

Graphical abstract
Abbreviations: C-ion, carbon ion; CPP, cell-penetrating peptide; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; HIMAC, Heavy Ion Medical Accelerator in Chiba; SOBP, spread-out Bragg peak; TBI, total body irradiation; TUNEL, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling
Keywords: Carbon ion radiotherapy, FGF1, Cellular internalization, Intestinal adverse effects, Radioprotector, Pancreatic carcinoma
Highlights
-
•
C-ion radiotherapy is applied to pancreatic carcinoma in the abdominal cavity.
-
•
The FGF1/CPP-C chimeric protein has an intracellular signaling mode.
-
•
FGF1/CPP-C protects against C-ion-induced intestinal damage.
-
•
FGF1/CPP-C inhibits the proliferation and metastasis of pancreatic carcinoma cells.
-
•
FGF1/CPP-C may be useful for C-ion radiotherapy against pancreatic cancer.
Abstract
Background and purpose
Carbon ion (C-ion) beams are concentrated to irradiate pancreatic carcinoma in the upper abdomen; however, this radiotherapy potentially causes adverse reactions in the gastrointestinal tract. FGF1 is a candidate radioprotector for radiation-induced intestinal damage, but may promote the malignancy of pancreatic cancer. An FGF1/CPP-C chimeric protein was created to enhance the intracellular signaling mode of FGF1 instead of FGFR signaling. The present study investigated the effects of FGF1/CPP-C on the intestinal adverse reactions of C-ion radiotherapy as well as its influence on the malignancy of pancreatic cancer.
Materials and methods
FGF1/CPP-C was administered intraperitoneally to BALB/c mice without heparin 12 h before total body irradiation (TBI) with low-LET C-ion (17 keV/μm) at 6–8 Gy. Several radioprotective effects were examined in the jejunum. The invasion and migration of the human pancreatic carcinoma cell lines MIAPaCa-2 and PANC-1 were assessed using Boyden chambers after cultures with FGF1/CPP-C.
Results
The FGF1/CPP-C treatment promoted crypt survival after C-ion irradiation at 7–8 Gy significantly more than the FGF1 treatment. FGF1/CPP-C also inhibited C-ion radiotherapy-induced apoptosis and reduced γH2AX foci in crypt cells more than FGF1. However, FGF1/CPP-C inhibited the downstream signaling pathways of FGFRs and suppressed the activation of cell-cycle regulatory molecules in the intestine until 4 h after TBI. Furthermore, IEC6 cells were arrested in G2M after cultures with FGF1/CPP-C or FGF1, suggesting that DNA repair after irradiation is promoted by FGF1/CPP-C-induced G2M arrest. In contrast, FGF1/CPP-C appeared to be internalized into MIAPaCa-2 and PANC-1 cells more efficiently than FGF1. Therefore, FGF1/CPP-C reduced the in vitro proliferation, invasion, and migration of MIAPaCa-2 and PANC-1 cells significantly more than FGF1 through the cellular internalization of FGF1.
Conclusion
These results suggest that the intracellular signaling mode of FGF1/CPP-C attenuates the intestinal adverse effects of C-ion radiotherapy without enhancing the malignancy of pancreatic carcinoma.
1. Introduction
Carbon ion (C-ion) radiotherapy is applied to control localized pancreatic cancer. The surrounding normal gastrointestinal (GI) organs are radiosensitive, whereas pancreatic cancer is radioresistant; therefore, radiotherapy is applied at high doses, which increases the risk of adverse reactions. In contrast, C-ion beams are concentrated to sufficiently irradiate the target while minimizing exposure of the surrounding tissues; therefore, C-ion radiotherapy is associated with fewer GI adverse reactions, such as anorexia, nausea, and vomiting, than conventional radiotherapy [1], [2]. However, if high-intensity doses of C-ion irradiation affect focal areas of the GI mucosa, C-ion radiotherapy potentially causes ulcers, bleeding, and perforation of the GI tract as adverse reactions, which limits radiation doses. Therefore, radioprotectors against radiation-induced intestinal damage are considered to be useful for increasing the clinical application of abdominal C-ion radiotherapy.
Several fibroblast growth factors (FGFs) have been shown to protect against radiation-induced intestinal damage [3], [4]. However, aberrant FGF signaling has been reported to promote tumor development by enhancing cell proliferation, cell survival, and tumor angiogenesis [5]; therefore, FGF radioprotectors may promote the progression and metastasis of tumors. On the other hand, FGF signaling has tumor suppressive functions under specific conditions [5]. Thus, the influence of FGFs on the malignancy of each cancer needs to be clarified in order to apply FGF radioprotectors to cancer radiotherapy.
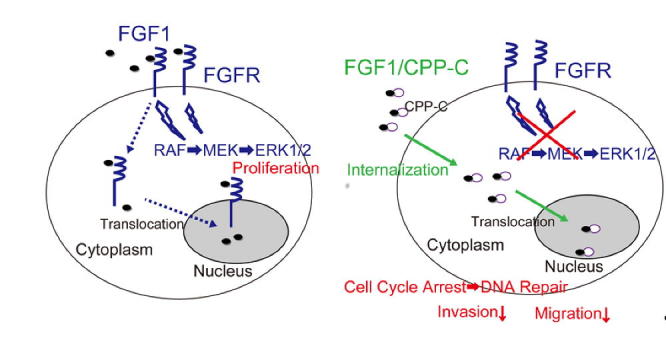
FGF has two signaling modes: a signaling pathway via cell surface FGF receptors (FGFRs) and intracellular signaling by internalized FGF. We previously reported that FGF12 is internalized into cells, and this process depends on two novel cell-penetrating peptide (CPP) domains of FGF12 (CPP-M and CPP-C) [6]. CPP-C, composed of approximately 10 amino acids, is a specific domain of the FGF11 subfamily (FGF11-FGF14) in the C-terminal region. FGF1 shares structural similarities with FGF12; however, FGF1 is internalized into cells markedly less than FGF12 because it lacks the corresponding CPP-C domain. Since CPP-C delivers FGFs into cells independently of FGFRs, the FGF1/CPP-C chimeric protein (FGF1/CPP-C) is internalized into cells more efficiently than wild-type FGF1 [6] (Fig. 1 and E1). The mitogenic activity of FGF1/CPP-C through FGFR1c or 2b was previously shown to be markedly weaker than that of FGF1 [7]. Nevertheless, FGF1/CPP-C promoted anti-apoptotic effects and crypt regeneration in the intestines after γ-irradiation more strongly than FGF1 [7]. Therefore, FGF1/CPP-C is expected to protect against adverse reactions after radiation therapy without enhancing the malignancy of tumors.
Fig. 1.

FGF1/CPP-C reacts with all FGFR subtypes more weakly than FGF1. (A) The structure of the FGF1/CPP-C fusion protein is shown. (B) In addition to the signaling pathway of FGF through cell surface receptors, the cellular internalization of FGF induces other signaling pathways. The potential signaling pathways by FGF1/CPP-C are shown. (C) The BaF3 transfectant cell line expressing each FGFR subtype was cultured for 42 h with FGF1 or FGF1/CPP-C at the indicated concentrations in the presence of 5 μg/ml heparin. Cell numbers were estimated from optical absorbance at 450 nm (ABS450) using WST-1 reagent. All values are means ± SD (n = 4). *P < 0.05; ***P < 0.001.
The present study investigated the influence of FGF1/CPP-C on C-ion-induced intestinal damage and the malignancy of human pancreatic cancer cell lines, and the results obtained demonstrated that FGF1/CPP-C exerted protective effects against C-ion-induced intestinal damage with the inhibition of the metastatic capabilities of human pancreatic cancer cell lines through the cellular internalization of FGF1/CPP-C.
2. Materials and methods
2.1. Cell culture
The rat intestinal epithelial cell line IEC6 was provided by the RIKEN BioSource Center (Tsukuba, Japan). IEC6 cells were maintained in medium consisting of Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 5% fetal calf serum (FCS) (HyClone, Logan, UT, USA) and 4 μg/ml insulin. The human pancreatic cancer cell lines MIAPaCa-2 and Panc-1 were purchased from ATCC (Manassas, VA, USA) and cultured in DMEM supplemented with 10% FCS.
2.2. Mice
Seven-week-old male BALB/c mice weighing between 23 and 27 g were obtained from Japan Clea (Tokyo, Japan). Mice were housed in an air-conditioned room in a specific-pathogen-free facility and irradiated at 8 weeks old with C-ion beams from the Heavy Ion Medical Accelerator in Chiba (HIMAC) at the National Institute of Radiological Sciences. All protocols complied with the Guidelines on Animal Experiments from the National Institutes for Quantum and Radiological Science and Technology and were approved by the Laboratory Animal Safety and Ethics Council of National Institutes for Quantum and Radiological Science and Technology. Crypt and TUNEL assays were performed on paraffin-embedded sections after total body irradiation (TBI) with C-ion, as described in the Supplementary Materials and Methods.
2.3. Cell growth assay
The proliferation of BaF3 transfectants was assessed using the tetrazolium salt WST-1 according to the manufacturer’s protocol (Roche Diagnostics, Mannheim, Germany) (Supplementary Materials and Methods).
2.4. Invasion and migration assays
The invasive capabilities of PANC-1 and MIAPaCa-2 cells were examined using transwell chambers coated with matrigel (BD Biosciences, Redwood City, MA, USA) and the migration speeds of MIAPaCA-2 and PANC-1 cells were assessed by the wound healing assay using IncuCyte (Essen Bioscience), as described previously (Supplementary Materials and Methods).
2.5. Western blot assay
The expression of MAPKs and a cell cycle regulator in the jejunum was analyzed by Western blotting as described in the Supplementary Materials and Methods. The experimental conditions for each antibody are listed in Supplementary Table 1.
2.6. Microarray analysis
The mRNA expression profiles of the jejunum were obtained by a DNA microarray analysis 4 h after C-ion irradiation, as described in the Supplementary Materials and Methods. Microarray data were submitted to NCBI GEO (Accession no.: GSE113641).
2.7. Irradiation
C-ions were accelerated to 290 MeV per nucleon beam using the HIMAC synchrotron at the National Institute of Radiological Sciences and spread out to a width of 6 cm. The desired linear energy transfer (LET) was obtained by inserting a given thickness. The desired irradiation field was obtained by the simultaneous use of an iron collimator and brass collimator. C-ions were obtained with a dose-averaged LET of 20 keV/μm at the entrance of the plateau, whereas C-ions were located with a dose-averaged LET of 80 keV/μm within the spread-out Bragg peak (SOBP). Mice were irradiated with a plateau of 6-cm SOBP beams without an energy reducer with a dose-averaged linear energy transfer (LET) of 17 keV/μm. Pancreatic cancer cell lines were irradiated with an LET value of 80 keV/μm, corresponding to a monoenergetic beam with a narrow Bragg peak at a depth of 10 cm.
2.8. Statistical analysis
All values represent the mean ± standard deviation of results, and values in each group were compared using ANOVA and Fisher’s protected least significant difference test (*P < 0.05; **P < 0.01; ***P < 0.001).
3. Results
3.1. FGF1/CPP-C is internalized into cells more efficiently than FGF1, but exhibits less mitogenic activity
An FGF1/CPP-C fusion protein was created to match the sequence of the FGF12 C-terminal region (Fig. 1A) (Supplementary Materials and Methods). Since FGF1 has no sequence corresponding to the C terminus of FGF12, which includes CPP-C (Fig. 1A), it was internalized into cells markedly less than FGF12. Therefore, the fusion of CPP-C to FGF1 enhanced the cellular internalization of the FGF1 protein (Fig. 1B and Supplementary Fig. 1) [6]. FGF1/CPP-C activated all FGFR subtypes, namely, FGF1/CPP-C shared the same receptor specificity as wild-type FGF1 (Fig. 1C). However, the reactivity of FGF1/CPP-C with FGFRs was significantly weaker than that of FGF1 (Fig. 1C); therefore, the addition of CPP-C to FGF1 reduced the mitogenic activity of FGF1.
3.2. FGF1/CPP-C protects the intestines against C-ion RT-induced damage more than FGF1
Ten micrograms of FGF1/CPP-C or wild-type FGF1 in the absence of heparin was intraperitoneally administered to BALB/c mice 12 h before TBI with a plateau of the SOBP beam of C-ion (17 keV/μm) at 6, 7, or 8 Gy. FGF1 and FGF1/CPP-C both appeared to effectively promote crypt survival. After C-ion irradiation at 8 Gy, FGF1/CPP-C increased the relative crypt number significantly more than saline and FGF1 (Fig. 2AB). FGF1/CPP-C also increased this number significantly more than saline after TBI with C-ion at 7 Gy. In addition, FGF1/CPP-C strongly inhibited radiation-induced apoptosis in the crypts of the jejunum 24 h after TBI, while 10 and 100 μg of FGF1/CPP-C resulted in reductions of approximately 20 and 45%, respectively (Fig. 2C and D). In contrast, 100 μg of FGF1 reduced radiation-induced apoptosis by only approximately 20% from that with saline, whereas 10 μg of FGF1 did not. The repair of double-strand DNA breaks (DSBs) was estimated by staining paraffin-embedded sections of the jejunum with an anti-γH2AX antibody (Fig. 2E). FGF1/CPP-C tended to reduce γH2AX+ cell numbers after TBI, which were significantly lower than those of the irradiated control 2, 4, and 6 h after irradiation; however, FGF1 did not reduce γH2AX+ cell numbers 4 and 6 h after irradiation (Fig. 2F). These results suggested that FGF1/CPP-C increased DNA repair and the survival of intestinal crypt cells, thereby promoting recovery from C-ion-induced intestinal damage.
Fig. 2.

FGF1/CPP-C protects against intestinal adverse effects induced by C-ion therapy. Each FGF without heparin was administered intraperitoneally to three BALB/c mice 12 h before total body irradiation (TBI) with Carbon ion (C-ion) beams. (A, B) The relative number of surviving crypts in the jejunum of mice treated with 10 μg of FGF was assessed 3.5 days after TBI at 6, 7, or 8 Gy. All values are means ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001. (C, D) TUNEL assays were performed using paraffin-embedded sections to evaluate apoptosis in the crypts of the jejunum treated with 10 or 100 μg of FGF 24 h after TBI at 8 Gy. Representative histological images from mice treated with 100 μg of FGF are shown. All values are means ± SD (n = 3). *P < 0.05 significantly different from the irradiated control; **P < 0.01 significantly different from the irradiated control. (E, F) The jejunum of mice treated with 10 μg of FGF was removed 2, 4, or 6 h after TBI at 8 Gy. Staining with an anti-γH2AX antibody was performed using paraffin-embedded sections and representative histological images from mice 4 h after irradiation are shown. The number of γH2AX+ cells was assessed in each crypt by screening more than 200 crypts. All values are means ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001.
3.3. FGF1/CPP-C suppresses invasive and migration capabilities of pancreatic carcinoma cells
In order to estimate the involvement of FGF1/CPP-C in the metastasis of pancreatic carcinoma, the in vitro invasiveness of the human pancreatic carcinoma cell lines, MIAPaCa-2 and PANC-1, was examined using an invasion assay after the culture with FGF1/CPP-C. FGF-1/CPP-C reduced the number of MIAPaCa-2 and PANC-1 cells that invaded through Matrigel-coated membranes (Fig. 3A). Although wild-type FGF1 also inhibited the invasion of MIAPaCA-2 and PANC-1 cells, FGF-1/CPP-C reduced the invasion of pancreatic carcinoma cells significantly more than FGF1 (Fig. 3B). The migration of pancreatic carcinoma cells was tracked by the wound healing assay in order to assess their migration rate (Fig. 3C). FGF1/CPP-C significantly reduced the migration speed of MIAPaCa-2 cells 24 and 48 h after the culture, whereas FGF1 only reduced it 24 h after the culture. In contrast, the migration speed of PANC-1 cells was decreased by FGF1/CPP-C 48 h after the culture. These results suggested that FGF1/CPP-C decreased the invasive and migration capabilities of pancreatic carcinoma cells.
Fig. 3.

FGF1/CPP-C inhibits metastatic capabilities of pancreatic carcinoma cell lines. (A) The invasiveness of MIAPaCa-2 and PANC-1 cells was examined by invasion assays 24 h after the incubation in Matrigel-coated transwells with 100 ng/ml of FGF1 or FGF1/CPP-C in the presence of 5 μg/ml heparin. Invading cells on the transwell membrane are shown. (B) The number of invading cells was assessed using the public domain ImageJ program (NIH, Bethesda, MD) and the ratio of invading cells was obtained by dividing them by the total number of seeded cells. All values are means ± SD (n = 4). **P < 0.01; ***P < 0.001. (C) The migration of MIAPaCA-2 and PANC-1 cells was tracked by the wound healing assay using IncuCyte (Essen Bioscience, Ann Arbor, MI, USA) during the culture in a 96-well-plate with 100 ng/ml of FGF1 or FGF1/CPP-C. The migration rate of cells was assessed after 24- and 48-h cultures. All values are means ± SD (n = 3). *P < 0.05.
3.4. FGF1/CPP-C is internalized into pancreatic carcinoma cells without mitogenic effects
The transcript levels of FGFRs were assessed in MIAPaCa-2 and PANC-1 cells using DNA microarrays (Fig. 4A). MIAPaCa-2 and PANC-1 cells strongly expressed FGFR3 transcripts, and the expression of FGFR1 transcripts was also observed in PANC-1 cells. However, both of the cell lines weakly expressed the FGFR2 and FGFR4 transcripts. FGF12B was efficiently internalized into MIAPaCa-2 and PANC-1 cells; therefore, FGF1/CPP-C was internalized into MIAPaCa-2 and PANC-1 cells more than FGF1 because FGF1/CPP-C was composed of FGF1 and CPP-C derived from FGF12 (Fig. 4B). Although MIAPaCa-2 and PANC-1 cells expressed the subfamily of FGFRs, with which FGF1 and FGF1/CPP-C react, neither FGF1 nor FGF1/CPPC stimulated the growth of MIAPaCa-2 or PANC-1 cells (Fig. 4C). FGF1/CPP-C did not significantly inhibit the proliferation of MIAPaCa-2 or PANC-1 cells (Fig. 4C). Moreover, FGF1/CPP-C did not affect the survival abilities of pancreatic carcinoma cells, when FGF1/CPP-C treatment was combined with C-ion irradiation (Supplementary Fig. 2). FGF1/CPP-C also did not enhance tumor formation by MIAPaCa-2 and PANC-1 cells in the xenograft mouse model, whereas FGF1 promoted the proliferation of PANC-1 cells (Supplementary Fig. 3). These results suggested that FGF1/CPP-C did not promote the proliferation of pancreatic carcinoma cells by inducing intracellular signaling through the cellular internalization of FGF1/CPP-C.
Fig. 4.

FGF1/CPP-C reduces the proliferation of pancreatic cancer cells. (A) The levels of FGFR1-4 transcripts were quantified in PANC-1 and MIAPaCa-2 cells using DNA microarrays. (B) The fluorescence levels of PANC-1 and MIAPaCa-2 cells were evaluated by flow cytometry after a 24-h culture with Alexa Fluor 568-labeled recombinant FGFs in order to examine the internalization of each FGF into cells. (C) PANC-1 or MIAPaCa-2 cells were cultured in 12-well plates at a density of 5 × 104 cells per well for 24 h, and further incubated for 24 h with FGF1 or FGF1/CPP-C at the indicated concentrations in the presence of 5 μg/ml of heparin. Cell numbers were counted by microscopy. All values are means ± SD (n = 3). ns: not significantly different from the control.
3.5. FGF1/CPP-C inhibits the downstream signaling pathways of FGFRs and G2M transition
The downstream signaling pathways of FGFRs were examined using a Western blot analysis after C-ion irradiation with a 12-h FGF1/CPP-C pretreatment. FGF1 activated the signaling of FGFRs through the pathways of Raf-MEK-Erk1/2, JNK, and p38 until 4 h after TBI (Fig. 5A and Supplementary Fig. 4). However, FGF1/CPP-C significantly inhibited the activation of these MAPK signaling pathways until 4 h after TBI (Fig. 5A and Supplementary Fig. 4). In order to screen for molecules contributing to the effects of FGF1/CPP-C on radiation-induced intestinal damage, the mRNA expression profile of the irradiated jejunum was obtained using a DNA microarray analysis 4 h after C-ion irradiation with the 12-h FGF1/CPP-C pretreatment (Fig. 5B). C-ion irradiation reduced the transcript levels of G2M transition genes and increased those of G2M inhibition genes. In contrast, the transcript levels of the G1 and G1S transition genes were increased by irradiation, suggesting that C-ion irradiation induced cell-cycle arrest at G2/M in the intestines because G2/M transition is tightly regulated by the cdc2/cyclin B complex [8]. However, neither FGF1 nor FGF1/CPP-C significantly affected the transcript levels of cell-cycle regulatory genes after irradiation (Fig. 5B). On the other hand, FGF1/CPP-C inhibited the expression and activation of cell-cycle regulatory molecules, whereas FGF1 promoted them (Fig. 5C). The FACS analysis showed that FGF1/CPP-C and FGF1 slightly induced G2/M arrest in the rat intestinal cell line IEC6. (Fig. 5D and Supplementary Fig. 5) (Supplementary Materials and Methods). These results suggested that FGF1/CPP-C suppressed the downstream signaling pathways of FGFRs and inhibited G2/M transition, which was consistent with the weak mitogenic activity of FGF1/CPP-C (Fig. 1C).
Fig. 5.

FGF1/CPP-C inhibits downstream signaling pathways of FGFRs and G2M transition. Each FGF without heparin was administered intraperitoneally to BALB/c mice 12 h before total TBI with C-ion at 8 Gy. (A) Ten or one hundred micrograms of each FGF was administered to mice, and the activation of MAPK genes in the jejunum was examined by a Western blot analysis 2 h after TBI. (B) Ten micrograms of FGF was administered to mice, and the expression levels of each cell cycle regulatory gene in the jejunum were measured 4 h after TBI using DNA microarrays. These values in the FGF-treated jejunum relative to those in the non-irradiated saline control were shown in the heat map (n = 2). (C) The levels of activation of cell-cycle regulatory genes were assessed in the jejunum treated with 10 μg of FGF by a Western blot analysis 4 h after TBI. (D) The cell-cycle distribution of the rat intestinal epithelial cell line IEC6 was examined 24 h after the treatment with 100 ng/ml of FGF1 or FGF1/CPP-C in the absence of heparin by propidium iodide (PI) staining using flow cytometry. All values are means ± SD (n = 3). ns: not significant.
4. Discussion
C-ion radiation therapy is used for the local treatment of deep-seated tumors in the body because of physically and biologically effective dose localization [9], [10]. In addition, C-ion beams, which are high-LET radiation, produce dense ionization along their trajectories to cause irreparable DSBs, resulting in the death of cancer cells [11], [12]. A tumor mass is critically targeted with the high-LET portion, and, thus, the surrounding normal tissue is exposed to the low-LET portion outside the spread-out Bragg peak, but not the high-LET portion [9]. Therefore, the adverse reactions of C-ion radiotherapy may be potentially ameliorated by some treatments. A previous study showed that FGF1/CPP-C protected against photon-induced intestinal damage [7], and, in the present study, the adverse effects of C-ion irradiation were also attenuated by the FGF1/CPP-C treatment (Fig. 2).
FGF1 exerted potent radioprotective effects against radiation-induced intestinal damage [3], [4]. The receptor specificity of FGF is important for this role. FGF1/CPP-C reacted with all of the FGFR subtypes, such as FGF1 (Fig. 1C); therefore, FGF1/CPP-C had the same receptor specificity as FGF1. However, the mitogenic activity of FGF1/CPP-C through each FGFR was at least 10-fold weaker than that of FGF1 (Fig. 1C), and FGF1/CPP-C significantly inhibited the activation of the downstream signaling of FGFRs in the intestines after C-ion irradiation (Fig. 5A and Supplementary Fig. 4), suggesting that the biological activity of FGF1/CPP-C through FGFRs was weaker than that of FGF1. Although the well-established signaling mode of FGF consists of FGFR binding and activation of the cytoplasmic receptor kinase domain [13], the cellular internalization of FGF is also part of its functional mode of signaling because endocytosed receptor-bound FGF1 reaches the nucleus via a nuclear localization signal, leading to DNA synthesis and cell proliferation [14], [15] (Fig. 1B). In addition, a pathway analysis showed that the expression of genes in the renin-angiotensin system was down-regulated significantly more by FGF1/CPP-C than by FGF1 after C-ion irradiation (Supplementary Table 2). The renin-angiotensin system is known to contribute to radiation-induced tissue damage and is a potential target for preventing and treating damage [16]. Thus, the intracellular signaling mode of FGF independently of FGFR signaling may contribute to the radioprotective effects of FGF1/CPP-C [7].
DSBs are the most frequently induced and harmful type of DNA damage caused by irradiation, and must be repaired by non-homologous end joining or homologous recombination through the assembly of DNA repair molecules [17] for the regeneration of tissues. FGF1/CPP-C significantly reduced γH2AX foci in the jejunum after C-ion irradiation (Fig. 2EF). Although C-ion irradiation increased the expression of homologous recombination genes (Supplementary Figs. 6 and 7), FGF1/CPP-C by itself did not increase the protein expression of Rad51 in the intestines (Supplementary Fig. 7). Several FGFs, including FGF1, FGF2, and FGF18, are known to contribute to G2 arrest [18], [19], [20]. Wild-type FGF1 induced G2M arrest in the intestinal cell line (Fig. 5D), but increased ERK signaling to promote cell proliferation (Fig. 5A). On the other hand, FGF1/CPP-C inhibited the expression and activation of G2M transition molecules (Fig. 5C), and the addition of FGF1/CPP-C to a culture of intestinal cells appeared to increase their G2M population (Supplementary Fig. 5). Moreover, FGF1/CPP-C significantly inhibited the activation of the MAPK signaling pathways including ERK (Fig. 5A). Therefore, the FGF1/CPP-C treatment induced G2 arrest in the jejunum in order to provide more time for DNA repair.
FGFR1b expression in human pancreatic cancer cells inhibited single-cell movement, in vitro invasion, and in vivo tumor formation and growth, whereas FGFR1c expression in non-malignant pancreatic ductal cells resulted in cellular transformation and in vivo tumor formation [21]. Therefore, some FGF signaling pathways may suppress the malignancy of tumors [5]. FGF1/CPP-C inhibited the invasive and migration capabilities of the pancreatic cancer cell lines, PANC-1 and MIAPaCa-2 more strongly than wild-type FGF1 (Fig. 3). However, PANC-1 cells expressed FGFR1, whereas MIAPaCa-2 cells did not (Fig. 4A); therefore, other signaling pathways besides FGFR1 may be responsible for inhibiting the malignancy of pancreatic cancer cell lines by FGF1/CPP-C. FGF12 also suppressed the in vitro invasiveness of PANC-1 and MIAPaCa-2 cells [7] and FGF1/CPP-C was internalized into these cells (Fig. 4B), suggesting that the cellular internalization of FGF1/CPP-C inhibited the metastasis of pancreatic cancer cells. Moreover, FGF1/CPP-C did not promote the in vitro proliferation rates or in vivo tumor formation in SCID mice of PANC-1 or MIAPaCa-2 cells (Fig. 4C and supplementary Fig. 3). There is also no evidence to indicate that the radioresistance of pancreatic carcinoma against C-ion radiotherapy developed after FGF1/CPP-C treatment (Supplementary Fig. 2). These results suggest that FGF1/CPP-C mediates the inhibition of pancreatic cancer cell metastasis via its intracellular functions through cellular internalization; therefore, the clinical usage of the FGF1/CPP-C radioprotector does not exacerbate the malignancy of pancreatic carcinoma.
5. Conclusions
FGF1/CPP-C increased DNA repair and the survival of intestinal crypt cells, resulting in the promotion of recovery from C-ion-induced intestinal damage. In contrast, FGF1/CPP-C decreased the proliferative, invasive, and migration capabilities of pancreatic carcinoma cells. FGF1/CPP-C suppressed the downstream signaling pathways of FGFRs and inhibited G2/M transition, which was consistent with the weak mitogenic activity of FGF1/CPP-C. Accordingly, the FGF1/CPP-C chimera protein, which is characterized by the intracellular signaling mode of FGF1, has the potential to prevent and treat adverse reactions in the GI tract after C-ion radiotherapy for pancreatic carcinoma in the abdomen.
Conflict of interest
Fumiaki Nakayama, Mayumi Fujita, Takeshi Yasuda, and Takashi Imai have a patent, JP-5818977, issued to the National Institute of Radiological Sciences.
Acknowledgments
This work was partially supported by a grant for Radiation Emergency Medical Preparedness from the National Institutes for Quantum and Radiological Science and Technology, Japan, and JSPS KAKENHI, Japan (Grant Numbers: JP17K10498 and JP16K10407). We wish to express our gratitude to Ms. S. Umeda and Ms. K. Takahashi for their technical assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ctro.2018.10.004.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Shinoto M., Yamada S., Terashima K., Yasuda S., Shioyama Y., Honda H. Carbon ion radiation therapy with concurrent gemcitabine for patients with locally advanced pancreatic cancer. Int J Radiat Oncol Biol Phys. 2016;95:498–504. doi: 10.1016/j.ijrobp.2015.12.362. [DOI] [PubMed] [Google Scholar]
- 2.Shinoto M., Yamada S., Yasuda S., Imada H., Shioyama Y., Honda H. Phase 1 trial of preoperative, short-course carbon-ion radiotherapy for patients with resectable pancreatic cancer. Cancer. 2013;119:45–51. doi: 10.1002/cncr.27723. [DOI] [PubMed] [Google Scholar]
- 3.Okunieff P., Mester M., Wang J., Maddox T., Gong X., Tang D. In vivo radioprotective effects of angiogenic growth factors on the small bowel of C3H mice. Radiat Res. 1998;150:204–211. [PubMed] [Google Scholar]
- 4.Hagiwara A., Nakayama F., Motomura K., Asada M., Suzuki M., Imamura T. Comparison of expression profiles of several fibroblast growth factor receptors in the mouse jejunum: suggestive evidence for a differential radioprotective effect among major FGF family members and the potency of FGF1. Radiat Res. 2009;172:58–65. doi: 10.1667/RR1570.1. [DOI] [PubMed] [Google Scholar]
- 5.Turner N., Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 6.Nakayama F., Yasuda T., Umeda S., Asada M., Imamura T., Meineke V. Fibroblast growth factor-12 (FGF12) translocation into intestinal epithelial cells is dependent on a novel cell-penetrating peptide domain: involvement of internalization in the in vivo role of exogenous FGF12. J Biol Chem. 2011;286:25823–25834. doi: 10.1074/jbc.M110.198267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakayama F., Umeda S., Yasuda T., Fujita M., Asada M., Meineke V. Cellular internalization of fibroblast growth factor-12 exerts radioprotective effects on intestinal radiation damage independently of FGFR signaling. Int J Radiat Oncol Biol Phys. 2014;88:377–384. doi: 10.1016/j.ijrobp.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 8.Smits V.A., Medema R.H. Checking out the G2/M transition. Biochim Biophys Acta. 2001;1519:1–12. doi: 10.1016/s0167-4781(01)00204-4. [DOI] [PubMed] [Google Scholar]
- 9.Kanai T., Furusawa Y., Fukutsu K., Itsukaichi H., EguchiKasai K., Ohara H. Irradiation of mixed beam and design of spread-out Bragg peak for heavy-ion radiotherapy. Radiat Res. 1997;147:78–85. [PubMed] [Google Scholar]
- 10.Tsujii H., Kamada T. A review of update clinical results of carbon ion radiotherapy. Jpn J Clin Oncol. 2012;42:670–685. doi: 10.1093/jjco/hys104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fokas E., Kraft G., An H.X., Engenhart-Cabillic R. Ion beam radiobiology and cancer: time to update ourselves. Bba-Rev Cancer. 2009;1796:216–229. doi: 10.1016/j.bbcan.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Hamada N. Recent insights into the biological action of heavy-ion radiation. J Radiat Res (Tokyo) 2009;50:1–9. doi: 10.1269/jrr.08070. [DOI] [PubMed] [Google Scholar]
- 13.Mason I.J. The ins and outs of fibroblast growth factors. Cell. 1994;78:547–552. doi: 10.1016/0092-8674(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 14.Wiȩdłocha A., Falnes P.O., Rapak A., Klingenberg Ø., Muñoz R., Olsnes S. Translocation of cytosol of exogenous, CAAX-tagged acidic fibroblast growth factor. J Biol Chem. 1995;270:30680–30685. doi: 10.1074/jbc.270.51.30680. [DOI] [PubMed] [Google Scholar]
- 15.Imamura T., Engleka K., Zhan X., Tokita Y., Forough R., Roeder D. Recovery of mitogenic activity of a growth factor mutant with a nuclear translocation sequence. Science. 1990;249:1567–1570. doi: 10.1126/science.1699274. [DOI] [PubMed] [Google Scholar]
- 16.Pinter M., Kwanten W.J., Jain R.K. Renin-angiotensin system inhibitors to mitigate cancer treatment-related adverse events. Clin Cancer Res. 2018 doi: 10.1158/1078-0432.CCR-18-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodarzi A.A., Jeggo P.A. The repair and signaling responses to DNA double-strand breaks. Adv Genet. 2013;82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 18.Tran T., Kolupaeva V., Basilico C. FGF inhibits the activity of the cyclin B1/CDK1 kinase to induce a transient G2 arrest in RCS chondrocytes. Cell Cycle. 2010;9:4379–4386. doi: 10.4161/cc.9.21.13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salotti J., Dias M.H., Koga M.M., Armelin H.A. Fibroblast growth factor 2 causes G2/M cell cycle arrest in ras-driven tumor cells through a Src-dependent pathway. PLoS One. 2013;8:e72582. doi: 10.1371/journal.pone.0072582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawano M., Umeda S., Yasuda T., Fujimura T., Ishikawa A., Imamura T. FGF18 signaling in the hair cycle resting phase determines radioresistance of hair follicles by arresting hair cycling. Adv Radiat Oncol. 2016;1:170–181. doi: 10.1016/j.adro.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z., Neiss N., Zhou S., Henne-Bruns D., Korc M., Bachem M. Identification of a fibroblast growth factor receptor 1 splice variant that inhibits pancreatic cancer cell growth. Cancer Res. 2007;67:2712–2719. doi: 10.1158/0008-5472.CAN-06-3843. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.