

Abstract
Paclitaxel is a powerful drug against restenosis and many forms of cancer. However, its clinical application hinges on the ability to achieve suitable stabilized drug concentrations in an aqueous suspension while hindering drug crystallization. To engineer such formulations, it is imperative to understand paclitaxel’s partitioning and crystallization within the carrier matrix. Lipid-polymer hybrid films have been recently shown to accommodate large paclitaxel loads and suppress crystallization. Additionally, such hybrid materials promote synergistic drug release compared to the pure constituents. Here, we leverage the composition sensitive photo-thermal induced resonance (PTIR) technique to study paclitaxel partitioning within hybrid films at the nanoscale. PTIR data reveal that paclitaxel nano-crystals segregate from lipid-only films but are well dispersed in polymer-only films. Remarkably, lipid-polymer hybrid films show enhanced partitioning of paclitaxel at the lipid-polymer phase boundaries, but still stifle crystallization, thus paving the way towards compositional and microstructural engineering of small-drug delivery systems.
Introduction
Paclitaxel is an effective chemotherapeutic agent against a broad range of cancers1–3and is used in drug eluting stents against restenosis.4 However, paclitaxel’s low solubility and tendency to crystallize in aqueous enviornments5,6 has hampered its efficacious clinical use. Traditional methods to formulate paclitaxel based on ethanol and polyoxyethylated triglycerides can cause severe inflammatory responses,6,7 motivating the development of alternative formulations. A promising strategy is to encapsulate paclitaxel in drug carriers such as polymer or lipid membranes.8,9 Drug carriers provide satisfactory solutions to the poor aqueous solubility, however paclitaxel still tends to crystallize and often segregates out of these membranes.9,10 Exploiting paclitaxel’s full potential remains an enduring challenge which requires engineering carriers that concurrently provide control over drug loading and suppress crystallization. The hybridization of lipids and polymers within a single membrane by self-assembly has enabled the emergence of new materials. Hybrid membranes combine benefits of lipids and polymers while having unique structural and dynamical properties making them very promising for sensing and small-molecule delivery applications.11–14 For example, lipid-polymer hybrid membranes composed of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC, Fig. 1a) and poly(butadiene-b-ethylene oxide) block polymer (PBD-b-PEO, Fig. 1a) have shown potential as superior paclitaxel delivery media.15 Notably, at parity of initial drug loading, the hybrid membranes microstructure effectively suppresses paclitaxel crystallization and synergistically augments the release of paclitaxel (Fig. 1a) with respect to single component films (see schematic in Fig. 1b).15 X-ray scattering, and atomic force microscopy (AFM) showed respectively that the DPPC/PBD-b-PEO films self-assemble into domains with chemically affine layers stacked perpendicular to the substrate (Fig. 1c),15 with heterogeneities spanning from the micro- to the nano-scale and an abundance of interfacial regions (Fig. 1d). Since the polymer-rich, lipid-rich and the interfacial regions, have typically unknown compositions, and because their microstructure and properties can be altered by paclitaxel incorporation, film engineering requires methods capable of assessing their structure and chemical composition with high spatial resolution and specificity.
Fig. 1.
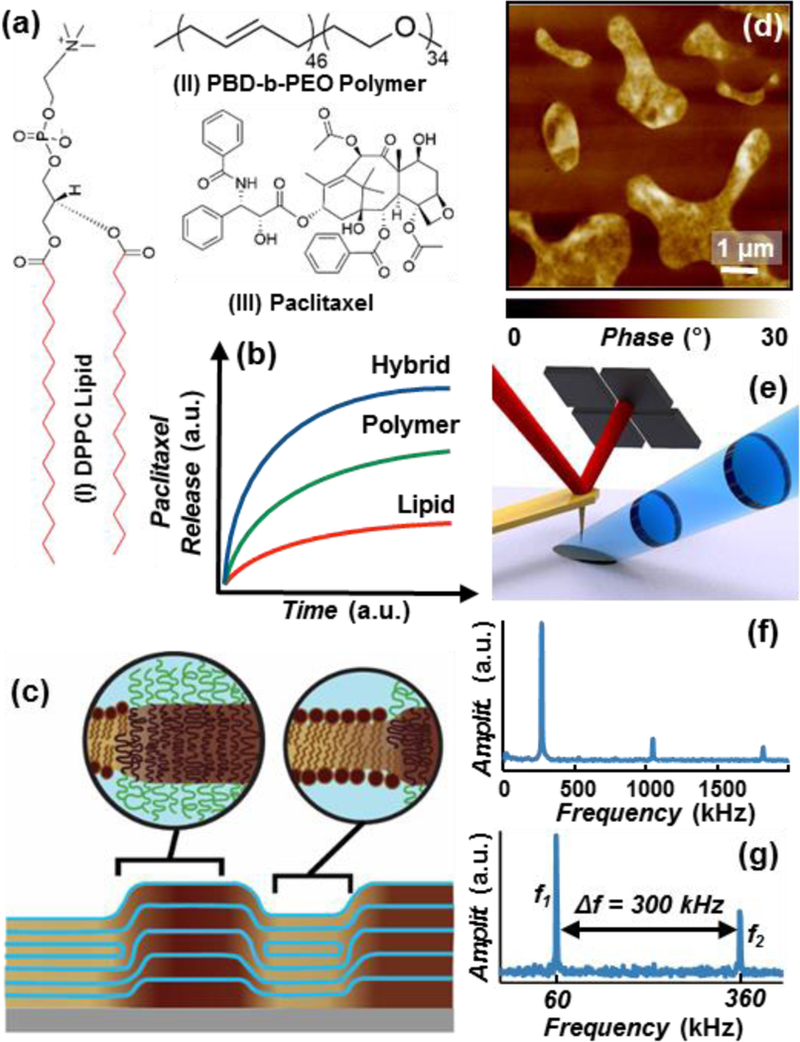
(a) Chemical structures of: (I) 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) lipid; the red bonds identify the deuterium substituted positions on the partially deuterated DPPC (d-DPPC), (II) poly(butadiene-b-ethylene oxide) block polymer (PBD-b-PEO) and (III) Paclitaxel. (b) Schematic of the paclitaxel cumulative release profiles from DPPC/PBD-b-PEO hybrid (blue), PBD-b-PEO (green), and DPPC (red) at parity initial paclitaxel load.15 (c) Schematic of the multi-layered DPPC/PBD-b-PEO hybrid films on solid substrate, depicting the self-assembled chemically-affine domains and domain boundaries perpendicular to the substrate. (d) Representative AFM phase image depicting drug-loaded DPPC/PBD-b-PEO hybrid films heterogeneity (phase separation). (e) PTIR Schematic: a pulsed tunable IR laser (blue) illuminates the portion of the sample centered around the gold-coated AFM probe. Light absorption in the sample induces local thermal expansion. The AFM cantilever transduces the expansion of the sample and provides nanoscale resolution. (f) In contact-mode PTIR experiments (i.e. spectra) the laser repetition rate matches the frequency of oscillation to the first cantilever mode (≈ 270 kHz). (g) In tapping-mode PTIR experiments (i.e. maps) the laser repetition rate matches the frequency difference (Δf) between the second (f2) and first (f1) modes of the cantilever.
The photothermal induced resonance (PTIR) technique,16,17 also known as AFM-IR, combines the spatial resolution of AFM with the chemical specificity of infrared (IR) spectroscopy. Amongst nanoscale chemical imaging methods,16,18 PTIR stands out because (a) its signal’s proportionality to the sample’s local absorption coefficient19,20 enables material identification,17 and (b) it probes the composition of up to a few µm of the sample thickness,20,21 rather than merely of the top layer.16,18 Initial PTIR experiments (in contact-mode) suggested that the increased paclitaxel release from DPPC/PBD-b-PEO hybrid membranes may be related to the higher paclitaxel concentration along the phase boundaries of lipid-rich and polymer-rich domains.21 However, the strong adhesion between the AFM tip and the single component films previously precluded a direct comparison between hybrid and single component films. Furthermore, the strong IR spectral overlap between DPPC and PBD-b-PEO has hindered the assessment of their intermixing.
Here, we leverage the novel capability to acquire PTIR images in tapping-mode, to study DPPC/PBD-b-PEO hybrid and single component films loaded with paclitaxel. The data reveal paclitaxel partitioning details in both hybrid and single component films with unprecedented resolution. Importantly, selective lipid deuteration enables assessing the width of the phase boundaries as well as the quantification of the lipid content (< 5 % molar fraction) within the polymer-rich domains in hybrid films.
Results and discussion
In PTIR a pulsed wavelength-tunable laser illuminates a small spot on the sample (≈ 50 µm diameter) centered around an AFM tip that serves as a near-field mechanical detector (Fig. 1e). The alignment between the AFM-tip and laser spot is maintained through the experiments because only the sample stage moves. Initial implementation of the technique used total internal reflection illumination through the substrate;22,23 however, in this work the sample is illuminated from the air side (Fig. 1e).24 The absorption of a light pulse in the sample, induces a rapid thermal expansion that kicks the cantilever in oscillation like a struck tuning fork with an amplitude (measured by the AFM detector) proportional to the energy absorbed in the sample.19,20 The PTIR mechanical detection scheme, works in principle at all wavelengths and its operating range has been recently extended from the mid-IR to the visible.25,26 PTIR’s working principles16,17 and its applications in photovoltaics,27,28 polymer science,29–32 pharmaceutics,33,34 plasmonics,35–37 2D materials,38,39 medicine,40,41 biology,40,42 geology43 have been reviewed recently. For example, PTIR has been applied to study the nanoscale distribution of drug-polymer blends33,34 and to study the trans-membrane protein conformation in purple bacterial membranes.44 Recent PTIR innovations, such as resonance-enhanced operation,24 ultra-sensitive nanosized probes,45 and most recently, the development of tapping-mode PTIR, have improved the technique sensitivity and throughput. PTIR operation in water has also been recently demonstrated.46
In this work, all PTIR measurements exploit a resonance enhanced excitation scheme to increase the sensitivity of the cantilever transduction.24 PTIR spectra were obtained by keeping the cantilever fixed in contact with the sample (contact-mode) while sweeping the laser wavelength and by tuning the laser repetition rate to match the frequency of one of the cantilever bending modes (≈ 270 kHz, Fig. 1f). PTIR images were obtained by scanning the AFM probe in tapping-mode (i.e. oscillating over the sample) while illuminating the sample with a fixed wavelength and leveraging a heterodyne detection scheme that enables non-linear mixing of the cantilever oscillation modes. This was achieved by setting the laser repetition rate (Δf ≈ 300 kHz) to match the difference between the second (f2 ≈ 360 kHz, demodulation frequency) and first (f1 ≈ 60 kHz, tapping frequency) bending modes of the AFM cantilever (Fig. 1g).
For reference, the Fourier-transform infrared (FTIR) spectra of the pure paclitaxel drug, PBD-b-PEO block copolymer, DPPC and partially deuterated DPPC (d-DPPC) lipids are reported in the supporting information (Fig. S1). The paclitaxel spectrum shows a few characteristic bands that do not spectrally overlap with the polymer or the lipid bands.21 Particularly, the amide I peak (≈ 1645 cm−1) was used as paclitaxel marker band in the subsequent PTIR experiments. Because DPPC and PBD-b-PEO have very similar IR spectra21 (Fig. S1); here d-DPPC\PBD-b-PEO films were fabricated to enable spectroscopic differentiation. Deuteration is a common spectroscopic trick to overcome IR spectral overlap since the C-D stretching vibrations occur in a very characteristic spectral region (from 2000 cm−1 to 2300 cm−1).47 For example, the d-DPPC spectrum shows two peaks at 2189 cm−1 and 2213 cm−1 due to symmetric and asymmetric CD2 stretching which were used as lipid marker bands in the drug-loaded hybrid membranes.
The PTIR spectrum of a paclitaxel single crystallite (Fig. 2c) shows good correlation with the corresponding macroscale FTIR spectrum, as expected, and was used as reference for determining the paclitaxel distribution in both single component films (DPPC and PBD-b-PEO) (Fig. 2 d-i) and hybrid films (Fig. 3,4). Small differences, between ATR FTIR (unpolarized) and PTIR (polarized) spectra can be attributed to the effect of light polarization and sampling differences, as recently discussed in detail elsewhere.21 As mentioned, (see schematic in Fig. 1b) at parity of initial drug loading, the paclitaxel cumulative release from the hybrid films outperforms the release from the polymer-only films (intermediate performance) and lipid-only films (worst performance). The AFM topography (Fig. 2d) and PTIR spectra (Fig. 2f) of a paclitaxel-loaded polymer film show that this sample is topographically and compositionally uniform. Particularly, the PTIR spectra display the prominent, amide I peak (≈ 1650 cm−1) characteristic of paclitaxel and two peaks (1724 cm−1, and 1448 cm-1) characteristic of the polymer.
Fig. 2.
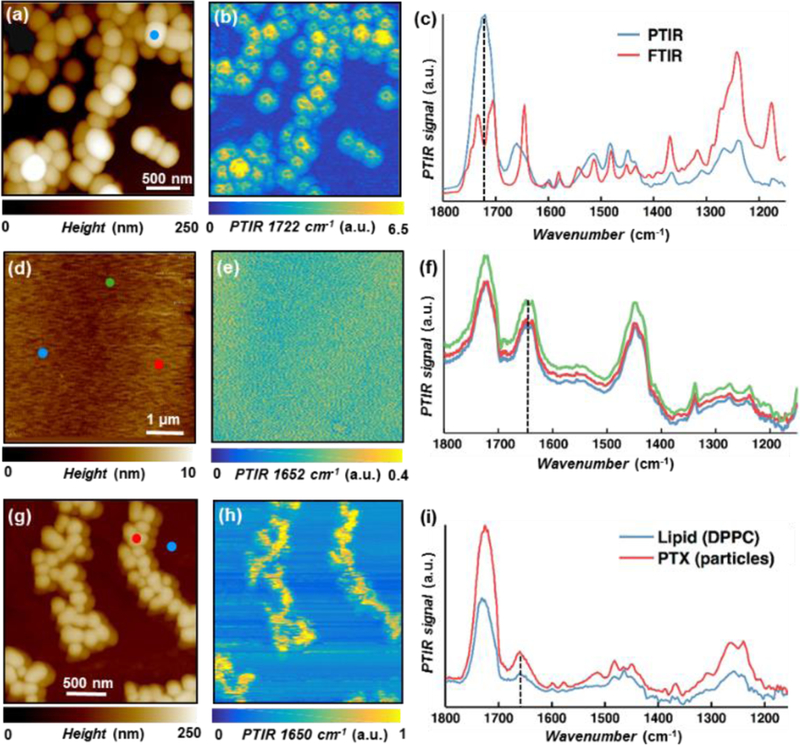
(a) AFM topography map, and (b) PTIR map (1722 cm−1, carboxylic acid stretching) of paclitaxel microcrystals. (c) Comparison between macroscale FTIR (red) and nanoscale PTIR spectra [blue, from marked position in panel (a)] of paclitaxel microcrystals, showing good agreement. (d) AFM topography map, (e) PTIR map (1652 cm−1, paclitaxel amide I), and (f) PTIR spectra [from the color-coded positions in panel (d)] of a paclitaxel loaded PBD-b-PEO film highlighting the film homogeneity. (g) AFM topography map, (h) PTIR map (1650 cm−1, paclitaxel amide I) and (i) PTIR spectra [from the color-coded positions in panel (g)] of a paclitaxel loaded DPPC film showing the partial segregation of paclitaxel nanocrystals from the DPPC film.
Fig. 3.
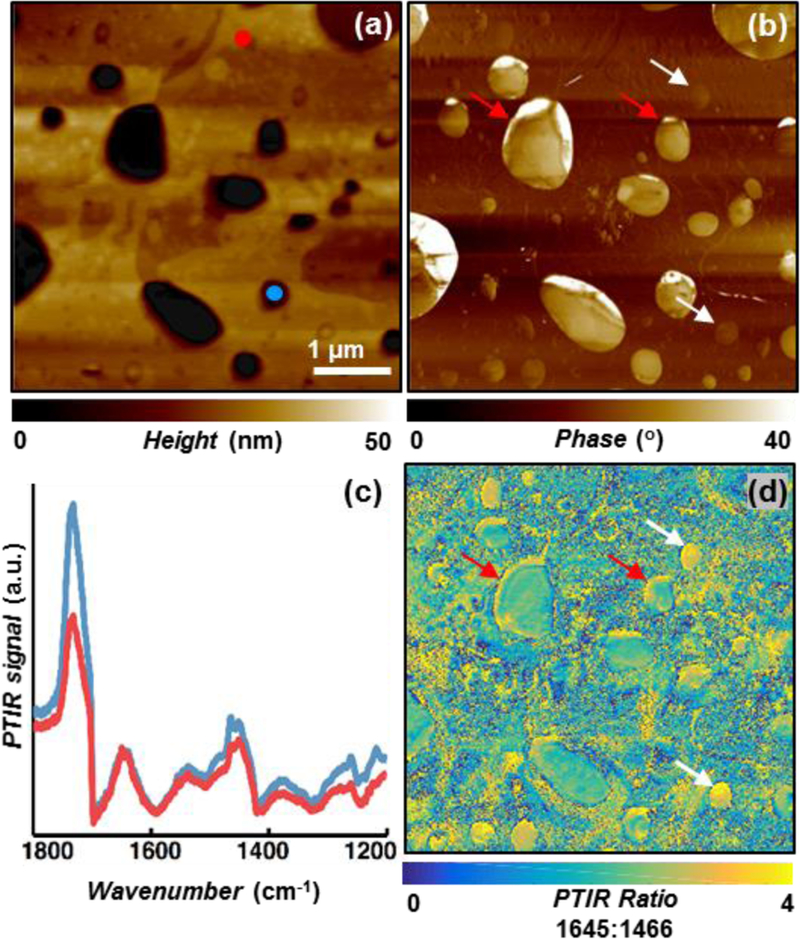
(a) AFM topography, and (b) phase map of paclitaxel loaded DPPC/PBD-b-PEO hybrid films showing strong heterogeneity. (c) PTIR spectra obtained at the marked locations in panel-(a) showing very similar spectra from the polymer-rich and lipid-rich domains. (d) PTIR ratio image obtained by dividing the paclitaxel amide I map (1645 cm−1) with respect to the polymer and lipid band (1466 cm−1) showing the complex and heterogeneous distribution of paclitaxel. Higher paclitaxel concentration is observed along several lipid-polymer phase boundaries (red arrows) and throughout some smaller domains (white arrows).
Fig. 4.
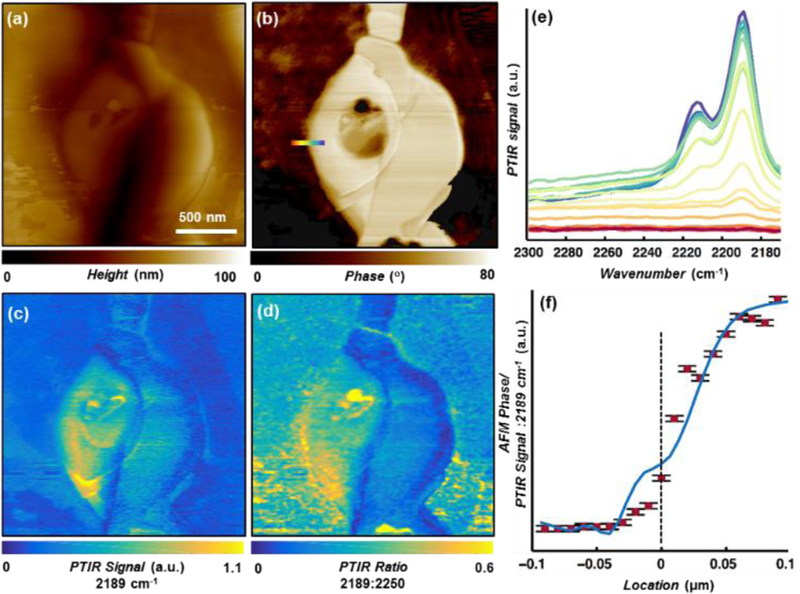
(a) AFM topography map, and (b) phase map of paclitaxel loaded d-DPPC/PBD-b-PEO hybrid films. (c) PTIR absorption map (2189 cm−1, d-DPPC symmetric CD2 stretching), and (d) PTIR ratio image obtained by dividing the d-DPPC symmetric CD2 stretching (2189 cm−1) map with respect to non-specific background absorption (2250 cm−1). (e) PTIR spectra obtained at the color-coded locations in panel-(b). (f) AFM phase line profile (blue) and 2189 cm−1 spectral intensity [red dots, derived from spectra in panel-(e)] across the color-coded location in panel-(b) showing an abrupt change in chemical composition. The line at “0” μm marks the edge of the lipid-rich domain, as observed in the topography image in panel-(a). Error bars represent a single standard deviation in the determination of the PTIR signal intensity.
In contrast, in the drug-loaded lipid film, the AFM topography (Fig. 2g) and PTIR image (1650 cm−1, Fig. 2h) due to paclitaxel amide I absorption reveal features resembling paclitaxel crystals in Fig. 2a-c, suggesting paclitaxel segregation and crystallization in the film. Although the IR spectra of DPPC and paclitaxel are partially overlapped, the PTIR spectra (Fig. 2h) obtained at the color-coded locations in Fig. 2g, confirm this interpretation due to the stronger paclitaxel marker bands in the regions with elevated AFM topography (drug crystallites). However, since the drug peaks can also be observed, albeit with weaker intensity, in the smooth area of the film, a fraction of the initially loaded drug is still finely dispersed in the lipid matrix. Consequently, the PTIR data suggests that paclitaxel partially aggregates and precipitates out of the hydrophobic DPPC membranes, reducing its bioavailability and efficacy,9,10 in agreement with small angle X-ray scattering data reported previously.15
Having justified the larger paclitaxel release from PBD-b-PEO polymer films than from DPPC films, we next investigate the origin of the even greater release from PBD-b-PEO/DPPC hybrid films (Fig. 1c). The AFM topography (Fig. 3a) and phase (Fig. 3b) images of the paclitaxel-loaded hybrid film show several micro- and nano- domains. In previous works, the regions displaying lower topography and higher phase have been assigned to a lipid-rich phase, whereas the areas with higher topography and lower phase have been assigned to a polymer-rich phase. 14,15 The PTIR ratio map (Fig. 3d) of the PTIR intensity at 1645 cm−1 (paclitaxel amide I) with respect to the PTIR intensity at 1466 cm−1 (polymer and lipid band) reveals the complex and heterogeneous distribution of paclitaxel in these films. The calculation of image ratios, is a common post-acquisition practice that mitigates the possible influence of the sample stiffness heterogeneity on the PTIR signal amplitude,48 because at any given location such effects are wavelength independent. In agreement with our previous work,21 a stronger signal (i.e. higher paclitaxel concentration) is observed along several lipid-polymer phase boundaries (for example, see red arrows in Fig. 3). In addition, several smaller domains (see white arrows in Fig. 3) show strong intensity (high paclitaxel concentration) throughout the whole domain. These domains (hereafter drug-rich domains) are also characterized by intermediate phase values (Fig. 3b) with respect to the polymer-rich and lipid-rich phases, suggesting that in these regions the polymer and lipid molecules may be strongly intermixed. Additionally, since these drug-rich domains do not display the large topographic protrusions (as observed in paclitaxel-loaded DPPC film, Fig. 2a) and because the drug release from the hybrid films is enhanced with respect to the single component films, we rule out the possibility that such domains could consist of crystallized paclitaxel. Although paclitaxel neither crystallizes in PBD-b-PEO nor in hybrid films, the superior performance of the hybrid films is attributed to their micro- and nano-structure. In fact, since paclitaxel preferentially partitions in the lipid-polymer boundary regions, and the domain boundaries run throughout the film thickness (like-domains are stacked, Fig. 1b) 11 such structure provides a confined pathway through-which drug permeation is facilitated. At parity of total drug loading, another characteristic that favors the enhanced release from the hybrid films with respect to PBD-b-PEO films is the heterogeneity in the local drug concentration that should lead to increased release both initially (higher driving force due to higher concentration) and at intermediate times (due to the lower film density in the regions that have already expelled the drug).
Because the lipid and the polymer have similar IR spectra (Fig. S1, 3c) we fabricated paclitaxel-loaded d-DPPC/PBD-b-PEO hybrid films using the partially deuterated d-DPPC lipid (Fig. 4) to assess their intermixing in hybrid films. Following previous work,14,15 the bright regions in the phase image (Fig. 4b) should correspond to lipid-rich regions; an assignment that is confirmed, in first approximation, by the PTIR image at 2189 cm−1 (d-DPPC CD2 symmetric stretching, Fig. 4c). However, the PTIR ratio map (Fig. 4d) of the d-DPPC CD2 symmetric stretching (2189 cm−1) with respect to the non-specific background absorption (2250 cm−1) clearly suggests that not all the regions displaying a high phase values in Fig. 4b are lipid-rich, as proposed previously.15
Consequently, relying only on AFM phase imaging to distinguish between lipid-rich and polymer-rich domains should be done with caution and chemically sensitive methods, such as PTIR, should be used whenever possible. Finely spaced (10 nm) PTIR spectra in Fig. 4e (from the color-coded positions shown in Fig. 4b) show that the lipid peaks (2189 cm−1 and 2213 cm-1) decay as we move away from the lipid-rich area, as expected, and indicate that the lipid content in the polymer-rich phase is either negligible or below the PTIR limit of detection (≈ 5 % mole fraction). The PTIR spectral intensity with respect to position (Fig. 4f) suggests that the extent of the lipid-polymer interfacial region is ≈ 70 nm (also corroborated by the AFM phase line profile).
Experimental Methods
Materials
All chemicals and materials were purchased from commercial sources and used as received without further purification. The nominal average molecular weight (Mn) of the PBD and PEO blocks was 2500 g·mol−1 and 1500 g·mol−1, respectively; leading to an overall Mn = 4000 g·mol−1 for PBD-b-PEO copolymer (1.06 polydispersity). All solvents used were high performance liquid chromatography grade.
Preparation of multi-lamellar membranes
Multi-lamellar membranes composed of phospholipids (DPPC), block copolymers (PBD-b-PEO), and drug molecules (Paclitaxel) and their hybrids were prepared by spin-coating on small (≈ 1 cm2) cleaved Si wafers. First, DPPC and PBD-b-PEO were dissolved in chloroform (25 mg/ml) and paclitaxel was dissolved in ethanol (5 mg/ml, 0.05 molar fraction). Later, a mixed solution with DPPC and PBD-b-PEO (1:1 molar ratio) paclitaxel was prepared to have 4:1 volume ratio of chloroform and ethanol (20 mg/ml) and spin-coated (16.7 Hz) for 30 s. To ensure complete solvent evaporation, the samples were placed into a vacuum desiccator overnight. Driven by solvent evaporation, the amphiphilic nature of lipid and polymer molecules yields well-organized multi-lamellar self-assembled membranes.15
PTIR Measurements
PTIR spectra and images were obtained with a commercial PTIR instrument interfaced with a quantum cascade laser with wavelength tunable from 2349 cm−1 (4.26 m) to 2165 cm−1 (4.62 m), and from 1934 cm−1 (5.17 µm) to 1126 cm−1 (8.88 µm). The laser was focused on the sample to a ≈ 50 µm spot size, with the light beam impinging from a ≈ 20° angle with respect to the sample plane. All PTIR experiments, were obtained using p-light polarization and commercially available gold-coated silicon AFM probes (225 µm nominal length and with nominal spring constant between 1 N/m and 7 N/m, as specified by the vendor). All PTIR measurements were obtained leveraging the resonant excitation of the cantilever either in contact mode (for spectra acquisition only) or tapping mode (for imaging only) using the same cantilever type. In contact mode, resonant excitation is achieved by matching the laser repetition rate (tunable from 0.1 kHz to 500 kHz) to one of the cantilever resonance frequencies (typically 270 kHz, Fig. 1f) using a 5 % duty cycle (≈ 160 ns pulse length). PTIR spectra were obtained in contact mode by sweeping the laser (2 cm−1 intervals) and averaging up to four spectra at each tip location. Tapping-mode AFM and PTIR images were acquired simultaneously by illuminating the sample with a fixed wavelength and by leveraging a heterodyne detection scheme where the laser repetition rate matches the frequency difference (Δf ≈ 300 kHz) between the second and first eigen-modes of the AFM cantilever (≈ 360 kHz, and ≈ 60 kHz, respectively) see Fig. 1g. The PTIR images were labelled with the laser wavelength used in the experiments, typically close but not always corresponding exactly to the IR peak maximum. All PTIR, phase and topography images were obtained at 0.5 Hz scan rate, with a 5 nm pixel size in both the horizontal and vertical directions.
Conclusions
In conclusion, PTIR’s high-resolution compositional sensitivity provides a detailed observation window on the chemical and structural complexity of paclitaxel loaded lipid-hybrid polymer films. We elucidate, for the first time, drug partition with nanoscale resolution in these materials and conjecture that this directly impacts the observed enhanced drug release properties. The partial segregation and crystallization of paclitaxel nanocrystals in lipid films limits their utility as paclitaxel delivery media with respect to polymer films. Although, paclitaxel crystallization is suppressed in both polymer-only and lipid-polymer hybrid films, the superior drug release capabilities of hybrid films, is attributed to paclitaxel preferential localization along the vertically aligned lipid-polymer phase-boundaries. The partial deuteration of the lipid component in hybrid films enables estimating the width of the lipid polymer interfacial region (≈ 70 nm) and reveals that the molar fraction of the lipid in the polymer-rich phase is below the PTIR limit of detection (estimated < 5 %). We believe that our work will foster the development of new lipid-polymer hybrid films their applications in drug delivery.
Supplementary Material
Acknowledgements
M.T. acknowledges support under the Cooperative Research Agreement between the University of Maryland, and the National Institute of Standards and Technology Center for Nanoscale Science and Technology, Award 70NANB14H209, through the University of Maryland. M.K. and C.L. acknowledge support from the National Science Foundation under grant no. DMR-1554435 and the National Institutes of Health under grant no. 1DP2EB024377–01.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Rowinsky EK, Cazenave LA and Donehower RC, J. Natl. Cancer Inst, 1990, 82, 1247–1259. [DOI] [PubMed] [Google Scholar]
- 2.Rowinsky EK, Onetto N, Canetta RM and Arbuck SG, Semin. Oncol, 1992, 19, 646–662. [PubMed] [Google Scholar]
- 3.Rowinsky EK and Donehower RC, New Engl. J. Med, 1995, 332, 1004–1014. [DOI] [PubMed] [Google Scholar]
- 4.Slavin L, Chhabra A and Tobis JM, Cardiol. Rev, 2007, 15, 1–12. [DOI] [PubMed] [Google Scholar]
- 5.Mastropaolo D, Camerman A, Luo Y, Brayer GD and Camerman N, Proc. Natl. Acad. Sci. U.S.A, 1995, 92, 6920–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gelderblom H, Verweij J, Nooter K and Sparreboom A, Eur. J. Cancer, 2001, 37, 1590–1598. [DOI] [PubMed] [Google Scholar]
- 7.Sparreboom A, van Tellingen O, Nooijen WJ and Beijnen JH, Cancer Res, 1996, 56, 2112–2115. [PubMed] [Google Scholar]
- 8.Ma P and Mumper RJ, J. Nanomed Nanotechnol, 2013, 4, 1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koudelka Š and Turánek J, J. Controlled Release, 2012, 163, 322–334. [DOI] [PubMed] [Google Scholar]
- 10.J. S., Tapia LV, R. A., C. A. and P. A., in Current Cancer Treatment - Novel Beyond Conventional Approaches, ed. Ozdemir O, InTech, 2011. [Google Scholar]
- 11.Schulz M, Olubummo A and Binder WH, Soft Matter, 2012, 8, 4849. [DOI] [PubMed] [Google Scholar]
- 12.Le Meins J-F, Schatz C, Lecommandoux S and Sandre O, Mater. Today, 2013, 16, 397–402. [Google Scholar]
- 13.Schulz M and Binder WH, Macromol. Rapid Commun, 2015, 36, 2031–2041. [DOI] [PubMed] [Google Scholar]
- 14.Kang M, Tuteja M, Centrone A, Topgaard D and Leal C, Adv. Funct. Mater, 2018, 28, 1704356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang M, Lee B and Leal C, Chem. Mater, 2017, 29, 9120–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Centrone A, Annu. Rev. Anal. Chem, 2015, 8, 101–126. [DOI] [PubMed] [Google Scholar]
- 17.Dazzi A and Prater CB, Chem. Rev, 2017, 117, 5146. [DOI] [PubMed] [Google Scholar]
- 18.Xiao L and Schultz ZD, Anal. Chem, 2018, 90, 440–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dazzi A, Glotin F and Carminati R, J. Appl. Phys, 2010, 107, 124519. [Google Scholar]
- 20.Lahiri B, Holland G and Centrone A, Small, 2013, 9, 439–445. [DOI] [PubMed] [Google Scholar]
- 21.Ramer G, Aksyuk VA and Centrone A, Anal. Chem, 2017, 89, 13524–13531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dazzi A, Prazeres R, Glotin F and Ortega JM, Opt. Lett., OL, 2005, 30, 2388–2390. [DOI] [PubMed] [Google Scholar]
- 23.Katzenmeyer AM, Aksyuk V and Centrone A, Anal. Chem, 2013, 85, 1972–1979. [DOI] [PubMed] [Google Scholar]
- 24.Lu F, Jin M and Belkin MA, Nat. Photonics, 2014, 8, 307–312. [Google Scholar]
- 25.Strelcov E, Dong Q, Li T, Chae J, Shao Y, Deng Y, Gruverman A, Huang J and Centrone A, Sci. Adv, 2017, 3, e1602165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katzenmeyer AM, Holland G, Kjoller K and Centrone A, Anal. Chem, 2015, 87, 3154–3159. [DOI] [PubMed] [Google Scholar]
- 27.Chae J, Dong Q, Huang J and Centrone A, Nano Lett, 2015, 15, 8114–8121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon Y, Chae J, Katzenmeyer AM, Yoon HP, Schumacher J, An S, Centrone A and Zhitenev N, Nanoscale, 2017, 9, 7771–7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morsch S, Liu Y, Lyon SB and Gibbon SR, ACS Appl. Mater. Interfaces, 2016, 8, 959–966. [DOI] [PubMed] [Google Scholar]
- 30.Tri PN and Prud’homme RE, Macromolecules, 2018, 51, 181–188. [Google Scholar]
- 31.Ghosh S, Kouamé NA, Ramos L, Remita S, Dazzi A, Deniset-Besseau A, Beaunier P, Goubard F, Aubert P-H and Remita H, Nat. Mater, 2015, 14, 505–511. [DOI] [PubMed] [Google Scholar]
- 32.Katzenmeyer AM, Canivet J, Holland G, Farrusseng D and Centrone A, Angew. Chem. Int. Ed, 2014, 53, 2852–2856. [DOI] [PubMed] [Google Scholar]
- 33.Purohit HS and Taylor LS, Mol. Pharm, 2015, 12, 1623–1635. [DOI] [PubMed] [Google Scholar]
- 34.Van Eerdenbrugh B, Lo M, Kjoller K, Marcott C and Taylor LS, Mol. Pharm, 2012, 9, 1459–1469. [DOI] [PubMed] [Google Scholar]
- 35.Khanikaev AB, Arju N, Fan Z, Purtseladze D, Lu F, Lee J, Sarriugarte P, Schnell M, Hillenbrand R, Belkin MA and Shvets G, Nat. Commun, 2016, 7, 12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chae J, Lahiri B and Centrone A, ACS Photonics, 2016, 3, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lahiri B, Holland G, Aksyuk V and Centrone A, Nano Lett, 2013, 13, 3218–3224. [DOI] [PubMed] [Google Scholar]
- 38.Brown LV, Davanco M, Sun Z, Kretinin A, Chen Y, Matson JR, Vurgaftman I, Sharac N, Giles AJ, Fogler MM, Taniguchi T, Watanabe K, Novoselov KS, Maier SA, Centrone A and Caldwell JD, Nano Lett, 2018, 18, 1628–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenberger MR, Wang MC, Xie X, Rogers JA, Nam S and King WP, Nanotechnology, 2017, 28, 355707. [DOI] [PubMed] [Google Scholar]
- 40.Perez-Guaita D, Kochan K, Batty M, Doerig C, Garcia-Bustos J, Espinoza S, McNaughton D, Heraud P and Wood BR, Anal. Chem, 2018, 90, 3140–3148. [DOI] [PubMed] [Google Scholar]
- 41.Paluszkiewicz C, Piergies N, Chaniecki P, Rekas M, Miszczyk J, and Kwiatek WM, J. Pharmaceut. Biomed, 2017, 139, 125–132. [DOI] [PubMed] [Google Scholar]
- 42.Giliberti V, Baldassarre L, Rosa A, de Turris V, Ortolani M, Calvani P and Nucara A, Nanoscale, 2016, 8, 17560–17567. [DOI] [PubMed] [Google Scholar]
- 43.Hassenkam T, Andersson MP, Dalby KN, Mackenzie DMA and Rosing MT, Nature, 2017, 548, 78–81. [DOI] [PubMed] [Google Scholar]
- 44.Giliberti V, Badioli M, Nucara A, Calvani P, Ritter E, Puskar L, Aziz EF, Hegemann P, Schade U, Ortolani M and Baldassarre L, Small, 2017, 13, 1701181. [DOI] [PubMed] [Google Scholar]
- 45.Chae J, An S, Ramer G, Stavila V, Holland G, Yoon Y, Talin AA, Allendorf M, Aksyuk VA and Centrone A, Nano Lett, 2017, 17, 5587–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin M, Lu F and Belkin MA, Light Sci. Appl, 2017, 6, e17096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Centrone A, Ying H, Jackson AM, Zerbi G, Stellacci F, Small, 2007, 3, 814–817. [DOI] [PubMed] [Google Scholar]
- 48.Barlow DE, Biffinger JC, Cockrell-Zugell AL, Lo M, Kjoller K, Cook D, Lee WK, Pehrsson PE, Crookes-Goodson WJ, Hung C-S, Nadeau LJ and Russell JN, Analyst, 2016, 141, 4848–4854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.