

Abstract
Purpose of review
In spite of significant morbidity and mortality associated with venous thromboembolism, the underlying pathogenesis remains poorly understood.
Recent findings
Clues to operant pathogenic mechanisms are found in the unique morphology and composition of these thrombi, which have substantial red blood cell and fibrin content. Recent studies have revealed biochemical and biophysical mechanisms that dictate fibrin structure in venous thrombi and promote retention of red blood cells within the contracted clots. These mechanisms include newly-recognized contributions of fibrin network structure and factor XIII(a)-mediated fibrin crosslinking to venous thrombus composition, size, and stability.
Summary
Continued work to elucidate mechanisms by which fibrin(ogen), factor XIII, and red blood cells contribute to venous thrombus formation, structure, and stability may expose novel molecular targets and strategies for reducing thrombosis and thrombotic complications in certain at-risk patients.
Keywords: Venous thrombosis, fibrinogen, factor XIII, red blood cells
INTRODUCTION
Venous thrombosis (VT) with or without pulmonary embolism (PE), collectively venous thromboembolism (VTE), is a major public health concern. VTE affects 1–2 individuals per 1000 each year in the United States and in Europe.(1–3) VTE is associated with a high mortality rate, estimated between 10–30%. Deaths usually occur within the first 30 days of diagnosis and are typically associated with PE. Even in surviving patients, up to 50% develop long-term sequelae, including post-thrombotic syndrome or chronic thromboembolic pulmonary hypertension, which dramatically decrease quality-of-life.(1–3) All current antithrombotic drugs used to reduce primary or recurrent VTE are associated with an increased risk of bleeding. Moreover, a subset of patients experience thrombosis recurrence in spite of aggressive anticoagulation therapy. Thus, efforts to understand the pathophysiology of VTE and advance discoveries into novel therapeutic strategies are important.
Although cellular, molecular, and biophysical mechanisms leading to VTE remain poorly understood, recent studies have revealed specific roles for fibrinogen, and fibrin clot formation, structure, and crosslinking in the development and composition of venous thrombi. These findings extend our understanding of the mechanisms contributing to VTE, and suggest fibrin(ogen) and factor XIII (FXIII) are new potential therapeutic targets for reducing development of venous thrombi and accelerating their resolution. Herein, we review these discoveries and their implications for basic and translational research into the causes and treatment of VTE.
PATHOGENESIS OF VTE
The current conceptual model of events leading to VTE includes pathologic interactions between dysregulated blood coagulation pathways, vascular function, and blood flow (stasis). This combination of mechanisms is known as “Virchow’s Triad.” Briefly, VTE occurs in the veins and is thought to initiate in the pocket region behind venous valves. Within this region, non-laminar blood flow promotes endothelial cell activation. Activated endothelial cells express cell adhesion molecules, leading to the accumulation of leukocytes. Together, these activated leukocytes and endothelial cells initiate the coagulation cascade, which is propagated by blood flow- and red blood cell (RBC)-mediated accumulation of platelets within the venous valve pocket.(4) These processes produce a burst of thrombin, culminating in conversion of fibrinogen to fibrin, which traps resident proteins and blood cells within the growing and contracting thrombus. Cumulatively, this process results in the formation of an intravascular mass that occludes blood flow.
Venous thrombus composition
In contrast to arterial clots, which are primarily composed of aggregated platelets, venous thrombi are rich in fibrin and RBCs. Accordingly, when compared to relatively platelet-rich arterial “white” clots, venous thrombi are often referred to as “red clots.” Transmission electron microscopy reveals a remarkable venous thrombus ultrastructure in which RBCs lose their characteristic discoid shape and become distorted.(5) These compressed RBC shapes, termed “polyhedrocytes,” are indicative of substantial forces that occur within the thrombus during platelet-mediated clot contraction.(6) Polyhedrocytes often appear arranged among fibrin layers in a “brick-and-mortar” fashion within the thrombus (sometimes called “Lines of Zahn”), suggesting an ordered and cyclical process leads to venous thrombus growth within the vessel. The proximity and arrangement of RBCs and fibrin within the clot ultrastructure implies a mechanistic relationship that has only recently been identified.
FIBRIN FORMATION AND FUNCTION
During coagulation, fibrin is generated by thrombin-mediated proteolytic cleavage of fibrinogen. This reaction releases N-terminal fibrinopeptides from the fibrinogen Aα- and Bβ-chains, exposes “knobs” at the N-termini of the α- and β- chains, and permits assembly of half-staggered fibrin monomers into fibrin protofibrils. Subsequent lateral aggregation and branching of protofibrils results in the formation of an insoluble 3-dimensional fibrin network. The molecular mechanisms governing this process have been extensively reviewed.(7–10) Abnormalities in circulating fibrinogen level and/or function, including congenital and acquired hypofibrinogenemia, afibrinogenemia, and dysfibrinogenemia, are associated with increased risk of bleeding and/or thrombosis.(11, 12)
Thrombin also proteolytically converts the (pro)transglutaminase FXIII to activated FXIII (FXIIIa). In contrast to other coagulation enzymes that proteolytically cleave their substrates, FXIIIa catalyzes the formation of new covalent bonds. These bonds are generated between lysyl residues in an amine donor molecule and glutamyl residues in an amine acceptor molecule, producing novel ε-N-[γ-glutamyl]-lysyl crosslinks. The primary substrate for FXIIIa is fibrin, where it crosslinks γ-chains and α-chains into γ-γ dimers and high molecular weight α-α and γ-α polymers. FXIIIa also crosslinks anti-fibrinolytic proteins (e.g., α2-antiplasmin) to the fibrin network. Studies have demonstrated essential contributions of these crosslinks to fibrin clot mechanical stability and resistance to fibrinolysis.(13)
Several findings are particularly noteworthy with regard to fibrin network formation, crosslinking, and function. First, the concentration of thrombin present during fibrin formation profoundly influences not only the rate of fibrinogen conversion to fibrin, but also the structure and function of the resulting fibrin network. Low thrombin concentrations produce thick fibrin fibers arranged in coarse, permeable networks with large pores, and these networks are relatively susceptible to fibrinolysis. In contrast, high thrombin concentrations produce thin fibrin fibers arranged in less permeable, dense networks with small pores, and these networks are relatively resistant to fibrinolysis.(7–10) Second, although FXIIIa crosslinking of fibrin has little-to-no influence on fibrin network density, it increases fibrin fiber compaction and stiffness by promoting protofibril coupling within individual fibers.(14–18) Fiber compaction also decreases the size of pores within individual fibers, with potentially important effects on diffusion of molecules like tissue-type plasminogen activator (tPA) through fibers and consequently, on a clot’s resistance to fibrinolysis. Thus, fibrin clot structural properties are not only interesting from an observational standpoint, but also appear to have direct functional implications for hemostasis and thrombosis.
FIBRIN FORMATION, STRUCTURE, AND FUNCTION IN VT
Abnormal ex vivo fibrin clot structure and VT
Several studies have documented abnormal fibrin formation, structure, and stability in patients with a history of VTE.(19) Compared to healthy controls, VTE patients generally have lower plasma clot permeability, lower clot compaction, and prolonged clot lysis times. Similar, though blunted, findings have also been observed in apparently healthy relatives of VTE patients, suggesting shared genetic or environmental causes of the abnormal clot structure.(20) Moreover, following discontinuation of anticoagulation after first DVT, faster formation of less permeable fibrin networks and prolonged clot lysis time is associated with increased risk of DVT recurrence (odds ratio 15.8; confidence interval 7.5–33.5).(21) Abnormal clot properties, including reduced plasma clot permeability and susceptibility to fibrinolysis are also associated with increased risk of developing long-term sequelae, including chronic thromboembolic pulmonary hypertension, post-thrombotic syndrome, and persistent venous obstruction.(22–24) Notably, although these observations are of platelet-poor plasma clots formed ex vivo, their association with VTE risk suggests these clot properties recapitulate aspects of venous thrombus (dys)function in vivo. Thus, abnormal fibrin clot structure, permeability, and resistance to fibrinolysis are not only powerful potential biomarkers for thromboembolic disease, but may also reveal fundamental aspects of the pathophysiology underlying intravascular clot formation and impact.
Plasma hypercoagulability, abnormal clot properties, and VT
Given the close relationship between thrombin generation and fibrin formation and crosslinking, it is perhaps not surprising that abnormal levels and/or function of proteins that regulate thrombin generation or activity are associated with both formation of abnormal clots and increased VTE risk. These abnormalities include elevated levels of factor VIII, prothrombin, or fibrinogen, presence of the factor VLeiden mutation, or reduced levels of antithrombin or proteins C or S.(2) For example, presence of the G20210A mutation is associated with elevated levels of circulating prothrombin and increased risk of DVT.(25, 26) During coagulation, elevated prothrombin enhances thrombin generation, with substantial effects on the fibrin network: formation of dense fibrin networks composed of thin fibrin fibers, and decreased clot permeability.(27, 28) Hyperprothrombinemic mice have increased fibrin deposition in thrombi, and increased thrombus weight.(29) Intriguingly, venous thrombi isolated from hyperprothrombinemic mice also have increased RBC content (MM Aleman & AS Wolberg, unpublished observation), suggesting fibrin network properties also influence the cellular content of venous thrombi formed in vivo.
Fibrin network density and FXIIIa-mediated fibrin crosslinking promote RBC retention in VT
Following the observation that thrombi isolated from hyperprothrombinemic mice showed increased RBC content, a second major clue to understanding the structure and composition of venous thrombi arose during experiments to analyze fibrin(ogen) function during VT. Using both genetic and pharmacologic approaches, we discovered that FXIII promotes RBC retention in venous thrombi.(30) Clots generated from whole blood isolated from F13a−/− mice demonstrate dramatic loss of RBCs during platelet-mediated clot contraction, resulting in the formation of abnormally small clots. Using an inferior vena cava ligation model of VT, we demonstrated that this mechanism also occurs in vivo.(30, 31) Using plasma from a patient with FXIII-deficiency and transglutaminase inhibitors in normal whole blood, we translated this observation to human blood, and showed a dose-dependent effect of FXIII on RBC retention.(30, 32) These findings are reminiscent of observations of “excessive RBC fallout” previously noted during contraction of whole blood clots in a family of FXIII-deficient patients.(33) Collectively, these observations expose a previously unrecognized role for FXIII activity during clot formation and VT.
Work to interrogate the operant mechanism by which fibrin(ogen) and FXIII promote RBC retention in thrombi has revealed at least two independent mechanisms. First, experiments to examine the contribution of fibrin network density showed that RBCs are effectively “trapped” by the fibrin network, and that clots with increased fibrin network density trap higher numbers of RBCs.(32) This observation may provide a mechanism linking plasma hypercoagulability and the formation of dense fibrin networks to increased thrombosis risk. For example, elevated levels of FVIII and prothrombin that enhance thrombin generation, fibrin deposition, and network density(27, 29, 34) may generate fibrin networks that trap higher numbers of RBCs in clots during clot contraction and produce larger thrombi.
Notably, however, since FXIII has little-to-no effect on fibrin network density(14, 15), the effects of FXIII on RBC retention in clots appear to be independent of fibrin network density. Experiments with recombinant fibrinogen variants capable of undergoing either α-chain or γ-chain crosslinking but not both, as well as the detection of crosslinked fibrin species in clots treated with crosslinking inhibitors, associated the effect of FXIII activity on RBC retention with the formation of high molecular weight fibrin species containing crosslinked α-chains.(32) This observation invokes intriguing findings that crosslinking – and particularly α-chain crosslinking – has significant functional consequences for fibrin biophysical and biomechanical properties.(14–18, 35–38) Crosslinking increases fibrin fiber elastic modulus (stiffness) and elasticity (3.75- and 1.2-fold, respectively), and decreases fiber extensibility (1.2-fold). Notably, α-chain crosslinking, even in the absence of γ-chain crosslinking, recapitulates a substantial portion of these effects, including a 2.5-and 1.5-fold increase in fiber stiffness and elasticity, respectively.(38) Given the considerable forces generated by contracting platelets(39), these studies suggest fibrin crosslinking enables the fibrin network to resist deformation and retain RBCs in clots during clot contraction. Thus, these observations may provide a (patho)physiologically-relevant rationale for the remarkable biophysical characteristics of fibrin in hemostasis and thrombosis.
Plasma FXIII promotes RBC retention in VT
FXIII is present in both plasma and cellular forms. Plasma FXIII is composed of 2 catalytic A subunits (FXIII-A2) and 2 non-catalytic B subunits (FXIII-B2) arranged as a tightly-associated heterotetramer (FXIII-A2B2).(40) FXIII-A2B2 circulates in plasma bound to fibrinogen at residues γ-chain 390–396 and possibly also to α-chain residue E396.(30, 41, 42) During coagulation, thrombin rapidly cleaves N-terminal peptides from the FXIII A-subunits. Calcium then induces a conformational change in FXIII, which promotes release of the FXIII-B subunits and yields fully activated FXIII-A2* (FXIIIa). Cellular FXIII is a homodimer consisting only of the catalytic A subunits (FXIII-A2). FXIII-A2 is found in the cytoplasm of bone marrow-derived cells, including megakaryocytes, monocytes, macrophages, osteoblasts, and platelets. Cellular FXIII-A2 can be activated non-enzymatically by elevated intracellular calcium concentrations that occur during platelet activation.(43) Following platelet activation, FXIII-A2* is exposed on the platelet surface(44); however, cellular mechanisms leading to its externalization in platelets and other cells are unknown. Although some studies have suggested platelet FXIII is essential for platelet contraction(45), we and others have detected only moderate or no influence of platelet FXIII on platelet contractile events.(31, 46, 47)
Since both plasma and platelet FXIII possess catalytic A subunits, both are capable of catalyzing fibrin crosslinking. Mitchell et al detected platelet FXIII-mediated crosslinking in vitro, but effects were only apparent when levels of plasma FXIII were less than 10% of normal levels.(44) Interestingly, however, plasma FXIII, but not platelet FXIII, promotes RBC retention in contracted clots.(31) This function is associated with an apparent need for fibrin α-chain crosslinking prior to initiation of platelet-mediated clot contraction.(31) Since plasma FXIII circulates bound to fibrin, it is rapidly converted to FXIIIa in concert with fibrin formation and can fulfill this temporal requirement for fibrin crosslinking.(41) In contrast, slower exposure of platelet FXIII likely happens after clot contraction – too late to mechanically stabilize fibrin fibers and enable them to retain RBCs during clot contraction.
FXIII as a therapeutic target
Given the roles of FXIII(a) in determining fibrin network permeability, resistance to fibrinolysis and mechanical disruption, and thrombus composition and size, it is intriguing to consider that FXIII(a) and fibrin crosslinking may be therapeutic targets for reducing VTE. This approach may offer certain advantages over anticoagulants that reduce plasma concentrations of vitamin K-dependent proteins (vitamin K antagonists) or directly target enzymes within the coagulation cascade (factor Xa, thrombin - the so-called “direct oral anticoagulants” or DOACs). Although each of these anticoagulants significantly reduce the incidence of VT, all are associated with increased bleeding risk.(2) Notably, all of these antithrombotics target molecules that lead to thrombin generation and therefore, reduce fibrin formation. A strategy that permits fibrin formation in situations necessitating hemostasis, but reduces the stability of thrombotic networks might mitigate this risk. In this regard, targeting FXIII(a) and/or fibrin crosslinking might offer advantages not realized with other anticoagulants. Unlike vitamin K antagonists or DOACs, a FXIII(a) inhibitor would not be expected to interfere with thrombin generation or fibrin deposition (Figure). Indeed, genetic reduction of FXIII in mice does not alter thrombin generation in plasma or prolong the time to hemostatic clot formation in saphenous vein puncture or tail transection bleeding models.(31) These findings are consistent with clinical observations indicating that although patients with <4% FXIIIa activity have a significantly increased risk of bleeding, individuals with ≥30% FXIIIa activity, including those with acquired FXIII deficiency, are usually asymptomatic.(48) Thus, FXIII(a) antagonists may enjoy a wide therapeutic range and could be used alone or in concert with conventional anticoagulants. Moreover, association of FXIII impact on thrombus RBC retention and size with plasma, but not platelet, FXIII(31) may facilitate development of antagonists that would only need to inhibit the plasma compartment. Strategies that target plasma FXIII but leave platelet FXIII intact may allow sufficient residual activity to prevent major bleeds associated with antithrombotics that target hemostatic proteins.
Figure. Coagulation cascade illustrating different points of intervention of current anticoagulants and a potential factor XIII/fibrin crosslinking inhibitor.
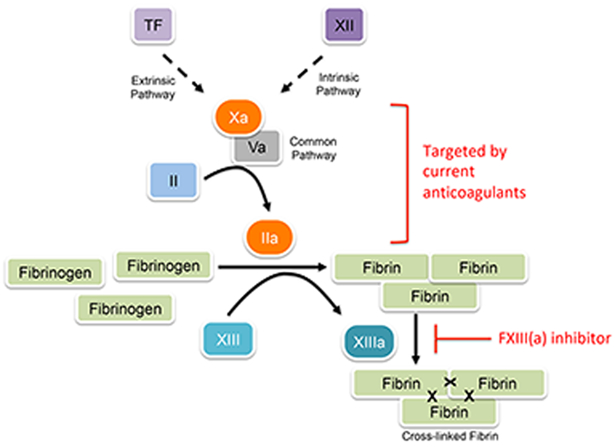
Current anticoagulants target enzymes leading to thrombin generation and therefore, interfere with fibrin formation. Blocking factor XIII(a) activity would permit normal fibrin formation, but reduce fibrin stability, red blood cell retention in thrombi, and thrombus size. In this way, factor XIII(a) inhibition would be mechanistically different from existing approaches. TF, tissue factor.
CONCLUSION and FUTURE DIRECTIONS
Collectively, these studies have identified newly-recognized mechanisms by which fibrin, FXIII activation, and fibrin crosslinking promote RBC retention in thrombi and mediate venous thrombus size. These discoveries have enhanced our understanding of VTE pathogenesis, but also revealed knowledge gaps that must be filled to advance these findings into clinical applications.
First, fibrin(ogen) has pleiotropic roles in hemostasis and thrombosis, including functions that influence inflammation, host defense, and thrombus resolution. Continued studies are necessary to understand these functions and identify antagonists with minimal effects on homeostatic effects of fibrinogen and fibrin deposition. Second, RBCs have biochemical and biophysical properties that may influence their incorporation into thrombi and their subsequent effects on thrombus stability and resolution. For example, RBCs with abnormal deformability as in sickle cell disease or spherocytosis may lead to increased retention in thrombi and therefore, result in formation of larger, more occlusive thrombi.(49) RBCs can also induce platelet accumulation and activation within clots in the presence of flow(4), and RBC compaction within contracted thrombi can decrease thrombus permeability and accessibility of fibrinolytic enzymes.(6) Thus, the relative impact of altering RBC presence in thrombi warrants careful evaluation. Third, identification of polyhedrocytes within venous thrombi demonstrates the presence and function of activated platelets within thrombi. Surprisingly, however, the contribution of platelets to VTE is poorly understood. Platelet antagonists that target platelet contraction may have important effects on thrombus development and function. Fourth, currently available transglutaminase inhibitors lack specificity for FXIII and have short half-lives not amenable to prophylactic dosing strategies. Moreover, the FXIII active site is structurally superficial, complicating efforts to design specific antagonists to this molecule. Advanced screens and sophisticated medicinal chemistry will be essential to identify and develop compounds with the necessary pharmacologic properties for in vivo use. Finally, it is clear that many biologic functions of FXIII have not been thoroughly elucidated. In addition to coagulation, FXIII may have roles in arthritis(50), adipogenesis(51), and cardiac repair(52, 53). It will be important to understand the impact of FXIII inhibition on these situations before the benefit of a FXIIIa antagonist can be realized in the clinic.
KEY POINTS.
The fibrin(ogen)-factor XIII-red blood cell axis mediates venous thrombus formation and composition.
Thrombin generation promotes venous thrombosis, in part, by altering fibrin network structure.
Fibrin network density and factor XIII influence red blood cell retention in venous thrombi through independent mechanisms.
Plasma, but not platelet, factor XIII promotes fibrin α-chain crosslinking and red blood cell retention in clots.
Fibrin and factor XIII(a)-mediated crosslinking may be therapeutic targets for reducing venous thromboembolism.
ACKNOWLEDGEMENTS
The author thanks Natalie R. Harris for reading the manuscript.
Footnotes
FINANCIAL SUPPORT
ASW is supported by funding from the National Institutes of Health (R01HL126974).
CONFLICTS OF INTEREST
Dr. Wolberg receives research funding from Novo Nordisk.
REFERENCES
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Thrombosis: a major contributor to the global disease burden. J Thromb Haemost 2014. October;12(10):1580–90. [DOI] [PubMed] [Google Scholar]
- 2.Wolberg AS, Rosendaal FR, Weitz JI, Jaffer IH, Agnelli G, Baglin T, et al. Venous thrombosis. Nat Rev Dis Pri. [Primer] 2015. May/07/online;1(15006):http://www.nature.com/articles/nrdp20156. [DOI] [PubMed] [Google Scholar]
- 3.Heit JA, Spencer FA, White RH. The epidemiology of venous thromboembolism. J Thromb Thrombolysis. 2016. January;41(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehmann M, Schoeman RM, Krohl PJ, Wallbank AM, Samaniuk JR, Jandrot-Perrus M, et al. Platelets drive thrombus propagation in a hematocrit and glycoprotein VI-dependent manner in an in vitro venous thrombosis model. Arterioscler Thromb Vasc Biol 2018. February 22. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study used a novel microfluidic device to examine the effects of valve geometry on hemodynamics and accrual of cellular material and fibrin in the vein valve pocket. The findings demonstrate inter-dependent roles for fibrin deposition, blood flow, red blood cell dynamics, and platelet function in venous thrombus formation.
- 5.Aleman MM, Walton BL, Byrnes JR, Wolberg AS. Fibrinogen and red blood cells in venous thrombosis. Thromb Res 2014. May;133 Suppl 1:S38–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cines DB, Lebedeva T, Nagaswami C, Hayes V, Massefski W, Litvinov RI, et al. Clot contraction: compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood. 2014. March 6;123(10):1596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haemost 2007. July;5 Suppl 1:116–24. [DOI] [PubMed] [Google Scholar]
- 8.Lord ST. Fibrinogen and fibrin: scaffold proteins in hemostasis. Curr Opin Hematol 2007. May;14(3):236–41. [DOI] [PubMed] [Google Scholar]
- 9.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev 2007. May;21(3):131–42. [DOI] [PubMed] [Google Scholar]
- 10.Chandrashekar A, Singh G, Jonah G, Sikalas N, Labropoulos N. Mechanical and biochemical role of fibrin within a venous thrombus. Eur J Vasc Endovasc Surg 2018. January 12;55(3):417–24. [DOI] [PubMed] [Google Scholar]
- 11.Hoffman M Alterations of fibrinogen structure in human disease. Cardiovasc Hematol Agents Med Chem 2008. July;6(3):206–11. [DOI] [PubMed] [Google Scholar]
- 12.Ariëns RA. Fibrin(ogen) and thrombotic disease. J Thromb Haemost 2013. June;11 Suppl 1:294–305. [DOI] [PubMed] [Google Scholar]
- 13.Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev 2011. July;91(3):931–72. [DOI] [PubMed] [Google Scholar]
- 14.Ryan EA, Mockros LF, Stern AM, Lorand L. Influence of a natural and a synthetic inhibitor of factor XIIIa on fibrin clot rheology. Biophys J 1999. November;77(5):2827–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hethershaw EL, Cilia La Corte AL, Duval C, Ali M, Grant PJ, Ariëns RA, et al. The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. J Thromb Haemost 2014. February;12(2):197–205. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen VG, Gurley WQ Jr., Burch TM. The impact of factor XIII on coagulation kinetics and clot strength determined by thrombelastography. Anesth Analg. 2004. Jul;99(1):120–3. [DOI] [PubMed] [Google Scholar]
- 17.Schroeder V, Kohler HP. Thrombelastographic studies on factor XIII. Thromb Haemost 2010. December;104(6):1277–9. [DOI] [PubMed] [Google Scholar]
- 18.Kurniawan NA, Grimbergen J, Koopman J, Koenderink GH. Factor XIII stiffens fibrin clots by causing fiber compaction. J Thromb Haemost 2014. October;12(10):1687–96. [DOI] [PubMed] [Google Scholar]
- 19.Undas A Prothrombotic fibrin clot phenotype in patients with deep vein thrombosis and pulmonary embolism: a new risk factor for recurrence. BioMed research international. 2017;2017:8196256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Undas A, Zawilska K, Ciesla-Dul M, Lehmann-Kopydlowska A, Skubiszak A, Ciepluch K, et al. Altered fibrin clot structure/function in patients with idiopathic venous thromboembolism and in their relatives. Blood. 2009. November 5;114(19):4272–8. [DOI] [PubMed] [Google Scholar]
- 21.Cieslik J, Mrozinska S, Broniatowska E, Undas A. Altered plasma clot properties increase the risk of recurrent deep vein thrombosis: a cohort study. Blood. 2018. February 15;131(7):797–807. [DOI] [PubMed] [Google Scholar]; * This study demonstrated that decreased clot permeability and prolonged clot lysis time are associated with a high risk of recurrent deep vein thrombosis. These findings suggest clot quality is both a mechanistic determinant of thrombosis and a prognostic biomarker with potential clinical implications.
- 22.Undas A, Ciesla-Dul M, Drazkiewicz T, Sadowski J. Altered fibrin clot properties are associated with residual vein obstruction: effects of lipoprotein(a) and apolipoprotein(a) isoform. Thromb Res 2012. September;130(3):e184–7. [DOI] [PubMed] [Google Scholar]
- 23.Mazur P, Gaweda B, Natorska J, Zabczyk M, Undas A, Sadowski J, et al. Fibrin structure in organized thrombotic material removed during pulmonary artery endarterectormy: the effect of vessel calibre. J Thromb Thrombolysis. 2016. August;42(2):212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siudut J, Grela M, Wypasek E, Plens K, Undas A. Reduced plasma fibrin clot permeability and susceptibility to lysis are associated with increased risk of postthrombotic syndrome. J Thromb Haemost 2016. April;14(4):784–93. [DOI] [PubMed] [Google Scholar]
- 25.Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3’-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88(10):3698–703. [PubMed] [Google Scholar]
- 26.Eichinger S, Minar E, Hirschl M, Bialonczyk C, Stain M, Mannhalter C, et al. The risk of early recurrent venous thromboembolism after oral anticoagulant therapy in patients with the G20210A transition in the prothrombin gene. Thromb Haemost 1999. January;81(1):14–7. [PubMed] [Google Scholar]
- 27.Wolberg AS, Monroe DM, Roberts HR, Hoffman M. Elevated prothrombin results in clots with an altered fiber structure: a possible mechanism of the increased thrombotic risk. Blood. 2003;101(8):3008–13. [DOI] [PubMed] [Google Scholar]
- 28.Janion-Sadowska A, Natorska J, Siudut J, Zabczyk M, Stanisz A, Undas A. Plasma fibrin clot properties in the G20210A prothrombin mutation carriers following venous thromboembolism: the effect of rivaroxaban. Thromb Haemost 2017. August 30;117(9):1739–49. [DOI] [PubMed] [Google Scholar]
- 29.Aleman MM, Walton BL, Byrnes JR, Wang JG, Heisler MJ, Machlus KR, et al. Elevated prothrombin promotes venous, but not arterial, thrombosis in mice. Arterioscler Thromb Vasc Biol 2013. May 30;33(8):1829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aleman MM, Byrnes JR, Wang J-G, Tran R, Lam WA, Di Paola J, et al. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest 2014. August 1;124(8):3590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kattula S, Byrnes JR, Martin SM, Cooley BC, Flick MJ, Wolberg AS. Plasma-, but not platelet-factor XIII promotes red blood cell retention in contracted clots and mediates clot size during venous thrombosis. Blood Advances. 2017;2(1):25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byrnes JR, Duval C, Wang Y, Hansen CE, Ahn B, Mooberry MJ, et al. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood. 2015. October 15;126(16):1940–8. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study demonstrated that factor XIIIa-mediated crosslinking of fibrin alpha-chains promotes red blood cell retention in contracting whole blood clots. This finding exposes a (patho)physiologic context for fibrin’s remarkable biophysical characteristics, with interesting implications for effects of fibrin(ogen) modifications on fibrin formation and function.
- 33.Hanna M Congenital deficiency of factor 13: report of a family from Newfoundland with associated mild deficiency of factor XII. Pediatrics. 1970. October;46(4):611–9. [PubMed] [Google Scholar]
- 34.Machlus KR, Lin F-C, Wolberg AS. Procoagulant activity induced by vascular injury determines contribution of elevated factor VIII to thrombosis and thrombus stability in mice. Blood. 2011. October 6;118(14):390–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collet JP, Shuman H, Ledger RE, Lee S, Weisel JW. The elasticity of an individual fibrin fiber in a clot. Proc Natl Acad Sci U S A 2005. June 28;102(26):9133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu W, Carlisle CR, Sparks EA, Guthold M. The mechanical properties of single fibrin fibers. J Thromb Haemost 2010. May;8(5):1030–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Houser JR, Hudson NE, Ping L, O’Brien ET 3rd, Superfine R, Lord ST, et al. Evidence that alphaC region is origin of low modulus, high extensibility, and strain stiffening in fibrin fibers. Biophys J 2010. November 3;99(9):3038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helms CC, Ariëns RA, Uitte de Willige S, Standeven KF, Guthold M. alpha-alpha cross-links increase fibrin fiber elasticity and stiffness. Biophys J 2012. January 4;102(1):168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lam WA, Chaudhuri O, Crow A, Webster KD, Li TD, Kita A, et al. Mechanics and contraction dynamics of single platelets and implications for clot stiffening. Nature materials. 2011. January;10(1):61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katona E, Penzes K, Csapo A, Fazakas F, Udvardy ML, Bagoly Z, et al. Interaction of factor XIII subunits. Blood. 2014. March 13;123(11):1757–63. [DOI] [PubMed] [Google Scholar]
- 41.Byrnes JR, Wilson C, Boutelle AM, Brandner CB, Flick MJ, Philippou H, et al. The interaction between fibrinogen and zymogen FXIII-A2B2 is mediated by fibrinogen residues 390–396 and the FXIII-B subunits. Blood. 2016. October 13;128(15):1969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith KA, Adamson PJ, Pease RJ, Brown JM, Balmforth AJ, Cordell PA, et al. Interactions between factor XIII and the alphaC region of fibrinogen. Blood. 2011. March 24;117(12):3460–8. [DOI] [PubMed] [Google Scholar]
- 43.Muszbek L, Haramura G, Polgar J. Transformation of cellular factor XIII into an active zymogen transglutaminase in thrombin-stimulated platelets. Thromb Haemost 1995. April;73(4):702–5. [PubMed] [Google Scholar]
- 44.Mitchell JL, Lionikiene AS, Fraser SR, Whyte CS, Booth NA, Mutch NJ. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. 2014. December 18;124(26):3982–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kasahara K, Souri M, Kaneda M, Miki T, Yamamoto N, Ichinose A. Impaired clot retraction in factor XIII A subunit-deficient mice. Blood. 2010. February 11;115(6):1277–9. [DOI] [PubMed] [Google Scholar]
- 46.Tutwiler V, Litvinov RI, Lozhkin AP, Peshkova AD, Lebedeva T, Ataullakhanov FI, et al. Kinetics and mechanics of clot contraction are governed by the molecular and cellular composition of the blood. Blood. 2016. January 07;127(1):149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rijken DC, Abdul S, Malfliet JJ, Leebeek FW, Uitte de Willige S. Compaction of fibrin clots reveals the antifibrinolytic effect of factor XIII. J Thromb Haemost 2016. July;14(7):1453–61. [DOI] [PubMed] [Google Scholar]
- 48.Menegatti M, Palla R, Boscarino M, Bucciarelli P, Muszbek L, Katona E, et al. Minimal factor XIII activity level to prevent major spontaneous bleeds. J Thromb Haemost 2017. September;15(9):1728–36. [DOI] [PubMed] [Google Scholar]; * This prospective study analyzed data from patients with factor XIII deficiency and characterized associations between factor XIII activity level and bleeding severity. The findings suggest 15 IU dl-1 factor XIII activity is a target level for starting prophylaxis to prevent major bleeds.
- 49.Byrnes JR, Wolberg AS. Red blood cells in thrombosis. Blood. 2017. October 19;130(16):1795–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raghu H, Cruz C, Rewerts CL, Frederick MD, Thornton S, Mullins ES, et al. Transglutaminase factor XIII promotes arthritis through mechanisms linked to inflammation and bone erosion. Blood. 2015. October 21;125(3):427–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mousa A, Cui C, Song A, Myneni VD, Sun H, Li JJ, et al. Transglutaminases factor XIII-A and TG2 regulate resorption, adipogenesis and plasma fibronectin homeostasis in bone and bone marrow. Cell death and differentiation. 2017. May;24(5):844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nahrendorf M, Aikawa E, Figueiredo JL, Stangenberg L, van den Borne SW, Blankesteijn WM, et al. Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular remodelling: implications for FXIII therapy and antithrombin use in myocardial infarction. Eur Heart J 2008. February;29(4):445–54. [DOI] [PubMed] [Google Scholar]
- 53.Gemmati D, Zeri G, Orioli E, Mari R, Moratelli S, Vigliano M, et al. Factor XIII-A dynamics in acute myocardial infarction: a novel prognostic biomarker? Thromb Haemost 2015. June 30;114(1):123–32. [DOI] [PubMed] [Google Scholar]