

Abstract
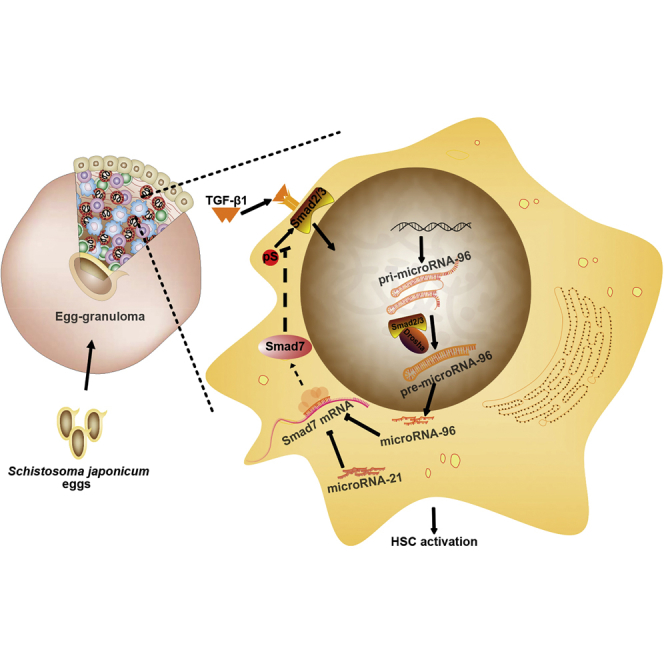
Infection with Schistosoma causes aberrant expression of host microRNAs (miRNAs), and normalizing the levels of dysregulated miRNAs can attenuate pathology. Here, we show that the host miRNA, miR-96, is markedly upregulated during the progression of hepatic schistosomiasis. We demonstrate that elevation of miR-96 induces hepatic fibrosis in infected mice by suppressing the expression of its target gene, Smad7. We show that infection with Schistosoma induces the expression of transforming growth factor β1 (TGF-β1), which in turn upregulates the expression of miR-96 through SMAD2/3-DROSHA-mediated post-transcriptional regulation. Furthermore, inhibition of miR-96 with recombinant adeno-associated virus 8 (rAAV8)-mediated delivery of Tough Decoy RNAs in mice attenuated hepatic fibrosis and prevented lethality following schistosome infection. Taken together, our data highlight the potential for rAAV8-mediated inhibition of miR-96 as a therapeutic strategy to treat hepatic schistosomiasis.
Keywords: schistosomiasis, microRNA, TGF-β/Smad7, hepatic stellate cell
Graphical Abstract
Introduction
Schistosomiasis, also known as bilharzia, is a serious parasitic disease that is common in tropical and subtropical regions. The schistosome parasite infects approximately 210 million people worldwide, resulting in 120,000–200,000 deaths annually.1, 2, 3 Schistosoma japonicum is endemic to Southeast Asia, where about 50 million people are at risk for infection.4 The schistosome parasite undergoes a complex life cycle involving multiple developmental stages. Following infection, the main pathology is granulomatous inflammation and hepatic fibrosis caused by the host response to schistosome eggs trapped in the liver. Hepatic fibrosis and the resulting portal hypertension are the primary causes of host mortality.
The liver is composed of hepatocytes and resident non-parenchymal cells such as hepatic stellate cells (HSCs) and Kupffer cells.5 The activation of HSCs is a key step in liver fibrosis.6 Upon activation, HSCs gradually transform into proliferative, contractile, and fibrogenic myofibroblasts. They secrete excess extracellular matrix (ECM) that is deposited around damaged sites in the liver undergoing constant wound repair, ultimately leading to fibrosis.7, 8 It has been shown that schistosome infection activates HSCs distributed around the periphery of egg-induced granulomas.4 Activated HSCs, in turn, produce IL13 and transforming growth factor β1 (TGF-β1), two major mediators of schistosomiasis hepatic fibrosis.9, 10, 11
MicroRNAs (miRNAs) play important roles in maintaining cellular homeostasis under both normal and diseased conditions. Dysregulation of miRNA expression is involved in fibrosis of multiple organ systems, including the vasculature (pulmonary fibrosis), liver, and kidney.12, 13, 14 Our previous study showed that increased expression of miR-21 in HSCs in a murine model of schistosome infection activated Smad7, resulting in the induction of hepatic fibrosis.15 Moreover, inhibition of miR-21 in infected mice with recombinant adeno-associated virus-8 (rAAV8)-mediated delivery of Tough Decoy RNAs (rAAV8-anti-miR-21-TuD) attenuated hepatic fibrosis by relieving the inhibitory effect of SMAD7 during SMAD signaling.15 However, reduction of miR-21 only partially mitigates hepatic fibrosis, suggesting at the involvement of additional regulators. Here, we report that host expression of miR-96 also plays a key role in schistosomiasis hepatic fibrosis by targeting smad7 in the TGF-β1/SMAD pathway. Furthermore, recombinant AAV8-mediated inhibition of miR-96 effectively attenuated this pathology, providing protection against lethal infection of Schistosoma in the well-studied murine model of the disease.
Results
Identification of Hepatic Fibrosis-Relevant miRNAs and Their Putative Targets
To identify host miRNAs that are involved in promoting schistosomiasis hepatic fibrosis, we assessed the expression profile of selected miRNAs based on our previous data15 and those previously reported to be involved in fibrogenesis of S. japonicum-infected liver tissues.14, 16, 17 We identified 29 upregulated (>2-fold) and 4 downregulated (>2-fold) miRNAs from among 52 mouse miRNAs in infected versus normal livers (Figure S1). Considering the key role of HSCs in the progression of liver fibrosis, we specifically assessed the expression of these 29 miRNAs in the HSCs of normal and S. japonicum-infected livers. As shown in Figure S2, miR-96 and three other miRNAs, miR-146, miR-184, and miR-2137, were highly upregulated in HSCs of infected livers (>4-fold). We next searched for putative targets of the highly upregulated miRNAs using miRNA target prediction programs (http://www.microrna.org/microrna/home.do). We found that SMAD7, a major inhibitory regulator of TGF-β/SMAD signaling that is involved in fibrogenesis, contained multiple putative target sites for miR-96 (Figure S3). Using a dual-luciferase reporter assay, we found that synthetic miR-96 mimics targeted the most conserved site in smad7, leading to 73% reduction (relative luciferase activity [RLA] = 27.02 ± 6.14) compared with the negative control (scrambled mimics, RLA = 100 ± 2.93). Conversely, when the target site was mutated, there was no reduction of luciferase activity by miR-96 mimics (Figure S4). Thus, miR-96 was selected for further investigation into its potential role in schistosomiasis hepatic fibrosis.
Downregulation of miR-96 Prevents the Lethality of Mice Following Schistosomiasis Infection
In an effort to treat schistosomiasis hepatic fibrosis in vivo by reducing levels of miR-96, we used the highly hepatotropic rAAV8 vector to express TuDs for the sustained inhibition of miR-96. The potency and specificity of the TuD-miR-96 constructs were validated in HEK293 cells (Figure S5). Mice were infected with a lethal dose of cercaria of S. japonicum and then injected intravenously with the rAAV8-anti-miR-96-TuD vector, the control vector, or PBS at 10 days post-infection (dpi) (Figure 1A). All mice receiving a single dose of rAAV8-anti-miR-96-TuD vector (n = 5) survived for the duration of the study (110 days), whereas in the control groups (n = 5 each), three mice receiving the control vector and four mice receiving PBS died within 90 dpi (Figure 1B). These results indicate that a single dose of rAAV8-anti-miR-96-TuD significantly protected infected mice from the lethal effects of schistosomiasis.
Figure 1.

Downregulation of miR-96 Protects Mice from Lethal Schistosome Infection
(A) Time schedule for schistosome infection and intravenous injections of AAV8-anti-miRNA-TuD vectors or PBS. Mice were infected percutaneously with 35 cercariae of S. japonicum at day 0 and treated with various vectors at a dose of 1 × 1012 viral genomes, or PBS by tail-vein injection at 10 dpi. The animals were subjected to a 110-day survival study. (B) Kaplan-Meier survival curves were plotted for all groups as indicated.
Repression of miR-96 Mediated by rAAV8-anti-miR-96-TuD Attenuates Hepatic Fibrosis
To investigate the mechanism of rAAV8-anti-miR-96-TuD-mediated protection against schistosomiasis-induced lethality, we exposed mice to a mild dose of S. japonicum cercaria and then injected them with rAAV8-anti-miR-96-TuD or controls as described above (Figure 2A). A mild dose was used to achieve slower disease progression and prolonged survival to allow for pertinent experimental observations to be carried out. We found that miR-96 levels were significantly reduced in the rAAV8-anti-miR-96-TuD-treated group compared with those in the control groups at 50 dpi (Figure 2B). We also measured several markers of hepatic fibrosis, including hydroxyproline content, collagen deposition, and liver granulomas. Hydroxyproline content was reduced by 36.76% ± 2.24% and 34.61% ± 8.28% in the anti-miR-96-TuD-treated group compared with those of the PBS and scramble control groups at 50 dpi (Figure 2C). Fibrosis as measured by Masson’s trichrome staining of collagen deposition in liver sections was also significantly reduced in the rAAV8-anti-miR-96-TuD-treated livers (5.05 ± 1.73) compared with those of the PBS (8.55 ± 1.93) and scramble (8.50 ± 2.24) control groups (Figures 2D and 2E). Interestingly, liver granulomas were also significantly smaller in the rAAV8-anti-miR-96-TuD group (110.76 × 103 ± 9.1 × 103 μm2) as compared with those in the PBS (150.46 × 103 ± 11.73 × 103 μm2) and scramble (140.14 × 103 ± 5.68 × 103 μm2) control groups (Figure 2F). However, when we examined the worm and egg burden recovered from infected mice, we found no significant differences among these groups, suggesting that the rAAV-delivered TuD treatments had no effect on the development and reproduction of the parasite. These data indicated that the rAAV8-anti-miR-96-TuD-mediated inhibition of miR-96 attenuates the pathological progression of hepatic fibrosis in the host, but has no effect on the life cycle of the parasite.
Figure 2.

Downregulation of miR-96 Mediated by rAAV8-Anti-miR-96-TuD Attenuated Schistosomiasis-Induced Hepatic Fibrosis
(A) Time schedule for schistosome infection and intravenous injections of viral vectors and sample withdraw. Mice were percutaneously infected with 16 cercariae of S. japonicum at day 0 or remained uninfected. The infected mice received various vectors at a dose of 1012 viral genomes or PBS at 10 dpi. Liver samples were collected at 50 dpi. (B) qRT-PCR analysis of miR-96, miR-214, miR-150, and miR-705 expression in liver samples at 50 dpi. (C) Collagen content of livers as determined by hydroxyproline content. (D) Fibrosis scores measured from Masson’s trichrome staining of liver sections. (E) H&E staining of liver sections (upper row). Masson’s trichrome staining of collagen in liver sections (bottom line). Scale bar, 100 μm. (F) Mean area of granuloma measured from Mayer’s H&E staining of liver sections using a calibrated measuring eyepiece. Data are represented as mean ± SD. *p < 0.05; **p < 0.01, compared between indicated groups.
TGF-β1 Promotes miR-96 Expression in HSCs through Post-transcriptional Regulation
To investigate how schistosome infection causes upregulation of miR-96 expression, we first profiled expression levels of miR-96 in different cell types within the infected liver. As shown in Figure 3A, elevated expression of miR-96 was observed in all cell types tested, but primarily in hepatocytes (∼20-fold; p = 0.002) and HSCs (∼15-fold; p = 0.003).
Figure 3.

TGF-β1 Elevates miR-96 Expression through Post-transcriptional Regulation
(A) qRT-PCR analysis of miR-96 expression in isolated hepatocytes, HSCs, and Kupffer cells from mouse livers at 0 and 50 dpi, and the miR-96 expression in HSCs of the wild-type mice at 0 and 50 dpi. (B) qRT-PCR analysis of tgf-β1 and il13 expression in the HSCs of infected mice. (C) qRT-PCR analysis of miR-96 and miR-214 expression in primary HSCs treated in vitro for 24 hr with IL10 (25 ng/mL), interferon (IFN)-γ (25 ng/mL), TGF-β1 (100 ng/mL), and IL13 (200 ng/mL). (D) qRT-PCR analysis of miR-96 expression in primary HSCs treated with different concentrations of TGF-β1 as indicated. (E) qRT-PCR analysis of miR-96 expression in primary HSCs treated with TGF-β1 (8 ng/mL) for various times. (F) qRT-PCR analysis of pri-miR-96 and pre-miR-96 expression levels in isolated HSCs from mice at 0 and 50 dpi. (G) qRT-PCR analysis of pri-miR-96 and pre-miR-96 expression in primary HSCs treated with TGF-β1 (10 ng/mL) for various times. (H) RNA-binding protein immunoprecipitation (RIP) analysis of post-transcriptional processing of miR-96. The cell lysates of HSCs isolated from infected (50 dpi) and uninfected (0 dpi) mice were immunoprecipitated with anti-SMAD2+SMAD3, anti-DROSHA, or normal rabbit IgG and subjected to qRT-PCR analysis. (I) In vitro RIP analysis of post-transcriptional processing of miR-96 and miR-21 maturation. qRT-PCR analysis of miR-96 and miR-21 expression in primary HSCs treated with TGF-β1 (100 ng/mL) or IL13 (200 ng/mL) for 24 hr. HSC lysates were immunoprecipitated and subjected to qRT-PCR analysis as described above. Data are represented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared between indicated groups.
Previous studies have identified both TGF-β1 and IL13 as major mediators of schistosomiasis hepatic fibrosis by activating HSCs.9, 10, 11 Our previous study demonstrated that both TGF-β1 and IL13 upregulated miR-21 expression in mouse primary HSCs in vitro through SMAD signaling.15 We detected significantly elevated expression of these cytokines in primary HSCs isolated from infected livers (Figure 3B). The simultaneous elevation of miR-96 in infected livers prompted us to ask whether TGF-β1 and IL13 also mediate elevated miR-96 expression. We found that exogenous expression of TGF-β1 in mouse primary HSCs significantly increased expression of miR-96 in a concentration- and time-dependent manner, whereas the other cytokines had no effect on miR-96 expression (Figures 3C–3E). It is worth mentioning that IL13, a major mediator for miR-21 upregulation,14 resulted in only a slight and insignificant elevation of miR-96 expression.
We next investigated the regulation of miR-96 biogenesis. It is well-known that miRNA expression is controlled at both transcriptional and post-transcriptional levels.18, 19 We first profiled pri-miR-96 and pre-miR-96 expression in infected (50 dpi) versus uninfected (0 dpi) mouse HSCs. We found that the relative levels of both pri- and pre-miR-96 in infected HSCs were higher (∼2- and ∼4-fold, respectively) than those in uninfected HSCs (Figure 3F). We stimulated mouse primary HSCs in vitro with TGF-β1 and measured the relative levels of pri- and pre-miR-96 at various time points post-stimulation. Starting at 12 hr after stimulation, the levels of pre-miR-96 were significantly increased over those of pri-miR-96 (Figure 3G). These data suggest that elevation of miR-96 in infected mice is achieved mainly through post-transcriptional processing rather than transcriptional regulation.
An earlier report indicated that miR-21 is upregulated through post-transcriptional regulation involving a protein complex consisting of DROSHA and activated SMAD 2/3 proteins.19 Our previous study showed that SMAD proteins were activated in HSCs upon schistosome infection.15 To test whether the SMAD2/3-DROSHA protein complex assembles on miR-96 in infected HSCs, we performed an RNA-binding protein immunoprecipitation (RIP) analysis.20 As shown in Figure 3H, both miR-21 and miR-96 were significantly enriched by anti-DROSHA and anti-SMAD2/3 antibodies in HSCs of infected mice (50 dpi) and uninfected HSCs, but not by non-specific IgG. By contrast, miR-214 was not detected regardless of the antibody used. These results indicated that the elevation of miR-96 is mainly through post-transcriptional regulation mediated by the SMAD2/3-DROSHA protein complex.
We further investigated whether TGF-β1 regulates miR-96 levels through a post-transcriptional step that is mediated by the SMAD2/3-DROSHA protein complex. Consistent with our previous data, stimulation of mouse primary HSCs with either TGF-β1 or Il13 resulted in the specific enrichment of miR-21 using anti-DROSHA and anti-SMAD2/3 antibodies (Figure 3I). However, only stimulation with TGF-β1 led to significant enrichment of miR-96 (Figure 3I), suggesting that TGF-β1, rather than IL13, is the primary mediator regulating cellular levels of miR-96. This finding is consistent with the results described above (Figure 3C). As expected, stimulation with both cytokines did not result in enrichment of miR-214. Taken together, these results indicate that TGF-β1 stimulates the elevation of miR-96 levels by mainly activating Smad proteins and inducing the formation of a complex composed of SMAD2/3, pri-miR-96, and subunits of the microprocessor complex such as DROSHA.
miR-96 Induces Hepatic Fibrosis by Targeting smad7
SMAD7 is a major inhibitor of the fibrogenesis-inducing TGF-β1/SMAD signaling pathway. As described above, SMAD7 contains multiple putative target sites for miR-96 (Figures S3 and S4). Transfection of the HSC-T6 cell line with a synthetic miR-96 inhibitor resulted in a significant 2-fold increase in SMAD7 expression as detected by qRT-PCR (Figure S6A) and Western blot analysis (Figure S6B), indicating that miR-96 is a regulator of Smad7 expression. We used the miR-96 inhibitor to investigate the effect of elevated smad7 expression on the production of col1 and col3, important biomarkers of fibrosis. In our previous study, we demonstrated that upregulation of miR-21 drives elevated expression of col1 and col2.14 Here, we used an miR-21 inhibitor as a positive control. We found that transfection with the miR-96 inhibitor significantly upregulated smad7 expression in the HSC-T6 cell line by ∼4-fold and significantly reduced the expression of col1 by 3-fold (Figure S6C). Levels of col3 were unchanged. These results indicate that inhibition of miR-96 upregulates smad7 expression and reduces expression of at least one fibrotic biomarker.
To further investigate whether downregulation of miR-96 in vivo can reduce the expression of both col1 and col3 in HSCs, we infected mice with parasites and then administered either rAAV8-anti-miR-96-TuD or the scramble control vector, as described above (Figure 2A). At 50 dpi, we isolated HSCs of infected mice and quantified the transcript levels of miR-96, smad7, col1, and col3. Similar to our results above, rAAV8-anti-miR-96-TuD-mediated intervention significantly reduced miR-96 expression in isolated HSCs (Figure 4A). Importantly, the reduction in miR-96 led to significantly increased Smad7 expression, as detected by qRT-PCR (Figure 4B, rAAV8-anti-miR-96-TuD group) and by Western blot analysis (Figure 4C, lane 2). These changes were also concomitant with decreases in col1 and col3 expression (Figures 4D and 4E, rAAV8-anti-miR-96-TuD group).
Figure 4.

Downregulation of miR-96 De-represses Smad7, Reducing the Deposition of ECM in Schistosome-Infected Livers
(A and B) RT-PCR analysis of miR-96 (A) and smad7 (B) expression in HSCs isolated from uninfected and infected mice administered with various rAAV8-TuD vectors as indicated at 50 dpi. (C) Western blot analysis of SMAD7 expression in HSCs isolated from mice as described above. (D and E) RT-PCR analysis of (D) col1 and (E) col3 expression in HSCs isolated from mice as described above. Data are represented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001, compared between indicated groups.
miR-96 and miR-21 Have an Additive Inhibitory Effect on Expression of Smad7
We next searched the Smad7 gene for miR-96 and miR-21 binding sites. We identified two highly conserved binding motifs for miR-21 and two highly conserved motifs for miR-96 in the 3′ UTR of Smad7. Two additional poorly conserved motifs for miR-96 were also identified elsewhere in the gene (Figure S3). Thus, we hypothesized that the two miRNAs have additive effects on regulating Smad7 expression. We first transfected mouse primary HSCs with an miR-21 mimic, an miR-96 mimic, or both mimics together. As expected, each miRNA mimic reduced smad7 expression when tested individually (Figure 5A). Importantly, co-transfection with both mimics led to an additive reduction in expression of smad7 (Figure 5A). Consistent with this observation, when we co-transfected HSC-T6 cells with inhibitors of both miR-21 and miR-96, we observed an additive effect on the upregulation of SMAD7 expression by Western blot analysis (Figure 5B). Finally, we investigated the additive effect of both miR-21 and miR-96 on Smad7 expression in vivo through rAAV8-anti-96/21-TuD intervention. For this experiment, mice were infected with a mild dose of S. japonicum cercaria and then injected intravenously with either the rAAV8-anti-miR-96-TuD vector, the rAAV8-anti-miR-21-TuD vector, or with both vectors. As shown in Figures 5C and 5D, the expression of Smad7 was increased in infected mice treated with either anti-miR-21-TuD or anti-miR-96-TuD vectors, but not in those treated with the anti-SCR-TuD vector or PBS. Importantly, the additive effects on the elevated expression of Smad7 were detected when infected mice were treated with both rAAV8-anti-miR-96-TuD and rAAV8-anti-miR-21-TuD vectors in combination.
Figure 5.

miR-96 and miR-21 Have an Additive Effect on the Inhibition of Smad7 In Vitro
(A) RT-PCR analysis of smad7 expression in mouse primary HSCs transfected with mimics of miR-21, miR-96, the combination of miR-21 and miR-96, or controls. (B) Western blot analysis of SMAD7 in the HSC-T6 cell line transfected with inhibitors of miR-21, miR-96, the combination of miR-21 and miR-96, or controls. (C) Western blot analysis of SMAD7 expression in liver samples from uninfected and infected mice treated with the rAAV8-anti-miR-96-TuD vector, the rAAV8-anti-miR-21-TuD vector, the combination of both vectors, or control vectors. (D) RT-PCR analysis of smad7 expression in mouse primary HSCs isolated from uninfected and infected mice treated with the rAAV8-anti-miR-96-TuD vector, the rAAV8-anti-miR-21-TuD vector, the combination of both vectors, or the control vector. Data are represented as mean ± SD. *p < 0.05; **p < 0.01, compared between indicated groups.
Discussion
There is increasing evidence that dysregulation of certain miRNAs is concomitant with tissue fibrosis. For example, our previous study showed that miR-21 regulates schistosomiasis hepatic fibrosis through the SMAD pathway.15 Infection by the schistosome parasite significantly upregulates miR-21 expression and elicits fibrogenesis by relieving the inhibitory effects of SMAD7.15 In this study, we demonstrated that miR-96 also promotes schistosomiasis hepatic fibrosis by a similar mechanism. Infection with schistosome induces TGF-β1 expression and thereby upregulates miR-96 expression. Elevated miR-96 suppresses Smad7 expression by binding to three seed regions in the 3′ UTR of smad7. Interestingly, we also show an additive effect of miR-96 and miR-21 on expression of Smad7 in mice and cell lines, and by transfecting mimics or inhibitors of miR-96 and miR-21. In addition, we also found that the upregulation of miR-96 in activated HSCs takes place through post-transcriptional regulation that is mediated by the SMAD2/3-DROSHA protein complex. Elevated TGF-β1 that results from schistosome infection leads to the phosphorylation and nuclear import of SMAD2/3. Phospho-SMAD2/3 complexes with DROSHA and promotes the maturation of miR-96. These findings provide new insights into the mechanism of schistosomiasis hepatic fibrosis through the alteration of host miRNAs.
miR-96 has been reported to be elevated in different malignancies including hepatocellular carcinoma,21 colon cancer,22 urothelial cancer,23 esophageal cancer,24 breast cancer, and prostate cancer.25, 26 It plays an important role in tumor progression by targeting tumor suppressors such as FOXO1 and AKT1S1,26, 27, 28 thus promoting cell proliferation and invasion. miR-96 is also a sensory organ-related miRNA. It is involved in the functional differentiation of hair cells, and mutations in miR-96 cause progressive hearing loss.29 We now report that miR-96 controls hepatic fibrosis in the murine model of human schistosomiasis by targeting the 3′ UTR of smad7. Importantly, rAAV8-mediated inhibition of miR-96 in infected mice resulted in elevated Smad7 expression and attenuation of hepatic fibrosis. Despite our previous report that other host miRNAs such as miR-21, miR-203, and miR-351 play important roles in the progression of schistosomiasis hepatic fibrosis, the novelty of this study is unique from our previous work: (1) we demonstrated that schistosome infection upregulates the expression of miR-96 via SMAD2/3-DROSHA-mediated post-transcriptional regulation in TGF-β1 signaling; (2) the elevated expression of miR-96 promotes liver fibrosis by targeting Smad7, an important inhibitor of TGF-β1 signaling; and (3) both miR-96 and miR-21 target the 3′ UTR of smad7 and have an additive effect on regulating Smad7 expression in vitro and in vivo. It is clear that schistosome infection causes the dysregulation of multiple host miRNAs, governing hepatic fibrosis through targeting various fibrosis-related genes and their signal pathways.
Previous studies have revealed the mechanism of the TGF-β1/SMAD canonical pathway for modulating miRNA expression. Activated SMAD proteins directly facilitate pri-miRNA transcription by binding SMAD binding element (SBE) motifs in the promoters of miRNAs.30 The SMAD2/3-DROSHA protein complex also promotes the maturation of miRNAs via post-transcriptional processing, increasing levels of pre-miRNAs. Davis et al.19 reported that TGF-β1 induces expression of miR-21 through promoting the processing of pri-miR-21 into pre-miR-21. A recent study showed that TGF-β1 upregulated miR-96 expression in adenocarcinoma cell lines through direct interaction of the SMAD2/3/4 protein complex with the promoter of pri-miR-96.26 In our current study, TGF-β1 had a limited effect on miR-96 transcription. Instead, we demonstrate that post-transcriptional processing mediated by the SMAD2/3-DROSHA protein complex is a key regulator of miR-96 expression in the HSCs of schistosomiasis-infected mice. Our result is consistent with the TGF-β1-mediated post-transcriptional upregulation of miR-21 that was previously reported.15, 19
TGF-β1 is a key activator of HSCs in the progression of hepatic fibrosis.31 In the case of schistosomiasis infection, IL13 is an additional important mediator of HSC activation.32 We demonstrated in our previous study that TGF-β1 and IL13 upregulate miR-21 expression through the SMAD pathway, and that both cytokines have an additive effect on miR-21 upregulation. By contrast, in our current study, we show that TGF-β1, but not IL13, significantly elevates the expression of miR-96 in mouse primary HSCs. In Figures 3C and 3I, we show that TGF-β1 is the main factor that upregulates the expression of miR-96 instead of IL13. It is known that TGF-β1 induces SMAD2/3 to form a complex with DROSHA, whereas IL13 induces SMAD1/2 to form a complex with DROSHA. Therefore, we propose that the SMAD2/3 complex, but not the SMAD1/2 complex, could bind to the promoter of miR-96.
SMAD7 is a key inhibitory regulator of SMAD signaling. Decreased expression of Smad7 is associated with tissue fibrosis, whereas elevated expression of Smad7 through gene transfer can block fibrogenesis.33, 34 Our previous study indicated that elevated miR-21 upregulates schistosomiasis fibrogenesis in mice through binding the 3′ UTR of smad7.15 In the current study, we show that infection with schistosome parasite induces expression of TGF-β1, which elevates miR-96 expression in HSCs mainly through post-transcriptional regulation mediated by the SMAD2/3-Drosha protein complex. Elevated miR-96 induces hepatic fibrogenesis in the infected liver by targeting the 3′ UTR of smad7 via binding sites that are distinct from those of miR-21. Importantly, the additive effects of miR-21 and miR-96 on relieving the inhibitory effect of Smad7 were detected in both in vitro cell lines and an in vivo animal model. Thus, inhibition of both miR-96 and miR-21 should be an attractive strategy to treat hepatic fibrosis, including schistosomiasis hepatic fibrosis. Recombinant AAV has shown great promise for in vivo gene transfer applications due mainly to its long-term episomal persistence, broad tissue tropism, and low immunogenicity.35, 36 We chose AAV8-based vectors for our current study based on its strong liver tropism, its safety, and efficacy that has been displayed in multiple preclinical and clinical studies.37, 38 Importantly, we demonstrated that a single administration of two vectors, rAAV8-anti-miR-96-TuD and rAAV8-anti-miR-21-TuD, provided efficient downregulation of both miR-96 and miR-21 expression, leading to an additive increase in Smad7 expression. Our data highlight the utility of using rAAV8-mediated delivery of miRNA inhibitors as an adjuvant therapy for fibrosis.
Materials and Methods
Mice and Parasite Infections
Six-week-old male BALB/c mice were purchased from the Experimental Animal Center of Second Military Medicine University (Shanghai, China). For infections, mice were injected percutaneously with 16–30 cercariae of S. japonicum shed from lab-infected snails (Oncomelania hupensis) obtained from the National Institute of Parasitic Disease, Chinese Center for Disease Control and Prevention. All procedures performed on animals within this study were conducted in accordance with and by approval of Second Military Medicine University.
Cell Culture and Transfection
HSC-T6, HEK293, and mouse primary HSCs were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) plus 50 μg/mL streptomycin and 50 IU penicillin, and transfected using the Lipofectamine 2000 Transfection Reagent (Life Technologies).
Isolation of Primary Mouse HSCs, Hepatocytes, and Kupffer Cells
Isolation of primary mouse HSCs, hepatocytes, and Kupffer cells was conducted as described previously.39 In brief, after anesthetizing a mouse, the hepatic portal vein was cannulated; then the line and pump was connected for in situ perfusion. The inferior vena cava was cut off gently and sequentially perfused with warmed 37°C EGTA buffer, pronase solution, and collagenase solution. The digested liver was excised and minced thoroughly on a Petri dish, then filtered through a 100-μm nylon strainer to achieve a single-cell solution. Cells were centrifuged and washed several times with Gey’s balanced salt solution B (GBSS/B) buffer at 50 × g, 5 min at 4°C, to eliminate the majority of hepatocytes. After hepatocytes were pelleted, the supernatant containing nonparenchymal cells was further centrifuged at 580 × g for 10 min at 4°C. Nonparenchymal cells were washed several times with GBSS/B solution until the supernatant was clear. The supernatant was discarded, and nonparenchymal cells were resuspended with 2 mL GBSS/B buffer and loaded on the top layer of an appropriate OptiPrep solution (11.2% OptiPrep solution for HSC isolation and 19% OptiPrep solution for Kupffer cells isolation), then centrifuged at 1,400 × g for 20 min. After centrifugation, the HSCs or Kupffer cells are separated between two layers of liquid. HSCs were further purified using negative selection with magnetic CD11b antibody beads and magnetic columns (MACS; Miltenyi, Auburn, CA). Kupffer cells were purified using positive selection with magnetic CD11b antibody beads.
RNA Extraction and Analysis
Total RNA was extracted from cells using the TriPure isolation system (Cat. no. 11667165001; Roche). The relative quantities of miR-96 and other markers such as smad7, col1, and col3 were measured from 500 ng of total RNA by qRT-PCR using the LightCycler 480 SYBR Green I Master (Cat. 04887352001; Roche) detection kit and the LightCycler 480II (Roche) instrument, and determined by the comparative cycle threshold (CT) method using snRNA U6 levels for normalization as recommended in the manufacturer’s instructions.
Immunoblot Assays
Immunoblot assays were performed on total cell lysates (50 μg) using anti-SMAD7 monoclonal and polyclonal antibodies (catalog [Cat.] no. Ab124890; Abcam). The anti-GAPDH antibody (Cat. no. Ab181602; Abcam) was used for normalization. IRDye 800CW goat anti-rabbit IgG (Cat. no. 926-32211; LI-COR) was used as a secondary antibody, and blots were visualized by the ODYSSEY Sa (LI-COR) instrument.
Liver Histopathology and Fibrosis Measurements
Liver histopathology and fibrosis measurements were conducted as described previously.40 In brief, for measuring hydroxyproline, we used the hydroxyproline assay kit (A030-3; Jiancheng Nanjing) following procedures for 80- to 100-mg liver samples. The percentage of hydroxyproline was calculated using the following formula: Hyp (μg/mg) = (ODsamples − ODblank/ODstandard − ODblank) × 5 μg/mL × (10 mL/sample weight), where OD is optical density. To calculate granuloma sizes for each H&E slice, we measured the area of 25 granulomas. Liver histopathology and fibrosis were detected by H&E staining and Masson’s staining. To evaluate the level of fibrosis, we calculated a fibrosis score using the following formula: fibrosis score = degree of granuloma size × degree of blue color; both degree values range from 1 to 4, and the fibrosis scores range from 1 to 16.
RIP Assay
Primary HSCs were isolated from uninfected and S. japonicum-infected mouse livers and assayed with the EZ-Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Cat. 17-701; Millipore). To obtain concentrated cell lysates, at least 2 × 107 cells are required for 100 μL of RIP Lysis Buffer. The assay was performed following the manufacturer’s protocol with anti-SMAD2+SMAD3 (Cat. no. ab65847; Abcam), anti-DROSHA antibodies (Cat. no. ab12286; Abcam), and normal rabbit IgG as a negative control antibody (Cat. no. PP64B; Millipore).
Dual-Luciferase Reporter Assay
The 3′ UTR of smad7 was cloned into a modified pGL-3 control vector, with the 3′ UTR placed downstream of the luciferase coding sequence. The transfection mixtures contained 100 ng of firefly luciferase reporter and 80 pM of miR-96 mimics. pRL-TK (Promega) was transfected as an internal control. Firefly luciferase activities were assayed using the dual-luciferase assay (Cat. no. E1910; Promega) 48 hr after transfection.
Construction of Plasmids and Vectors
The β-Gal gene was PCR-amplified from the pAAV2.1TBGLacZ plasmid using primer claczF with a NheI site and claczR with a XhoI site. The PCR fragment was cut with these two restriction enzymes and cloned into NheI and XhoI sites within the psiCHECK plasmid. Three copies of perfectly complementary miR-96 sites were designed based on the annotated miR-96 sequence and inserted between the XhoI and NotI restriction sites 3′ of the β-Gal gene to make the pmiCHECK-96 sensor plasmid. To express miR-96, we amplified a pri-miR-96 fragment from mouse genomic DNA using primers pri-miR-96F and pri-miR-96R and cloned into the PpuMI site within the pAAVscCBPI-pGluc plasmid. BamHI-digested anti-miR-96-TuD fragment (synthesized from GenScript) was cloned into the BamHI site in pAAVscCBPI-TuD-let-7p-Gluc following removal of anti-let-7-TuD. rAAV8 vectors used in the study were produced, purified, and titered as described previously.41
Statistical Analysis
Results are reported as mean ± SD and compared between groups using two-tailed Student’s t test, one-way ANOVA, or the Kaplan-Meier method. Data were considered statistically significant for p values less than 0.05.
Author Contributions
W.P., G.G., and X.L. designed the research; X.L. and D.Z. performed experiments and analyzed the data; J.X., Q.S., X.H., and R.B. performed experiments; W.P., G.G., and X.L. wrote the paper.
Conflicts of Interest
The authors declare no competing financial interests.
Acknowledgments
We thank the staff of the National Institute of Parasitic Disease, Chinese Center for Disease Control and Prevention for their help with parasite infections. We thank Dr. Phillip Tai of UMass Medical School for critical review, edit, and comment on the paper. This study was supported by the National Natural Science Foundation of China (grant 81430051) and the National Basic Research Program (973 Program; grant 2007CB513100) in China, a grant from the NIH (UL1RR031982 to G.G.), and an internal grant from the University of Massachusetts Medical School (to G.G.).
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.omtm.2018.10.002.
Contributor Information
Guangping Gao, Email: guangping.gao@umassmed.edu.
Weiqing Pan, Email: wqpan0912@aliyun.com.
Supplemental Information
References
- 1.Fenwick A. The global burden of neglected tropical diseases. Public Health. 2012;126:233–236. doi: 10.1016/j.puhe.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Thétiot-Laurent S.A.L., Boissier J., Robert A., Meunier B. Schistosomiasis chemotherapy. Angew. Chem. Int. Ed. Engl. 2013;52:7936–7956. doi: 10.1002/anie.201208390. [DOI] [PubMed] [Google Scholar]
- 3.Lozano R., Naghavi M., Foreman K., Lim S., Shibuya K., Aboyans V., Abraham J., Adair T., Aggarwal R., Ahn S.Y. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gryseels B., Polman K., Clerinx J., Kestens L. Human schistosomiasis. Lancet. 2006;368:1106–1118. doi: 10.1016/S0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- 5.Zhao C.-Q., Zhou Y., Ping J., Xu L.-M. Traditional Chinese medicine for treatment of liver diseases: progress, challenges and opportunities. J. Integr. Med. 2014;12:401–408. doi: 10.1016/S2095-4964(14)60039-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman S.L. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreira R.K. Hepatic stellate cells and liver fibrosis. Arch. Pathol. Lab. Med. 2007;131:1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- 8.Bataller R., Brenner D.A. Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin. Liver Dis. 2001;21:437–451. doi: 10.1055/s-2001-17558. [DOI] [PubMed] [Google Scholar]
- 9.Loverde P.T., Osman A., Hinck A. Schistosoma mansoni: TGF-β signaling pathways. Exp. Parasitol. 2007;117:304–317. doi: 10.1016/j.exppara.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mentink-Kane M.M., Cheever A.W., Thompson R.W., Hari D.M., Kabatereine N.B., Vennervald B.J., Ouma J.H., Mwatha J.K., Jones F.M., Donaldson D.D. IL-13 receptor alpha 2 down-modulates granulomatous inflammation and prolongs host survival in schistosomiasis. Proc. Natl. Acad. Sci. USA. 2004;101:586–590. doi: 10.1073/pnas.0305064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreira Rde.C., Montenegro S.M., Domingues A.L., Bandeira A.P., Silveira C.A., Leite L.A., Pereira Cde.A., Fernandes I.M., Mertens A.B., Almeida M.O. TGF beta and IL13 in schistosomiasis mansoni associated pulmonary arterial hypertension; a descriptive study with comparative groups. BMC Infect. Dis. 2014;14:282. doi: 10.1186/1471-2334-14-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu G., Friggeri A., Yang Y., Milosevic J., Ding Q., Thannickal V.J., Kaminski N., Abraham E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010;207:1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chau B.N., Xin C., Hartner J., Ren S., Castano A.P., Linn G., Li J., Tran P.T., Kaimal V., Huang X. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003205. 121ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roderburg C., Urban G.-W., Bettermann K., Vucur M., Zimmermann H., Schmidt S., Janssen J., Koppe C., Knolle P., Castoldi M. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 15.He X., Xie J., Zhang D., Su Q., Sai X., Bai R., Chen C., Luo X., Gao G., Pan W. Recombinant adeno-associated virus-mediated inhibition of microRNA-21 protects mice against the lethal schistosome infection by repressing both IL-13 and transforming growth factor beta 1 pathways. Hepatology. 2015;61:2008–2017. doi: 10.1002/hep.27671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai P., Piao X., Liu S., Hou N., Wang H., Chen Q. MicroRNA-gene expression network in murine liver during Schistosoma japonicum infection. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0067037. e67037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han H., Peng J., Hong Y., Zhang M., Han Y., Liu D., Fu Z., Shi Y., Xu J., Tao J., Lin J. MicroRNA expression profile in different tissues of BALB/c mice in the early phase of Schistosoma japonicum infection. Mol. Biochem. Parasitol. 2013;188:1–9. doi: 10.1016/j.molbiopara.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Lee Y., Jeon K., Lee J.-T., Kim S., Kim V.N. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis B.N., Hilyard A.C., Lagna G., Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain R., Devine T., George A.D., Chittur S.V., Baroni T.E., Penalva L.O., Tenenbaum S.A. RIP-Chip analysis: RNA-binding protein immunoprecipitation-microarray (Chip) profiling. Methods Mol. Biol. 2011;703:247–263. doi: 10.1007/978-1-59745-248-9_17. [DOI] [PubMed] [Google Scholar]
- 21.Ladeiro Y., Couchy G., Balabaud C., Bioulac-Sage P., Pelletier L., Rebouissou S., Zucman-Rossi J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–1963. doi: 10.1002/hep.22256. [DOI] [PubMed] [Google Scholar]
- 22.Sarver A.L., French A.J., Borralho P.M., Thayanithy V., Oberg A.L., Silverstein K.A.T., Morlan B.W., Riska S.M., Boardman L.A., Cunningham J.M. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer. 2009;9:401. doi: 10.1186/1471-2407-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kriebel S., Schmidt D., Holdenrieder S., Goltz D., Kristiansen G., Moritz R., Fisang C., Müller S.C., Ellinger J. Analysis of tissue and serum microRNA expression in patients with upper urinary tract urothelial cancer. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0117284. e0117284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia H., Chen S., Chen K., Huang H., Ma H. MiR-96 promotes proliferation and chemo- or radioresistance by down-regulating RECK in esophageal cancer. Biomed. Pharmacother. 2014;68:951–958. doi: 10.1016/j.biopha.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J., Kong X., Li J., Luo Q., Li X., Shen L., Chen L., Fang L. miR-96 promotes tumor proliferation and invasion by targeting RECK in breast cancer. Oncol. Rep. 2014;31:1357–1363. doi: 10.3892/or.2013.2934. [DOI] [PubMed] [Google Scholar]
- 26.Siu M.K., Tsai Y.-C., Chang Y.-S., Yin J.J., Suau F., Chen W.-Y., Liu Y.N. Transforming growth factor-β promotes prostate bone metastasis through induction of microRNA-96 and activation of the mTOR pathway. Oncogene. 2015;34:4767–4776. doi: 10.1038/onc.2014.414. [DOI] [PubMed] [Google Scholar]
- 27.Yu J.-J., Wu Y.-X., Zhao F.-J., Xia S.-J. miR-96 promotes cell proliferation and clonogenicity by down-regulating of FOXO1 in prostate cancer cells. Med. Oncol. 2014;31:910. doi: 10.1007/s12032-014-0910-y. [DOI] [PubMed] [Google Scholar]
- 28.Gao F., Wang W. MicroRNA-96 promotes the proliferation of colorectal cancer cells and targets tumor protein p53 inducible nuclear protein 1, forkhead box protein O1 (FOXO1) and FOXO3a. Mol. Med. Rep. 2015;11:1200–1206. doi: 10.3892/mmr.2014.2854. [DOI] [PubMed] [Google Scholar]
- 29.Kuhn S., Johnson S.L., Furness D.N., Chen J., Ingham N., Hilton J.M., Steffes G., Lewis M.A., Zampini V., Hackney C.M. miR-96 regulates the progression of differentiation in mammalian cochlear inner and outer hair cells. Proc. Natl. Acad. Sci. USA. 2011;108:2355–2360. doi: 10.1073/pnas.1016646108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morikawa M., Koinuma D., Tsutsumi S., Vasilaki E., Kanki Y., Heldin C.-H., Aburatani H., Miyazono K. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 2011;39:8712–8727. doi: 10.1093/nar/gkr572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bataller R., Brenner D.A. Liver fibrosis. J. Clin. Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wynn T.A., Thompson R.W., Cheever A.W., Mentink-Kane M.M. Immunopathogenesis of schistosomiasis. Immunol. Rev. 2004;201:156–167. doi: 10.1111/j.0105-2896.2004.00176.x. [DOI] [PubMed] [Google Scholar]
- 33.Li Q., Zhang D., Wang Y., Sun P., Hou X., Larner J., Xiong W., Mi J. MiR-21/Smad 7 signaling determines TGF-β1-induced CAF formation. Sci. Rep. 2013;3:2038. doi: 10.1038/srep02038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung A.C.K., Dong Y., Yang W., Zhong X., Li R., Lan H.Y. Smad7 suppresses renal fibrosis via altering expression of TGF-β/Smad3-regulated microRNAs. Mol. Ther. 2013;21:388–398. doi: 10.1038/mt.2012.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie J., Ameres S.L., Friedline R., Hung J.-H., Zhang Y., Xie Q., Zhong L., Su Q., He R., Li M. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat. Methods. 2012;9:403–409. doi: 10.1038/nmeth.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li M., Tang Y., Wu L., Mo F., Wang X., Li H., Qi R., Zhang H., Srivastava A., Ling C. The hepatocyte-specific HNF4α/miR-122 pathway contributes to iron overload-mediated hepatic inflammation. Blood. 2017;130:1041–1051. doi: 10.1182/blood-2016-12-755967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grieger J.C., Samulski R.J. Adeno-associated virus as a gene therapy vector: vector development, production and clinical applications. Adv. Biochem. Eng. Biotechnol. 2005;99:119–145. [PubMed] [Google Scholar]
- 38.Gernoux G., Guilbaud M., Dubreil L., Larcher T., Babarit C., Ledevin M., Jaulin N., Planel P., Moullier P., Adjali O. Early interaction of adeno-associated virus serotype 8 vector with the host immune system following intramuscular delivery results in weak but detectable lymphocyte and dendritic cell transduction. Hum. Gene Ther. 2015;26:1–13. doi: 10.1089/hum.2014.070. [DOI] [PubMed] [Google Scholar]
- 39.Mederacke I., Dapito D.H., Affò S., Uchinami H., Schwabe R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015;10:305–315. doi: 10.1038/nprot.2015.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiaramonte M.G., Donaldson D.D., Cheever A.W., Wynn T.A. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J. Clin. Invest. 1999;104:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao G.-P., Alvira M.R., Wang L., Calcedo R., Johnston J., Wilson J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.