

ABSTRACT
Ubiquitin A-52 residue ribosomal protein fusion product 1 (Uba52), a ubiquitin-ribosomal fusion gene, is a major source of ubiquitin protein for covalent modification of proteinaceous substrates recycled by ubiquitin-proteasome system (UPS). Its role in early embryo development has not been studied. Using the CRISPR/Cas9 gene editing tool, the objective of this study was to determine if UBA52 protein is required for mammalian embryogenesis. Matured metaphase II porcine oocytes were injected with CRISPR Cas9+guide RNAs (Uba52 gRNA) or Cas9 without gRNAs as control, followed by in vitro fertilization (IVF) and embryo culture to day 7. Injection of Cas9+gRNAs affected embryo development. On day 4 of embryo culture, the proportion of 2-, 4- and 8-cell stage embryos was significantly different between the Uba52 gRNA and control group (P<0.05), with more 8-cell stage embryos in the control and more 4- and 2-cell stage embryos in the Uba52g RNA group. This delay in the development of Uba52 gRNA embryos occurred at the transition from the 4- to 8-cell stages, around the time of major zygotic genomic activation. The percentage of blastocyst formation on day 7 and the cell number per blastocyst were significantly lower in the Uba52 gRNA group than in the control (P<0.05). Genotyping by PCR and DNA gel electrophoresis analysis showed that 91.8% of embryos that failed to develop to blastocyst had either a monoallelic or a biallelic modification of the Uba52 gene. In comparison, only 24.4% of embryos that reached blastocyst had a monoallelic modification and biallelic editing was not found in any of the blastocysts. Based on immuno-labeling intensity, both UBA52 and proteasome protein levels on days 4 and 7 of culture were significantly lower in the Uba52 gRNA group than in the control (P<0.05), in agreement with UBA52 western blotting-densitometry of day 4 embryos. Morphological examination of blastomere nuclei revealed abnormal nuclear structure in the Uba52 gRNA group, such as reduced size, irregular shapes, nucleus fragmentation and uneven DNA distribution at all stages of embryo development. Nuclear morphology studies of embryos injected with Cas9+gRNAs and co-injected with plasmid DNA encoding nuclear localized GFP further supported these observations. In conclusion, our data indicate that the Uba52 gene is essential in early embryogenesis.
KEY WORDS: Uba52, Development, Embryo, Ribosomal, Ubiquitin
Summary: Ubiquitin A-52 residue ribosomal protein fusion product 1 (Uba52), a ubiquitin-ribosomal fusion gene, is a major source of ubiquitin protein for covalent modification of proteinaceous substrates recycled by ubiquitin-proteasome system. Using CRISPR/cas9 gene editing tool, we demonstrate that that Uba52 gene is essential in early embryogenesis in mammals.
INTRODUCTION
Ubiquitin (UBB/UBC/UBD) is a 76-amino acid small protein with molecular mass of 8.5 kDa. It is a highly conserved protein in eukaryotes, with a fundamental role in selective protein degradation by the ubiquitin–proteasome system (UPS). In this pathway, ubiquitin molecules are attached covalently to the substrate proteins, a process called ubiquitination, and mediated by a multi-enzymatic complex including ubiquitin-activating enzymes E1 (UBA1), ubiquitin-conjugating enzymes E2 (e.g. UBE2A, UBE2B, UBE2C), ubiquitin ligases E3 (e.g. UBE3A and others), and ubiquitin chain elongation/ubiquitination factors enzymes E4 (e.g. UBE4A/UBE4B), which work sequentially in a cascade (Sutovsky, 2003). Ubiquitinated substrates are most commonly degraded by the 26S proteasome (Ciechanover and Schwartz, 1994). Besides cellular homeostasis/protein turnover, the UPS has been implicated in the pathogenesis of many diseases (Hershko and Ciechanover, 1998), as well as in the physiological events of mammalian fertilization and embryogenesis (Sutovsky, 2003), sperm function (Sutovsky et al., 2001) and the control of mitochondria inheritance (Song et al., 2016). In addition to its role in protein degradation, the non-proteolytic consequences of protein ubiquitination also play an important role in cellular functions such as signaling, cell cycle control, transcriptional regulation and apoptosis (Komander and Rape, 2012).
Ubiquitin is encoded by multiple genes including monomeric UB-ribosomal fusion genes, Uba52 and RPS27A, that encode one UB unit fused to a ribosomal protein, and polyubiquitin genes, UBB, UBC and UBD that harbor up to ten tandem repeats of monoubiquitin coding units (Baker and Board, 1991; Finley et al., 1989; Wiborg et al., 1985). While the polyubiquitin genes play a key role in stress-responses such as heat shock, starvation (Finley et al., 1987; Fornace et al., 1989), DNA damage, oxidative stress (Vihervaara et al., 2013) and heavy metal cytotoxicity response (Lee et al., 2015), the ubiquitin-ribosomal fusion genes are stably expressed to satisfy the demand for ubiquitin under basal conditions (Bianchi et al., 2015). The Uba52 gene has been identified as a housekeeping gene (Warrington et al., 2000) and the transcript has been used as a reference in rhesus monkey (Ahn et al., 2008), starfish (Sadritdinova et al., 2014) and bovine tissues (Schoen et al., 2015), to quantify gene expression. Through cDNA microarray analysis of gene expression in porcine oocytes and early preimplantation embryos, Uba52 expression in the blastocyst stage embryo was six times higher than the metaphase II oocytes (Whitworth et al., 2005). A similar expression pattern was observed in rhesus monkey (Mtango and Latham, 2007) and mouse (Zeng et al., 2004) embryos. This suggests that Uba52 has a functional role in early embryogenesis. Indeed, the Uba52 and other UPS gene expression was dysregulated in the aberrant rhesus monkey embryos with reduced developmental potential (Mtango and Latham, 2007). In spite of gene expression profiles, there have been no studies to determine its roles in early embryogenesis. We thus hypothesized that this ubiquitin fusion protein was essential in mammalian embryo development.
To study the specific roles of a gene, gene mutation approaches have been used. Recently, the bacterial clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated system (Cas9), has become increasingly popular for creating gene edits in both the somatic cells and embryos to study gene function (Cho et al., 2013; Cong et al., 2013). The CRISPR/Cas9 system has been used to efficiently generate genetically modified animals via zygotic injections of Cas9 and guide RNA (gRNA) in many species, such as the mouse (Wang et al., 2013), pig (Hai et al., 2014; Whitworth et al., 2017) and monkey (Niu et al., 2014), indicating its versatility and universality. High specificity and efficiency of gene editing as well as low cost and ease of application has helped to promote its widespread use in biomedical research, including the increasingly important domestic pig model. Moreover, injecting multiple CRISPR guides at the same time can increase the possibility of gene editing (Spate et al., 2016).
The purpose of this study was to determine if Uba52 gene expression is required for early embryogenesis by injecting Cas9 and guide RNAs into metaphase II arrested porcine oocytes, and examining embryo development up to and including the blastocyst stage. To our knowledge, this is the first report that Uba52 gene modification/knockout causes development lethal arrest prior to embryo implantation, indicating its importance in mammalian embryogenesis.
RESULTS
Comparison of embryo development between the Uba52 gRNA and control group
The objective was to compare the developmental potential of control and gene-edited embryos. There were two groups: the Uba52 gRNA group injected with CRISPR/Cas9+gRNAs and the sham control group injected with CRISPR/Cas9 without gRNAs. A total of 1934 injected oocytes (control: 784; Uba52 gRNA: 1150) from twelve replicates were used for embryo development study. The percentage of embryos cleaved on day 2 (Fig. 1A) was not different between the Uba52 gRNA group and control (59.2±4.2 versus 60.6±4.2%, respectively). However, on day 4 of culture, the distribution of embryonic developmental stages was different between the two groups. Compared to the sham control, the Uba52 gRNA group had more embryos at 2-cell (19.2±8.2 versus 9.4±8.2%) and 4-cell stage (51.8±8.5 versus 36.9±8.5%), and fewer reaching the 8-cell stage (29.0±9.9% versus 58.7±9.9; P<0.05). The micro-manipulated control group without gRNA was not different from the non-manipulated in vitro fertilization (IVF) control, and both groups were different from the Uba52 gRNA group (Fig. 1B). Thus, the delay in the development of Uba52 gRNA embryos to 8-cell stage was accrued at the transition from the 4-cell to 8-cell stage, which in the domestic pig coincides with the degradation/depletion of maternally stored proteins, and major zygotic genome activation.
Fig. 1.

Embryo development on days 2, 4 and 7 of culture. (A) Per cent of embryos cleaved on day 2 of culture (Day of IVF=0; n=784 for the control group and 1150 for the Uba52 gRNA, in 12 replicates). (B) Distribution of 2-, 4- and 8-cell stage embryos on day 4 in the non-manipulated IVF embryos (n=86 from three replicates), manipulated control and Uba52gRNA group (same embryos from day 2, as in A). The distribution of embryos at each stage was not different between IVF and sham groups. The Uba52 gRNA group was significantly different from both IVF and sham control by X2 test. (C) Blastocyst formation at day 7. Per cent of blastocysts formed in the Uba52gRNA group was lower than that in the sham control (P<0.01). (D) Cell number per blastocyst from 70 control and 17 Uba52 gRNA blastocysts of three replicates. NS, not significant; ** indicates difference between the Uba52gRNA and control groups at P<0.01.
Blastocysts were morphologically evaluated on day 7. Embryos that had cavitated and possessed a normal or thinning zona pellucida, were considered to be blastocysts. The developmental arrest observed on day 4 in the Uba52 gRNA group resulted in a significantly lower percentage of blastocyst formation compared to the sham injected controls (4.0±1.0 versus 23.9±1.3%, P<0.01) on day 7 (Fig. 1C). In addition, the average number of nuclei in the Uba52 gRNA embryos that did develop to blastocyst on day 7 (Fig. 1D) was lower than that in the control group (28.8±6.5 and 49.2±2.8 for the Uba52 gRNA and control group, respectively; P<0.01).
Genotyping of Uba52 gene in day 7 blastocysts and arrested embryos
To determine the Uba52 gene modifications in early embryos, a total of 90 day 7 embryos, including 41 blastocysts and 49 arrested 4-cell- to morula-stage embryos were assayed by PCR and gel electrophoresis (Fig. 2A,B). Of the 41 blastocysts, thirty-one embryos (75.6%) only had wild-type (WT) bands and classified as WT/WT. Ten blastocysts (24.4%) had one WT band and one lower band (deletion), signifying monoallelic modification (WT/Mod). No biallelic modification of Uba52 gene (classified as Mod/Mod) was found in day 7 blastocysts. To confirm that there was no Uba52 gene modifications in the wild-type bands of day 7 Uba52 gRNA embryos, four WT/WT blastocysts were further Topo cloned and Uba52 gene was sequenced. There was no modification of Uba52 gene detected in such blastocysts, confirming the genotyping results of Uba52 by PCR and gel electrophoresis.
Fig. 2.

Genotyping of Uba52 gene in day 7 blastocysts and arrested embryos. Genotyping of Uba52 gene by DNA gel electrophoreses (A), distributions of WT/WT, WT/Mod, and Mod/Mod genotypes in the day 7 Uba52 gRNA blastocysts (B) and arrested embryos (C). Bl, blastocyst. * and ** indicate monoallelic (WT/Mod) and biallelic (Mod/Mod) modification of Uba52 gene, respectively. Control and manipulated WT/WT blastocysts had a single band at 305 bp (arrow in A).
Among the 49 arrested day 7 embryos, four embryos showed only wild type (8.2%), 45 embryos (91.8%) had either monoallelic (26 embryos, 53.0%; indicated by one star in Fig. 2A) or biallelic modification (19 embryos, 38.8%; indicated by two stars in Fig. 2A). Again, the Uba52 modified embryos identified by DNA gel electrophoresis were sequenced and confirmed to be Uba52 modification (Fig. 3). These data suggest that one allele of the Uba52 gene was sufficient to support development to blastocyst in some embryos, but biallelic modified embryos could not develop. Thus, Uba52 expression appears to be essential for embryo development.
Fig. 3.

Topo cloning and sequencing of Uba52 gene in the modified embryos. (A) Location of guides flanking exon 1 of the Uba52 gene and potential cutting sites indicated by PAM (protospacer adjacent motif). (B) Sequencing of six pooled modified embryos to show cutting sites. (C). Sequencing of individual embryos to confirm monoallelic (individuals 1 and 3) and biallelic (individual 2) modifications.
Decreased UBA52 and proteasome subunit content in the Uba52 gRNA embryos
The objective of this experiment was to evaluate the UBA52 and proteasomal subunit protein content in the control and Uba52 gRNA embryos on day 4 and 7 of culture by immunostaining. Both Uba52 and proteasomal subunit gene expression changes have been associated with developmental potential of mammalian oocytes and embryos (Mtango and Latham, 2007). UBA52 protein was localized in both the cytoplasm and nuclei of embryo blastomeres (Fig. 4A-D). The intensity of immune-reactive UBA52 in the Uba52 gRNA group was significantly lower on days 4 and 7, compared with control (Fig. 4E,F; P<0.01). To confirm the specificity of UBA52 immuno-labeling, western blotting (WB) was carried out on day 4 embryos to determine their UBA52 protein content. The density of UBA52 band, which migrated consistently at the predicted size of ∼14.5 kDa, was lower in the Uba52gRNA group than in sham control (band density was normalized against residual protein load, with background subtraction: 25.5 versus 12.0, arbitrary units, for the sham and Uba52gRNA groups, respectively; lanes 2 and 3 in Fig. 4G), which agrees with immuno-labeling analysis.
Fig. 4.

Immunoreactive UBA52 protein in day 4 and day 7 embryos. (A-D) Representative images of day 4 and 7 embryos immuno-labeled for UBA52 (green) and DNA (blue). (E,F) Quantification of fluorescence intensity of UBA52 on day 4 (E) and 7 (F). Relative intensity values were adjusted so the average of the control group was equal to 1. Fluorescence intensity of UBA52 was reduced by the Cas9+gRNA injection (***P<0.001). Bar lines are LS-means±s.e.m. (G) Western blotting of day 4 embryos confirmed the reduction of UBA52 band in Cas9+gRNA injected embryos (lane 3 versus 2). Lanes 1, 2, 3 and 4 represent 62 MII oocytes, 45 control embryos, 45 Uba52 gRNA embryos and a total of 100,000 unspecified fibroblast cells. Arrows indicate predicted Uba52 bands.
Being previously tied to developmental potential/competence of mammalian oocytes and embryos, the localization (both nuclear and cytoplasmic) and relative abundance of proteasomal subunit proteins was evaluated with immunocytochemistry with a previously characterized antibody (Sutovsky et al., 2004; Zigo et al., 2018) recognizing multiple 20S proteasomal core subunits (Fig. 5). As would be expected, the labeling intensity of proteasomes was stronger inside the nucleus than the cytoplasm (Fig. 5A-D). The combined nuclear and cytoplasmic intensity of immune-reactive proteasomes in the Uba52 gRNA group was significantly lower (P<0.01) than control, both on day 4 and day 7 (Fig. 5E,F), with a reduction by 20% on day 4 and more than 50% on day 7 in the Uba52 gRNA versus control.
Fig. 5.

Quantification of proteasomal subunit proteins in the day 4 and day 7 embryos. (A-D) Representative images of embryos that were immuno-labeled for proteasome (green) and counter stained for DNA (blue). (E-F) Scatter plots of proteasome labeling intensity for day 4 (E) and day 7 (F) embryos. *** shows that immunoreactive proteasome was lower in the Cas9+gRNAs group than the control (P<0.001). Relative intensity values were corrected so the average of the control group was equal to 1. Bar lines represent LS-means±s.e.m.
Taken together, these experiments showed that CRISPR/Cas9+Uba52 gRNAs injection efficiently lowered the content of UBA52 protein and proteasomal subunits in the embryos, and disrupted embryo development on both day 4 and day 7.
Nuclear morphology abnormalities in the Uba52g RNA embryos
To understand the importance of the Uba52 gene and protein for the maintenance of nuclear structure, the morphology of blastomere nuclei was examined in live 4- and 8-cell stage embryos (day 4) after DNA staining with Hoechst 33342. Invariably, the blastomere nuclei of 42 sham-injected embryos had a regular, oval shape (Fig. 6A). In contrast to the control embryos, the Uba52 gRNA embryos (a total of 55 from three replicates) displayed various morphological abnormalities, ranging from large sized (compared to control), oval-shaped nuclei with densely aggregated chromatin and uneven DNA distribution, to small, disorganized and irregularly shaped nuclei (arrows in Fig. 6B,C). The small nuclear size in the Uba52 gRNA embryos could be either the results of degeneration and fragmentation of previously regular blastomere nuclei or it could originate from single, detached mitotic chromosomes making karyomeres/micronuclei after entering interphase.
Fig. 6.
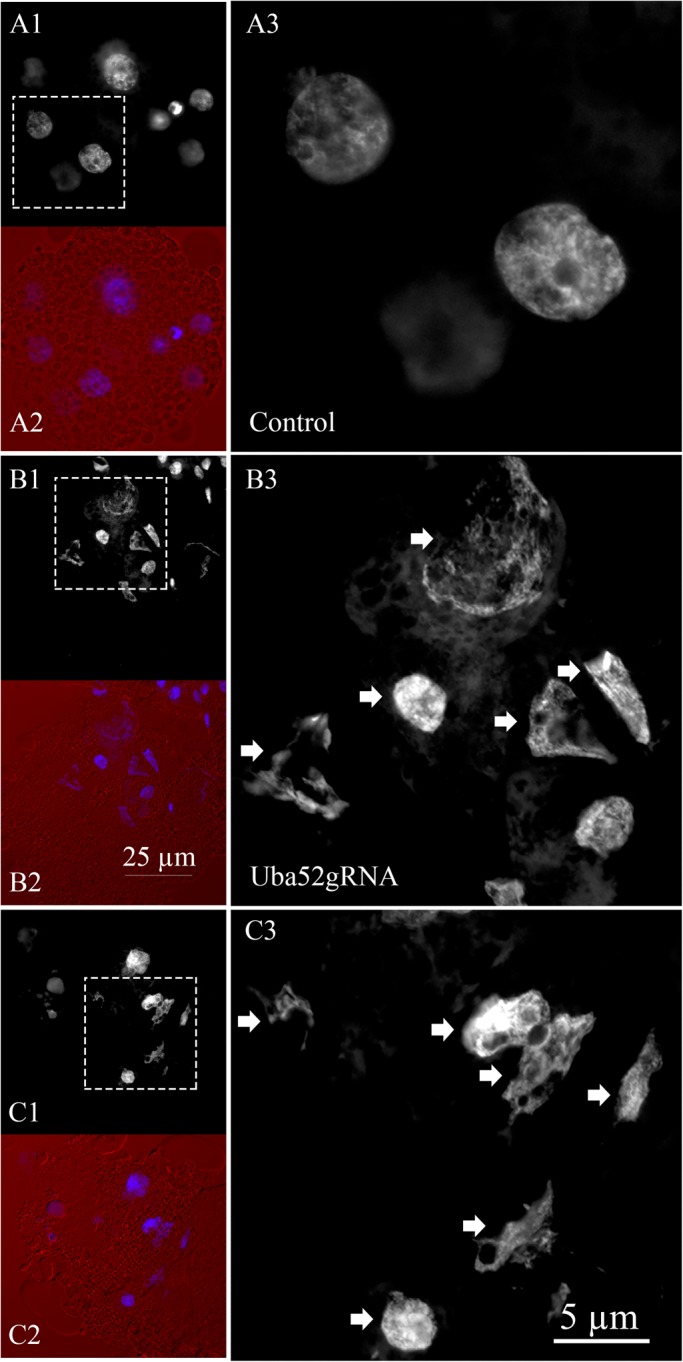
Nuclear morphology of day 4 embryo stained with Hoechst 33342. (A) A control 8-cell embryo. (B,C) Representative Uba52 gRNA embryos. Grayscale panels show DNA staining, pseudo-colored panels show overlay of DNA (blue) and bright field (red) images. A3, B3 and C3 are four times magnified rectangles traced in panels A1, B1 and C1. Compared to control, all nuclei in the Uba52 gRNA embryos show morphological abnormalities, indicated by the arrows.
GFP coinjection with Cas9+gRNAs confirmed nuclear morphological abnormalities
The green fluorescent protein (GFP) is often used in many studies to determine the subcellular localization of other proteins by analyzing fusion proteins. A similar approach to examine the nuclear morphology of embryo blastomeres was used by injecting Cas9+gRNAS with plasmid DNA encoding nuclear-import signal GFP construct before fertilization. The embryos were collected on day 4 of culture, fixed, and stained with DNA stain DAPI. The nuclei of control group (Cas9+GFP without gRNAs) were morphologically normal, with regular size and oval shape (Fig. 7A). In the Uba52gRNA group, various blastomere-nuclear abnormalities were observed at the developmental stages ranging from 2-cell to 8-cell (Fig. 7B,C), including irregular shapes, smaller size and uneven hyper-condensed chromatin. Lower intensity of the nuclear GFP fluorescence was noticeable in the 8-cell Uba52gRNA embryos, compared to 8-cell control, and 2- to 6-cell control and Uba52gRNA embryos. The nuclear perimeter of in the Uba52 gRNA embryos was often uneven, resulting in a patched appearance of GFP. These results confirm that Uba52 gene modification resulted in embryonic nuclear abnormalities, which may have contributed to the observed developmental arrest.
Fig. 7.

Representative images of control (Cas9+GFP) and Cas9+gRNA+GFP co-injected embryos on day 4 of culture, showing nuclear-imported GFP (green) and DNA counter-staining with DAPI (blue). (A) 8-cell stage control embryo. (B,C) Cas9+gRNAs+GFP injected embryos showing abnormal nuclear morphology (arrows) at 6- and 8-cell stage, respectively.
DISCUSSION
Modification of Uba52 gene by CRISPR/Cas9 technology had a dramatic effect on the development of porcine embryos in vitro. There was a significant reduction in the number of blastocysts formed on day 7 of embryo culture and the genotyping of the genetically edited embryos injected with Uba52 gRNA confirmed that over 91% of them carried monoallelic or biallelic modifications of the Uba52 gene. Furthermore, the blastomere nuclei of the Uba52 gRNA embryos displayed abnormal nuclear morphology and highly variable size, often being much smaller than the sham control nuclei. Proper timing of key developmental processes during the early preimplantation period is very important for the attainment of blastocyst stage. The genome-wide sweep of maternal mRNAs and proteins, DNA replication, chromatin remodeling and major zygotic genome all have to be precisely coordinated. Many of these early processes are actually regulated by UPS (Gilberto and Peter, 2017; Moreno and Gambus, 2015). The timely onset of the expression of key cell cycle regulators is essential for normal development, and aberrations can lead to apoptosis and developmental arrest (Hara et al., 2006). Correct expression of cell cycle regulators during preimplantation development is critical in order to sustain developing embryo response to DNA damage, stress and other adverse conditions by activating either survival and repair mechanisms, or apoptotic processes. Modification of Uba52 gene in the present study likely represents a multipronged insult to several key functions of the blastomere, resulting in a lethal developmental arrest. Accordingly, mutation of Uba52 in mice is embryo lethal, affecting embryo ubiquitin levels, ribosome assembly, cell cycle progression and overall protein synthesis (Kobayashi et al., 2016). While the Uba52 deficient mouse fetuses die before embryonic day 10.5, it is possible that they develop to blastocyst and implant. In contrast, porcine zygotes in the present study became arrested as early as the 4-8 cell stage of preimplantation development and no blastocyst with biallelic Uba52 modification were found. This discrepancy could be due to species difference but also to compensate for related ubiquitin-ribosomal fusion protein genes which may occur when the gene is deleted completely, but not when a truncated dysfunctional protein is being produced due to a gene modification (present study). Mutation of Uba52 homologs in the Saccharomyces cerevisiae showed that the UBI1 and UBI2 double mutant is not viable (Finley et al., 1989); UBI1 and UBI2 genes in yeast encode identical (at amino acid sequence level) ubiquitin-tail ribosomal protein.
Ubiquitin is the most abundant protein in the Eukaryotic cells, representing up to 5% of total protein. However, the free unconjugated ubiquitin pool is surprisingly small, which means that, despite its pervasive roles in many cell functions, ubiquitin is not produced in excess. The embryonic genome becomes activated at 4-cell stage in the pig. During normal development from zygote to 4-cell stage embryo, transcripts involved in protein catabolic processes are highly abundant at both the 2- and 4-cell stages. Many of them are directly linked to ubiquitin (Østrup et al., 2013). However, when Uba52 gene was modified mono- or bi-allelically in the present study, residual ubiquitin and maternal Uba52 mRNA pools may have been sufficient to support embryo development for a short time, enabling cell cycle progressing, mitosis, RNA translation and processing (including ribosomal machinery), protein catabolism and chromatin remodeling (Østrup et al., 2013). Thus, the gene modified embryos still could cleave and develop to 4-cell and even to 8-cell stage as we observed. However, after 4 days of culture, there was a statistically significant reduction of the UBA52 protein content in the Uba52 gRNA group compared to control. Such a reduction would represent a major insult to many critical cell functions, and result in a development delay leading to a failure of blastocyst formation in the biallelically modified embryos. Since Uba52 is a housekeeping gene, it may have to be activated along with other housekeeping genes in advance of the major genome activation to support essential cellular functions (Latham and Schultz, 2001). As UBA52 protein contributes both to free ubiquitin pool and to ribosomal protein complex, it is reasonable to assume that many cell key functions such as cell cycle control and sustaining nuclear structures could have been impaired by UBA52 deficiency. In the present study, the majority of embryos arrested at 8-cell stage, i.e. within one cell cycle after the zygotic genome activation, indicating that the genome activation itself may also be affected. The observed reduction of embryo proteasome content would further aggravate the effect of Uba52 obliteration on protein synthesis and turnover.
As demonstrated in normal cell division (Amin et al., 2008), major changes in nuclear structure are required to allow segregation of duplicated chromosomes to the daughter cells during mitosis. However, for the porcine embryo, it takes over 12 h to complete each cell cycle between fertilization and the 8-cell stage (Mateusen et al., 2005). The nuclei in all the control embryos had evenly distributed DNA/heterochromatin. In the early control embryo blastomeres, the complex spatial arrangements in the nucleus are maintained by attachments to a nuclear matrix consisting of the nuclear lamina and an internal fibrogranular network made of nuclear matrix proteins and RNA (Nickerson, 2001). Ubiquitin regulates the cell cycle both by proteolytic and non-proteolytic mechanisms (Gilberto and Peter, 2017). Not only does ubiquitin impact all stages of DNA replication, particularly by protecting DNA from insults (García-Rodríguez et al., 2016; Moreno and Gambus, 2015), but it also drives cell cycle progression (e.g. cyclin ubiquitination and proteasomal degradation at metaphase/anaphase transition). Furthermore, ubiquitin plays a critical role in regulating the dynamics of nucleosomal chromatin structure (Gilberto and Peter, 2017) wherein nucleosome histones must be evicted from DNA and deposited in a semi-conservative manner onto new DNA strands, the gaps being filled with newly synthesized histones. Thus, to maintain genome integrity, nucleosome assembly during S-phase necessitates an adequate histone supply (Alabert and Groth, 2012), which is regulated by the processing factor stem-loop binding protein (SLBP). Interestingly, histone mRNA processing is activated by CRL4WDR23 through multi-monoubiquitination of SLBP (Brodersen et al., 2016). Cells lacking SLBP exhibit severe DNA replication defects. After S-phase, SLBP is rapidly degraded by SCF cyclin F complexes (Dankert et al., 2016), and this proteolytic degradation is critical for genome maintenance upon genotoxic stress. Thus, the non-proteolytic and proteolytic modes of regulation of SLBP by ubiquitin cooperate in space and time to restrict histone synthesis to S phase and thereby maintain genome stability. These mechanisms may also provide explanations for the misshaped nuclear structure, small sized nucleus and fragmented blastomere nucleus in Cas9 gRNA injected embryos in the current study. If so, Uba52 gene knockout would reduce cellular ubiquitin content and cause a severe insult to DNA and cell functions, resulting in abnormal nuclear morphology and developmental arrest. With Uba52 being a fusion gene, the UBA52 protein is formed by co-translation of full length 76-amino acid (AA) mono-ubiquitin linked through its C-terminus to the N-terminus of 52 AA ribosomal/ribonucleo-protein (RNP) CEP52 (Redman and Burris, 1996). The UBA52 as one of the ubiquitin-tail fusion RNPs is involved in the ribosomal biogenesis (Finley et al., 1989) and their relative abundance in cells correlates with the cellular content of assembled ribosomes (Redman and Burris, 1996). Therefore, it was very possible that ribosomal biogenesis was likely affected as well.
In previous studies, CRISPR/Cas9 gRNA was injected into zygote/1-cell stage porcine embryos to modify genes that are not essential for pre- or post-implantation development (Whitworth et al., 2017). However, Uba52 is classified as a housekeeping gene (Ahn et al., 2008; Sadritdinova et al., 2014; Schoen et al., 2015) and is likely activated before major embryonic genome activation (Latham and Schultz, 2001; Østrup et al., 2013). Thus, the injection time window had to be moved to metaphase II stage oocyte, i.e. prior to fertilization and oocyte activation, to assure timely modification of the Uba52 prior to first embryo mitosis. High efficiency of such a gene modification indicates that the current method was efficient to interfere with development.
In conclusion, the present study demonstrated that the CRISPR/Cas9 of Uba52 gene significantly reduced (and completely prevented in bi-allelic conformation) porcine blastocyst formation in vitro, decreasing UBA52 and proteasomal subunit protein content, and causing developmental arrest at 4-cell to 8-cell stage and blastomere nuclear deformation. We thus conclude that Uba52 plays an essential role in the pre-implantation embryo development.
MATERIALS AND METHODS
Reagents and antibodies
All chemicals used in this study were purchased from Sigma Chemical Co. (St. Louis, USA) unless otherwise stated. Rabbit polyclonal antibody against the 20S proteasomal core subunits was purchased from Enzo Life Sciences Inc. (catalog #PW8155, Farmingdale, USA) and validated in previous studies by western blotting and immunocytochemistry (Zigo et al., 2018). Two anti-UBA52 antibodies were used in the present study. The UBA52 monoclonal antibody was from Abcam (catalog #ab109227, Cambridge, USA) and UBA52 polyclonal antibody from Thermo Fisher Scientific (catalog #PA5 23685, Rockford, USA). Affinity purified goat anti-rabbit IgG FITC secondary antibody was obtained from Invitrogen.
Design of gRNAs to build specific CRISPRs
Five 17–20 bp guides were designed to target the sequence located adjacent to an S. pyogenes (Spy) protospacer adjacent motif (PAM) (Ran et al., 2015) within exon 1 of Uba52 gene. The targets were selected by the following method. Repeat Masker (Smit and Green, 1996) (‘Pig’ repeat library) was used to identify any repetitive elements in the Uba52 genomic sequence and these areas were not used as potential targets. Specificity of each potential guide was confirmed by searching for similar porcine sequences in GenBank (https://www.ncbi.nlm.nih.gov/genbank/). If guides and the adjacent PAM sequence had similarity to other areas of the genome, they were removed from subsequent analysis. In addition, structural analysis of the 20 bp guide with the CRISPR RNA (crRNA) and the trans-activating crRNA (tracrRNA) (Hsu et al., 2013) was evaluated for potential disruption of gRNA structure by mFold (http://unafold.rna.albany.edu). If potential guides were predicted to form an appropriate ‘handle’ to interact with Cas9 and were not predicted to form a tight hairpin that could potentially prevent interaction with the genome, they were added to the finalized list of potential guides. Five guides were chosen for the experiment based on the criteria listed above. The five guides and the PAM sequences (Fig. 3A) were: Guide 1, ATCTTTGTGAAGACCCTGACGG; Guide 2, GATAAGGAGGGTGAGTTGGG; Guide 3, CCAACTCACCCTCCTTATCCTGG; Guide 4, ACATTCTCAATGGTATCACTGGG and Guide 5, TATCACTGGGCTGACCTCAGGG.
In vitro transcription of single guide RNAs for the CRISPR/Cas9 system
Template guide DNA was first synthesized by Integrated DNA Technologies in the form of a gBlock. A T7 promoter sequence was added upstream of the guide for in vitro transcription. Each gBlock was diluted to final concentration 0.1 ng/μl and PCR amplified with a gBlock F primer (ACTGGCACCTATGCGGGACGAC) and a gBlock R primer (AAAAGCACCGACTCGGTGCCAC) with Q5 (New England Biolabs, Ipswich, MA) following standard protocol. PCR conditions consisted of an initial denaturation of 98°C for 1 min followed by 35 cycles of 98°C (10 s), 68°C (30 s) and 72°C (30 s). Each PCR amplified gBlock was purified by using a QIAGEN (Valencia, USA) PCR purification kit following standard protocol. Purified gBlock amplicons were used as template for in vitro transcription by standard protocol with the MEGAshortscript T7 transcription kit (Ambion; Thermo Fisher Scientific) followed by purification using the MEGAclear T7 clean-up kit (Ambion). Quality of the synthesized RNAs were visualized on a 2.0% RNA-free agarose gel and concentrations 260:280 ratios were determined via spectrophotometry. Polyadenylated Cas9 mRNA containing 5-methylcytidine and pseudouridine modifications was used (TriLink Biotechnologies). Five gRNAs were mixed together then with Cas9 mRNA and diluted in nuclease-free water at a final concentration of 20 and 20 ng/μl, respectively. Prepared RNA was divided into 5 μl aliquots and stored at −80°C until oocyte injection.
Porcine oocyte collection and in vitro maturation
Detailed procedures for oocyte collection and in vitro maturation have been described previously (Abeydeera et al., 1998). Briefly, ovaries from pre-pubertal gilts were collected at a local slaughterhouse and transported to the laboratory in a warm box (25–30°C). Cumulus-oocyte complexes (COCs) were aspirated from antral follicles (3–6 mm in diameter). Oocytes with uniform ooplasm and compact cumulus were collected, and washed three times in HEPES-buffered Tyrode lactate (TL-HEPES) medium containing 0.1% (w/v) polyvinyl alcohol (PVA) and one time with the maturation medium (Abeydeera et al., 1998). Batches of fifty COCs were transferred to 500 μl of the maturation medium that had been covered with mineral oil in a 4-well plate (Nunc) and equilibrated at 38.5°C, 5% CO2 in air. Oocyte maturation medium was tissue culture medium (TCM) 199 (Mediatech, Manassas, USA) supplemented with 0.1% PVA, 3.05 mM D-glucose, 0.91 mM sodium pyruvate, 0.57 mM cysteine, 0.5 μg/ml LH, 0.5 μg/ml FSH, 10 ng/ml EGF, 10% porcine follicular fluid, 75 μg/ml penicillin G and 50 μg/ml streptomycin. After 40 h in vitro maturation, cumulus cells were removed with 0.1% hyaluronidase in TL-HEPES-PVA medium and the oocytes were washed three times and transferred into TL-HEPES-PVA medium (pH 7.4) for microinjections.
Cytoplasmic injection of metaphase II oocytes with CRISPR/Cas9+gRNAs
Microinjection was performed on the heated stage of a Zeiss Axiovert-35 inverted microscope (Zeiss, Jena, Germany) fitted with Eppendorf micromanipulators and Femtojet 5247 injector (Eppendorf, Hauppauge, USA). Glass micropipettes with an outer diameter of 1.0 mm and an inner diameter of 0.78 mm were pulled to a fine point of <1.0 μm (Sutter Instrument, Navato, USA). The mixture of CRISPR RNA (crRNA, 100 ng/µl) and 20 ng/µl of gRNAs was microinjected into cytoplasm of oocyte (designated as Uba52gRNA group). crRNA (100 ng/μl) without gRNAs was injected as a control. Surviving oocytes were washed three times in fertilization medium and used for IVF.
In vitro fertilization, embryo culture and assessment of development
Injected oocytes were placed into 100 μl drops of a modified Tris-buffered medium (mTBM) containing caffeine and BSA, covered with mineral oil, which had been equilibrated for 48 h at 38.5°C in 5% CO2 in air as described (Mao et al., 2012). The dishes were kept in a CO2 incubator until spermatozoa were added for fertilization. Sperm-rich ejaculate fraction from a boar of known fertility was collected weekly on Tuesdays and used for IVF on Wednesdays and Thursdays. The semen was checked for motility (minimum 80%) right after collection, and kept at room temperature (24°C). For IVF, 4 ml of semen was centrifuged at 600 g for 10 min to remove the seminal plasma. The supernatant was discarded, and the sperm pellet was re-suspended at room temperature in the BTS extender (Minitube, Delavan, USA) after sperm concentration was adjusted to 1×108 cells/ml. On fertilization day, semen was diluted in mTBM. The processed semen was added in to oocyte-containing fertilization droplets at a final sperm concentration of 1×104 cells/ml. Oocytes were co-incubated with spermatozoa for 5–6 h at 38.5°C, 5% CO2 in air, at which time point the putative zygotes were washed three times and transferred to four-well plates containing 500 ml of zygote culture medium (Mao et al., 2012) for additional incubation at 38.5°C, in 5% CO2 in air. The number of embryos cleaved on day 2, the number of 2-, 4- and 8-cell stage embryos on day 4, and the number of blastocysts formed on day 7 after fertilization (IVF day=0), were recorded under a stereomicroscope. Only embryos with blastomeres of equal size were counted to determine the numbers of 2-, 4- and 8-cell stage embryos.
PCR screening for insertions and deletions
PCR assay was designed to assess the presence of Uba52 gene edits including insertions and deletions (INDELs) in the resulting embryos with an amplicon size of 305 bp. Sense and antisense primer sequences were: Uba52F, AGGCATAGGGCTGGCAGTCT and Uba52R, TCCGTCCACACAGGACAGCA.
INDELs were determined by PCR amplification of the UBA52 gene in the Exon 1 region flanking the projected cutting site introduced by the CRISPR/Cas9 system. PCR conditions of the INDELs assay consisted of an initial denaturation of 94°C for 1 min followed by 37 cycles of 94°C (30 s), 52°C (30 s) and 68°C (15 s) finishing with a final extension at 72°C for 2 min 30 s. Resulting amplicons were then visualized by electrophoresis using a 4% agarose gel.
TOPO cloning and DNA sequencing
The resulting PCR products were Sanger DNA sequenced at the University of Missouri DNA Core facility. PCR amplicons from each embryo were TOPO cloned using the TOPO TA kit (Thermo Fisher Scientific) by following standard protocol. Clones were propagated on Luria–Bertani (LB) agarose plates containing 50 μg/ml kanamycin and resistant recombinants were selected. Plasmids containing the Uba52 amplicon were identified by EcoRI digestion, and subsequent DNA agarose gel electrophoresis. Plasmids that contained the Uba52 amplicon were DNA sequenced at the University of Missouri DNA core by using the Uba52F oligonucleotide. Sequences were aligned to the wild-type Uba52 gene and INDELS were examined.
Immunofluorescence
A standard immunofluorescence procedure for labeling embryos against Uba52 (Sutovsky et al., 2005) was performed as follows. Immediately after embryo collection, zona pellucida was removed by a short, 5 s incubation in acidic PBS (pH 1.79). Embryos were fixed in 2% (v/v) formaldehyde for 40 min at room temperature, washed, permeabilized in phosphate-buffered saline (PBS) containing 0.1% (v/v) Triton X-100, and blocked for 25 min in 0.1 M PBS containing 5% normal goat serum (NGS) and 0.1% Triton X-100. Samples were incubated overnight at 4°C with primary antibody diluted at 1:200 in 0.1 M PBS containing 1% NGS and 0.1% Triton X-100. On the following day, after a wash in PBS, the primary antibodies were detected by a mixture of goat anti-rabbit IgG-FITC diluted 1:100 and DNA stain DAPI (4,6-diamidino-2-phenylindole, 2.5 μg/ml), incubated at room temperature for 40 min. Negative controls were obtained by the replacement of primary antibody with normal rabbit serum at the immunoglobulin concentration matching that of the relevant specific antibody. Embryos were mounted on slides using Vectashield anti-fade mounting medium (Vector Laboratories Inc., Burlingame, USA), and observed with a 40× and 60× infinity-corrected objectives using a Nikon Eclipse 800 microscope (Nikon Instruments, Melville, USA). Images of individual embryos were acquired using a high-resolution Cool Snap CCD camera (Roper Scientific, Tucson, USA) and MetaMorph software (v7.1, Universal Imaging, Downington, USA) with a fixed setting for all images. Images were cropped, sized and arranged into panels using Photoshop CC version 2017 (Adobe Systems). Quantification of intensity of labeling in the equatorial plane of entire embryos was performed using Image Studio Lite software (v5.2, LI-COR Biotechnology, Lincoln, USA). The circumference of the embryo was selected and the mean intensity obtained using the analysis tool of Image Studio. Background intensity was obtained from the area surrounding the embryo using the same technique and the value subtracted from embryo intensity.
Number of nuclei in embryos
The number of nuclei in the fixed embryos was determined after counter staining with DAPI as described above. Live embryos used for DNA preparation and PCR assays were stained with 20 mM Hoechst 33342 dye (2′-[4-ethoxyphenyl]-5-[4-methyl-1-piperazinyl]-2,5′-bi-1H-benzimidazole trihydrochloride trihydrate) and mounted on slides in culture medium. The number of nuclei was used as an estimate of the number of cells in an embryo.
SDS/PAGE and western blotting-densitometry
Western blotting method described previously (Miles et al., 2013; Zigo et al., 2018) was used to determine the UBA52 protein levels in embryos with a little modification. After the zona pellucida was removed, embryos were washed three times in PBS and boiled with loading buffer [50 mM TRIS (pH 6.8), 150 mM NaCl, 2% SDS, 20% glycerol, 5% β-mercaptoethanol, 0.02% bromophenol blue]. Gel electrophoresis was performed on 4-20% gradient gels (PAGEr Gels; Cambrex Bio Science, Rockland, USA), followed by transfer to PVDF membranes (Millipore, Bedford, USA) using an Owl wet transfer system (Fisher Scientific) at a constant 50 V for 4 h. The membranes were incubated sequentially with 10% non-fat milk for 1 h at room temperature, primary antibody (#ab109227, Abcam) at 1:1000 dilution overnight at 4°C, and HRP-conjugated goat anti-rabbit antibody (1:10,000 dilution) for 40 min at room temperature. The membranes were reacted with chemiluminescent substrate (Luminata Crescendo Western HRP Substrate; Millipore). Blots were screened with ChemiDoc Touch Imaging System (Bio-Rad) to visualize the protein bands and analyzed by Image Lab Touch Software (Bio-Rad). Unless otherwise specified, procedures were carried out at room temperature. Residual gels and membranes after chemiluminescence detection were stained with Coomassie Brilliant Blue (CBB) R-250 (both Thermo Scientific) for protein normalization (Zigo et al., 2018). The UBA52 band intensity was normalized based on both the band density of Coomassie staining of residual gel and the number of embryos used preferentially to normalization on actin/tubulin, the quantities of which are affected by cell number and developmental competence. Negative control was obtained by the replacement of primary antibody with a non-immune rabbit serum.
Statistical analysis
Dependent variables were analyzed for normality by using the Wilk–Shapiro test (SAS, 2014). Data for the dependent variables, percentage of zygotes cleaved on day 2 (cleavage rate), and percentage of zygotes forming an apparent blastocysts (blastocyst formation rate) were arcsine-transformed. Data on per cent of zygotes cleaved on day 2, per cent of blastocysts formed on day 7, number of nuclei in day 7 blastocyst and relative intensity of immunolabeling of UBA52 and proteasome were analyzed by analysis of variance using the PROC GLM procedure of SAS (SAS, 2014). The proportion of 2-, 4- and 8-cell stage embryos on day 4 of embryo culture, as a function determination of embryonic developmental potential between the Uba52 gRNA and control, was analyzed by FREQ procedure of SAS with X2 as an option (SAS, 2014). For all variables, treatments were fixed effects and replicate was considered a random effect. A value of P<0.05 was considered statistically significant. In the results, the least-squares means and the standard errors of means are presented.
Acknowledgements
The authors would like to thank Dr Randall Prather and his laboratory members and the staff of the National Swine Resource and Research Center for help with ovary transportation and boar semen collection.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: K.D.W., P.S.; Methodology: J.M., C.O., M.S., M.Z., K.D.W., P.S.; Validation: K.D.W., P.S.; Formal analysis: J.M., M.S., M.Z., K.D.W., P.S.; Investigation: C.O., M.S., M.Z., P.S.; Resources: C.O., P.S.; Data curation: J.M., K.D.W., P.S.; Writing - original draft: J.M., K.D.W., P.S.; Writing - review & editing: C.O., M.S., M.Z., K.D.W., P.S.; Visualization: J.M., M.Z.; Supervision: K.D.W., P.S.; Project administration: P.S.; Funding acquisition: P.S.
Funding
Funding was provided by Agriculture and Food Research Initiative Competitive Grant no. 2015-67015-23231 from the USDA National Institute of Food and Agriculture to P.S. and seed funding from the Food for the 21st Century Program of the University of Missouri to P.S.
References
- Abeydeera L. R., Wang W.-H., Prather R. S. and Day B. N. (1998). Maturation in vitro of pig oocytes in protein-free culture media: fertilization and subsequent embryo development in vitro. Biol. Reprod. 58, 1316-1320. 10.1095/biolreprod58.5.1316 [DOI] [PubMed] [Google Scholar]
- Ahn K., Huh J.-W., Park S.-J., Kim D.-S., Ha H.-S., Kim Y.-J., Lee J.-R., Chang K.-T. and Kim H.-S. (2008). Selection of internal reference genes for SYBR green qRT-PCR studies of rhesus monkey (Macaca mulatta) tissues. BMC Mol. Biol. 9, 78 10.1186/1471-2199-9-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C. and Groth A. (2012). Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 13, 153-167. 10.1038/nrm3288 [DOI] [PubMed] [Google Scholar]
- Amin M. A., Matsunaga S., Uchiyama S. and Fukui K. (2008). Depletion of nucleophosmin leads to distortion of nucleolar and nuclear structures in HeLa cells. Biochem. J. 415, 345-351. 10.1042/BJ20081411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker R. T. and Board P. G. (1991). The human ubiquitin-52 amino acid fusion protein gene shares several structural features with mammalian ribosomal protein genes. Nucleic Acids Res. 19, 1035-1040. 10.1093/nar/19.5.1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M., Giacomini E., Crinelli R., Radici L., Carloni E. and Magnani M. (2015). Dynamic transcription of ubiquitin genes under basal and stressful conditions and new insights into the multiple UBC transcript variants. Gene 573, 100-109. 10.1016/j.gene.2015.07.030 [DOI] [PubMed] [Google Scholar]
- Brodersen M. M. L., Lampert F., Barnes C. A., Soste M., Piwko W. and Peter M. (2016). CRL4(WDR23)-mediated SLBP ubiquitylation ensures histone supply during DNA replication. Mol. Cell 62, 627-635. 10.1016/j.molcel.2016.04.017 [DOI] [PubMed] [Google Scholar]
- Cho S. W., Kim S., Kim J. M. and Kim J.-S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230-232. 10.1038/nbt.2507 [DOI] [PubMed] [Google Scholar]
- Ciechanover A. and Schwartz A. L. (1994). The ubiquitin-mediated proteolytic pathway: mechanisms of recognition of the proteolytic substrate and involvement in the degradation of native cellular proteins. FASEB J. 8, 182-191. 10.1096/fasebj.8.2.8119489 [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A. et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819-823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankert J. F., Rona G., Clijsters L., Geter P., Skaar J. R., Bermudez-Hernandez K., Sassani E., Fenyö D., Ueberheide B., Schneider R. et al. (2016). Cyclin F-mediated degradation of SLBP limits H2A.X accumulation and apoptosis upon genotoxic stress in G2. Mol. Cell 64, 507-519. 10.1016/j.molcel.2016.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D., Özkaynak E. and Varshavsky A. (1987). The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation, and other stresses. Cell 48, 1035-1046. 10.1016/0092-8674(87)90711-2 [DOI] [PubMed] [Google Scholar]
- Finley D., Bartel B. and Varshavsky A. (1989). The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature 338, 394-401. 10.1038/338394a0 [DOI] [PubMed] [Google Scholar]
- Fornace A. J. Jr, Alamo I. Jr, Hollander M. C. and Lamoreaux E. (1989). Ubiquitin mRNA is a major stress-induced transcript in mammalian cells. Nucleic Acids Res. 17, 1215-1230. 10.1093/nar/17.3.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Rodríguez N., Wong R. P. and Ulrich H. D. (2016). Functions of ubiquitin and SUMO in DNA replication and replication stress. Front. Genet. 7, 87 10.3389/fgene.2016.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilberto S. and Peter M. (2017). Dynamic ubiquitin signaling in cell cycle regulation. J. Cell Biol. 216, 2259-2271. 10.1083/jcb.201703170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T., Teng F., Guo R., Li W. and Zhou Q. (2014). One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res. 24, 372-375. 10.1038/cr.2014.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K., Nakayama K. I. and Nakayama K. (2006). Geminin is essential for the development of preimplantation mouse embryos. Genes Cells 11, 1281-1293. 10.1111/j.1365-2443.2006.01019.x [DOI] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425-479. 10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G. and Zhang F. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31, 827-832. 10.1038/nbt.2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M., Oshima S., Maeyashiki C., Nibe Y., Otsubo K., Matsuzawa Y., Nemoto Y., Nagaishi T., Okamoto R., Tsuchiya K. et al. (2016). The ubiquitin hybrid gene UBA52 regulates ubiquitination of ribosome and sustains embryonic development. Sci. Rep. 6, 36780 10.1038/srep36780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D. and Rape M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203-229. 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- Latham K. E. and Schultz R. M. (2001). Embryonic genome activation. Front. Biosci. 6, D748-D759. 10.2741/A639 [DOI] [PubMed] [Google Scholar]
- Lee J.-Y., Tokumoto M., Fujiwara Y. and Satoh M. (2015). Involvement of ubiquitin-coding genes in cadmium-induced protein ubiquitination in human proximal tubular cells. J. Toxicol. Sci. 40, 901-908. 10.2131/jts.40.901 [DOI] [PubMed] [Google Scholar]
- Mao J., Whitworth K. M., Spate L. D., Walters E. M., Zhao J. and Prather R. S. (2012). Regulation of oocyte mitochondrial DNA copy number by follicular fluid, EGF, and neuregulin 1 during in vitro maturation affects embryo development in pigs. Theriogenology 78, 887-897. 10.1016/j.theriogenology.2012.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateusen B., Van Soom A., Maes D. G. D., Donnay I., Duchateau L. and Lequarre A.-S. (2005). Porcine embryo development and fragmentation and their relation to apoptotic markers: a cinematographic and confocal laser scanning microscopic study. Reproduction 129, 443-452. 10.1530/rep.1.00533 [DOI] [PubMed] [Google Scholar]
- Miles E. L., O'Gorman C., Zhao J., Samuel M., Walters E., Yi Y.-J., Sutovsky M., Prather R. S., Wells K. D. and Sutovsky P. (2013). Transgenic pig carrying green fluorescent proteasomes. Proc. Natl. Acad. Sci. USA 110, 6334-6339. 10.1073/pnas.1220910110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S. P. and Gambus A. (2015). Regulation of unperturbed DNA replication by ubiquitylation. Genes (Basel) 6, 451-468. 10.3390/genes6030451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtango N. R. and Latham K. E. (2007). Ubiquitin proteasome pathway gene expression varies in rhesus monkey oocytes and embryos of different developmental potential. Physiol. Genomics 31, 1-14. 10.1152/physiolgenomics.00040.2007 [DOI] [PubMed] [Google Scholar]
- Nickerson J. (2001). Experimental observations of a nuclear matrix. J. Cell Sci. 114, 463-474. [DOI] [PubMed] [Google Scholar]
- Niu Y., Shen B., Cui Y., Chen Y., Wang J., Wang L., Kang Y., Zhao X., Si W., Li W. et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836-843. 10.1016/j.cell.2014.01.027 [DOI] [PubMed] [Google Scholar]
- Østrup O., Olbricht G., Østrup E., Hyttel P., Collas P. and Cabot R. (2013). RNA profiles of porcine embryos during genome activation reveal complex metabolic switch sensitive to in vitro conditions. PLoS ONE 8, e61547 10.1371/journal.pone.0061547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A., Cong L., Yan W. X., Scott D. A., Gootenberg J. S., Kriz A. J., Zetsche B., Shalem O., Wu X., Makarova K. S., Koonin E. V., Sharp P. A. and Zhang F. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186-191. 10.1038/nature14299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman K. L. and Burris G. W. (1996). The cDNA for the ubiquitin-52-amino-acid fusion protein from rat encodes a previously unidentified 60 S ribosomal subunit protein. Biochem. J. 315, 315-321. 10.1042/bj3150315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadritdinova A. F., Dmitriev A. A., Snezhkina A. V., Belenikin M. S., Krasnov G. S., Manylov O. G., Kudryavtsev A. A., Melnikova N. V., Speranskaya A. S., Darii M. V. et al. (2014). A new reliable reference gene UBA52 for quantitative real-time polymerase chain reaction studies in pyloric cecal tissues of the starfish Asterias rubens. Genet. Mol. Res. 13, 3972-3980. 10.4238/2014.May.23.8 [DOI] [PubMed] [Google Scholar]
- SAS. (2014). SAS/STAT User's Guide. Cary, NC: Statistical Analysis Systems Institute, Inc. [Google Scholar]
- Schoen K., Plendl J., Gabler C. and Kaessmeyer S. (2015). Identification of stably expressed reference genes for RT-qPCR data normalization in defined localizations of cyclic bovine ovaries. Anat. Histol. Embryol. 44, 200-211. 10.1111/ahe.12128 [DOI] [PubMed] [Google Scholar]
- Spate A. M., Whitworth K. M., O'Gorman C. W., Byrne A. K., Prather R. S. and Wells K. D. (2016). The use of paired CRISPR guide RNA and the cas9 system does not always produce site specific deletions of gene sequence in porcine cell and embryo culture. Reprod Fertil. Dev. 29, 120 10.1071/RDv29n1Ab26 [DOI] [Google Scholar]
- Song W.-H., Yi Y.-J., Sutovsky M., Meyers S. and Sutovsky P. (2016). Autophagy and ubiquitin-proteasome system contribute to sperm mitophagy after mammalian fertilization. Proc. Natl. Acad. Sci. USA 113, E5261-E5270. 10.1073/pnas.1605844113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A. F. and Green P. (1996) RepeatMasker. http://www.repeatmasker.org 10.1002/jemt.10319 [DOI] [Google Scholar]
- Sutovsky P. (2003). Ubiquitin-dependent proteolysis in mammalian spermatogenesis, fertilization, and sperm quality control: killing three birds with one stone. Microsc. Res. Tech. 61, 88-102. 10.1002/jemt.10319 [DOI] [PubMed] [Google Scholar]
- Sutovsky P., Moreno R., Ramalho-Santos J., Dominko T., Thompson W. E. and Schatten G. (2001). A putative, ubiquitin-dependent mechanism for the recognition and elimination of defective spermatozoa in the mammalian epididymis. J. Cell Sci. 114, 1665-1675. [DOI] [PubMed] [Google Scholar]
- Sutovsky P., Manandhar G., McCauley T. C., Caamaño J. N., Sutovsky M., Thompson W. E. and Day B. N. (2004). Proteasomal interference prevents zona pellucida penetration and fertilization in mammals. Biol. Reprod. 71, 1625-1637. 10.1095/biolreprod.104.032532 [DOI] [PubMed] [Google Scholar]
- Sutovsky P., Manandhar G., Laurincik J., Letko J., Caamano J. N., Day B. N., Lai L., Prather R. S., Sharpe-Timms K. L., Zimmer R. et al. (2005). Expression and proteasomal degradation of the major vault protein (MVP) in mammalian oocytes and zygotes. Reproduction 129, 269-282. 10.1530/rep.1.00291 [DOI] [PubMed] [Google Scholar]
- Vihervaara A., Sergelius C., Vasara J., Blom M. A. H., Elsing A. N., Roos-Mattjus P. and Sistonen L. (2013). Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc. Natl. Acad. Sci. USA 110, E3388-E3397. 10.1073/pnas.1305275110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F. and Jaenisch R. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910-918. 10.1016/j.cell.2013.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington J. A., Nair A., Mahadevappa M. and Tsyganskaya M. (2000). Comparison of human adult and fetal expression and identification of 535 housekeeping/maintenance genes. Physiol. Genomics 2, 143-147. 10.1152/physiolgenomics.2000.2.3.143 [DOI] [PubMed] [Google Scholar]
- Whitworth K. M., Agca C., Kim J.-G., Patel R. V., Springer G. K., Bivens N. J., Forrester L. J., Mathialagan N., Green J. A. and Prather R. S. (2005). Transcriptional profiling of pig embryogenesis by using a 15-K member unigene set specific for pig reproductive tissues and embryos. Biol. Reprod. 72, 1437-1451. 10.1095/biolreprod.104.037952 [DOI] [PubMed] [Google Scholar]
- Whitworth K. M., Benne J. A., Spate L. D., Murphy S. L., Samuel M. S., Murphy C. N., Richt J. A., Walters E., Prather R. S. and Wells K. D. (2017). Zygote injection of CRISPR/Cas9 RNA successfully modifies the target gene without delaying blastocyst development or altering the sex ratio in pigs. Transgenic Res. 26, 97-107. 10.1007/s11248-016-9989-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiborg O., Pedersen M. S., Wind A., Berglund L. E., Marcker K. A. and Vuust J. (1985). The human ubiquitin multigene family: some genes contain multiple directly repeated ubiquitin coding sequences. EMBO J. 4, 755-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng F., Baldwin D. A. and Schultz R. M. (2004). Transcript profiling during preimplantation mouse development. Dev. Biol. 272, 483-496. 10.1016/j.ydbio.2004.05.018 [DOI] [PubMed] [Google Scholar]
- Zigo M., Kerns K., Sutovsky M. and Sutovsky P. (2018). Modifications of the 26S proteasome during boar sperm capacitation. Cell Tissue Res. 372, 591-601. 10.1007/s00441-017-2786-6 [DOI] [PMC free article] [PubMed] [Google Scholar]