

Abstract
Background:
Multiple studies have implicated Protease Activated Receptor-1 (PAR-1), a G-protein coupled receptor activated by proteolytic cleavage of its N-terminus, as one target coupling thrombin-mediated proteolysis to tumor progression.
Objective:
We analyzed the role of PAR-1 in the setting of two distinct spontaneously developing tumor models in mice.
Methods:
We interbred PAR-1-deficient mice with TRAMP (Transgenic Adenocarcinoma of the Mouse Prostate) mice, which spontaneously develop prostate tumors, and adenomatous polyposis coli Min (APCMin/+) mice, which spontaneously develop intestinal adenomas.
Results:
Analyses of TRAMP mice with advanced disease (30 weeks) revealed that PAR-1 deficiency resulted in significantly larger and more aggressive prostate tumors. Prostates collected at an earlier time point (12 weeks of age) revealed that PAR-1 promotes apoptosis in transformed epithelia. In vitro analyses of TRAMP-derived cells revealed that activated protein C-mediated PAR-1 cleavage can induce tumor cell apoptosis, suggesting tumor cell intrinsic PAR-1 functions can limit tumor progression. Paralleling results in TRAMP mice, PAR-1-deficient APCMin/+ mice developed 3-fold more adenomas than PAR-1 expressing mice, and the adenomas that formed were significantly larger. Moreover, loss of PAR-1 expression was shown to limit apoptosis in transformed intestinal epithelial cells.
Conclusions:
Together, these results demonstrate a previously unrecognized role for PAR-1 in impeding tumor progression in vivo. These results also offer a cautionary note suggesting that long-term PAR-1 inhibition could increase malignancy risk in some contexts.
INTRODUCTION
Protease activated receptor-1 (PAR-1) is a G-protein coupled receptor belonging to a family of receptors whose defining feature is irreversible proteolytic activation. Proteolytic cleavage exposes a tethered ligand of the N-terminal extracellular domain, leading to intracellular signaling. Thrombin-mediated PAR-1 cleavage is a critical mechanism of platelet activation in humans [1], but PAR-1 is not expressed by murine platelets [2]. PAR-1 is expressed by multiple other cell types in both mice and humans [3], and PAR-1 functions have been linked to proliferation, survival, cytokine/growth factor secretion, and motility [3]. PAR-1 can be activated by several proteases in addition to thrombin, including activated protein C, matrix metalloproteases (MMPs), kallikreins, plasmin, and the tissue factor/fVIIa/fXa complex [4]. The tethered ligand exposed following proteolytic cleavage of PAR-1 is variable depending on the activating protease, leading to differential or opposing cell signaling pathways [5–7].
Consistent with the fact that PAR-1 is widely expressed and coupled to numerous cellular processes, studies in mice show that PAR-1 plays a major role in numerous disease processes [8]. Several experimental studies have led to a widely-held consensus that PAR-1 activation drives cancer progression, and PAR-1 activation is proposed as one of the mechanisms coupling thrombin-mediated proteolysis to cancer progression [8]. While much of the available data supporting this conclusion is based on in vitro analyses [9–14], there are ample in vivo analyses based on either specific PAR-1 inhibitors or genetic manipulation of PAR-1 in tumor cells or stromal cells [15–21]. However, the majority of the in vivo data regarding the role of PAR-1 in malignancy is based on analyses of fully-transformed tumor cells. Analyses of in situ tumor progression in a murine breast cancer model showed no apparent role for PAR-1, but analyses of other spontaneous tumor models are lacking [22].
To define the role of PAR-1 in tumor progression in the context of malignancies developing in situ, we studied PAR-1-deficient mice in two spontaneously developing murine tumor models: TRAMP (Transgenic Adenocarcinoma of the Mouse Prostate) mice, and mice carrying the Adenomatous Polyposis Coli Multiple Intestinal Neoplasia allele (Apcmin/+), which spontaneously develop intestinal adenomas. Surprisingly, the genetic elimination of PAR-1 resulted in more aggressive tumor progression in both experimental settings, indicating that PAR-1 can function as an inhibitor of tumor progression in some contexts.
METHODS
Murine tumor models and statistical analysis
All mice were backcrossed at least 8 generations onto a C57Bl/6 background. Female mice carrying the TRAMP transgene [23] were interbred with PAR-1-deficient mice (F2r−/−) [24], hereafter referred to as PAR-1−/−. Urogenital tracts from male TRAMP+/PAR-1−/− and TRAMP+/PAR-1+/+ mice were harvested at 12 or 30 weeks of age. Mice carrying the Adenomatous Polyposis Coli Multiple Intestinal Neoplasia allele (Apcmin/+) [25] were interbred with PAR-1−/− mice to generate APCMin/+ mice with and without PAR-1 expression. Intestinal tissue was harvested from male and female mice at 12 weeks of age. All mice used in these studies were generated in house and co-housed throughout the study. The Cincinnati Children’s Hospital Research Foundation Institutional Animal Care and Use Committee approved all animal studies. Statistical analyses were done using Prism Graphpad software. Comparisons of continuous variables between genotypes were made using a Mann-Whitney U test. Nominal variables were compared using a Fisher’s exact test.
Histology and immunohistochemistry
Hematoxylin and eosin (H&E) staining (5 μm sections) and immunohistochemistry were performed on formalin-fixed, paraffin-embedded tissue and imaged using a Nikon Ni-E microscope. For immunohistochemistry, antigen retrieval was achieved by treating deparaffinized tissue sections in citrate buffer heated with a pressure cooker. Sections were permeabilized with 0.3% Triton X-100 in PBS and blocked with 3% goat serum in PBS. Synaptophysin (SYP) staining was performed using a monoclonal antibody (H-8; Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:25 in a diluent prepared from the mouse-on-mouse kit (M.O.M.; Vector Laboratories; Burlingame, CA). M.O.M. antigen blocking and application of the biotinylated anti-mouse secondary antibody were performed per manufacturer’s specifications. Secondary antibody was detected with the Vector Red alkaline phosphatase kit (Vector Laboratories). Ki-67 (1:100 dilution; Neomarkers; Fremont, CA; #RB1510PO) and CC3 (1:100 dilution; Cell Signaling; Danvers, MA; #9661s) staining areas were measured by morphometric analysis using the Nikon NIS-Elements program (Version 4.5) by a blinded observer.
mRNA expression analyses
Gene expression was measured on RNA isolated from prostate tissue using Trizol, and cDNA was prepared with a High Capacity RNA-to-cDNA kit (Applied Biosystems; Waltham, MA). Real-time PCR analyses of synaptophysin were performed using the Taqman method with probe/primer sets (Mouse Syp: Mm00436850_m1; Mouse B2m: Mm00437762_m1; Applied Biosystems). The SV40 Large T Antigen probe/primer was obtained by a customized preparation from Applied Biosystems. Expression analyses of the genes listed in the Table S1 were performed using PowerUp SYBR Green PCR Master Mix (#A25776; Thermofisher) according to the manufacturer’s instructions.
Please see Data S1 for details regarding in vitro analyses.
RESULTS
PAR-1 deficiency enhances prostate tumor progression and promotes the formation of neuroendocrine tumors in TRAMP mice.
To define the role of PAR-1 in the setting of a spontaneously occurring prostate tumor developing in situ, PAR-1 deficient (PAR-1−/−) mice were interbred with TRAMP mice expressing the SV40 Large T antigen under control of the prostate epithelial-specific Probasin promoter [23]. Prostate tissue was harvested from male TRAMP+/PAR-1−/− and TRAMP+/PAR-1+/+ mice at 30 weeks of age, a time point when control animals are expected to have advanced disease. The vast majority (> 95%) of TRAMP+ male mice survived to 30 weeks of age, regardless of PAR-1 genotype. As expected, urogenital tracts (UGTs) harvested from TRAMP+/PAR-1+/+ were ~6-fold larger than those from mice not expressing the TRAMP transgene. Surprisingly, UGTs harvested from PAR-1-deficient TRAMP mice were almost 2-fold larger than those harvested from PAR-1 expressing mice (Fig. 1A.) The difference in UGT mass was not due to a genotype-dependent tendency toward prostatic hypertrophy in PAR-1−/− mice, as UGTs from 30 week old TRAMP−/PAR-1−/− mice were slightly smaller than age matched UGTs from TRAMP−/PAR-1+/+ animals (Fig. 1A). This modest PAR-1-dependent difference in UGT mass did not manifest as a difference in function. Fecundity of 8 aged-matched PAR-1−/− males (1.0 ± 0.23 litters/month) breeding over a 6 – 20 month time frame was not different from that of PAR-1+/+ males (1.2 ± 0.4 litters/month; P = 0.34; Mann Whitney U test).
Figure 1. PAR-1 impedes prostate tumor growth.

(A) Urogenital tracts (UGTs) harvested from 30 week old TRAMP mice with and without PAR-1 expression revealed that loss of PAR-1 significantly increased tumor size. This was not due to any inherent propensity for prostate hyperplasia in PAR-1−/− mice as UGTs harvested from TRAMP−/PAR-1−/− were smaller than those harvested from PAR-1+/+ animals at the same age. Horizontal bars represent medians. (B) Histological examination of prostate tumors from 30 week old TRAMP mice revealed that the majority of PAR-1+/+ mice had epithelial (EP) appearing prostate adenocarcinoma. In contrast, the NE appearing phenotype was more common in tumors harvested from PAR-1−/− mice. Note the complete loss of glandular structure and invasion of acinar connective tissue in NE tumors of both genotypes. Also note that prostate tissue from PAR-1+/+ and PAR-1−/− without the TRAMP transgene were similar in appearance. Size bars represent 25 μm.
It was also notable that the tumor mass in the TRAMP+/PAR-1−/− animals appeared bimodal, with 12 animals (41%) carrying tumors that were dramatically larger (>4 gms) than those carried by the majority of control animals, where only 5 animals (16%) had tumors comparable in size to this larger subset of TRAMP+/PAR-1−/− UGTs (Fig. 1A). Analysis of H&E stained sections from all UGTs harvested at 30 weeks of age revealed that the majority of prostate tumors in PAR-1+/+ mice were composed of highly transformed, epithelial-appearing cells (Fig. 1B). While the prostatic glandular structure in these animals was clearly dysmorphic, the glandular nature was still evident. The largest tumors harvested from TRAMP+/PAR-1+/+ mice displayed a more aggressive and invasive phenotype, consisting of sheets of highly-transformed malignant cells without any visible glandular structure, consistent with a previously described neuroendocrine (NE) phenotype (Fig. 1B), which occurs in 10–20% of C57BL/6-derived TRAMP mice [26]. NE morphology was also readily evident in the very large tumors (>4 grams) harvested from PAR-1−/− mice, suggesting that loss of PAR-1 promotes the development of this more aggressive phenotype (Fig. 1B).
To specifically determine if the genotype-dependent differences in the number of large tumors was due to differences in NE differentiation, we tested for NE transformation with the established neuroendocrine marker Synaptophysin (SYP). Sections stained for H&E and SYP were evaluated by a blinded investigator. Consistent with initial qualitative observations, tumors displaying a predominantly NE-phenotype correlated with the largest tumors and were significantly more common in PAR-1−/− mice (13 of 29) than control mice (5 of 32) (Fig. 2). NE-appearing tumors harvested from PAR-1+/+ and PAR-1−/− mice were similar in size (Fig. 2C). While the relative difference in the frequency of neuroendocrine tumors was a major contributor to the overall genotype dependent difference in tumor mass, it is notable that genotype-dependent comparisons of epithelial tumors revealed that PAR-1 deletion results in increased tumor size even in the absence of neuroendocrine differentiation (Fig. 2C). This suggests that loss of PAR-1 might promote prostate tumor growth through mechanism(s) independent of neuroendocrine differentiation.
Figure 2. PAR-1 deletion is associated with a neuroendocrine phenotype in TRAMP mice.

(A) Shown are typical examples of histological sections of prostate tumors from 30 week TRAMP mice immunohistochemically stained for synaptophysin (SYP, red staining). The epithelial phenotype (EP) more common in PAR+/+ mice had no evidence of SYP staining. Neuroendocrine (NE) appearing tumors, which were more common in PAR-1−/− mice, stained strongly for SYP. Size bars represent 25 µm. (B) The frequency of the NE phenotype was much higher in prostate tumors harvested from PAR-1−/− mice relative to PAR-1+/+ mice (P value generated with a Fisher’s exact test). (C) Comparison of UGT weights from 30 week old TRAMP mice with epithelial tumors (i.e., excluding NE tumors) revealed that tumors harvested from PAR-1−/− mice were significantly larger. There was no genotype dependent difference in UGT mass when NE tumors were compared.
PAR-1 promotes apoptosis in transformed prostate epithelia
To define the role of PAR-1 in early TRAMP tumor progression, we investigated prostate tumor development at 12 weeks of age; a time point well before the neuroendocrine switch. Similar to what was observed in 30 week old mice, UGTs harvested from PAR-1−/− mice tended to be smaller than those harvested from control mice in the absence of the TRAMP transgene, but this did not reach statistical significance. In contrast to 30 week old mice, UGT harvested from PAR-1+/+ and PAR-1−/− mice expressing the TRAMP transgene were similar in mass at this early time point, and similar to UGTs harvested from TRAMP–/PAR-1+/+ mice (Fig. 3A). Notably, UGTs from TRAMP+/PAR-1−/− mice were significantly larger than those from TRAMP−/PAR-1−/−, while no difference was discernable between UGTs from TRAMP+ and TRAMP−/PAR-1+/+ mice (Fig. 3A). These findings imply that tumor growth rates are accelerated by systemic loss of PAR-1 even at this early stage of tumor progression. Measurement of SV40-T mRNA from UGTs harvested at this time point revealed no PAR-1-dependent differences (data not shown), indicating that any PAR-1-dependent differences observed in TRAMP mice were not simply due to genotype-dependent differences in expression of the TRAMP transgene.
Figure 3. PAR-1 induces apoptosis in transformed prostate epithelium.

(A) In the absence of the TRAMP transgene, the mass of UGTs harvested from 12 week old PAR-1+/+ mice tended to be larger than those harvested from PAR1−/− mice, but this did not reach statistical significance. UGTs harvested from 12 week old PAR-1+/+ and PAR1−/− mice carrying the TRAMP transgene were indistinguishable in size. More importantly, UGTs harvested from PAR-1+/+ mice were similar in size regardless of the presence or absence of the TRAMP transgene, whereas UGTs harvested from TRAMP+/PAR-1−/− mice were significantly larger than those harvested from TRAMP−/PAR-1−/− animals. These findings imply that tumor growth rates are accelerated by systemic loss of PAR-1 even at this early stage of tumor progression. Horizontal bars represent medians. (B) Shown are typical examples of the histological appearance of prostates harvested at 12 weeks from PAR-1+/+ and PAR-1−/− mice, including immunohistochemical analyses of markers of proliferation (Ki-67, arrowheads showing positive nuclei) and apoptosis (cleaved caspase 3, CC3, dark staining). Size bars represent 25 µm (C) Consistent with the early time point of evaluation, there was no evidence of neuroendocrine differentiation in either genotype based on quantitation of synaptophysin mRNA. The level of synaptophysin mRNA was similar in UGTs harvested from PAR-1+/+ (n = 12) and PAR-1−/− (n = 9) TRAMP+ mice, and not different than the level of expression in TRAMP− UGTs. Note that some synaptophysin expression would be expected due to the presence of neuronal tissue. (D) Quantitation of Ki-67+ nuclei was indistinguishable between PAR-1 genotypes (n = 9 and 5). (E) Quantitative analysis of the percentage of CC3 staining epithelial cells in early transformed prostate epithelium from 12 week old mice demonstrated diminished apoptosis in prostates harvested from PAR-1−/− mice (n = 9) relative to PAR-1+/+ controls (n = 8). Approximately 1500 prostate epithelial cells were evaluated from each mouse by a researcher blinded to genotype.
Histological analyses of prostate tissue harvested from 12 week old TRAMP+ mice demonstrated that all the animals had clear signs of prostatic intraepithelial neoplasia (PIN) regardless of PAR-1 genotype. Based on H&E analysis, there were no obvious histological differences between cohorts (Fig. 3B). The prostate epithelia from TRAMP+ mice consisted of irregular, atypical-appearing epithelial cells with large nuclei, frequent nucleoli, and loss of polarity. Although there was evidence of crowding and a multilayer appearance, the abnormal epithelia retained a glandular morphology, and remained confined to the acinus without evidence of invasion. Based on SYP staining, none of the prostates harvested at this early time point had any histological evidence of NE differentiation, regardless of PAR-1 genotype (data not shown). Consistent with this, synaptophysin mRNA expression was similar between UGTs harvested from PAR-1+/+ and PAR-1−/− mice carrying the TRAMP transgene (Fig. 3C).
To determine the impact of PAR-1 on the proliferative capacity of prostate epithelia in TRAMP+ mice at this early time point, prostate sections from PAR-1+/+ and PAR-1−/− mice were stained for the proliferative marker Ki-67. As expected, Ki-67+ cells were relatively common in areas of PIN, and rare in areas of non-transformed prostate or TRAMP− prostates, regardless of genotype. When calculated as a percentage of Ki-67+ nuclei relative to total nuclei in areas of prostate epithelium, the frequency of proliferative cells was similar between PAR-1 genotypes (Fig. 3B & D). We next analyzed the effect of PAR-1 deletion on the survival of transformed prostate epithelia by immunohistochemically staining for the apoptosis marker cleaved caspase 3 (CC3). Genetic elimination of PAR-1 resulted in significantly less cell death as evidenced by decreased CC3 staining relative to control mice (Fig. 3B & E). Only rare CC3 staining was observed in non-transformed prostate epithelia from TRAMP– mice, regardless of PAR-1 genotype (data not shown).
Activated Protein C-mediated PAR-1 activation promotes TRAMP tumor cell apoptosis.
In order to define the tumor cell-intrinsic potential of PAR-1 activation to promote apoptosis in TRAMP prostate tumor cells, we exposed well-established TRAMP-C1 cells [27] to peptide mimetics of activated protein C-mediated (TR47), or thrombin-mediated (TR42) PAR-1 activation [7] and measured CC3 activity. Analyses 30 minutes and 24 hours after TR47 exposure revealed that activated protein C-mediated PAR-1 activation induces apoptosis in TRAMP-C1 (Fig. 4A), whereas TR42 exposure had no effect on CC3 activity. We also evaluated the potential of these peptide mimetics to promote phosphorylation of ERK1/2 in TRAMP-derived cells. Here, TR42 exposure resulted in a significant increase in ERK phosphorylation relative to TR47 (Fig. 4B). Increased in vitro PAR-1 mediated apoptosis is consistent with our in vivo findings of increased apoptosis in PAR-1 sufficient prostate tumors. Collectively, these data support the working concept that thrombin and activated protein C have opposing effects on prostate tumor cells, and that activated protein C-mediated activation of PAR-1 expressed by transformed prostate epithelial could promote apoptosis thereby limiting tumor progression.
Figure 4. Activated protein C-mediated activation of PAR-1 induces apoptosis in TRAMP-derived tumor cells.

(A) Cleaved caspase 3 (CC3) activity was measured in TRAMP-C1 cells following exposure to 10 μM of scrambled peptide (ScTR47, SCR), TR47 (activated protein C mimetic) peptide and TR42 (thrombin mimetic) peptide. TR47 exposure significantly increased CC3 activity, whereas TR42 exposure did not. (B) Western blot analyses of ERK1/2 expression and phosphorylation in TRAMP-C1 cells treated with 10 μM of SCR, TR47, or TR42 showed that thrombin-mediated PAR-1 activation significantly increases phospho-ERK relative to activated protein C-mediated activation. Also shown is a densitometry analyses of the Western Blot relative to the β-actin control based on 3 separate Western blots with similar results. (*P < 0.05, **P < 0.005)
Previous studies have shown that PAR-2 promotes tumor progression [22, 28], and that PAR-1 and PAR-2 play cooperative roles in some contexts [29, 30]. In order to determine the impact of PAR-1 deletion on the expression of other PARs, as well as key cellular components of the hemostatic system, we compared the expression of PAR-2, −3, −4, tissue factor (TF), thrombomodulin (THBD), and endothelial protein C receptor (EPCR) in prostates harvested from 12 week old TRAMP mice with and without PAR-1 deletion. Prostates harvested from PAR-1−/− TRAMP mice demonstrated diminished expression of PAR-2 and −3 relative to PAR-1+/+ mice (Fig. S1). No genotype dependent differences were observed in the expression of PAR-4, TF, THBD or EPCR (Fig. S1). Analyses of PAR expression in non-transformed (TRAMP negative) prostate tissue revealed similar diminutions in PAR-2 and −3 in PAR-1−/− mice (Fig. S1), indicating that these differences were not related to the TRAMP transgene or downstream transformation events.
PAR-1 deficiency enhances spontaneous intestinal adenoma formation
To determine the role of PAR-1 in an experimental setting of in situ tumorigenesis without NE differentiation, PAR-1−/− mice were interbred with mice heterozygous for a tumor suppressor mutation in the adenomatous polyposis coli gene (APCMin/+). APCMin/+ mice develop multiple intestinal adenomas throughout the intestinal tract with 100% penetrance. PAR-1+/+ and PAR-1−/− mice carrying the APCMin/+ allele were harvested at 12 weeks of age, a time point where multiple adenomas are expected. The entire small and large intestine was harvested, prepared as Swiss rolls, and evaluated histologically for evidence of adenoma formation.
As expected, mice carrying the APCMin/+ allele developed multiple adenomas, primarily in the small intestine. Adenomas generally consisted of nodular thickening of the mucosa characterized by elongated glands, nuclear pleomorphism, loss of polarity, and loss of cellular orientation with some glands characterized by epithelial cell piling (Fig. 5A). Adenomas harvested from APCMin/+/PAR-1−/− were similar in overall morphology compared to those harvested from PAR-1 expressing mice (Fig. 5A). While qualitatively similar, loss of PAR-1 led to a ~3-fold increase in the number of gastrointestinal (GI) adenomas (Fig. 5B). Data stratified by GI tract regions (jejunem, ileum and colon) showed that loss of PAR-1 did not affect the relative location of developing adenomas (Fig. 5C). Consistent with previous reports [25], the majority of adenomas formed in the ileum, followed by the jejunum, and only a small fraction was evident in the colon, regardless of PAR-1 genotype. Although the overall architecture of adenomas was similar between genotypes, adenomas harvested from PAR-1−/− mice generally appeared larger than those harvested from PAR-1+/+ animals. This was confirmed by morphometric analyses of adenoma surface area visible in histological cross sections. Paralleling the observations in prostate tumors, adenomas harvested from PAR-1−/− mice were ~2-fold larger than those harvested from control mice (Fig. 5A & D). The non-adenomatous intestinal tissue from these animals appeared healthy and was indistinguishable between genotypes (Fig. 5E). Note that roughly equal numbers of male and female APCMin/+ mice were enrolled in these studies. Separate analyses revealed that the PAR-1 dependence on adenoma number and size was similar in males and females and therefore not dependent on sex (data not shown).
Figure 5. PAR-1 impedes intestinal adenoma formation.

(A) Shown are representative H&E stained tissue sections demonstrating the typical appearance and relative size of intestinal adenomas from PAR-1+/+ and PAR-1−/− mice. (B) Quantitation of adenomas from histological sections of the entire intestinal tract prepared from 12 week old APCMin/+/PAR-1+/+ and APCMin/+/PAR-1−/− mice. PAR-1 deficiency markedly increased the number of adenomas observed. Horizontal bars represent medians. (C) The relative frequency of adenomas in each section of the intestine was not significantly different between genotypes. (P = N.S., Chi-square test) (D) The area of adenomas in intestines from APCMin/+ mice was measured by morphometric analysis of histological sections. Adenomas from PAR-1−/− mice were significantly larger in area compared to adenomas from PAR-1+/+ animals. The data shown represent individual adenomas measured from 12 PAR-1+/+ mice and 11 PAR-1−/− mice. Horizontal bars represent medians. (E) Nonadenometous intestinal tissue from APCMin/+ mice appeared intact and healthy and was indistinguishable between PAR-1 genotypes.
PAR-1 promotes apoptosis of transformed epithelia in APCMin/+ mice
In light of the observation that PAR-1 induces apoptosis but not cellular proliferation in transformed prostate epithelium, we next tested the proliferative/cell death indices of adenometous intestinal epithelium from APCMin/+ mice. Staining for the nuclear proliferation marker Ki-67 revealed that the overwhelming majority of intestinal epithelial cells in adenomatous tissue stained strongly positive for Ki-67, regardless of PAR-1 genotype (Fig. 6A). Proliferation in non-adenomatous intestinal epithelial tissue was limited to the proximal third of the intestinal crypts, and was also similar between PAR-1 genotypes (Fig. 6A).
Figure 6. PAR-1 induces apoptosis in intestinal adenomas.
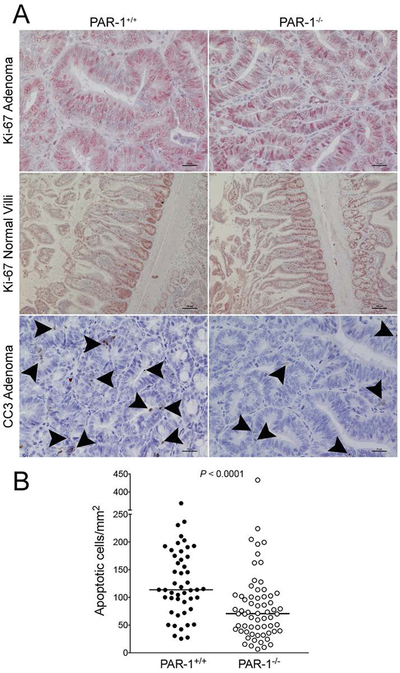
(A) Shown are representative immunohistochemical stains of intestinal tissue from APCMin/+ mice with and without PAR-1 deletion. In adenomatous tissue, the vast majority of nuclei were Ki-67 positive (brown), regardless of PAR-1 genotype. Ki-67 staining of intestinal tissue in nonadenometous intestinal tissue was localized to intestinal crypts, with no observable differences between PAR-1 genotypes. The frequency of cells staining positive for cleaved caspase 3 (CC3, dark staining noted by arrowheads) was higher in adenomatous tissue from PAR-1+/+ mice relative to PAR-1−/− mice. (B) Quantitation of CC3+ staining cells as a function of adenoma surface area confirmed that loss of PAR-1 significantly limited apoptosis in transformed intestinal epithelia. Note that each data point represents an individual adenoma. Data was collected from 8 representative mice of each genotype. The horizontal bars represent medians.
In order to determine if PAR-1 promotes the apoptosis of transformed intestinal epithelia, intestinal tissue was tested for the presence of the apoptosis marker CC3 in transformed and healthy-appearing tissue. Paralleling observations in TRAMP mice, the genetic elimination of PAR-1 resulted in a significant decrease in the number of CC3-positive staining cells in intestinal adenomas. As a function of apoptotic CC3-positive cells per unit area, adenomas from APCMin/+/PAR-1−/− mice demonstrated ~35% fewer apoptotic foci compared to adenomas from APCMin/+/PAR-1+/+ mice (Fig. 6A & B). Also paralleling results in TRAMP mice, the genotype-dependent difference in CC3 foci was limited to transformed epithelia. In healthy appearing non-tumorous epithelial tissue, the frequency of CC3-positive cells was not different between PAR-1 genotypes (data not shown).
DISCUSSION
These studies establish that PAR-1 functions can limit tumor progression in two distinct settings in vivo; one driven by oncogene expression (TRAMP mice), and one by the loss of a tumor suppressor gene (APCMin/+ mice). These studies show that one mechanism by which PAR-1 appears to limit tumor progression is by promoting the apoptosis of transformed cells at a relatively early stage in tumor development, thereby limiting tumor growth potential. Although the mechanistic details remain to be fully resolved, these studies reveal a previously unrecognized function of PAR-1 in suppressing tumor growth.
The finding presented here are surprising in that the preponderance of the literature suggests that PAR-1 promotes tumor progression. This conclusion is based on in vitro analyses, which may be limited by factors such as the lack of tumor/stromal cell interaction [9–14], as well as multiple detailed in vivo analyses based on highly-specific PAR-1 inhibitors [18], or genetic manipulation of PAR-1 in tumor cells or stromal cells [15–21, 31, 32]. Further complicating the picture, analyses of a murine model of spontaneously developing breast carcinoma demonstrated no impact of PAR-1 deletion on tumor progression [22]. Clearly, PAR-1 plays a context-dependent role in tumor pathogenesis. Notably, previous in vivo studies where PAR-1 was shown to promote tumor progression utilized transplantable, fully-transformed xenograft and congenic tumor cell lines, whereas the current study was based on spontaneous, in situ tumor development. It is conceivable that PAR-1 plays very different roles in earlier stages of tumorigenesis than in later stages when tumors are fully transformed. Future studies focused on targeting PAR-1 at different time-points in tumor development could better define its role at each stage of cancer progression. It is also conceivable that PAR-1 has a distinctly specific role (or no role whatsoever) in cancer progression depending on the tumor type. The studies presented here represent the first analyses of PAR-1 in prostate and intestinal tumorigenesis. It is entirely conceivable that analyses of PAR-1 in the progression of other tumor types may yield very different results.
A major finding regarding the role of PAR-1 in TRAMP mice was more frequent neuroendocrine prostate tumor differentiation in PAR-1−/− animals analyzed at later stages of cancer progression. Neuroendocrine differentiation is a rare occurrence in human prostate cancer, but is clinically relevant as it is associated with a dismal prognosis [33]. Determining whether PAR-1 plays a role in neuroendocrine differentiation in human prostate cancer will require further study. However, analyses at an early stage of disease, prior to neuroendocrine differentiation, showed that loss of PAR-1 also diminished apoptosis in transformed prostate epithelia. This finding was recapitulated in early stage intestinal adenomas from APCMin/+ mice, an experimental context without neuroendocrine differentiation. One straightforward explanation for the relative increase in neuroendocrine tumors in PAR-1-deficient TRAMP mice is that PAR-1 deletion increases the chances of individual tumor cell survival, thus leading a greater likelihood of a neuroendocrine switch.
The data presented here also suggest that activation of tumor cell-associated PAR-1 by activated protein C rather than thrombin promotes apoptosis in TRAMP-derived tumor cells. However, the extracellular region of PAR-1 can be cleaved at various amino acid residues by upwards of twelve different enzymes [34], making a firm identification of the PAR-1 activator in this context impossible. Activated protein C-mediated PAR1 signaling is generally considered to convey cytoprotective effects on endothelial and other cells [7, 35], thus increases of cleaved caspase 3 in the presence of the activated protein C-like PAR1 signaling mimetic, TR47, is unexpected. The PAR-1 interactome of cancer cells are likely to differ from that of endothelial cells. This illustrates that different cells, and especially cancer cells, may have adapted responses to natural stimuli, but the PAR-1 interactome on different cells remains poorly defined. Moreover, the precise role of activated protein C-mediated versus thrombin-mediated PAR-1 activation in these experimental contexts was not evaluated in vivo. The recent generation of mice carrying specific mutations in PAR-1 that bias signaling toward a particular protease [36] would be an invaluable tool in better resolving the mechanistic puzzle of which activator is driving specific individual phenotypes. Lastly, the potential of PAR-1 activation by proteases other than thrombin and activated protein C to promote apoptosis cannot be discounted [37].
Further complicating our understanding of PAR-1 biology is the fact that this receptor is expressed on multiple cell types in the tumor microenvironment in addition to tumor cells, including macrophages, endothelial cells, and fibroblasts [3]. The current studies point to at least one tumor cell intrinsic mechanism involving PAR-1 driven apoptosis, but cannot exclude the possibility of tumor cell extrinsic PAR-1 dependent mechanisms. The generation of mice with cell type-specific PAR-1 deletion would be useful in determining if tumor cell extrinsic mechanisms are at play in these experimental settings. It is also worth noting that several recent studies have shown that PAR-1 and PAR-2 play cooperative signaling roles in some contexts [29, 30], and PAR-2 has been shown to promote tumor progression [22, 28]. It remains to be determined whether alterations in PAR-2 signaling resulting from loss of PAR-1/PAR-2 heterodimers play any role in the phenotype observed in the studies presented here. However, it is notable that PAR-2 expression was significantly diminished in the prostates of PAR-1−/− deficient mice, regardless of the presence of the TRAMP transgene. The relative loss of PAR-2 expression in PAR-1−/− murine prostates makes it seem less likely that mechanisms related to PAR-2 driven tumor growth play a significant role here.
In summary, these data reveal a previously under-recognized role of PAR-1 in impeding tumor progression in the context of two independent murine models of cancer. Whether PAR-1 ever plays a similar role in humans malignancies remains to be determined. Previous studies showed a correlation between increased PAR-1 expression and poor prognosis in human prostate cancer [38], suggesting PAR-1 drives prostate cancer progression. However. it is conceivable that PAR-1 expressed by some cellular compartments promotes cancer progression, while PAR-1 in other compartments limits it. In this regard, it is notable that the preponderance of PAR-1 upregulation in human prostate cancer appears to be in the endothelial compartment [39]. Complete elimination of PAR-1 would reflect the summation of all PAR-1 activities in all cell types. Therefore, the current study may represent a cautionary note on the long-term use of PAR-1 inhibitors. The clinical use of PAR-1 inhibitors to prevent thromboembolic complications is relatively recent [40], making it impossible to know if long-term PAR-1 inhibition has any impact, positive or negative, on tumorigenesis in humans. Several studies have shown a correlation between long-term anticoagulation and a decreased incidence of multiple types of cancer, including prostate cancer [41–43], suggesting that thrombin may play a role in early events important in tumorigenesis. Indeed, this conclusion is supported by numerous studies in animal models [44], including studies showing that thrombin inhibition significantly limits prostate tumor progression in TRAMP mice [45]. However, thrombin has over a dozen recognized substrates, and the data presented here suggests that thrombin does not promote tumor progression in TRAMP mice via a mechanism coupled to PAR-1 activation. Further study is critically needed to better understand the mechanisms coupling PAR-1 functions to cancer progression and to determine whether there are contexts where long-term PAR-1 inhibition could pose a cancer risk.
Supplementary Material
ESSENTIALS.
Protease activated receptor-1 (PAR-1) has been proposed to drive cancer progression.
Surprisingly, PAR-1 deletion accelerated tumor progression in two distinct experimental settings.
PAR-1 deletion was shown to limit the apoptosis of transformed epithelial cells.
Thrombin- and activated protein C-mediated PAR-1 activation have unique effects on tumor cell biology.
ACKNOWLEDGEMENTS
This work was funded by grants from the National Institutes of Health (JSP, R01 CA204058 and CA207503) and the Pelotonia Fellowship Program (GNA). Any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect those of the Pelotonia Fellowship Program.
Footnotes
ADDENDUM
J. S. Palumbo designed and supervised the study, acquired and analyzed data, and revised the manuscript. G. N. Adams assisted in study design, acquired and analyzed data, and wrote the manuscript. K. A. Steinbrecher acquired/analyzed data, assisted with study design and interpretation of data, and reviewed/edited the manuscript. M. J. Flick and D. P. Witte assisted with study design and interpretation of data and reviewed/edited the manuscript. L. O. Mosnier provided critical reagents and reviewed/edited the manuscript. B. K. Sharma, M. Frederick, L. Rosenfeldt, and E Harmel-Laws generated data and offered critical technical and material support.
Disclosure of Conflict of Interest
L.O. Mosnier has a patent pending: Protease Activated Receptor-1 (PAR1) Derived Cytoprotective Polypeptides and Related Methods (PCT/US12/00546) with royalties paid to The Scripps Research Institute. The authors state that they have no conflict of interest.
REFERENCES
- 1.Coughlin SR. Protease-activated receptors and platelet function. Thrombosis and haemostasis. 1999; 82: 353–6. [PubMed] [Google Scholar]
- 2.Derian CK, Santulli RJ, Tomko KA, Haertlein BJ, Andrade-Gordon P. Species differences in platelet responses to thrombin and SFLLRN. receptor-mediated calcium mobilization and aggregation, and regulation by protein kinases. Thrombosis research. 1995; 78: 505–19. [DOI] [PubMed] [Google Scholar]
- 3.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacological reviews. 2001; 53: 245–82. [PubMed] [Google Scholar]
- 4.Koukos G, Sevigny L, Zhang P, Covic L, Kuliopulos A. Serine and metalloprotease signaling through PAR1 in arterial thrombosis and vascular injury. IUBMB life. 2011; 63: 412–8. 10.1002/iub.465. [DOI] [PubMed] [Google Scholar]
- 5.Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, Hooper JD. Structure, function and pathophysiology of protease activated receptors. Pharmacology & therapeutics. 2011; 130: 248–82. 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Mihara K, Ramachandran R, Renaux B, Saifeddine M, Hollenberg MD. Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). The Journal of biological chemistry. 2013; 288: 32979–90. 10.1074/jbc.M113.483123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012; 120: 5237–46. 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV. Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer metastasis reviews. 2015; 34: 775–96. 10.1007/s10555-015-9599-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darmoul D, Gratio V, Devaud H, Lehy T, Laburthe M. Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. The American journal of pathology. 2003; 162: 1503–13. 10.1016/s0002-9440(10)64283-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang E, Boire A, Agarwal A, Nguyen N, O’Callaghan K, Tu P, Kuliopulos A, Covic L. Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer research. 2009; 69: 6223–31. 10.1158/0008-5472.can-09-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fazzini A, D’Antongiovanni V, Giusti L, Da Valle Y, Ciregia F, Piano I, Caputo A, D’Ursi AM, Gargini C, Lucacchini A, Mazzoni MR. Altered protease-activated receptor-1 expression and signaling in a malignant pleural mesothelioma cell line, NCI-H28, with homozygous deletion of the beta-catenin gene. PloS one. 2014; 9: e111550 10.1371/journal.pone.0111550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi X, Gangadharan B, Brass LF, Ruf W, Mueller BM. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Molecular cancer research : MCR. 2004; 2: 395–402. [PubMed] [Google Scholar]
- 13.Bar-Shavit R, Turm H, Salah Z, Maoz M, Cohen I, Weiss E, Uziely B, Grisaru-Granovsky S. PAR1 plays a role in epithelial malignancies: transcriptional regulation and novel signaling pathway. IUBMB life. 2011; 63: 397–402. 10.1002/iub.452. [DOI] [PubMed] [Google Scholar]
- 14.Tellez C, Bar-Eli M. Role and regulation of the thrombin receptor (PAR-1) in human melanoma. Oncogene. 2003; 22: 3130–7. 10.1038/sj.onc.1206453. [DOI] [PubMed] [Google Scholar]
- 15.Villares GJ, Zigler M, Dobroff AS, Wang H, Song R, Melnikova VO, Huang L, Braeuer RR, Bar-Eli M. Protease activated receptor-1 inhibits the Maspin tumor-suppressor gene to determine the melanoma metastatic phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108: 626–31. 10.1073/pnas.1006886108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Queiroz KC, Shi K, Duitman J, Aberson HL, Wilmink JW, van Noesel CJ, Richel DJ, Spek CA. Protease-activated receptor-1 drives pancreatic cancer progression and chemoresistance. International journal of cancer. 2014; 135: 2294–304. 10.1002/ijc.28726. [DOI] [PubMed] [Google Scholar]
- 17.Yin YJ, Salah Z, Maoz M, Even Ram SC, Ochayon S, Neufeld G, Katzav S, Bar-Shavit R. Oncogenic transformation induces tumor angiogenesis: a role for PAR1 activation. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003; 17: 163–74. 10.1096/fj.02-0316com. [DOI] [PubMed] [Google Scholar]
- 18.Cisowski J, O’Callaghan K, Kuliopulos A, Yang J, Nguyen N, Deng Q, Yang E, Fogel M, Tressel S, Foley C, Agarwal A, Hunt SW 3rd, McMurry T, Brinckerhoff L, Covic L. Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. The American journal of pathology. 2011; 179: 513–23. 10.1016/j.ajpath.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villares GJ, Zigler M, Wang H, Melnikova VO, Wu H, Friedman R, Leslie MC, Vivas-Mejia PE, Lopez-Berestein G, Sood AK, Bar-Eli M. Targeting melanoma growth and metastasis with systemic delivery of liposome-incorporated protease-activated receptor-1 small interfering RNA. Cancer research. 2008; 68: 9078–86. 10.1158/0008-5472.can-08-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohen I, Maoz M, Turm H, Grisaru-Granovsky S, Maly B, Uziely B, Weiss E, Abramovitch R, Gross E, Barzilay O, Qiu Y, Bar-Shavit R. Etk/Bmx regulates proteinase-activated-receptor1 (PAR1) in breast cancer invasion: signaling partners, hierarchy and physiological significance. PloS one. 2010; 5: e11135 10.1371/journal.pone.0011135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kancharla A, Maoz M, Jaber M, Agranovich D, Peretz T, Grisaru-Granovsky S, Uziely B, Bar-Shavit R. PH motifs in PAR1&2 endow breast cancer growth. Nature communications. 2015; 6: 8853 10.1038/ncomms9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Versteeg HH, Schaffner F, Kerver M, Ellies LG, Andrade-Gordon P, Mueller BM, Ruf W. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer research. 2008; 68: 7219–27. 10.1158/0008-5472.can-08-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, Rosen JM. Prostate cancer in a transgenic mouse. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92: 3439–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darrow AL, Fung-Leung WP, Ye RD, Santulli RJ, Cheung WM, Derian CK, Burns CL, Damiano BP, Zhou L, Keenan CM, Peterson PA, Andrade-Gordon P. Biological consequences of thrombin receptor deficiency in mice. Thrombosis and haemostasis. 1996; 76: 860–6. [PubMed] [Google Scholar]
- 25.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science (New York, NY). 1990; 247: 322–4. [DOI] [PubMed] [Google Scholar]
- 26.Chiaverotti T, Couto SS, Donjacour A, Mao JH, Nagase H, Cardiff RD, Cunha GR, Balmain A. Dissociation of epithelial and neuroendocrine carcinoma lineages in the transgenic adenocarcinoma of mouse prostate model of prostate cancer. The American journal of pathology. 2008; 172: 236–46. 10.2353/ajpath.2008.070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer research. 1997; 57: 3325–30. [PubMed] [Google Scholar]
- 28.Svensson KJ, Kucharzewska P, Christianson HC, Skold S, Lofstedt T, Johansson MC, Morgelin M, Bengzon J, Ruf W, Belting M. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108: 13147–52. 10.1073/pnas.1104261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McEachron TA, Pawlinski R, Richards KL, Church FC, Mackman N. Protease-activated receptors mediate crosstalk between coagulation and fibrinolysis. Blood. 2010; 116: 5037–44. 10.1182/blood-2010-06-293126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaber M, Maoz M, Kancharla A, Agranovich D, Peretz T, Grisaru-Granovsky S, Uziely B, Bar-Shavit R. Protease-activated-receptor-2 affects protease-activated-receptor-1-driven breast cancer. Cellular and molecular life sciences : CMLS. 2014; 71: 2517–33. 10.1007/s00018-013-1498-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams GN, Rosenfeldt L, Frederick M, Miller W, Waltz D, Kombrinck K, McElhinney KE, Flick MJ, Monia BP, Revenko AS, Palumbo JS. Colon Cancer Growth and Dissemination Relies upon Thrombin, Stromal PAR-1, and Fibrinogen. Cancer research. 2015. 10.1158/0008-5472.can-15-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Auvergne R, Wu C, Connell A, Au S, Cornwell A, Osipovitch M, Benraiss A, Dangelmajer S, Guerrero-Cazares H, Quinones-Hinojosa A, Goldman SA. PAR1 inhibition suppresses the self-renewal and growth of A2B5-defined glioma progenitor cells and their derived gliomas in vivo. Oncogene. 2016; 35: 3817–28. 10.1038/onc.2015.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parimi V, Goyal R, Poropatich K, Yang XJ. Neuroendocrine differentiation of prostate cancer: a review. American journal of clinical and experimental urology. 2014; 2: 273–85. [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao P, Metcalf M, Bunnett NW. Biased signaling of protease-activated receptors. Frontiers in endocrinology. 2014; 5: 67 10.3389/fendo.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouwens EA, Stavenuiter F, Mosnier LO. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. Journal of thrombosis and haemostasis : JTH. 2013; 11 Suppl 1: 242–53. 10.1111/jth.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinha RK, Wang Y, Zhao Z, Xu X, Burnier L, Gupta N, Fernandez JA, Martin G, Kupriyanov S, Mosnier LO, Zlokovic BV, Griffin JH. PAR1 biased signaling is required for activated protein C in vivo benefits in sepsis and stroke. Blood. 2018; 131: 1163–71. 10.1182/blood-2017-10-810895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki T, Yamashita C, Zemans RL, Briones N, Van Linden A, Downey GP. Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-kappaB-, and p53-dependent pathway. American journal of respiratory cell and molecular biology. 2009; 41: 742–55. 10.1165/rcmb.2008-0157OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Liu D, Zhou W, Wang M, Xia W, Tang Q. Prognostic value of matrix metalloprotease-1/protease-activated receptor-1 axis in patients with prostate cancer. Medical oncology (Northwood, London, England). 2014; 31: 968 10.1007/s12032-014-0968-6. [DOI] [PubMed] [Google Scholar]
- 39.Kaushal V, Kohli M, Dennis RA, Siegel ER, Chiles WW, Mukunyadzi P. Thrombin receptor expression is upregulated in prostate cancer. The Prostate. 2006; 66: 273–82. 10.1002/pros.20326. [DOI] [PubMed] [Google Scholar]
- 40.Wang A Review of vorapaxar for the prevention of atherothrombotic events. Expert opinion on pharmacotherapy. 2015; 16: 2509–22. 10.1517/14656566.2015.1099629. [DOI] [PubMed] [Google Scholar]
- 41.Haaland GS, Falk RS, Straume O, Lorens JB. Association of Warfarin Use With Lower Overall Cancer Incidence Among Patients Older Than 50 Years. JAMA internal medicine. 2017; 177: 1774–80. 10.1001/jamainternmed.2017.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pengo V, Noventa F, Denas G, Pengo MF, Gallo U, Grion AM, Iliceto S, Prandoni P. Long-term use of vitamin K antagonists and incidence of cancer: a population-based study. Blood. 2011; 117: 1707–9. 10.1182/blood-2010-08-304758. [DOI] [PubMed] [Google Scholar]
- 43.Schulman S, Lindmarker P. Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. The New England journal of medicine. 2000; 342: 1953–8. 10.1056/nejm200006293422604. [DOI] [PubMed] [Google Scholar]
- 44.Remiker AS, Palumbo JS. Mechanisms coupling thrombin to metastasis and tumorigenesis. Thrombosis research. 2018; 164 Suppl 1: S29–s33. 10.1016/j.thromres.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 45.Hu L, Ibrahim S, Liu C, Skaar J, Pagano M, Karpatkin S. Thrombin induces tumor cell cycle activation and spontaneous growth by down-regulation of p27Kip1, in association with the up-regulation of Skp2 and MiR-222. Cancer research. 2009; 69: 3374–81. 10.1158/0008-5472.can-08-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.