

Abstract
Triple negative breast cancer (TNBC) is characterized by elevated locoregional recurrence risk despite aggressive local therapies. New tumor-specific radiosensitizers are needed. We hypothesized that the ATR inhibitor, VX-970, would preferentially radiosensitize TNBC. Non-cancerous breast epithelial and TNBC cell lines were investigated in clonogenic survival, cell cycle, and DNA damage signaling and repair assays. In addition, patient-derived xenograft (PDX) models generated prospectively as part of a neoadjuvant chemotherapy study from either baseline tumor biopsies or surgical specimens with chemoresistant residual disease were assessed for sensitivity to fractionated radiotherapy, VX-970, or the combination. To explore potential response biomarkers, exome sequencing was assessed for germ-line and/or somatic alterations in homologous recombination (HR) genes and other alterations associated with ATR inhibitor sensitivity. VX-970 preferentially inhibited ATR-Chk1-CDC25a signaling, abrogated the radiotherapy-induced G2/M checkpoint, delayed resolution of DNA double strand breaks and reduced colony formation after radiotherapy in TNBC cells relative to normal-like breast epithelial cells. In vivo, VX-970 did not exhibit significant single agent activity at the dose administered even in the context of genomic alterations predictive of ATR inhibitor responsiveness, but significantly sensitized TNBC PDXs to radiotherapy. Exome sequencing and functional testing demonstrated that combination therapy was effective in both HR proficient and deficient models. PDXs established from patients with chemoresistant TNBC were also highly radiosensitized. In conclusion, VX-970 is a tumor specific radiosensitizer for TNBC. Patients with residual TNBC after neoadjuvant chemotherapy, a subset at particularly high risk of relapse, may be ideally suited for this treatment intensification strategy.
Keywords: ATR inhibitor, VX-970, chemotherapy resistance, triple negative breast cancer
Introduction
Triple negative breast cancer (TNBC) is an aggressive subset of breast cancer that is associated with higher rates of recurrence compared to other subtypes. Further, these risks persist regardless of surgery choice (mastectomy or breast conservation) and the addition of radiotherapy (1–3). Although uncontrolled systemic disease is a significant problem in TNBC, emerging data suggests that failure to eradicate microscopic residual locoregional TNBC at diagnosis is an important source of systemic spread (4–6). Therefore, even modest improvements in locoregional therapy in TNBC could improve survival in these high-risk patients.
The cellular DNA damage response (DDR) is essential for maintaining cell viability and preventing disease. Loss of elements of the DDR is a common feature of TNBC and may be compensated for by increased activity of other overlapping DNA repair pathway elements (7). Enhanced levels of cellular DNA repair from these compensatory branches of the DDR are an important mechanism of resistance to DNA damaging therapy, including RT(8, 9). ATR (Ataxia Telangiectasia Mutated and Rad-3 Related protein kinase) is a critical component of the DDR that is activated by single stranded DNA such as can be produced during replication stress, double-strand break resection, or DNA crosslinks(10). Activation of ATR ultimately leads to G2/M and intra-S checkpoint arrest, slowing of origin firing, replication fork stabilization, and DNA repair. Many of the driving events in TNBC pathogenesis including loss of G1 checkpoint control, oncogene induced replication stress, and deficiency in homologous recombination (HR) repair increase dependence on ATR pathway signaling following DNA damage, including single and double strand breaks induced by RT(7, 11–19). Therefore, we hypothesized that the ATR inhibitor, VX-970, would be a tumor-specific radiosensitizer for TNBC.
The development of clinically relevant preclinical models that are representative of the TNBC biology encountered at the time of adjuvant radiotherapy is crucial for testing new strategies to optimize RT in this patient population that is at high risk of relapse and death from disease (20, 21). Here, we tested our hypothesis that ATR inhibition would radiosensitize TNBC in patient-derived xenograft (PDX) models established from primary tumors of patients enrolled onto MC1137, the Breast Cancer Genome Guided Therapy Study (BEAUTY) (22). As part of BEAUTY, tumor biopsy samples were obtained for exome sequencing, RNA sequencing, and implantation into immune-deficient mice for PDX models. Importantly, resistant residual tumor samples from patients who did not achieve a pathologic complete response were also obtained at the time of surgical resection following standard of care taxane and anthracycline-based neoadjuvant chemotherapy for characterization and PDX development. Patients with residual TNBC after neoadjuvant chemotherapy are at particularly high risk locoregional and distant relapse and may be ideally suited for clinical trials aimed at enhancing the efficacy of adjuvant RT. Our results in these highly clinically relevant models indicate that the ATR inhibition-radiotherapy combination is a promising tumor-specific intensification strategy for TNBC.
Materials and Methods
PDX generation
The details of BEAUTY, a prospective institutional review board-approved preoperative chemotherapy clinical trial, have previously been reported (23). In brief, patients age 18 years or older with stage I-III breast cancer, 1.5 cm in size or larger, and being recommended for preoperative chemotherapy by their treating physicians were eligible. Following informed written consent, patients received 12 weeks of weekly paclitaxel (with trastuzumab for human epidermal growth factor receptor 2-positive [HER2+] disease) followed by four cycles of an anthracycline-based regimen. Baseline ultrasound-guided percutaneous tumor biopsies and tumor tissue collected after preoperative chemotherapy from surgery were injected into six to eight week old female nonobese diabetic (NOD-SCID) (NOD.CB17-Prkdcscid/J) mice or NOD.Cg-PrkdcscidIl2rgtm1Wj1/SzJ (NSG) mice purchased from Jackson Laboratories (Bar Harbor, ME) for PDX generation, as previously described (22, 23). Histology was confirmed by hematoxylin and eosin and immunohistochemical stains.
Cell lines, Tissue culture, and ATR inhibitor
Established TNBC cell lines MDA-MB-231 and BT-549, and the human non-tumorigenic mammary gland epithelial cell line, MCF10A, were obtained from the American Type Culture Collection (ATCC). The HCC1806 TNBC cell line was a gift of Fergus Couch (Mayo Clinic, Rochester, MN). The cell lines were authenticated by short tandem repeat analysis (24) performed at the Mayo Clinic. MDA-MB-231 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) and 100 units/ml of penicillin/streptomycin. BT-549 and HCC1806 cell lines were cultured in RPMI 1640 Medium with 10% fetal bovine serum (FBS) and 100 units/ml of penicillin/streptomycin. MCF10A cells were cultured in Mammary Epithelial Cell Growth Medium (MEGM) supplemented with the MEGM Bulletkit (catalog no. CC-3150) from Lonza (Basel, Switzerland). All cell lines were kept at 37 °C under 5% CO2.
VX-970, an agent proprietary to EMD Serono (Darmstadt, Germany) was provided by the Division of Cancer Treatment and Diagnosis, National Cancer Institute, (Rockville, MD). VX-970 was dissolved in D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) as 1 mM stock solution.
To assess colony formation, appropriate numbers of cells were counted and seeded in six well plates, and treated with DMSO control or VX-970 (80 nM) 1 hour prior to RT. Cells were then incubated with and without the drug for an additional 17 hours, rinsed with phosphate buffered saline (PBS) and incubated in fresh media for 7–14 days to allow for colony formation. After staining with Coomassie® Blue, colonies with ≥50 cells were quantified, and survival analysis was performed (25). Cell survival was assessed for up to two logs of cell kill with RT alone to ensure accuracy of measurements. Cell survival curves were fitted with the linear quadratic model using GraphPad Prism.
Immunoblotting, immunofluorescence, and cell cycle analysis
(ab104306), Rad51(ab133534), Cdc25A (ab989) and PKAP (ab70369) antibodies were purchased from Abcam (Cambridge, Massachusetts); PChk-1 (Ser345) antibody (2348S) was purchased from Cell Signaling Technology (Beverly, Massachusetts); γH2AX antibody (05–636) was purchased from Merck Millipore (Billerica, Massachusetts).
For Western blot analysis, cells were first harvested and lysed with NETN buffer (150mM NaCl, 20mM Tris-HCl, pH 8.0, 0.5mM EDTA). After mixing with 6x Laemmli Loading Buffer (300mM Tris pH 6.8, 60% Glycerol, 12% SDS, 0.03% Bromophenol Blue), all samples were boiled at 95 °C for 10 minutes. Samples were then analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer to a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% milk, the membrane was incubated with indicated primary antibodies and horseradish peroxidase-conjugated secondary antibody followed by chemiluminescent detection.
For immunofluorescence, cells were cultured on coverslips, fixed with 3% paraformaldehyde, permeabilized with 0.5% triton x-100, and stained with the indicated primary antibodies. The cells were then stained with fluorescent conjugated secondary antibody for visualization and analysis.
For cell cycle analysis, logarithmically proliferating MDA-MB-231, BT-549, HCC1806, and MCF10A cells were incubated with VX-970 or vehicle one hour prior to 10 Gy or mock treatment and then incubated for an additional 23 hours. Cells were then released by trypsinization and cell cycle analysis was performed using fluorescence-activated cell sorting (FACS), as previously described (26).
In Vivo PDX studies
All procedures of animal studies were performed according to National Institutes of Health guidelines and approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC) and Biosafety Committee. PDX tumors grown to 1 cm in diameter were resected and manually disaggregated into a single cell suspension using a 1-cc syringe. Two million tumor cells suspended in 100 μL of matrigel and PBS were then injected into the hind-leg of 4–5 week old female athymic nude mice (Hsd: Athymic Nude-Foxn1nu from Envigo, Indianapolis, IN). The hind-leg was selected to minimize organ exposure from RT. Once the tumors reached 70 mm3 in size, mice were randomized into the following four groups: 1) sham radiotherapy (RT) + TPGS vehicle, 2) RT (2 Gy x 5 days) + TPGS vehicle, 3) VX970 (60 mg/kg) + sham RT, and 4) RT (2 Gy x 5 days) + VX-970 (60 mg/kg) administered one hour prior to RT daily for five days. VX-970 or vehicle was delivered by oral gavage. VX-970 or vehicle was administered one hour prior to each fraction. RT was delivered to the hind leg of anesthetized mice immobilized with a plastic restraint through a single Cs137 lateral beam while the remainder of the body was shielded with lead. Daily fractions of 2 Gy were administered in order to mimic the daily dose fractionation used in the clinic. All mice were observed daily and tumors were measured thrice weekly. Animals were monitored until the tumors became ulcerated, erythematous, impaired movement or reached 2000 mm3, at which time the mice were euthanized.
For pharmacodynamic assessments, mice with established tumors were randomized and treated as above (3 mice per group). Animals were then euthanized on the fifth day, four hours after the final RT fraction and PChk-1 (Ser345) was assessed using immunoblot.
Ex Vivo Functional Assessments in PDX models
After growing TNBC PDX tumors to 1 cm in diameter, tumors were resected and a single cell suspension was made. In order to assess the functional status of HR, cells were irradiated ex-vivo with 10 Gy or mock treated and four hours later were mounted on coverslips, fixed with 3% paraformaldehyde, permeabilized with 0.5% triton x-100, and stained with the RAD51 and Geminin primary antibodies noted above(11, 27). Geminin staining cell nuclei were analyzed for the formation of RAD51 foci. DNA double strand breaks (DSBs) are repaired by HR primarily in the S and G2 phases of the cell cycle when a sister chromatid is available to serve as a template for error-free repair. RAD51 foci formation were only assessed in nuclei that co-stained with geminin, which is expressed S and G2, in order to control for potential differences in tumor proliferation. In addition, staining with a human-specific antibody to geminin enabled avoidance of contamination of the analysis with murine cells(27). The formation and resolution of γH2AX was similarly assessed.
Statistical analyses
In vitro data are presented as the mean +/− standard error of the mean (SEM) from three or more experiments. Two-tailed Student t tests were used to measure statistical differences in percent of cells with more than 5 foci in immunofluorescence at each time point and cell cycle experiments at each phase of the cell cycle between the RT and VX970 + RT groups, the primary comparison of interest. For in vivo studies, estimates of time to tumor doubling were made using Kaplan-Meier survival method with group associations made using the log-rank test. All statistical tests were two-sided, and a p value of < 0.05 was considered statistically significant.
Results
The ATR inhibitor VX-970 preferentially sensitizes TNBC cells to radiotherapy
VX-970 is a potent inhibitor of ATR (Ki <200pM) that is highly selective over other phosphatidylinositol 3’kinase-related kinases (28). To begin testing our hypothesis that VX-970 would be an effective radiosensitizer of TNBC, we initially selected cell lines representing varying subtypes of TNBC (MDA-MB-231, HCC1806, and BT-549) for investigation in clonogenic survival assays (29). Administration of VX-970 one hour prior to RT significantly decreased the surviving fraction of all three TNBC cell lines, with the most robust effect noted in MDA-MB-231 (Fig. 1 A-C). In comparison, the non-cancerous human breast epithelial cell line, MCF10A, was sensitized to lesser extent (Fig. 1 D). Of note, little to no cell killing was observed when either TNBC or MCF10A cells were treated with VX-970 alone at the same dose that achieved significant radiosensitization (Supplementary Fig.1). These results suggested that ATR inhibition may be an attractive radiosensitizing strategy, with greater sensitization of TNBC over normal cells within the irradiated target volume, and little single agent cytotoxicity at the dose required for radiosensitization outside of the irradiated target volume.
Figure 1.
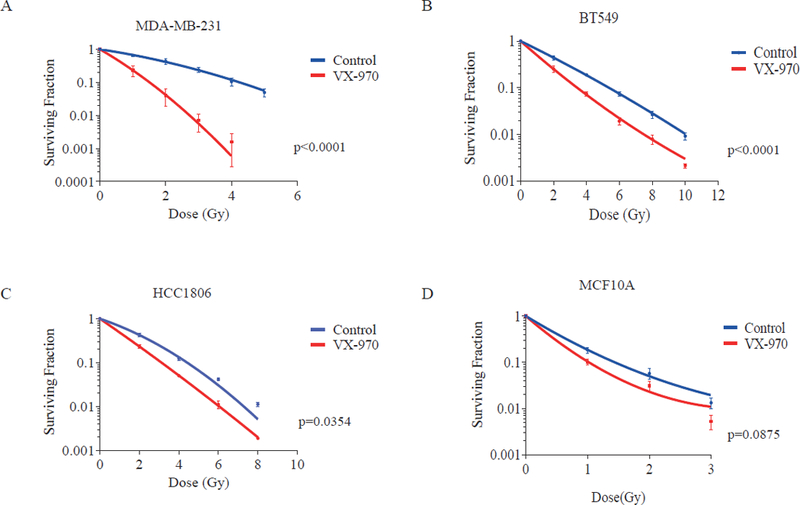
The ATR inhibitor, VX-970, preferentially sensitizes TNBC cells to RT. Clonogenic assays were used to assess the surviving fraction of TNBC cell lines MDA-MB-231 (A), BT-549 (B), and HCC1806 (C), and the normal breast epithelial cell line, MCF10A (D), following RT or RT plus VX-970 (80 nM). For clonogenic assays, cells were trypsinized, plated, allowed to adhere for 4 hours, and treated with vehicle or VX-970 1 hour prior to exposure to varying doses of RT. After 16 hours cells were washed and cultured for 14 days, after which the surviving fraction was assessed. Data are presented as the mean ± SEM from three independent experiments.
VX-970 delays resolution of RT-induced DNA DSBs in TNBC cells
The DNA DSB is the lethal lesion caused by RT, with a single unrepaired DSB potentially resulting in cell death. However, for every DSB, 25 single strand breaks are induced by RT (9). In proliferating cells, when replication forks encounter single strand DNA lesions, ATR plays a pivotal role in preventing fork collapse and subsequent conversion of single strand breaks to more lethal DNA DSBs. Phosphorylation of the histone H2AX (γH2AX) and recruitment of 53BP1 to DNA damage sites are key early events in the DDR that lead to the recruitment of a number of other mediators and effectors of DNA DSB repair. Ongoing DNA repair activity can be assessed indirectly by measuring the formation and resolution of γH2AX and 53BP1 foci, with delays in foci resolution correlating with decreased repair (30). As shown in figure 2A-B, we observed no significant difference in 53BP1 or γH2AX foci at 24 hours when MCF10A cells were pre-treated with VX-970 prior to RT, compared with control. However, significantly more γH2AX foci and 53BP1 foci persisted at 24 hours in BT-549 TNBC cells pre-treated with VX-970, indicative of persistent unrepaired DNA damage (Fig. 2C-D), suggesting that inhibition of ATR may have a greater functional impact on DNA DSB repair in TNBC cells compared with normal cells.
Figure 2.
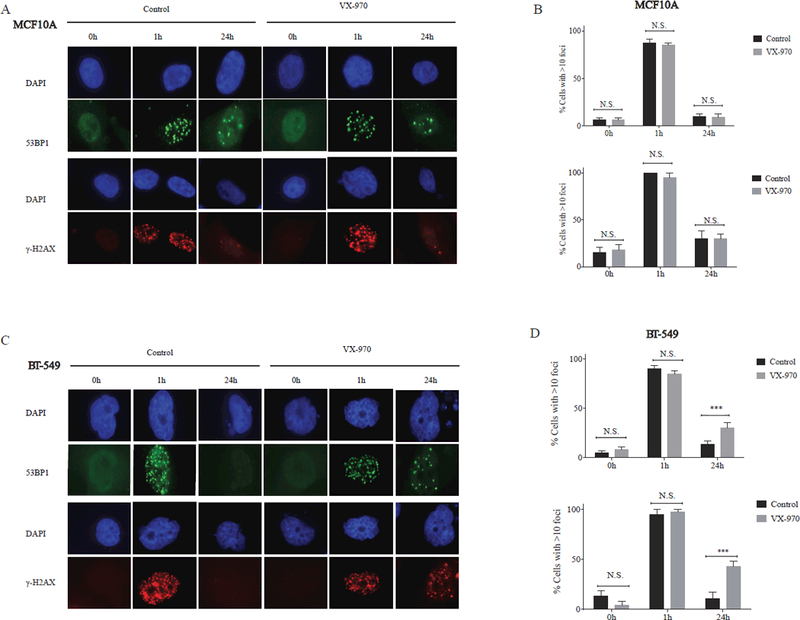
VX-970 delays resolution of RT-induced DNA double strand breaks in TNBC cells but not normal breast epithelial cells. MCF10A (A) and BT-549 (C) cells were pretreated with vehicle (DMSO) or VX-970 (80 nM) for one hour prior to exposure to 2 Gy and stained for ƔH2AX and 53 BP1 foci using immunofluorescence. (B, D) The percentage of cells with > 10 ƔH2AX and 53BP1 nuclear foci was quantified at 1 hour and 24 hours following 2 Gy. Bars represent the mean ± SEM.
VX-970 abrogates radiotherapy-induced cell-cycle arrest
We next tested whether VX-970 could override the cell-cycle arrest induced by RT in TNBC. As expected, RT caused an accumulation of the G2-M population in all TNBC cell lines and MCF10A normal breast epithelial cells (Fig. 3A-D). During oncogenesis, mutations may be acquired that enable cancer cells to bypass cell cycle checkpoints. For example, TNBC is characterized by a high frequency of inactivating p53 mutations, gain of MDM2, and Rb1 loss. These alterations result in deficient G1 checkpoint control, potentially leading to greater reliance on the ATR-mediated G2-M checkpoint in order to avoid premature mitotic entry and mitotic catastrophe after DNA damage (7, 31, 32). Here, the addition of VX-970 one hour prior to RT significantly reduced RT-induced G2-M accumulation in MDA-MB-231, BT-549, and HCC1806 TNBC cell lines (all p<0.01, Fig. 3A-C). The greatest absolute reduction in RT-induced G2-M accumulation by VX-970 was seen in MDA-MB-231 (Fig. 3A), the TNBC line that also exhibited the most robust ATR inhibitor-induced radiosensitizing effect (Fig. 1A). In comparison, VX-970 had the least impact on RT-induced cell cycle checkpoints in non-cancerous MCF10A breast epithelial cells (Fig. 3D). These results suggest that non-malignant cells may be less sensitive to the cell cycle checkpoint abrogating effects of ATR inhibition after RT than TNBC cells.
Figure 3.
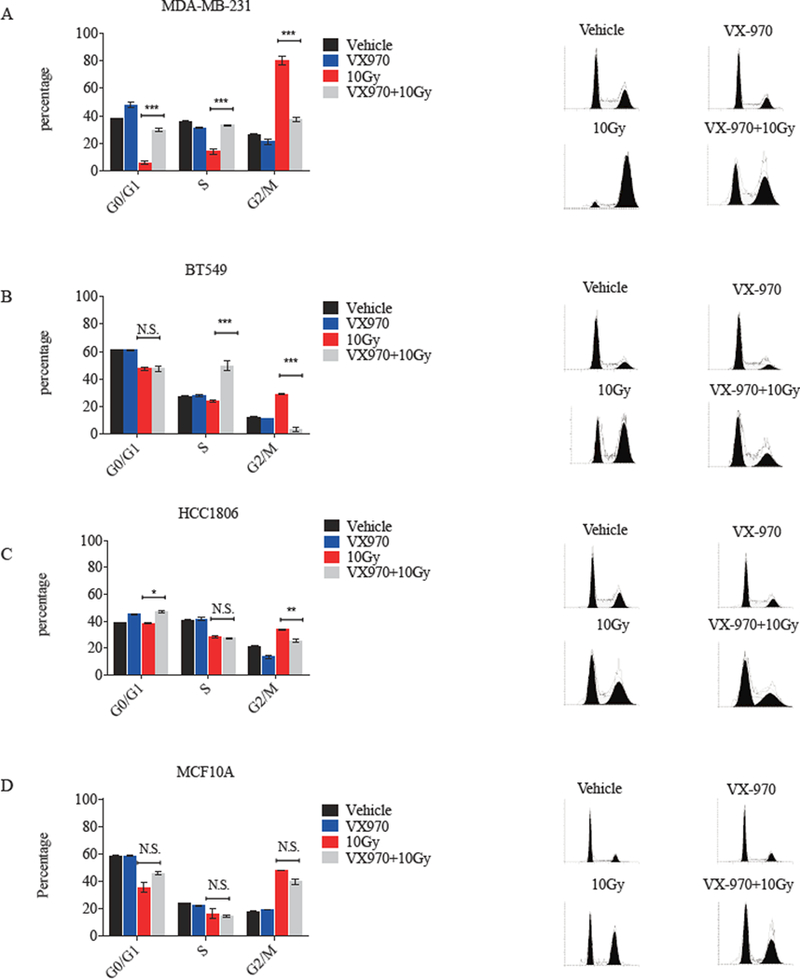
VX-970 disrupts RT-induced cell-cycle checkpoints in TNBC cells. MDA-MB-231 (A), BT-549 (B), HCC1806 (C), and MCF10A cells (D) were treated with vehicle, VX-970 (80 nM), 10 Gy, or VX-970 one hour prior to 10 Gy and analyzed by flow cytometry after 24 hours. Bars represent mean ± SEM. P values are shown for the comparison of RT alone versus VX-970 + RT.
VX-970 blocks ATR-mediated Chk1 phosphorylation and RT-induced CDC25A degradation
Chk1 is the primary substrate of ATR and the observation that VX-970 abrogates RT-induced cell cycle arrest in TNBC cells suggests that VX-970 is inhibiting the ATR-Chk1 pathway. To evaluate the impact of VX-970 on RT-induced ATR-Chk1 pathway signaling in TNBC and normal breast epithelial cells, we assessed ATR-mediated Chk1 phosphorylation (Ser345) and CDC25a levels, given that these have been proposed as potential biomarkers of response to ATR inhibition (33). As expected, RT induced the phosphorylation of Chk1 (Fig 4A-D). However, phosphorylation of Chk1 was induced to a lesser extent in the non-cancerous human breast epithelial cell line MCF10A relative to the three TNBC lines, suggesting that TNBC cells may have a greater dependence on the ATR-Chk1 pathway signaling for cell cycle checkpoint and DNA repair. When TNBC cells were treated with VX-970, there was a decrease in RT-induced phosphorylation of Chk1 but not total Chk1 protein levels, suggesting that ATR kinase activity was being inhibited in these lines (Fig. 4A-D).
Figure 4.

VX-970 disrupts ATR pathway DNA damage signaling. MDA-MB-231 (A), BT-549 (B), HCC1806 (C), and MCF10A cells (D) were pretreated with vehicle (DMSO) or VX-970 (80 nM) for one hour and either harvested (no IR) or exposed to IR (10 Gy) and harvested at the indicated time points. Cell lysates were then immunoblotted for the indicated antigens.
CDC25a is a substrate of Chk1 kinase that is targeted for degradation by the proteasome upon Chk-1 mediated phosphorylation. Elevated CDC25a expression has previously been reported to be associated with sensitivity to ATR inhibition (33). Here, we observed that CDC25a was expressed in MDA-MB-231, BT-549, HCC1806 and MCF10A cells (Fig. 4 A-D). As predicted by the RT-induced activation of Chk1, there was a decrease in CDC25a following RT in MDA-MB-231 cells (Fig. 4A). This RT-induced reduction in CDC25a expression was abrogated by VX-970, consistent with the observation that VX-970 causes progression through the G2/M checkpoint after RT (Fig. 3A) and radiosensitization (Fig. 1A) in that cell line. In BT-549 cells, VX-970 also increased CDC25a levels at 24 hours, but not at 4 hours after RT (Fig. 4B). In contrast, CDC25a levels were not markedly impacted by VX-970 in HCC1806 or MCF10A cells (Fig. 4C-D), consistent with the observation of less override of RT-induced G2-M arrest in these two lines (Fig. 3C-D).
VX-970 sensitizes PDX models established from chemosensitive and chemoresistant TNBC in the clinic to radiotherapy
We next evaluated four Mayo Clinic (MC) TNBC PDX models (MCTNBC1, MCTNBC2, MCTNBC3, and MCTNBC4) for sensitivity to VX-970, RT, or combination therapy, in vivo. MCTNBC1 and MCTNBC3 were previously generated from pre-treatment TNBC biopsy specimens of two patients enrolled on the preoperative chemotherapy clinical trial, BEAUTY, that went on to achieve a pathologic complete responses to doxorubicin, cyclophosphamide, and paclitaxel neoadjuvant chemotherapy. In contrast, MCTNBC2 and MCTNBC4 were generated from the chemoresistant surgical specimens of two other unique patients who had significant residual disease following the same standard of care neoadjuvant chemotherapy regimen. The treatment schema for the assessment of efficacy of VX-970, RT, or the combination is illustrated in figure 5A.
Figure 5.

VX-970 sensitizes TNBC PDXs to RT (A) Schema of PDX trial. RT (or sham treatment) was delivered in five daily fractions of 2 Gy and vehicle or VX-970 (60 mg/kg) was administered one hour prior to each fraction by oral gavage. (B) Mean tumor size over time is plotted for each group. (C) Survival without tumor doubling is plotted for each group. P-values were obtained using the log rank test and are shown for the comparison of all four groups and the primary comparison of interest, RT vs VX-970 + RT.
Consistent with our in vitro observations in established TNBC cell lines, there was no significant prolongation of tumor doubling time in vivo with VX-970 alone in MCTNBC1, MCTNBC2, or MCTNBC4 at the dose and schedule administered (Fig. 5B-C). For the paired comparison of VX-970 vs control, the median time to tumor doubling was 5 days vs. 5 days for MCTNBC1 (p = 0.353), 8 days vs. 10 days for MCTNBC2 (p = 0.308), and 4 vs 7 days for MCTNBC4 (p = 0.770). Compared with control, VX-970 prolonged the tumor doubling time of MCTNBC3 but the absolute effect was small (9 vs 12 days, p = 0.0235) and no longer significant when adjusted for multiple comparisons using the Bonferroni correction (p = 0.0705).
We next analyzed whole exome sequencing data from tumor specimens used to establish the four PDX models for previously proposed ATR inhibitor biomarkers of response. Single nucleotide variants, insertions and deletions, and copy number alterations identified are shown in supplementary tables 1-4. Surprisingly, the lack of single agent activity was observed despite the presence of alterations predictive of ATR inhibitor sensitivity in other models including mutations in p53 (MCTNBC2, MCTNBC3, MCTNBC4), ARID1A (MCTNBC4), gains in Myc (MCTNBC1, MCTNBC2, MCTNBC4), and gains in CCNE2 (MCTNBC4) (34).
In contrast, pre-treatment with VX-970 profoundly impacted the sensitivity of three of four TNBC PDXs to RT (Fig. 5). For MCTNBC1, MCTNBC2, and MCTNBC4 the median time to tumor doubling was 17 vs 42 days (p = 0.0018), 18 vs 38 days (p<0.0001), and 14 vs 56 days (p=0.013) for the RT and VX-970 + RT groups, respectively. MCTNBC3 was the most radiosensitive PDX model. In that model, the median time to tumor doubling was 46 vs 86 days for the RT and VX-970 + RT groups, respectively, but was not significantly different (p=0.697). Individual animal tumor volume data is displayed in supplementary figure 2. The combination of VX-970 + RT was well tolerated, with minimal impact on animal weight relative to vehicle gavage (Supplementary Fig.3).
Pharmacodynamic readouts and impact of HR
In order to assess the on-target effects of VX-970 in vivo, we harvested tumors of mice randomized and treated according to the schema in figure 5A four hours after the final treatment on day 5. As expected, tumors from animals randomized to fractionated RT exhibited greater phosphorylation of Chk1, whereas Chk1 phosphorylation was effectively suppressed in mice treated with the combination of VX-970 + RT (Fig 6A). Next, as a readout of DNA repair, we assessed the formation and resolution of γH2AX foci in TNBC PDX tumor cell nuclei, ex-vivo, after exposure to either RT alone or VX-970 + RT. Consistent with our findings in established TNBC cell lines, there was a significant delay in the resolution of γH2AX foci at 24 hours in irradiated PDX cells pre-treated with VX-970 (Fig. 6 B-C shown for MCTNBC2), suggesting that the presence of VX-970 leads to greater unrepaired DNA damage and greater efficacy of the combination observed in vivo (Fig. 5 B-C).
Figure 6.
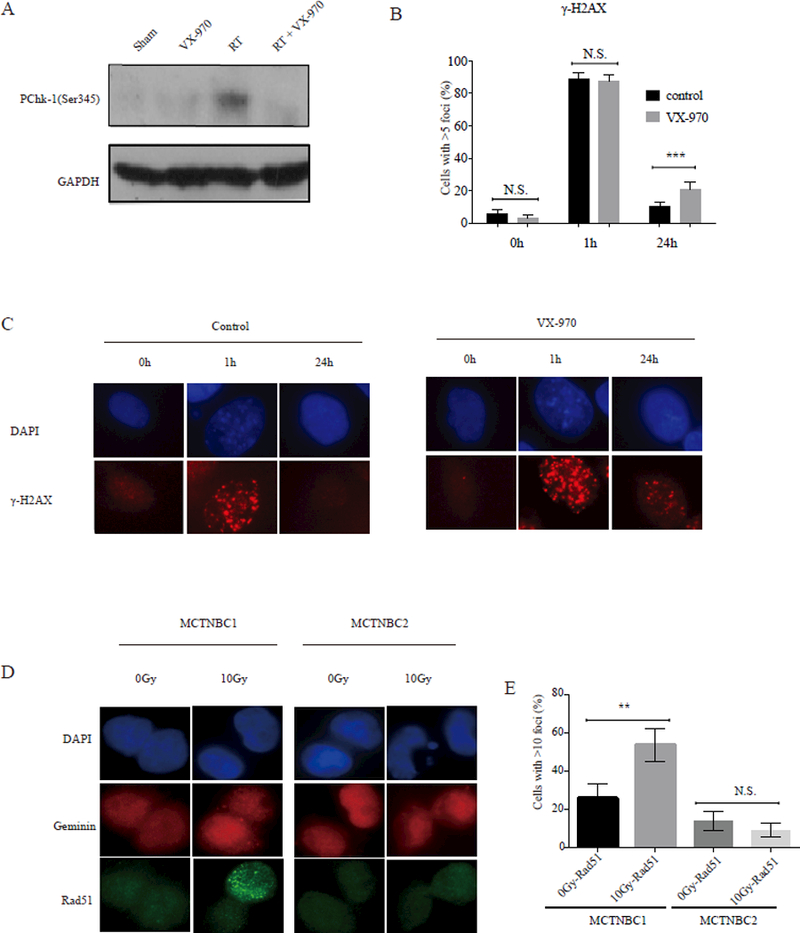
DNA damage signaling and repair in TNBC PDXs treated with VX-970, RT, or the combination (A) Western blotting analysis of phosphorylated Chk-1 (pChk-1) following treatment with either sham RT, RT (2 Gy x 5 days), VX970 (60 mg/kg daily for five days) or RT + VX-970 administered one hour prior to daily fractionated RT administered over five days, as shown in the schema in figure 5A. MCTNBC1 tumor samples were harvested four hours after the last radiation treatment on day five. Samples were pooled from three mice per group. (B, C) TNBC PDX cells were pretreated, ex-vivo, with vehicle (DMSO) or VX-970 (80 nM) for one hour prior to exposure to 2 Gy and stained for ƔH2AX using immunofluorescence at the indicated time points. (B) The percentage of nuclei with > 5 ƔH2AX foci was quantified at 1 hour and 24 hours following 2 Gy. Bars represent mean ± SEM. All statistical tests were two-sided. (D) MCTNBC2 cells harboring a BRCA1 mutation were irradiated ex-vivo with 0 or 10 Gy and geminin staining cell nuclei were analyzed four hours later for the formation of RAD51 foci. MCTNBC1 is also shown here as a positive control. (E) The percentage of geminin staining nuclei with > 5 RAD51 foci was quantified. Bars represent mean ± SEM.
Emerging evidence suggests that TNBC may be further classified based on the status of HR, with potentially important treatment implications (11, 35, 36). Therefore, we sought to characterize the status of HR as a response biomarker in the PDX models by analyzing the available exome data of the corresponding patient tumors for germ-line and/or somatic alterations in a list of 95 genes known to be effectors or regulators of HR and highly correlated with HR function in breast cancer (11). Of note, MCTNBC2 was identified as harboring a BRCA1 nonsense mutation (Q248*).
Because competent HR can be re-acquired in tumors with BRCA1/2 mutations via a number of mechanisms (37–43) we also employed an ex vivo RAD51 foci assay to assess the functional status of HR in that PDX model(11). The recruitment of RAD51 to DNA damage sites is a crucial step in HR that is dependent on BRCA1, BRCA2, and the integrity of the entire HR pathway (11). After generation of a single cell suspension, MCTNBC2 cells were irradiated or mock-treated and analyzed for the formation of RT-induced RAD51 foci. MCTNBC1, which harbored no HR gene alterations, was similarly treated as a positive control. As demonstrated in Figure 6 D-E, a substantially increased fraction of MCTNBC1 nuclei demonstrated RAD51 foci four hours following RT compared to baseline, suggesting proficiency of the HR pathway in that PDX line. In contrast, there was minimal RT-induced RAD51 foci formation in MCTNBC2 nuclei. These results suggested that HR function remained dysregulated in MCTNBC2, as predicted by the pathogenic BRCA1 mutation in the corresponding human tumor.
Collectively, these data suggested that the combination of VX-970 + RT is efficacious in both HR proficient and HR deficient PDX models.
Discussion
In this study, the selective ATR inhibitor, VX-970, preferentially sensitized TNBC cells to RT by abrogating RT-induced cell cycle checkpoints and inhibiting DNA DSB repair. In vivo, when administered one hour prior to RT, VX-970 significantly delayed tumor growth in TNBC PDX models previously established in the context of a NAC clinical study. Furthermore, this approach was not only highly effective in PDX models derived from pre-treatment biopsy specimens, but also PDXs derived from chemotherapy resistant TNBC (i.e. tumors that grew from a resected surgical specimen after 20 weeks of neoadjuvant chemotherapy), a patient population in whom the risk of locoregional recurrence is unacceptably high even after mastectomy and conventional postmastectomy RT (44).
The tumor-specific radiosensitization of TNBC cells, relative to normal breast epithelial cells, is consistent with prior studies demonstrating that common molecular features of TNBC increase dependence on ATR in the cellular response to DNA damage. Indeed, a recurring defect in TNBC is loss of G1 cell cycle checkpoint control resulting from mutations in TP53, the most frequently altered gene in TNBC, as well as MDM2 gain and Rb loss(7). The consequences of these alterations include greater reliance on ATR-mediated intra-S and G2/M checkpoints following DNA damage (13, 45, 46). Moreover, TNBC is characterized by high levels of oncogene induced replication stress resulting from frequent amplification of Myc and Cyclin E1, which was also reflected in copy number analyses of the TNBC PDX models examined here(7). Myc and Cyclin E1 amplification drive cells to enter S phase, even in the presence of DNA lesions such as RT-induced single strand breaks, resulting in replication fork stalling and ATR activation (47, 48). In the absence of ATR, stalled replication forks may collapse, leading to more dangerous replication-associated double strand breaks and cell death (48, 49). Consistently, three PDX models that benefitted from combination therapy in our study harbored mutations in p53, and three had gains in Myc.
Finally, emerging data suggests that up to 40% of TNBCs may have functional deficiencies in HR DNA double-strand break repair due to mutations in BRCA1, BRCA2, PALB2, ATM, and other genetic or epigenetic alterations in the HR pathway (11, 36, 50). HR status has already emerged as an important biomarker of response to DNA repair targeted therapy in breast cancer with the poly (ADP-ribose) polymerase (PARP) inhibitor, olaparib, recently being shown to improve outcomes compared with standard chemotherapy in patients with germline BRCA1 and BRCA2-associated metastatic breast cancer (51). HR is not only vital for DNA DSB repair, but also plays an important role in the cellular response to replication-associated DNA damage. Inhibition of HR results in increased replication stress and ATR-mediated signaling, and ATR inhibition has previously been reported to preferentially target HR deficient cells, enhancing their sensitivity to DNA damage (14, 52, 53). Of note, MCTNBC2, a PDX harboring a functional BRCA1 nonsense mutation, significantly benefitted from combination therapy. However, it is noteworthy that we also observed significant tumor growth delay when VX-970 was administered prior to RT in HR proficient models.
We did not observe marked activity of VX-970 monotherapy in vitro or in vivo at the doses utilized. However, we cannot rule out the possibility that other doses or treatment schedules would have resulted in greater single agent effects. Nevertheless, VX-970 administered at these doses significantly sensitized established TNBC cell lines and TNBC PDX models to RT. Collectively, our data suggest potential broad, tumor specific applicability of this combination in TNBC.
Recently, large prospective clinical trials in women with early stage breast cancer have revealed that the addition of regional nodal irradiation to whole breast or chest wall irradiation resulted in a greater absolute reduction in distant metastases than locoregional recurrences (4–6). The implication from these studies is that clinically significant locoregional recurrences may go undetected or be only detected after a distant relapse has already occurred in a subset of women with non-metastatic breast cancer. Therefore, a novel strategy that enhances the efficacy of radiotherapy may not only improve locoregional control, but also significantly reduce the risk of systemic dissemination and death from TNBC.
Neoadjuvant chemotherapy has ushered in a paradigm shift in breast cancer management given the ability to improve the likelihood of breast conserving surgery and the substantial long-term prognostic information obtained at the time of surgery. Standard taxane and anthracycline-based preoperative chemotherapy result in pathologic complete response (pCR) rates that approach 50% in TNBC (54). Furthermore, patients with a pCR exhibit excellent locoregional and distant control (55). In contrast, TNBC patients with residual disease after neoadjuvant chemotherapy (i.e. chemoresistant disease) have unacceptably high rates of locoregional recurrence and distant relapse despite aggressive subsequent local and systemic therapies with a risk that approaches 50% in some studies (44, 55, 56). These data suggest that residual TNBC identified at the time of surgery may be cross-resistant to RT and not adequately addressed with conventional adjuvant RT strategies. Thus, this subset of TNBC may be best suited for investigation of novel RT intensification strategies aimed at overcoming therapeutic resistance. Promisingly, MCTNBC2 and MCTNBC4, the two PDXs established from chemoresistant surgical specimens and potentially most representative of the disease biology encountered at the time of adjuvant radiotherapy in these patients, significantly benefitted from the ATR inhibitor/RT combination. Our plan is to test this novel strategy in a clinical trial of patients with residual TNBC after preoperative chemotherapy.
Supplementary Material
Acknowledgements
This work was supported in part by the American Society for Radiation Oncology, the National Cancer Institute of the National Institutes of Health under Award Number P50CA116201, K12 HD065987, HALT Cancer at X and the Lead Academic Participating Site (LAPS) program under Award Number 5U10CA180790 (RM).
Financial Support: Robert W. Mutter, the American Society for Radiation Oncology (ASTRO), Grant P50 CA116201 from the National Institutes of Health, K12 HD065987, HALT Cancer at X, and the Lawrence W. and Marilyn Matteson Fund in Cancer Research, Lead Academic Participating Site (LAPS) program under Award Number 5U10CA180790.
Footnotes
The authors have no conflicts of interest to disclose
References
- 1.Nguyen PL, Taghian AG, Katz MS, Niemierko A, Abi Raad RF, Boon WL, et al. Breast cancer subtype approximated by estrogen receptor, progesterone receptor, and HER-2 is associated with local and distant recurrence after breast-conserving therapy. J Clin Oncol. 2008;26(14):2373–8. [DOI] [PubMed] [Google Scholar]
- 2.Kyndi M, Sorensen FB, Knudsen H, Overgaard M, Nielsen HM, and Overgaard J. Estrogen receptor, progesterone receptor, HER-2, and response to postmastectomy radiotherapy in high-risk breast cancer: the Danish Breast Cancer Cooperative Group. J Clin Oncol. 2008;26(9):1419–26. [DOI] [PubMed] [Google Scholar]
- 3.Abdulkarim BS, Cuartero J, Hanson J, Deschenes J, Lesniak D, and Sabri S. Increased risk of locoregional recurrence for women with T1–2N0 triple-negative breast cancer treated with modified radical mastectomy without adjuvant radiation therapy compared with breast-conserving therapy. J Clin Oncol. 2011;29(21):2852–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thorsen LB, Offersen BV, Dano H, Berg M, Jensen I, Pedersen AN, et al. DBCG-IMN: A Population-Based Cohort Study on the Effect of Internal Mammary Node Irradiation in Early Node-Positive Breast Cancer. J Clin Oncol. 2016;34(4):314–20. [DOI] [PubMed] [Google Scholar]
- 5.Whelan TJ, Olivotto IA, and Levine MN. Regional Nodal Irradiation in Early-Stage Breast Cancer. N Engl J Med. 2015;373(19):1878–9. [DOI] [PubMed] [Google Scholar]
- 6.Poortmans PM, Collette S, Kirkove C, Van Limbergen E, Budach V, Struikmans H, et al. Internal Mammary and Medial Supraclavicular Irradiation in Breast Cancer. N Engl J Med. 2015;373(4):317–27. [DOI] [PubMed] [Google Scholar]
- 7.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaspers JE, Rottenberg S, and Jonkers J. Therapeutic options for triple-negative breast cancers with defective homologous recombination. Biochim Biophys Acta. 2009;1796(2):266–80. [DOI] [PubMed] [Google Scholar]
- 9.Ciccia A, and Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cimprich KA, and Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mutter RW, Riaz N, Ng CK, Delsite R, Piscuoglio S, Edelweiss M, et al. Bi-allelic alterations in DNA repair genes underpin homologous recombination DNA repair defects in breast cancer. J Pathol. 2017;242(2):165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campaner S, and Amati B. Two sides of the Myc-induced DNA damage response: from tumor suppression to tumor maintenance. Cell Div. 2012;7(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428–30. [DOI] [PubMed] [Google Scholar]
- 14.Huntoon CJ, Flatten KS, Wahner Hendrickson AE, Huehls AM, Sutor SL, Kaufmann SH, et al. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73(12):3683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18(6):721–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buisson R, Lawrence MS, Benes CH, and Zou L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017;77(17):4567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494(7437):366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cescon DW, Haibe-Kains B, and Mak TW. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc Natl Acad Sci U S A. 2015;112(9):2841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tokunaga E, Yamashita N, Tanaka K, Inoue Y, Akiyoshi S, Saeki H, et al. Expression of APOBEC3B mRNA in Primary Breast Cancer of Japanese Women. PLoS One. 2016;11(12):e0168090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017;17(4):254–68. [DOI] [PubMed] [Google Scholar]
- 21.Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell. 2016;30(1):183. [DOI] [PubMed] [Google Scholar]
- 22.Yu J, Qin B, Moyer AM, Sinnwell JP, Thompson KJ, Copland JA 3rd, et al. Establishing and characterizing patient-derived xenografts using pre-chemotherapy percutaneous biopsy and post-chemotherapy surgical samples from a prospective neoadjuvant breast cancer study. Breast Cancer Res. 2017;19(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goetz MP, Kalari KR, Suman VJ, Moyer AM, Yu J, Visscher DW, et al. Tumor Sequencing and Patient-Derived Xenografts in the Neoadjuvant Treatment of Breast Cancer. J Natl Cancer Inst. 2017;109(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capes-Davis A, Reid YA, Kline MC, Storts DR, Strauss E, Dirks WG, et al. Match criteria for human cell line authentication: where do we draw the line? Int J Cancer. 2013;132(11):2510–9. [DOI] [PubMed] [Google Scholar]
- 25.Franken NA, Rodermond HM, Stap J, Haveman J, and van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–9. [DOI] [PubMed] [Google Scholar]
- 26.Mesa RA, Loegering D, Powell HL, Flatten K, Arlander SJ, Dai NT, et al. Heat shock protein 90 inhibition sensitizes acute myelogenous leukemia cells to cytarabine. Blood. 2005;106(1):318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010;16(24):6159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014;5(14):5674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Surovtseva YV, Jairam V, Salem AF, Sundaram RK, Bindra RS, and Herzon SB. Characterization of Cardiac Glycoside Natural Products as Potent Inhibitors of DNA Double-Strand Break Repair by a Whole-Cell Double Immunofluorescence Assay. J Am Chem Soc. 2016;138(11):3844–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benada J, and Macurek L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules. 2015;5(3):1912–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kriegsmann M, Endris V, Wolf T, Pfarr N, Stenzinger A, Loibl S, et al. Mutational profiles in triple-negative breast cancer defined by ultradeep multigene sequencing show high rates of PI3K pathway alterations and clinically relevant entity subgroup specific differences. Oncotarget. 2014;5(20):9952–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz S, Mayor-Ruiz C, Lafarga V, Murga M, Vega-Sendino M, Ortega S, et al. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol Cell. 2016;62(2):307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stover EH, Konstantinopoulos PA, Matulonis UA, and Swisher EM. Biomarkers of Response and Resistance to DNA Repair Targeted Therapies. Clin Cancer Res. 2016;22(23):5651–60. [DOI] [PubMed] [Google Scholar]
- 35.Lord CJ, and Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20. [DOI] [PubMed] [Google Scholar]
- 36.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Afghahi A, Timms KM, Vinayak S, Jensen KC, Kurian AW, Carlson RW, et al. Tumor BRCA1 Reversion Mutation Arising during Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin Cancer Res. 2017;23(13):3365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng L, Fong KW, Wang J, Wang W, and Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288(16):11135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panier S, and Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15(1):7–18. [DOI] [PubMed] [Google Scholar]
- 40.Boersma V, Moatti N, Segura-Bayona S, Peuscher MH, van der Torre J, Wevers BA, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature. 2015;521(7553):537–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, and Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68(8):2581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–5. [DOI] [PubMed] [Google Scholar]
- 44.Yang TJ, Morrow M, Modi S, Zhang Z, Krause K, Siu C, et al. The Effect of Molecular Subtype and Residual Disease on Locoregional Recurrence in Breast Cancer Patients Treated with Neoadjuvant Chemotherapy and Postmastectomy Radiation. Ann Surg Oncol. 2015;22 Suppl 3:S495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127(5):582–95. [DOI] [PubMed] [Google Scholar]
- 46.Rundle S, Bradbury A, Drew Y, and Curtin NJ. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers (Basel). 2017;9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schoppy DW, Ragland RL, Gilad O, Shastri N, Peters AA, Murga M, et al. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J Clin Invest. 2012;122(1):241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halazonetis TD, Gorgoulis VG, and Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–5. [DOI] [PubMed] [Google Scholar]
- 49.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18(6):721–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turner N, Tutt A, and Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9. [DOI] [PubMed] [Google Scholar]
- 51.Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017. [DOI] [PubMed] [Google Scholar]
- 52.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31(3):318–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krajewska M, Fehrmann RS, Schoonen PM, Labib S, de Vries EG, Franke L, et al. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene. 2015;34(26):3474–81. [DOI] [PubMed] [Google Scholar]
- 54.Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, et al. Impact of the Addition of Carboplatin and/or Bevacizumab to Neoadjuvant Once-per-Week Paclitaxel Followed by Dose-Dense Doxorubicin and Cyclophosphamide on Pathologic Complete Response Rates in Stage II to III Triple-Negative Breast Cancer: CALGB 40603 (Alliance). J Clin Oncol. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Minckwitz G, Untch M, Blohmer JU, Costa SD, Eidtmann H, Fasching PA, et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol. 2012;30(15):1796–804. [DOI] [PubMed] [Google Scholar]
- 56.Masuda N, Lee SJ, Ohtani S, Im YH, Lee ES, Yokota I, et al. Adjuvant Capecitabine for Breast Cancer after Preoperative Chemotherapy. N Engl J Med. 2017;376(22):2147–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.