-glycosylated NIS after PTEN knockdown was 2.17 versus 1.77 (P=0.008; Supplementary Fig. S4B). Although the total NIS increased, the ratio of hyper- to hypo-glycosylated NIS decreased compared to control (1.71 versus 2.18, P<0.001). Importantly, we did not observe differences in NIS mRNA levels following knockdown (Fig. 2B and Supplementary Fig. S4C). The latter is particularly pertinent since the thyroid cancer cell line models we utilized do not express endogenous NIS, and the exogenous FL hNIS is driven by a CMV promoter. Hence, differences in NIS protein levels are independent of transcript expression and are almost certainly related to post-transcriptional phenomena.
Figure 2. PTEN siRNA knockdown increases NIS protein level in BCPAP-FL hNIS thyroid cancer cells.
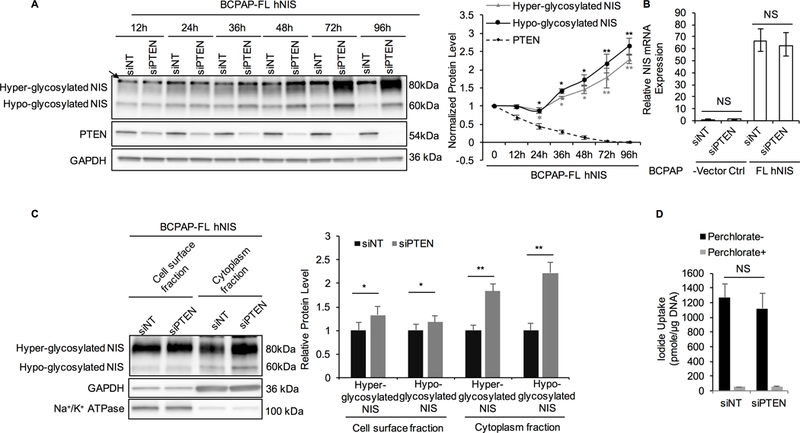
. A. Immunoblot of NIS and PTEN using total lysates extracted from BCPAP-FL hNIS cells transiently transfected with non-targeting siRNA (siNT) or PTEN siRNA (siPTEN) over different time points. GAPDH is used as a loading control for normalization. The graph represents normalized protein levels of hyper- and hypo-glycosylated NIS, and PTEN after PTEN knockdown normalized to siNT at each time point. Data represent means ± SEM of 3 independent experiments.B. Relative expression of NIS mRNA in BCPAP-FL hNIS and vector control cells transiently transfected with siNT or siPTEN for 72 hours. Data represent means ± SEM of 3 independent experiments.C. Immunoblot of NIS using cell surface fraction and cytoplasmic fraction from BCPAP-FL hNIS transiently transfected with siNT or siPTEN for 72 hours. Na+/K+ ATPase was used as a loading control for the cell surface fraction. For cytoplasmic NIS, GAPDH was used as a loading control. Bar chart shows the relative cell surface and cytoplasmic NIS protein levels in BCPAP-FL hNIS cells after siPTEN knockdown compared to control. Data represent means ± SEM of 3 independent experiments.D. Iodide uptake by BCPAP-FL hNIS transiently transfected with siNT or siPTEN siRNA knockdown for 72 hours. Non-NIS-mediated iodide uptake was examined by conducting parallel experiments in the presence of perchlorate. Data represent means ± SEM of 3 independent experiments.*P<0.05, **P<0.01, NS = not significant.
Because canonical NIS iodide-pump function is dependent on plasma membrane localization, we then investigate cell surface and cytoplasmic NIS protein levels following PTEN knockdown. In the cytoplasmic fraction, hyper- and hypo-glycosylated NIS levels were increased significantly after PTEN knockdown (1.84- and 2.22-fold, P=0.006 and 0.003, respectively; Fig. 2C). In the cell surface fraction, we also observed that hyper- and hypo-glycosylated NIS levels increased (1.33- and 1.18-fold, P=0.029 and 0.032, respectively). Importantly, even though NIS protein levels increased after PTEN knockdown, we observed no differences in cellular iodide uptake (Fig. 2D and Supplementary Fig. S4D). Our data indicate that PTEN loss results in increased exogenous NIS protein levels after 24 hours independent of NIS transcription. Moreover, cytoplasmic NIS increases more significantly compared to plasma membrane NIS, indicating that PTEN signaling might also affect NIS sub-cellular localization and function.
PTEN siRNA knockdown is associated with increased NIS protein stability in a LARG-dependent manner
Our data so far indicate that PTEN knockdown increases exogenous NIS protein levels independent of mRNA levels. Hence, we hypothesized that PTEN signaling might affect NIS protein stability. To test our hypothesis, we performed Cycloheximide (CHX) chase assay in BCPAP- and 8505C-FL hNIS cells transfected with siPTEN or siNT control. We observed that the hyper-glycosylated NIS is more stable, but the hypo-glycosylated NIS is comparatively unstable, with protein levels approaching zero at four hours after CHX treatment (Fig. 3A). PTEN knockdown increased the half-life of both hyper- and hypo-glycosylated NIS, from 60 to more than 72 hours and from 1.75 to 2.5 hours respectively (Fig. 3A). Overall, these data indicate that NIS protein stability could potentially explain the increased NIS protein levels in thyroid cancer cells after PTEN knockdown.
Figure 3. NIS increase after PTEN siRNA knockdown is due to increased protein stability and is LARG dependent.
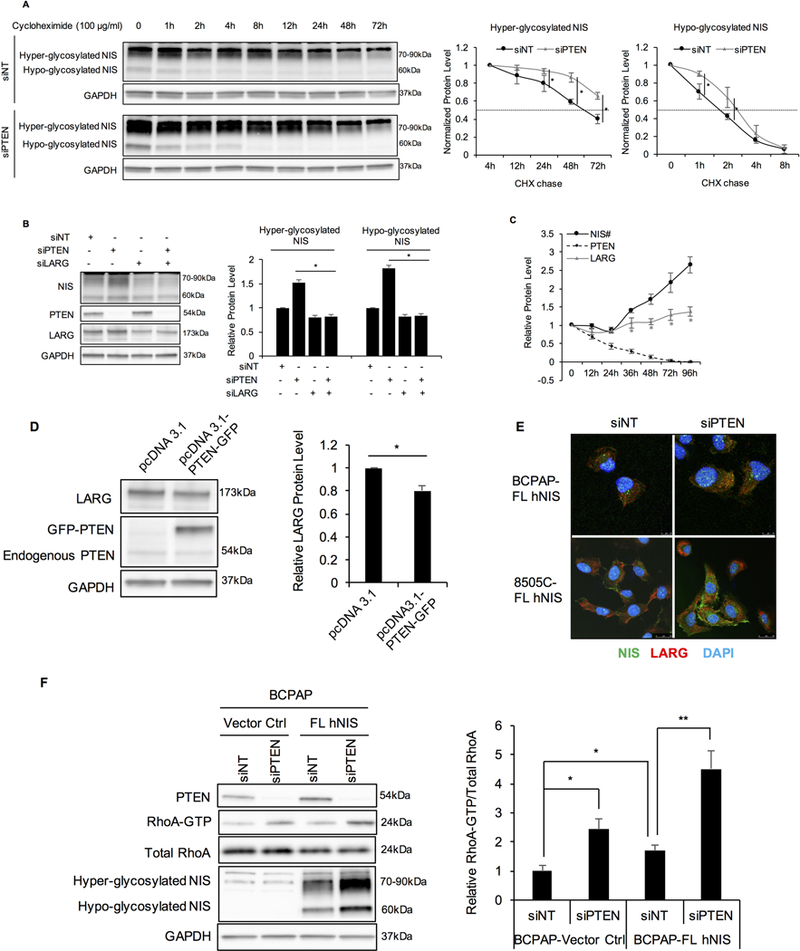
A. siPTEN increases NIS stability. 8505C-FL hNIS cells were transfected with siNT or siPTEN. At 48 hours after transfection, cells were treated with 100 μg/ml cycloheximide (CHX). Cells were harvested at 0, 1, 2, 4, 8, 12, 24, 48, and 72-hour-time points, and lysates were prepared to perform Western blot analysis for NIS protein levels. Hyper-glycosylated NIS in cells transfected with siNT continued to increase after treatment with CHX and reached the highest level at 4 hours. Hence, hyper-glycosylated NIS band intensity was normalized to GAPDH and then normalized to t=4 hour time point. Hypo-glycosylated NIS was normalized to t=0 controls. GAPDH is used as a loading control for normalization. Data represent means ±SEM of 3 independent experiments.B. NIS increase after PTEN knockdown is LARG dependent. Immunoblots of NIS, PTEN, and LARG in BCPAP-FL hNIS after transient transfection with siNT, siPTEN, siLARG and both siPTEN and siLARG. Bar charts, normalized NIS protein levels in BCPAP-FL hNIS after siPTEN knockdown in cells with or without LARG siRNA knockdown. GAPDH is used as a loading control for normalization. Data represent means ±SEM of 3 independent experiments. C. Relative PTEN, LARG and NIS protein levels in BCPAP-FL hNIS cells after PTEN siRNA knockdown over different time points. Data represent means ±SEM of 3 independent experiments. #NIS represents hypo-glycosylated NIS.D. Immunoblots of LARG and PTEN in BCPAP-FL hNIS transiently transfected with pcDNA3.1 empty vector or pcDNA3.1-PTEN-GFP for 48 hours. GAPDH is used as a loading control for normalization. Bar charts, Relative LARG protein levels in BCPAP-FL hNIS after overexpression of PTEN or empty vector. Data represent means ±SEM of 3 independent experiments. E. Confocal microscopy of NIS and LARG immunofluorescence staining in BCPAP- and 8505C-FL hNIS transiently transfected with siNT or siPTEN for 72 hours. Scale bar, 10 μm for BCPAP cells and 25 μm for 8505C cells. Data are representative of 3 sets of independent experiments.F. PTEN siRNA knockdown increases RhoA-GTP which is exaggerated by NIS. Left panel, BCPAP-Vector control and -FL hNIS cells transiently transfected with siNT or siPTEN for 72 hours. Immunoblots of PTEN, RhoA, NIS using total lysates, and RhoA after GST-Rhotekin pull-down. GAPDH is used as a loading control for normalization. Bar chart, normalized ratios of active to total RhoA levels. Data represent means ±SEM of 3 independent experiments.*P<0.05, **P<0.01.
To investigate whether LARG is involved in the increase of NIS protein levels resulting after PTEN knockdown, we studied NIS protein levels in BCPAP- and 8505C-FL hNIS cells transiently transfected with siNT, siPTEN, siLARG and both siPTEN and siLARG for 72 hours. Similar to our previous observations, NIS protein level was increased significantly after PTEN knockdown (Fig. 3B). In contrast, NIS protein decreased compared to control cells following LARG knockdown, regardless of PTEN knockdown. The fold change of hyper- and hypo-glycosylated NIS was 0.82 and 0.83, respectively, as compared to control (P=0.013 and 0.007, respectively; Fig. 3B). These data show that the increase of NIS protein levels following PTEN knockdown is LARG-dependent.
Furthermore, PTEN knockdown also resulted in increased LARG protein levels, with time points of LARG increments consistent with NIS increments (both starting from 36 hours after PTEN siRNA knockdown) (Fig. 3C and Supplementary Fig. S5). E.g., LARG was increased about 1.29-fold at 72 hours compared to control (P=0.012). Consistently, overexpression of PTEN in BCPAP-FL hNIS cells for 72 hours revealed 20% decrease in LARG protein levels compared to control (P=0.018; Fig. 3D). Taken together, our data indicate that PTEN loss increases LARG protein levels, which in turn interacts with NIS to increase NIS protein stability and hence, NIS protein levels in thyroid cancer cells. Immunofluorescence analysis corroborates these observations: following PTEN knockdown, immunofluorescence intensity of NIS and LARG increased, as well as the overlap of staining representing co-localization of both proteins (Fig. 3E).
Next, to investigate the role of PTEN and NIS together in RhoA activation, we performed RhoA activity assay in BCPAP-vector control and -FL hNIS cells transiently transfected with siNT or siPTEN for 72 hours. First, following transfection with siNT, BCPAP-FL hNIS cells displayed a higher level of activated RhoA-GTP than vector control cells, validating our previous observations. In both of vector control and FL hNIS cells, PTEN knockdown induced a significant increase in activated RhoA-GTP (Fig. 3F). Expectedly, the increase was more pronounced in cells with FL hNIS than in vector control cells. These data show that NIS exaggerates the increase of activated RhoA-GTP levels induced by loss-of-function of PTEN in thyroid cancer cell lines.
Inhibition of signaling downstream of PTEN increases cell surface NIS and is associated with NIS glycosylation state
Since we observed PTEN knockdown increases NIS protein levels, we then turned our attention to investigate if pharmacologic inhibition of signaling downstream of PTEN could also affect NIS protein levels or localization. To test our hypothesis, we used LY294002 (PI3Ki), MK-2206 (AKTi) and rapamycin (mTORi), and monitored NIS localization through immunofluorescence microscopy in BCPAP-FL hNIS cells. Treatment with LY294002, MK-2206, or rapamycin revealed increased fluorescence intensity of cell surface NIS compared to cells treated with DMSO (1.49-, 1.32- and 1.67-fold, P=0.029, 0.044 and 0.004, respectively; Fig. 4A). Furthermore, we observed decreased intracellular NIS after treatment with LY294002 and rapamycin, with a more significant decrease noted in cells treated with rapamycin (fold change=0.38, P<0.001) (Fig. 4A). We observed the same results in 8505C-FL hNIS cells after treatment (Supplementary Fig. S6).
Figure 4. Association between inhibition of signaling downstream of PTEN and NIS protein levels, glycosylation and localization.
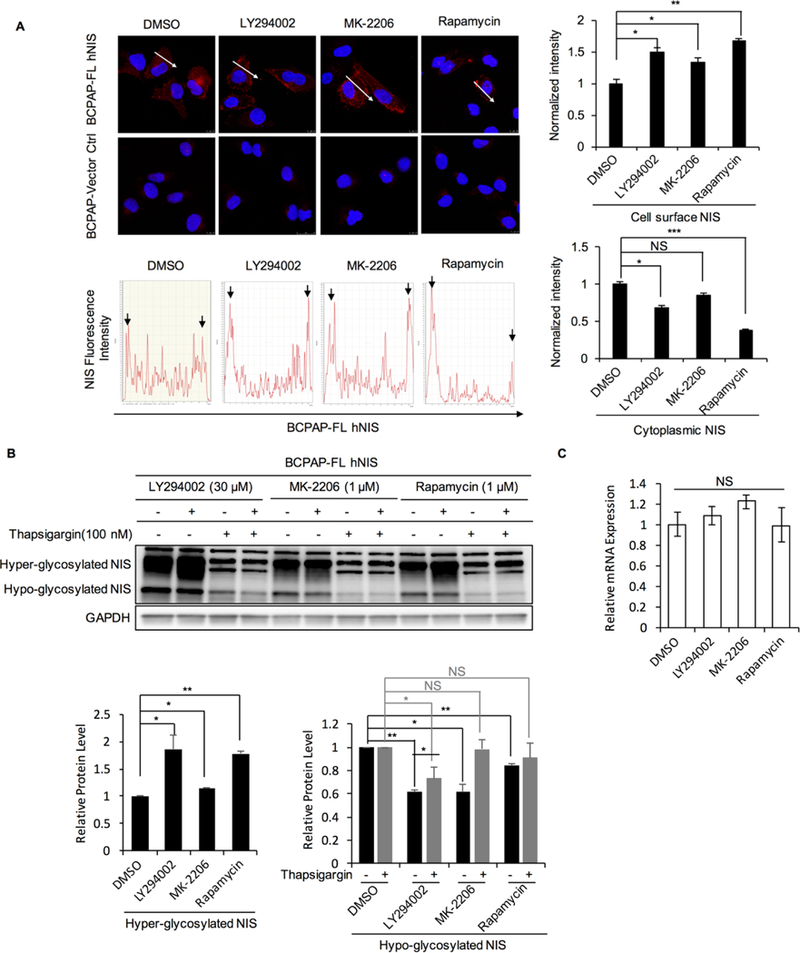
A. Inhibition of signaling downstream of PTEN increases cell surface NIS protein levelsImmunofluorescence of NIS in BCPAP-FL hNIS treated with DMSO, LY294002, MK-2206 or rapamycin. Immunofluorescence of Charts, intensity quantification of fluorescence along the arrows depicted in the microscopic images. Peaks on both sides represent cell surface NIS and peaks between these boundaries represent cytoplasmic NIS. Normalized cell surface and cytoplasmic NIS intensity were calculated by measuring the areas of the peaks representing each and then normalizing to distance. Bar chart shows the difference of normalized intensity of cell surface and cytoplasmic NIS between cells treated with DMSO and CHX. Data represent means ±SEM of 3 independent experiments. Scale bar, 10 μm. Arrows point at the peaks representing cell surface NIS.B. PI3K/AKT/mTOR inhibitors enhance the process of hypo-glycosylated NIS to be modified and matured into hyper-glycosylated NIS. Left panel, Immunoblots of NIS in BCPAP-FL hNIS cells treated with LY294002, MK-2206, rapamycin or DMSO for 48 hours with or without thapsigargin. Bar chart in the middle, hyper-glycosylated NIS was increased 1.85-, 1.15- and 1.78-folds after treatment with LY294002, MK-2206 and rapamycin compared to control, P=0.023, 0.031, 0.005 respectively. Bar chart on the right, hypo-glycosylated NIS decreased 0.61-, 0.61- and 0.84 folds respectively by LY294002, MK-2206 and rapamycin compared to control, P=0.001, 0.010, 0.005 respectively. After treatment with thapsigargin, hypo-glycosylated NIS decreased 0.73-fold (P=0.0095) in cells cotreated with LY294002 compared to control. Data represent means ±SEM of 3 independent experiments. C. Relative expression of NIS mRNA in BCPAP-FL hNIS after treatment with LY294002, MK-2206, rapamycin or DMSO. Data represent means ±SEM of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.001, NS = not significant.
Since the glycosylation state of NIS has been reported to affect NIS trafficking and membrane localization in thyroid cells (30,31,34), we next investigated whether these inhibitors could also affect NIS glycosylation. We observed that PI3K/AKT/mTOR inhibition increases hyper-glycosylated NIS levels, whereas that of hypo-glycosylated NIS decreases. In cells co-treated with thapsigargin, which inhibits hypo-glycosylated NIS modification into hyper-glycosylated NIS by inducing ER stress, we found NIS protein levels did not change in cells treated with MK-2206 or rapamycin, but still decreased in cells treated with LY294002 (Fig. 4B). Expectedly, the mRNA level of NIS did not change after treatment with these drugs compared to controls (Fig. 4C). These data provide circumstantial evidence that PI3K/AKT/mTOR inhibitors enhance the process of hypo-glycosylated NIS turning into the mature hyper-glycosylated NIS. Additionally, in cells treated with the LY294002, although NIS also decreased in cells co-treated with thapsigargin, the decrease was more pronounced in cells without thapsigargin (0.73-fold versus 0.61-fold, P=0.033; Fig. 4B). Moreover, we observed that LARG protein levels decreased after treatment with LY294002, but remained unchanged after MK-2206 or rapamycin treatments (Supplementary Fig. S7). These observations indicate that the PI3K inhibitor LY294002 might also decrease LARG protein levels to affect NIS protein levels.
Association between PTEN knockdown and NIS glycosylation
We next investigated whether PTEN knockdown also affects NIS glycosylation. Indeed, the direct evidence of this association was observed in CHX chase assay. After CHX exposure arresting NIS translation, we observed decreasing hypo-glycosylated NIS in cells with PTEN expression, yet a relative maintenance or increase in hyper-glycosylated NIS (Fig. 5). By 4 hours, hypo-glycosylated NIS was essentially null, while hyper-glycosylated NIS started to decrease because of degradation. However, after PTEN knockdown, we observed hyper-glycosylated NIS decreased immediately after treatment with CHX. The normalized hyper-glycosylated NIS at 4 hours in cells with siPTEN transfection was significantly decreased compared to siNT control (0.95-fold versus 1.46-fold normalized to 0-time point, P=0.036; Fig. 5). These data suggest that PTEN loss could be associated with inhibition of NIS glycosylation. We therefore postulate that PTEN knockdown may inhibit glycosylation after which cytoplasmic (non-/hypo-glycosylated and part of hyper-glycosylated) NIS interacts with LARG with this NIS-LARG interaction stabilizing NIS.
Figure 5. Association between PTEN knockdown and NIS glycosylation.
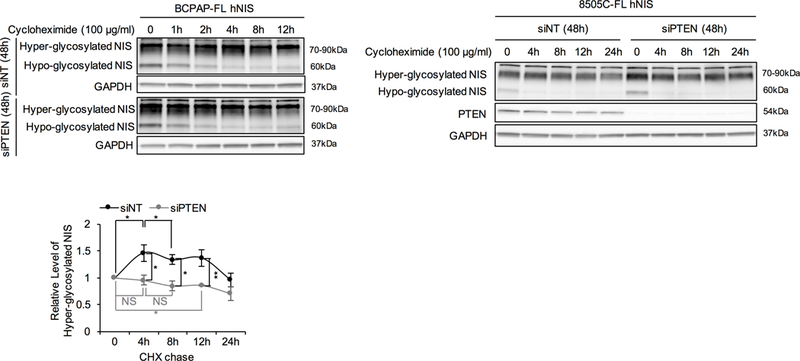
Association between PTEN knockdown and NIS glycosylation. Left and middle panel, Immunoblots of NIS using the lysates from the CHX chase assay in BCPAP- and 8505C-FL hNIS transiently transfected with siNT or siPTEN (detailed method described in Figure 3A). Right panel, hyper-glycosylated NIS band intensity normalized to GAPDH and then normalized to 0-time point controls in 8505C-FL hNIS. Data represent means ± SEM of 3 independent experiments. *P<0.05, **P<0.01.
Collaborative effect of PTEN and glycosylating enzyme DPAGT1 on glycosylation of NIS
We next investigated if the change of NIS glycosylation by altering PTEN signaling is through particular glycosylating enzymes. Since PI3K/AKT/mTOR inhibitors partly reversed the inhibition of tunicamycin (inhibitor of enzymes involved in the initial step of N-linked glycosylation) on NIS glycosylation (Supplementary Fig. S8), we hypothesized PTEN or downstream signaling could affect initial steps of NIS glycosylation such as through such glycosylating enzymes. After knockdown of UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase (encoded by DPAGT1 gene), an enzyme involved in the first step of N-linked glycosylation (35), we observed a band shift of hyper-glycosylated NIS (Fig. 6A), which is indicative of the de-glycosylation of NIS. In cells co-treated with LY294002, MK-2206 or rapamycin, the shift distances were decreased compared with controls (0.61-, 0.80-and 0.53-fold; P=0.008, 0.037 and 0.007 respectively); (8505C-FL hNIS cells; Fig. 6A). These data indicate that PI3K/AKT/mTOR inhibitors reverse the de-glycosylation of NIS caused by decreasing levels of DPAGT1. Interestingly, while DPAGT1 knockdown showed an effect on de-glycosylation of NIS, it also increased NIS protein levels, which can be counteracted by PI3K, AKT or mTOR inhibition (Fig. 6B). We also investigated NIS protein levels in BCPAP-FL hNIS cells transiently transfected with siNT, siPTEN, siDPAGT1 or both siPTEN and siDPAGT1 over different time points. Knockdown was confirmed by Western blot (Supplementary Fig. S9). At 24 hours after PTEN or DPAGT1 knockdown, we observed decreased NIS protein levels, with knockdown of both PTEN and DPAGT1 resulting in larger decrements (fold change of hypo-glycosylated NIS: 0.67 versus 0.63, P=0.009; Fig. 6C). Taken together, these data provide further supportive evidence that PTEN affects NIS glycosylation, and this effect could potentially be mediated through the glycosylating enzyme DPAGT1.
Figure 6. Collaborative effect of PTEN and glycosylating enzyme DPAGT1 on glycosylation of NIS.
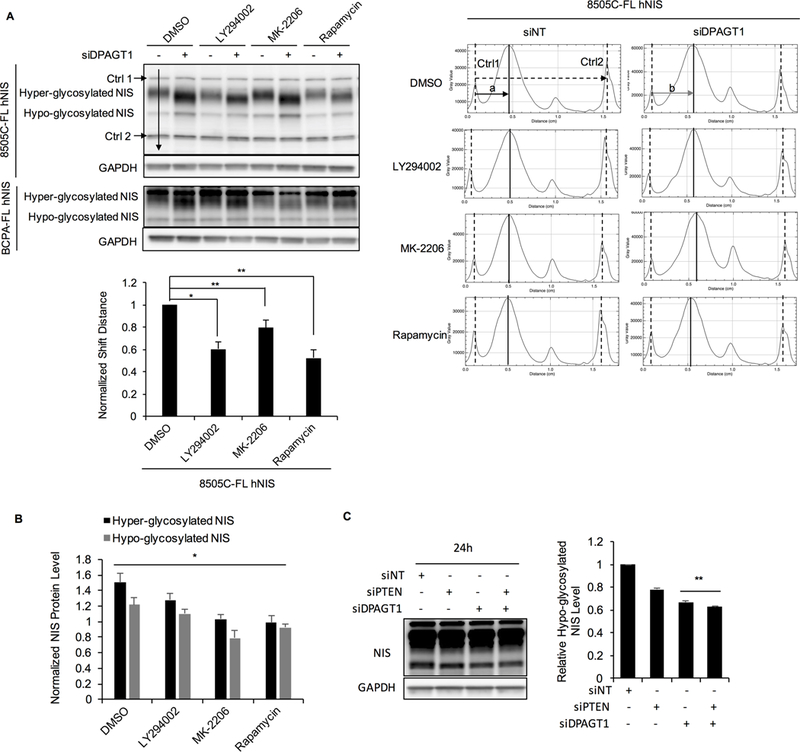
A. Immunoblots of NIS in 8505C-FL hNIS and BCPAP-FL hNIS cells transiently transfected with siNT or siDPAGT1 and then treated with LY294002, MK-2206, rapamycin or DMSO. Charts on the right represent the intensities along the representative arrow depicted on the immunoblot. We annotated the middle of the peaks as the position of each band. The top and bottom bands which are non-specific bands that are stable with different treatments and remain equidistant were set as controls (Ctrl1 and Ctrl2). The distances between Ctrl1 and Ctrl2 are the same in all of lanes. Distance (a) represents the distance between hyper-glycosylated NIS with Ctrl1 after siNT transfection, and distance (b) represents the distance after siDPAGT1 knockdown. Distance difference (a-b) represents the distance shift. Bar chart shows the distance shifts in cells treated with each drug normalized to DMSO. Data represent means ±SEM of 3 independent experiments. B. DPAGT1 knockdown increases NIS protein levels, which is counteracted by PI3K, AKT or mTOR inhibitors. Bar chart shows the hyper- and hypo-glycosylated NIS protein levels in BCPAP-FL hNIS treated with drugs after DPAGT1 knockdown normalized to siNT transfection. Data represent means ±SEM of 3 independent experiments. C. Immunoblot of NIS in BCPAP-FL hNIS transiently transfected with siNT, siPTEN, siDPAGT1 or both siPTEN and siDPAGT1 for 24 hours. GAPDH is used as a loading control for normalization. Bar chart, intensity of hypo-glycosylated bands normalized to GAPDH then normalized to siNT control. Data represent means ±SEM of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.001, NS = not significant.
Discussion
NIS, a plasma membrane protein classically mediating iodide uptake in thyroid tissue, is generally decreased in thyroid cancer cells, resulting in reduced radioiodide uptake. Several studies reported decreased NIS mRNA levels in differentiated thyroid cancer (DTC) compared to normal thyroid tissue (2,7–10), with the former induced by PI3K or MAPK signaling activation, which plays critical roles in thyroid cancer initiation and progression (36). Interestingly, we found that PTEN loss, which activates PI3K and MAPK signaling, results in increased NIS protein levels in our thyroid cancer cell line models. Importantly, since exogenous FL hNIS expression is driven by a CMV promoter, the effects on NIS protein levels are independent of transcript expression levels. Furthermore, our data suggest that increased NIS protein levels, preferentially in the cytoplasm, could have non-canonical pump-independent function associated with pro-tumorigenic phenotypes.
Besides its canonical iodide uptake function, the non-pump function of NIS has remained elusive in thyroid cancer. Our observation that NIS significantly enhances cell migration provides direct evidence for the pro-tumorigenic role of non-pumping NIS in thyroid cancer. Relatedly, it has been reported that the secondary-structure of NIS contains a putative density-95/discs large/zona occludens-1 (PDZ) class 1 target motif (T/S-X-V/L) at the COOH-terminus, endowing it with the potential for binding proteins through PDZ protein-protein interactions (37). For example, NIS has been reported to interact with pituitary tumor transforming gene (PTTG) binding factor (PBF) in the cytoplasm of COS-7 kidney and FRTL-5 thyroid cell lines (38). Importantly, a more recent study has shown that NIS interacts with leukemia associated RhoA guanine exchange factor (LARG) in breast and liver cancer cell lines (33). These studies further support that NIS has iodide-pump independent function, including in organs other than the thyroid. As relevant to thyroid cancer, our data show that the NIS-LARG interaction is valid and promotes NIS crosstalk with PTEN signaling.
Germline PTEN mutations cause Cowden syndrome (CS), characterized by a high risk of thyroid, breast and other cancers (18,20,21). Somatic PTEN mutations occur in a wide variety of sporadic human cancers (22). It is also well-established that PTEN mechanistically functions as a PIP3 (phophatidylinositol-3,4,5-triphosphate) 3’-phosphatase to reduce the levels of intracellular PIP3 and antagonize PI3K-AKT signaling (22). The Rho GTPase family of proteins, which are controlled by Rho guanine-nucleotide-exchange factors (Rho GEFs), play a key role in cancer cell motility (39). Interestingly, PIP3 has been reported to recruit Rho GEFs to then activate Rho GTPase (32). Our data show that PTEN loss increases LARG (a Rho GEF), which can interact with NIS, coupled with an increase in downstream activated RhoA-GTP.
It remained intriguing to investigate how PTEN loss increases NIS protein levels. We found that PTEN knockdown increases NIS protein stability and protein levels in a LARG-dependent manner. Furthermore, PTEN knockdown increases LARG protein levels. Hence, we speculate that the NIS-LARG interaction can increase NIS protein stability. Indeed, LARG is a protein with a long half-life (60% protein retained 72 hours post-knockdown), potentially explaining increased NIS stability as pertinent to the NIS-LARG interaction. Supportively, it has been reported that LARG binds to ATP-binding cassette transporter A1 (ABCA1) to then stabilize it (40). Moreover, we observed decreased NIS protein levels following treatment with the ROCK inhibitor Y-27632 (Supplementary Fig. S10), indicating that RhoA activation might also be involved in stabilizing NIS. Additionally, our data indicate that PTEN loss or downstream PI3K/AKT/mTOR signaling activation inhibit NIS glycosylation and its classical membrane localization. Several studies have reported that the glycosylation state of NIS affects its trafficking and membrane localization in thyroid cells (30,31,34), further supporting our observations. Taken together, we postulate that PTEN loss can inhibit NIS glycosylation to then increase cytoplasmic NIS. Cytoplasmic NIS is then stabilized by LARG through NIS-LARG protein-protein interaction. In parallel, PTEN loss also increases LARG protein levels and RhoA activation, further contributing to NIS stabilization and downstream effects. Notably, we also observed that the cell surface NIS increases slightly after PTEN knockdown, compared to the more pronounced increase in cytoplasmic NIS. This observation could be due to the interaction with LARG, which localizes at the sub-membrane region, with the intracellular C-terminal region of trans-membrane NIS to also stabilize it. Corroborating this observation, LARG has been reported to interact with a number of plasma membrane proteins such as the histamine-H1 receptor, the insulin-like growth factor-1 (IGF-1) receptor, and the semaphorin 4D/plexin-B1 receptor (41–43).
We utilized knockdown of DPAGT1, an enzyme involved in the initial step of N-linked glycosylation to further investigate a possible association between PTEN signaling and NIS glycosylation. We observed that PI3K/AKT/mTOR inhibitors can reverse the de-glycosylation process caused by reduced DPAGT1. In contrast, PTEN knockdown results in a larger effect on de-glycosylation through reduction of DPAGT1. However, whether PTEN is directly involved in regulating this and other glycosylating enzymes warrants further investigation. More interestingly, while NIS is de-glycosylated by DPAGT1 knockdown, protein levels of NIS increase and can be counteracted by PI3K/AKT/mTOR inhibitors. Since we believe that de-glycosylation should decrease NIS stability, we postulate that the subcellular localization could explain the NIS increase. However, this observation also warrants further exploration.
We utilized PI3K/AKT/mTOR inhibitors to investigate the effect of these pharmacologic drugs on NIS protein levels, glycosylation and membrane localization. Our data show that LY294002 (PI3Ki), MK-2206 (AKTi) and rapamycin (mTORi) increase the mature hyper-glycosylated NIS but decrease the hypo-glycosylated NIS, with consistent increase in cell surface NIS protein. Interestingly, LY294002 significantly induces NIS mRNA expression and iodide uptake in rat thyroid cells FRTL-5 and PCCL3, and LY294002 and Akti-1/2 also increase iodide uptake in exogenous NIS-expressing papillary thyroid cancer (PTC) cell lines (23). Relatedly, LY294002 and rapamycin increase NIS expression in PCCL3 cells, however, LY294002 and Akti-1/2 had little effect on cell surface or total NIS protein levels in HEK293 human kidney cells or COS7 monkey kidney cells expressing exogenous NIS (44). Thus, the modulation of NIS protein by PI3K/AKT/mTOR inhibitors appears to be cell-type and context-dependent.
Intriguingly, studies of NIS in breast cancer, another major CS component cancer, provide further supportive evidence that the activation of PTEN downstream signaling increases intracellular NIS. It has been reported that in the breast cancer MCF7 cell line, the activating mutation PI3K p110αCAAX increases under-glycosylated NIS, with predominant localization in the cytoplasm (45). Similarly, MEK activation has also been reported to be involved in maintaining NIS protein stability in human breast cancers (46). Importantly, a more recent study reported that NIS protein level in breast cancer is associated with poor prognosis and chemotherapy resistance (47). The association between NIS levels and prognosis in DTC remains inconclusive. Interestingly, we have previously reported that follicular thyroid cancer (FTC) is proportionately more prevalent in PTEN mutation positive patients compared to the more common PTC in the general population (19). FTC is known to have poorer prognosis than PTC, with increased development of lung and bone metastases (48). Whether the PTEN-related NIS localization and associated non-canonical function is involved in these invasive phenotypes needs further study.
The ability to concentrate radioiodide (as related to the pump-function of NIS) is generally considered to indicate a more differentiated thyroid cancer phenotype (49). Re-differentiation studies in thyroid cancers generally focus on increasing iodide uptake by using some compounds such as retinoic acid (RA), PPARγ agonists, and histone deacetylase (HDAC) inhibitors, which can increase endogenous NIS expression (49). However, recent clinical trials utilizing these compounds have shown disappointing results (49). As relevant to our observations, inhibition of mTOR signaling has been reported to induce NIS expression through upregulating the transcription factor TTF1 (50,51). Our data suggest that PTEN and downstream signaling have an impact on exogenous NIS protein levels and localization, mimicking a change-of-function effect. We speculate that these changes functionally resemble a less differentiated phenotype, at least characterized by increased cellular migration. Certainly, investigating a comprehensive set of differentiation markers will be key to ascertain or refute this hypothesis. Hence, as a master regulator of PI3K/AKT/mTOR signaling, it is plausible that PTEN also regulates the differentiation of thyroid cancer upstream of mTOR signaling. Relatedly, research efforts have also been focused on inducing exogenous NIS expression for the theoretical possibility of radioiodide therapy in refractory thyroid cancers and some non-thyroidal cancers (6). However, while exogenous NIS could increase radioiodide uptake in some cancer cells, the therapeutic effect varies in different cancer cell types (6), presumably due to differential radiosensitivity to I-131. Our data provide evidence that exogenous NIS expression increases cell migration in thyroid cancer cells. It would hence be of clinical interest to investigate if the efficacy of NIS-transfer facilitated I-131 therapy is associated with cytoplasmic NIS level and/or NIS-induced cell migration.
In summary, we found that in thyroid cancer cells, NIS interacts with LARG to subsequently crosstalk with signaling cascades downstream of PTEN to enhance cell migration. PTEN loss, a common somatic alteration and upregulation of PI3K/AKT/mTOR signaling pathway found in multiple cancer types, likely inhibits NIS glycosylation to impair NIS membrane localization, resulting in increased cytoplasmic NIS. Furthermore, PTEN loss increases LARG protein levels and RhoA activation, thus stabilizing NIS, especially the intracellular form, through NIS-LARG interaction (Fig. 7). Together, our data suggest that in thyroid cancers, NIS, particularly within the cytoplasm, has pump-independent pro-tumorigenic roles and crosstalks with the PTEN-PI3K/AKT/mTOR pathway, a major signaling cascade in cancer. We suspect that this crosstalk may be generalizable in extra-thyroidal malignancies, and suggest targeted therapies beyond attempting to increase NIS levels.
Figure 7.
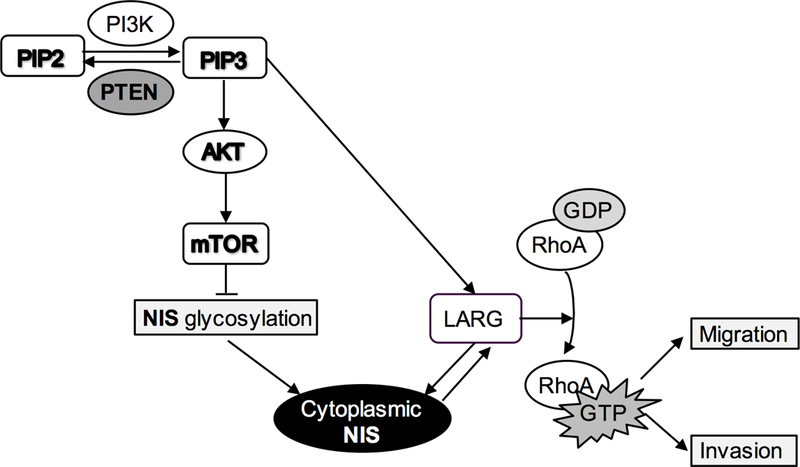
Proposed signaling model of crosstalk between NIS and PTEN signaling pathway
Supplementary Material
Acknowledgments
This work was supported, in part, by the National Cancer Institute (P01CA124570, R01CA118989) and the American Cancer Society (RPG-02–151-01-CCE, Clinical Research Professorship) [all to C. Eng]. We thank Dr. Ritika Jaini for helpful discussions, Todd Romigh and Tammy Sadler for assistance in cell culture and Western blot analysis, and Shikha Gautam for technical advice related to FL hNIS stable cell lines and NIS antibody. L. Yehia is an Ambrose Monell Foundation Cancer Genomic Medicine Fellow at the Cleveland Clinic Genomic Medicine Institute. C. Eng is the Sondra J. and Stephen R. Hardis Chair of Cancer Genomic Medicine at the Cleveland Clinic and an American Cancer Society Clinical Research Professor.
Abbreviations list:
- CS
Cowden syndrome
- DTC
Differentiated thyroid cancer
- FL hNIS
Full-length human NIS
- I-131
Radioiodine-131
- LARG
Leukemia associated RhoA guanine exchange factor
- NIS
Sodium iodide symporter
Footnotes
Disclosure of Potential Conflicts of Interest:
No potential conflicts of interest were disclosed.
References
- 1.Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature 1996;379:458–60 [DOI] [PubMed] [Google Scholar]
- 2.Smanik PA, Ryu KY, Theil KS, Mazzaferri EL, Jhiang SM. Expression, exon-intron organization, and chromosome mapping of the human sodium iodide symporter. Endocrinology 1997;138:3555–8 [DOI] [PubMed] [Google Scholar]
- 3.Cho JY, Leveille R, Kao R, Rousset B, Parlow AF, Burak WE Jr, et al. Hormonal regulation of radioiodide uptake activity and Na+/I- symporter expression in mammary glands. J Clin Endocrinol Metab 2000;85:2936–43 [DOI] [PubMed] [Google Scholar]
- 4.Semba RD, Delange F. Iodine in human milk: perspectives for infant health. Nutr Rev 2001;59:269–78 [DOI] [PubMed] [Google Scholar]
- 5.Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016;26:1–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kogai T, Brent GA. The sodium iodide symporter (NIS): regulation and approaches to targeting for cancer therapeutics. Pharmacol Ther 2012;135:355–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arturi F, Russo D, Schlumberger M, du Villard JA, Caillou B, Vigneri P, et al. Iodide symporter gene expression in human thyroid tumors. J Clin Endocrinol Metab 1998;83:2493–6 [DOI] [PubMed] [Google Scholar]
- 8.Ringel MD, Anderson J, Souza SL, Burch HB, Tambascia M, Shriver CD, et al. Expression of the sodium iodide symporter and thyroglobulin genes are reduced in papillary thyroid cancer. Mod Pathol 2001;14:289–96 [DOI] [PubMed] [Google Scholar]
- 9.Lazar V, Bidart JM, Caillou B, Mahe C, Lacroix L, Filetti S, et al. Expression of the Na+/I- symporter gene in human thyroid tumors: a comparison study with other thyroid-specific genes. J Clin Endocrinol Metab 1999;84:3228–34 [DOI] [PubMed] [Google Scholar]
- 10.Tavares C, Coelho MJ, Eloy C, Melo M, da Rocha AG, Pestana A, et al. NIS expression in thyroid tumors, relation with prognosis clinicopathological and molecular features. Endocr Connect 2018;7:78–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dohan O, Baloch Z, Banrevi Z, Livolsi V, Carrasco N. Rapid communication: predominant intracellular overexpression of the Na(+)/I(−) symporter (NIS) in a large sampling of thyroid cancer cases. J Clin Endocrinol Metab 2001;86:2697–700 [DOI] [PubMed] [Google Scholar]
- 12.Saito T, Endo T, Kawaguchi A, Ikeda M, Katoh R, Kawaoi A, et al. Increased expression of the sodium/iodide symporter in papillary thyroid carcinomas. J Clin Invest 1998;101:1296–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wapnir IL, van de Rijn M, Nowels K, Amenta PS, Walton K, Montgomery K, et al. Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J Clin Endocrinol Metab 2003;88:1880–8 [DOI] [PubMed] [Google Scholar]
- 14.Tazebay UH, Wapnir IL, Levy O, Dohan O, Zuckier LS, Zhao QH, et al. The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat Med 2000;6:871–8 [DOI] [PubMed] [Google Scholar]
- 15.Liu B, Herve J, Bioulac-Sage P, Valogne Y, Roux J, Yilmaz F, et al. Sodium iodide symporter is expressed at the preneoplastic stages of liver carcinogenesis and in human cholangiocarcinoma. Gastroenterology 2007;132:1495–503 [DOI] [PubMed] [Google Scholar]
- 16.Gainor DL, Chute DJ, Lorenz RR. Sodium Iodide Symporter Expression in Adenoid Cystic Carcinoma of the Head and Neck. JAMA Otolaryngol Head Neck Surg 2015;141:739–44 [DOI] [PubMed] [Google Scholar]
- 17.Navarra M, Micali S, Lepore SM, Cesinaro AM, Celano M, Sighinolfi MC, et al. Expression of the sodium/iodide symporter in human prostate adenocarcinoma. Urology 2010;75:773–8 [DOI] [PubMed] [Google Scholar]
- 18.Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012;18:400–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J Clin Endocrinol Metab 2011;96:E2063–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eng C PTEN: one gene, many syndromes. Hum Mutat 2003;22:183–98 [DOI] [PubMed] [Google Scholar]
- 21.Zbuk KM, Eng C. Cancer phenomics: RET and PTEN as illustrative models. Nat Rev Cancer 2007;7:35–45 [DOI] [PubMed] [Google Scholar]
- 22.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012;13:283–96 [DOI] [PubMed] [Google Scholar]
- 23.Kogai T, Sajid-Crockett S, Newmarch LS, Liu YY, Brent GA. Phosphoinositide-3-kinase inhibition induces sodium/iodide symporter expression in rat thyroid cells and human papillary thyroid cancer cells. J Endocrinol 2008;199:243–52 [DOI] [PubMed] [Google Scholar]
- 24.Vadysirisack DD, Shen DH, Jhiang SM. Correlation of Na+/I- symporter expression and activity: implications of Na+/I- symporter as an imaging reporter gene. J Nucl Med 2006;47:182–90 [PubMed] [Google Scholar]
- 25.Castro MR, Bergert ER, Beito TG, Roche PC, Ziesmer SC, Jhiang SM, et al. Monoclonal antibodies against the human sodium iodide symporter: utility for immunocytochemistry of thyroid cancer. J Endocrinol 1999;163:495–504 [DOI] [PubMed] [Google Scholar]
- 26.Suzuki M, Van Paesschen W, Stalmans I, Horita S, Yamada H, Bergmans BA, et al. Defective membrane expression of the Na(+)-HCO(3)(−) cotransporter NBCe1 is associated with familial migraine. Proc Natl Acad Sci U S A 2010;107:15963–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao SH, Wang WL, Chen CY, Chang YL, Wu YY, Wang YT, et al. Analysis of Protein Stability by the Cycloheximide Chase Assay. Bio Protoc 2015;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waltz F, Pillette L, Ambroise Y. A nonradioactive iodide uptake assay for sodium iodide symporter function. Anal Biochem 2010;396:91–5 [DOI] [PubMed] [Google Scholar]
- 29.Lakshmanan A, Scarberry D, Shen DH, Jhiang SM. Modulation of sodium iodide symporter in thyroid cancer. Horm Cancer 2014;5:363–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung T, Youn H, Yeom CJ, Kang KW, Chung JK. Glycosylation of Sodium/Iodide Symporter (NIS) Regulates Its Membrane Translocation and Radioiodine Uptake. PLoS One 2015;10:e0142984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy O, De la Vieja A, Ginter CS, Riedel C, Dai G, Carrasco N. N-linked glycosylation of the thyroid Na+/I- symporter (NIS). Implications for its secondary structure model. J Biol Chem 1998;273:22657–63 [DOI] [PubMed] [Google Scholar]
- 32.Amin E, Dubey BN, Zhang SC, Gremer L, Dvorsky R, Moll JM, et al. Rho-kinase: regulation, (dys)function, and inhibition. Biol Chem 2013;394:1399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lacoste C, Herve J, Bou Nader M, Dos Santos A, Moniaux N, Valogne Y, et al. Iodide transporter NIS regulates cancer cell motility and invasiveness by interacting with the Rho guanine nucleotide exchange factor LARG. Cancer Res 2012;72:5505–15 [DOI] [PubMed] [Google Scholar]
- 34.Beyer S, Lakshmanan A, Liu YY, Zhang X, Wapnir I, Smolenski A, et al. KT5823 differentially modulates sodium iodide symporter expression, activity, and glycosylation between thyroid and breast cancer cells. Endocrinology 2011;152:782–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eckert V, Blank M, Mazhari-Tabrizi R, Mumberg D, Funk M, Schwarz RT. Cloning and functional expression of the human GlcNAc-1-P transferase, the enzyme for the committed step of the dolichol cycle, by heterologous complementation in Saccharomyces cerevisiae. Glycobiology 1998;8:77–85 [DOI] [PubMed] [Google Scholar]
- 36.Xing M Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer 2013;13:184–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dohan O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, et al. The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr Rev 2003;24:48–77 [DOI] [PubMed] [Google Scholar]
- 38.Smith VE, Read ML, Turnell AS, Watkins RJ, Watkinson JC, Lewy GD, et al. A novel mechanism of sodium iodide symporter repression in differentiated thyroid cancer. J Cell Sci 2009;122:3393–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 2014;33:4021–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okuhira K, Fitzgerald ML, Tamehiro N, Ohoka N, Suzuki K, Sawada J, et al. Binding of PDZ-RhoGEF to ATP-binding cassette transporter A1 (ABCA1) induces cholesterol efflux through RhoA activation and prevention of transporter degradation. J Biol Chem 2010;285:16369–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfreimer M, Vatter P, Langer T, Wieland T, Gierschik P, Moepps B. LARG links histamine-H1-receptor-activated Gq to Rho-GTPase-dependent signaling pathways. Cell Signal 2012;24:652–63 [DOI] [PubMed] [Google Scholar]
- 42.Taya S, Inagaki N, Sengiku H, Makino H, Iwamatsu A, Urakawa I, et al. Direct interaction of insulin-like growth factor-1 receptor with leukemia-associated RhoGEF. J Cell Biol 2001;155:809–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aurandt J, Vikis HG, Gutkind JS, Ahn N, Guan KL. The semaphorin receptor plexin-B1 signals through a direct interaction with the Rho-specific nucleotide exchange factor, LARG. Proc Natl Acad Sci U S A 2002;99:12085–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu YY, Zhang X, Ringel MD, Jhiang SM. Modulation of sodium iodide symporter expression and function by LY294002, Akti-1/2 and Rapamycin in thyroid cells. Endocr Relat Cancer 2012;19:291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knostman KA, McCubrey JA, Morrison CD, Zhang Z, Capen CC, Jhiang SM. PI3K activation is associated with intracellular sodium/iodide symporter protein expression in breast cancer. BMC Cancer 2007;7:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z, Beyer S, Jhiang SM. MEK inhibition leads to lysosome-mediated Na+/I- symporter protein degradation in human breast cancer cells. Endocr Relat Cancer 2013;20:241–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chekhun VF, Andriiv AV, Lukianova NY. Significance of iodine symporter for prognosis of the disease course and efficacy of neoadjuvant chemotherapy in patients with breast cancer of luminal and basal subtypes. Exp Oncol 2017;39:65–8 [PubMed] [Google Scholar]
- 48.Sugino K, Ito K, Nagahama M, Kitagawa W, Shibuya H, Ohkuwa K, et al. Prognosis and prognostic factors for distant metastases and tumor mortality in follicular thyroid carcinoma. Thyroid 2011;21:751–7 [DOI] [PubMed] [Google Scholar]
- 49.Hong CM, Ahn BC. Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer for Reapplication of I-131 Therapy. Front Endocrinol (Lausanne) 2017;8:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tavares C, Eloy C, Melo M, Gaspar da Rocha A, Pestana A, Batista R, et al. mTOR Pathway in Papillary Thyroid Carcinoma: Different Contributions of mTORC1 and mTORC2 Complexes for Tumor Behavior and SLC5A5 mRNA Expression. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plantinga TS, Heinhuis B, Gerrits D, Netea MG, Joosten LA, Hermus AR, et al. mTOR Inhibition promotes TTF1-dependent redifferentiation and restores iodine uptake in thyroid carcinoma cell lines. J Clin Endocrinol Metab 2014;99:E1368–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.