

Abstract
Objective:
Approximately 30% of epilepsy patients are refractory to existing antiseizure drugs (ASDs). Given that the properties of these patients’ central nervous systems are likely to be altered due to their epilepsy, tissues from rodents that have undergone epileptogenesis might provide a therapeutically-relevant disease substrate for identifying compounds capable of attenuating pharmacoresistant seizures. To facilitate the development of such a model, this study describes the effects of classical glutamate receptor antagonists and 20 ASDs on recurrent epileptiform discharges (REDs) in brain slices derived from the kainate-induced status epilepticus model of temporal lobe epilepsy (KA-rats).
Methods:
Horizontal brain slices containing the medial entorhinal cortex (mEC) were prepared from KA-rats, and REDs were recorded from the superficial layers. CNQX, APV, TTX, or ASDs were bath applied for 20 minutes. Concentration-dependent effects and EC50 values were determined for RED duration, frequency, and amplitude.
Results:
ASDs targeting sodium and potassium channels (carbamazepine, eslicarbazepine, ezogabine, lamotrigine, lacosamide, phenytoin, and rufinamide) attenuated REDs at concentrations near their average therapeutic plasma concentrations. GABAergic synaptic transmission modulating ASDs (clobazam, midazolam, phenobarbital, stiripentol, tiagabine, and vigabatrin) attenuated REDs only at higher concentrations and, in some cases, prolonged RED durations. ASDs with other/mixed mechanisms of action (bumetanide, ethosuximide, felbamate, gabapentin, levetiracetam, topiramate, and valproate) and glutamate receptor antagonists weakly or incompletely inhibited RED frequency, increased RED duration, or had no significant effects.
Significance:
Taken together, these data suggest that epileptiform activity recorded from the superficial layers of the mEC in slices obtained from KA-rats are differentially sensitive to existing ASDs. The different sensitivities of REDs to these ASDs may reflect persistent molecular, cellular, and/or network level changes resulting from disease. These data are expected to serve as a foundation upon which future therapeutics may be differentiated and assessed for potentially translatable efficacy in refractory epilepsy patients.
Keywords: Antiepileptic Drugs, Pharmacoresistance, Brain Slices
1. Introduction
Epilepsy is a neurological disorder characterized by spontaneous recurrent seizures that affects one in 26 individuals worldwide.1 Despite considerable progress in the development of new antiseizure drugs (ASDs), approximately 30% of epilepsy patients are refractory to existing pharmacological treatments.2,3 This pharmacoresistance has been defined by the International League Against Epilepsy (ILEA) as “failure of adequate trials of two tolerated, appropriately chosen and used antiepileptic drug schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom”.4 The central nervous system (CNS) of an epilepsy patient that is resistant to existing ASDs is likely to be fundamentally different from that of an otherwise healthy individual due to a number of potential underlying mechanisms.5 Therefore, the discovery of optimal treatments for these medically refractory seizures is expected to be facilitated by the development and use of animal models that recapitulate this pharmacoresistance and its underlying mechanisms.
Although multiple in-vivo models of pharmacoresistant epilepsy have been developed and are in active use6, in-vitro models of pharmacoresistant seizures can be used to compliment these drug-discovery strategies and may offer several advantages over in-vivo models as it applies to drug screening. For these reasons, in-vitro brain slice models of spontaneous seizure-like events (SLEs) and recurrent epileptiform discharges (REDs), such as the low-Mg2+ induced epileptiform activity model, have been extensively studied.7–9 SLEs, and REDs in particular, in many of these models exhibit varying degrees of pharmacoresistance and altered sensitivities to specific ASDs depending on the model used. Perhaps most intriguing are models that employ brain slices obtained from animals that have undergone the enduring changes in CNS composition and function that occur during epileptogenesis, such as the pilocarpine and kainic acid (KA) models of temporal lobe epilepsy (TLE). Brain slices from these rodents often produce spontaneous SLEs and/or REDs under otherwise normal conditions (i.e. in the absence of hyperexcitable saline or chemoconvulsants)10,11, and they exhibit altered sensitivities to various ASDs and other pharmacological agents.11–18 Therefore, brain slices from rodents that have undergone epileptogenesis might provide an ideal model for the identification of novel compounds capable of attenuating pharmacoresistant seizures from a therapeutically-relevant disease substrate.
Previous experiments from our laboratories have specifically examined modified versions of the high potassium, picrotoxin, and low Mg2+ models where spontaneous SLEs and REDs were recorded from the superficial layers of the medial entorhinal cortex (mEC) in brain slice tissues collected from rats subjected to the multiple low-dose KA model of TLE (KA-rats).11,19 While this earlier work demonstrated that tissues from KA-rats had exaggerated resistances to several ASDs, a more comprehensive evaluation of the sensitivities or resistances to existing ASDs in this model is warranted. Therefore, the work presented here describes the concentration-dependent actions of 20 ASDs in this in-vitro slice model, most of these for the first time. While ASDs that target voltage-gated sodium channels (VGSCs) and potassium channels effectively attenuate REDs in this model, our results clearly show that most ASDs targeting GABAergic synaptic transmission and/or other molecular targets incompletely block REDs at concentrations well beyond their expected therapeutic ranges or have no effects at all. Thus, this in-vitro slice model may be useful for identifying and differentiating novel therapies for refractory epilepsy, and it is actively being used for these purposes in the National Institute of Neurological Disorders and Stroke (NINDS) funded Epilepsy Therapy Screening Program (ETSP: https://www.ninds.nih.gov/Current-Research/Focus-Research/Focus-Epilepsy/ETSP).
2. Methods
2.1. Animals
All experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Utah’s Animal Care and Use Committee. Principles outlined in the ARRIVE guidelines have been considered when planning experiments. Adult male Sprague–Dawley rats weighing 150–200 g (Charles River) were housed in an AAALAC accredited facility in groups no larger than 6 rats per plastic cage with free access to food and water. Animals were allowed to rest for 5–7 days prior to KA administration.
2.2. Kainic Acid administration
Animals were treated with KA (Tocris, 0222) following a modified protocol described previously.11,19 Seizure activity was scored throughout KA administration using the Racine scale.20 KA was administered via i.p. injection with an initial dose of 10 mg/kg. During the first hour, stage 1–3 seizures were generally observed and KA was reduced to 5.0 mg/kg (i.p.) and injected every 30 min until stage 4–5 seizures were first identified. Animals were required to display at least one stage 4–5 seizure every hour for 3.5 h to be included in experiments. After 3.5 h, animals were given an injection of 0.9% saline (1–2 ml) subcutaneously to prevent dehydration and aid in recovery. Animals were then individually housed due to aggressive behavioral characteristics. Animals were allowed to recuperate for 14 days prior to experimentation. As previously reported, 96% of all rats that are subjected to this protocol go on to develop spontaneous recurrent seizures within 15 weeks.21
2.3. Slice Preparation
Two-three weeks after KA treatment, rats were anesthetized using sodium pentobarbital (50 mg/kg, i.p.) and their brains were quickly removed and placed in ice cold, oxygenated 95% O2–5% CO2 sucrose-based Artificial Cerebral Spinal Fluid (ACSF) solution containing (in mM): sucrose (125.0), KCl (3.0), NaH2PO4 (1.2), MgSO4 (2.0), NaHCO3 (26.0), glucose (10.0), and CaCl2 (2.0). The brains were then surgically blocked and glued to the platform of a vibrating blade microtome (Vibratome 3000). Typically, 8–10 horizontal slices (350 µm) per rat were obtained in this fashion. Slices were incubated at room temperature 1–2 h prior to experiments in oxygenated ACSF which contained NaCl (126 mM) instead of sucrose and 10 μM glycine (pH = 7.33–7.38; osmolarity = 290–310 mOsm). Slices were then transferred to a submersion chamber (Slicemate, Scientifica, Inc. UK) and perfused with oxygenated ACSF at a rate of 2.0–2.5 ml/min and maintained at a constant temperature of 31 ± 1 °C. REDs were facilitated with ACSF where the K+ concentration was adjusted to 6 mM, 10 μM glycine was added, and the Mg2+ concentration was lowered to 0.1 mM.11,19
2.4. Field potential recordings and analysis
Extracellular field excitatory postsynaptic potentials (fEPSPs) were recorded from eight brain slices per rodent simultaneously using a Slicemaster recording system (Scientifica, Uckfield, East Sussex, United Kingdom). Borosilicate glass electrodes (World Precision Instruments TW-150) were filled with ACSF and pulled to 1.5–2.5 MΩ resistance using a flaming brown micropipette electrode puller (Sutter Instrument Company Model P 97). A twisted Nichrome/Formvar wire stimulating electrode was used to evoke fEPSPs. The stimulating electrode was placed in the angular bundle fibers while placing the recording electrode in layer II of the mEC. Stimulations were given at a rate of 1 stimulation every 5 seconds using Slice-ISO stimulators (npi electronic GmbH, Tamm, Germany), and fEPSPs were measured using Slice-2A amplifiers (npi electronic GmbH, Tamm, Germany). Spontaneous fEPSPs were recorded using a low pass 1 kHz filter, at a sampling rate of 10 kHz, and recorded in gap-free mode using a Digidata 1550 (Axon Instruments, Union City, CA). Data were acquired using pCLAMP 10.4.2 and analyzed using CLAMPFIT 10.4 software (Axon Instruments, Union City, CA). After 20 minutes of baseline recording of REDs, investigational compounds were applied via bath exchange for 20 minutes followed by a 20-minute washout to assess reversibility.
A graphical representation of the following analysis method is presented in Supporting Information Figure S1. Data were high-pass filtered before analysis (Bessel 8 pole filter, cutoff 5Hz). A threshold search method was used to identify spontaneous REDs (threshold set to no less than 2x baseline noise). CLAMPFIT 10.4 burst analysis with a burst-delimiting interval of 1000–1500 ms was then employed. Burst duration, frequency, and amplitude were averaged and binned in 60 second increments to assess baseline stability of these measures; individual slices that exhibit unstable baselines or drifted >20% over 20 minutes were excluded. The percent change caused by a 20-minute exposure to investigational compounds was assessed using a paired student t test (statistical significance set at p ≤ 0.05). Concentration-response curves were fit to data when the investigational compound was tested at a minimum of 4 concentrations and EC50s were calculated (slope constrained to 1, initial values constrained to 100%, GraphPad Prism version 6, La Jolla California USA, www.graphpad.com). Typically, all data at each concentration tested were derived from a minimum of 5 brain slices obtained from a minimum of 2 rats. When ≥2 concentrations of the same ASD were tested in tissues from the same rat, slices were randomly assigned. In some cases, relatively large data sets (N>12 brain slices) resulted from internal replication studies where results did not differ between independent time-separated experiments and were therefore combined (data not shown).
2.5. Drug preparation
For all experiments described, stock solutions in Dimethyl sulfoxide (DMSO) (0.01–0.10 M) were made fresh on the day of the first experiment, visually inspected for solubility when diluted into ACSF at working concentrations, frozen, and then reused for multiple experiments and applied via bath perfusion. The working concentration of DMSO was kept below 0.01% for each solution. Bumetanide, carbamazepine, clobazam, clonazepam, ethosuximide, felbamate, midazolam, phenobarbital, phenytoin, sodium valproate, and stiripentol were obtained from Sigma. Eslicarbazepine, gabapentin, levetiracetam, rufinamide, tiagabine, and topiramate were obtained from TCI America (Portland, OR, U.S.A.). Lacosamide and ezogabine was obtained from Axon Medchem (Groningen, The Netherlands). Lamotrigine was obtained from AK Scientific (Union City, CA, U.S.A.).
3. Results
Spontaneous REDs were recorded in layer II of the mEC of brain slices obtained from KA-rats. Data were collected from a total of 722 brain slices from 141 KA-rats. In a representative sample of 100 brain slices, REDs consisted of high-frequency population spike discharges superimposed on a slow, negative, field potential wave recurring at 0.29 ± 0.1 Hz. After isolating the high-frequency population spike component of these REDs, the mean durations of the REDs were found to be 566.0 ± 32.0 ms. These descriptive parameters were similar to the late recurrent discharges (LRDs) reported for other low-Mg2+ based models of in-vitro epileptiform activity in the mEC.7,8
3.1. AMPA/Kainate and NMDA receptor antagonists differentially affect REDs
The roles of non-NMDA and NMDA glutamate receptors in mediating REDs were examined by testing their selective antagonists, CNQX and APV, respectively. These data, and a description of the results, are presented in Supporting Information Figure S2.
3.2. Evaluation of the concentration-dependent effects of 20 ASDs on REDs
ASDs from various mechanistic classes were evaluated in order to comprehensively examine the sensitivities and/or resistances of REDs to these commonly used agents. A summary of the concentration-dependent effects of all ASDs tested here on RED duration, frequency, and amplitude is presented in Table 1. An examination of these data reveals that ASDs can produce a variety of qualitatively and quantitatively different effects on these RED parameters. To better illustrate these different effects, data from four representative ASDs with qualitatively different effects are presented in greater detail in Figures 1 through 4 and are described sequentially here.
Table 1.
Summary of ASD effects on RED duration, frequency, and amplitude in layer II of the mEC.
| Generic Name | Conc. (µM) | N | Duration ± SEM (%) |
Frequency ± SEM (%) |
Amplitude ± SEM (%) |
|---|---|---|---|---|---|
| Bumetanide | 10 | 8 | 108.5 ± 12.7 | 83.7 ± 4.3 ** | 102.6 ± 4.1 |
| 30 | 6 | 124.7 ± 20.4 | 78.1 ± 6.7 * | 103.6 ± 3.3 | |
| 100 | 6 | 98.4 ± 6.5 | 95.0 ± 5.1 | 103.0 ± 3.8 | |
| 300 | 7 | 164.3 ± 21.0 * | 74.4 ± 7.4 ** | 117.5 ± 4.5 ** | |
| Carbamazepine | 3 | 11 | 101.6 ± 5.3 | 108.9 ± 8.8 | 99.9 ± 1.8 |
| 10 | 7 | 76.9 ± 8.2 * | 95.9 ± 13.8 | 110.8 ± 3.1 * | |
| 30 | 7 | 37.3 ± 5.4 *** | 97.7 ± 13.5 | 120.9 ± 11.1 | |
| 100 | 6 | 13.4 ± 3.5 *** | 39.7 ± 12.1 ** | 150.2 ± 12.9 * | |
| Clobazam | 10 | 11 | 119.2 ± 17.0 | 110.6 ± 12.1 | 99.9 ± 7.2 |
| 30 | 8 | 408.0 ± 121.5 * | 65.5 ± 12.4 * | 121.1 ± 6.1 * | |
| 100 | 7 | 133.3 ± 31.6 | 74.7 ± 10.7 | 124.3 ± 11.1 | |
| 300 | 7 | 67.7 ± 26.7 | 52.4 ± 7.3 *** | 115.0 ± 10.9 | |
| Eslicarbazepine | 10 | 14 | 105.3 ± 5.7 | 91.7 ± 8.6 | 102.1 ± 3.3 |
| 30 | 10 | 120.4 ± 14.2 | 79.5 ± 6.1 ** | 106.4 ± 4.3 | |
| 100 | 8 | 83.8 ± 13.6 | 54.6 ± 12.2 ** | 114.2 ± 5.4 * | |
| 300 | 11 | 32.5 ± 13.7 *** | 7.9 ± 3.7 *** | 53.5 ± 17.5 * | |
| Ethosuximide | 10 | 5 | 118.5 ± 12.6 | 97.7 ± 3.3 | 103.3 ± 2.6 |
| 30 | 9 | 112.1 ± 8.2 | 110.0 ± 9.0 | 103.2 ± 1.9 | |
| 100 | 6 | 111.6 ± 11.7 | 93.5 ± 8.7 | 101.2 ± 4.0 | |
| 300 | 6 | 106.3 ± 9.2 | 89.8 ± 8.2 | 100.9 ± 1.1 | |
| 1000 | 7 | 133.1 ± 10.5 * | 93.2 ± 4.8 | 100.1 ± 2.8 | |
| Ezogabine | 0.3 | 6 | 91.6 ± 8.5 | 109.7 ± 5.7 | 100.3 ± 2.9 |
| 1 | 8 | 61.1 ± 8.9 ** | 107.1 ± 12.5 | 99.0 ± 5.3 | |
| 3 | 9 | 56.1 ± 10.1 ** | 91.6 ± 15.1 | 99.2 ± 2.7 | |
| 30 | 5 | 6.9 ± 2.8 *** | 4.2 ± 2.2 *** | 43.9 ± 12.1 ** | |
| 100 | 7 | 0.0 ± 0.0 *** | 0.0 ± 0.0 *** | 0.0 ± 0.0 *** | |
| Felbamate | 30 | 7 | 95.8 ± 7.3 | 102.7 ± 15.2 | 84.5 ± 5.6 * |
| 100 | 6 | 73.3 ± 10.5 | 102.3 ± 8.8 | 113.0 ± 4.8 | |
| 300 | 6 | 106.7 ± 24.5 | 95.9 ± 17.3 | 93.6 ± 2.5 | |
| Gabapentin | 30 | 7 | 88.0 ± 8.7 | 88.9 ± 13.0 | 98.3 ± 0.7 |
| 100 | 8 | 107.0 ± 12.3 | 102.6 ± 11.9 | 98.2 ± 3.5 | |
| 300 | 6 | 102.9 ± 10.0 | 87.3 ± 8.3 | 96.1 ± 1.5 | |
| 1000 | 9 | 89.1 ± 9.4 | 100.9 ± 9.6 | 98.5 ± 2.7 | |
| Lacosamide | 10 | 7 | 122.6 ± 14.8 | 74.0 ± 6.5 ** | 103.1 ± 3.7 |
| 30 | 6 | 98.7 ± 9.9 | 79.9 ± 4.7 ** | 100.4 ± 3.0 | |
| 100 | 8 | 70.0 ± 7.9 ** | 74.4 ± 4.8 *** | 103.4 ± 2.4 | |
| 300 | 8 | 56.2 ± 11.6 ** | 43.4 ± 12.8 ** | 126.3 ± 4.4 *** | |
| Lamotrigine | 10 | 8 | 95.2 ± 4.5 | 96.0 ± 4.4 | 97.6 ± 4.4 |
| 30 | 7 | 89.1 ± 7.5 | 77.9 ± 9.3 | 107.4 ± 2.9 * | |
| 100 | 23 | 42.6 ± 4.5 *** | 45.4 ± 4.2 *** | 94.1 ± 2.8 * | |
| 300 | 8 | 3.7 ± 2.4 *** | 12.7 ± 5.5 *** | 61.9 ± 18.8 | |
| Levetiracetam | 3 | 8 | 89.0 ± 7.9 | 96.8 ± 3.9 | 104.9 ± 5.2 |
| 10 | 11 | 94.8 ± 8.3 | 108.5 ± 8.4 | 92.2 ± 1.9 ** | |
| 30 | 7 | 105.7 ± 7.0 | 105.4 ± 4.9 | 91.9 ± 3.3 | |
| 100 | 9 | 102.0 ± 8.8 | 102.9 ± 11.0 | 103.0 ± 4.0 | |
| 300 | 8 | 121.6 ± 12.4 | 98.9 ± 14.0 | 98.6 ± 6.1 | |
| Midazolam | 3 | 8 | 102.1 ± 10.8 | 95.4 ± 11.3 | 102.1 ± 5.4 |
| 10 | 7 | 125.7 ± 19.8 | 108.5 ± 27.2 | 109.2 ± 3.7 * | |
| 30 | 6 | 137.6 ± 16.8 | 69.9 ± 11.5 * | 94.9 ± 6.5 | |
| Phenobarbital | 10 | 14 | 104.4 ± 5.5 | 90.7 ± 7.1 | 102.0 ± 3.0 |
| 30 | 12 | 107.9 ± 9.6 | 87.9 ± 5.8 | 113.7 ± 6.1 * | |
| 100 | 20 | 115.8 ± 10.3 | 84.6 ± 6.1 * | 111.3 ± 3.6 *** | |
| 300 | 14 | 27.5 ± 9.2 *** | 34.2 ± 9.6 *** | 95.7 ± 16.8 | |
| Phenytoin | 10 | 7 | 125.8 ± 35.2 | 80.7 ± 9.0 | 101.7 ± 1.3 |
| 30 | 8 | 124.6 ± 29.9 | 111.0 ± 12.9 | 106.0 ± 5.0 | |
| 100 | 16 | 76.4 ± 12.1 | 49.7 ± 6.6 *** | 100.2 ± 3.4 | |
| 300 | 7 | 41.4 ± 9.8 *** | 41.4 ± 10.6 ** | 106.9 ± 4.2 | |
| Rufinamide | 10 | 6 | 97.6 ± 5.6 | 104.2 ± 12.1 | 98.3 ± 2.4 |
| 30 | 14 | 104.2 ± 8.1 | 83.6 ± 9.6 | 103.4 ± 1.7 | |
| 100 | 10 | 66.2 ± 13.2 * | 88.8 ± 10.5 | 114.4 ± 3.6 ** | |
| 300 | 12 | 111.5 ± 24.6 | 62.1 ± 6.8 *** | 161.1 ± 15.0 ** | |
| Stiripentol | 100 | 7 | 124.6 ± 7.8 * | 86.9 ± 4.8 * | 105.9 ± 2.4 * |
| 300 | 9 | 139.4 ± 8.0 ** | 94.1 ± 13.0 | 110.6 ± 5.5 | |
| Tiagabine | 3 | 6 | 55.5 ± 4.5 *** | 97.7 ± 13.3 | 104.7 ± 3.7 |
| 10 | 9 | 30.8 ± 7.8 *** | 151.0 ± 32.2 | 102.8 ± 5.5 | |
| 30 | 9 | 45.3 ± 7.5 *** | 107.8 ± 8.1 | 98.9 ± 6.1 | |
| 100 | 6 | 53.4 ± 8.2 ** | 62.8 ± 12.2 * | 120.7 ± 7.0 * | |
| Topiramate | 10 | 8 | 104.5 ± 8.3 | 88.6 ± 9.3 | 94.0 ± 5.4 |
| 30 | 10 | 124.2 ± 10.2 * | 90.1 ± 3.0 | 103.3 ± 2.0 | |
| 100 | 9 | 140.9 ± 9.7 ** | 74.6 ± 4.8 *** | 105.1 ± 2.0 * | |
| 300 | 7 | 142.0 ± 16.7 * | 77.1 ± 5.4 ** | 106.3 ± 4.4 | |
| Valproate | 1000 | 6 | 97.2 ± 4.1 | 104.1 ± 6.7 | 100.6 ± 3.7 |
| Vigabatrin | 100 | 7 | 108.9 ± 5.9 | 97.0 ± 7.3 | 105.0 ± 3.1 |
| 300 | 8 | 119.7 ± 15.2 | 78.9 ± 5.3 ** | 103.0 ± 4.1 | |
| 1000 | 9 | 149.6 ± 21.4 * | 75.0 ± 10.3 * | 106.5 ± 1.7 ** | |
| 3000 | 9 | 201.0 ± 59.1 | 31.2 ± 5.5 *** | 105.5 ± 5.1 |
“Conc.” = concentration. All data are represented as mean ± SEM changes from baseline values after a 20 minute ASD exposure (% of baseline). “N” values represent number of independent brain slices from which data was quantified. A paired two-tailed student t-test was used to compare normalized baselines to ASD-mediated parameters after a 20 min drug exposure
= p<0.05
= p<0.01
= p<0.001).
Figure 1:
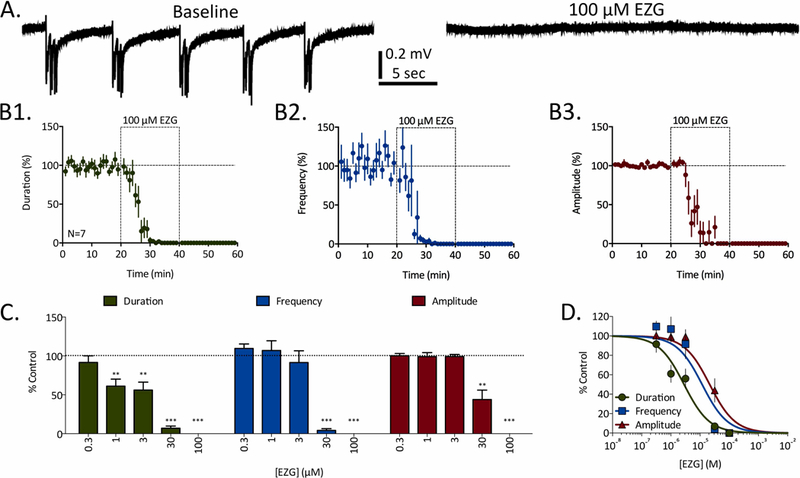
Concentration-dependent inhibition of REDs by ezogabine (EZG). A) Representative traces of REDs recorded before (baseline) and in the presence of 100 µM EZG. B) Time courses for 100 µM EZG’s effects of RED duration (B1, green), frequency (B2, blue), and amplitude (B3, red). Dashed-line boxes from 20 to 40 minutes represent the duration of EZG exposure. Data points represent mean ± SEM for RED parameters in 1-minute bins. C) Concentration-dependent effects of EZG on RED duration (green), frequency (blue), and amplitude (red). Bars represent mean ± SEM of N slices (see Table 1) and * = p<0.05, ** = p<0.01, *** = p<0.001. D) Concentration-response curves fit to data from panel C for EZG’s effects on RED duration (green), frequency (blue), and amplitude (red).
Ezogabine (EZG) was the only ASD of 20 tested that completely blocked REDs at the concentrations tested. Data from a series of experiments testing 0.3–100 µM EZG is presented in Figure 1. As can be observed in representative traces taken both before and after a 20-minute exposure to 100 µM EZG (Fig. 1A), REDs are entirely absent in the presence of this ASD at this concentration. The time courses of EZG’s effects on RED parameters are presented in Figure 1B. When average duration, frequency, and amplitude are quantified in 1-minute increments, the stability of these baseline RED parameters over time can be observed. Upon bath exchange with 100 µM EZG, all three RED parameters are reduced to 0% of their baseline values within 20 minutes (N=7). While lower concentrations produced smaller magnitude effects that were partially reversed after washing EZG from the slices, the complete block observed in the presence of 100 µM EZG did not reverse with washing for 20 minutes. Concentration-response and significance data for this series of experiments are presented in Figure 1C. When these data are fit with sigmoidal concentration-response curves (Fig. 1D), EC50 values can be calculated for all RED parameters; these values were 2.8 µM, 11.8 µM, and 21.4 µM for duration, frequency, and amplitude, respectively.
Although all other ASDs failed to demonstrate a complete block of REDs at the concentrations tested in these experiments, many of these same ASDs demonstrated a differential (e.g. duration versus frequency), and concentration-dependent, attenuation of RED parameters (e.g. carbamazepine, eslicarbazepine, lacosamide, lamotrigine, rufinamide, and phenytoin). The details of this differential attenuation of RED parameters can be observed in the example of carbamazepine (CBZ, Fig. 2). Representative traces taken both before and after exposure to 30 µM CBZ illustrate an attenuation of RED duration with minimal alteration in frequency (Fig. 2A). This differential effect on duration versus frequency can be more clearly seen in the averaged time course data (Fig. 2B); whereas 30 µM CBZ significantly and reversibly attenuated RED duration, no effects were observed on RED frequency over the 20-minute ASD exposure. Concentration-response and significance data presented in Figure 2C illustrates CBZ’s significant attenuation of RED duration at all concentrations ≥ 10 µM, while RED frequency is only significantly attenuated at 100 µM. Sigmoidal concentration-response curves fit to these data were used to calculate the following EC50 values: 28.5 µM, 128.7 µM, and 153.6 µM for duration, frequency, and amplitude, respectively.
Figure 2:
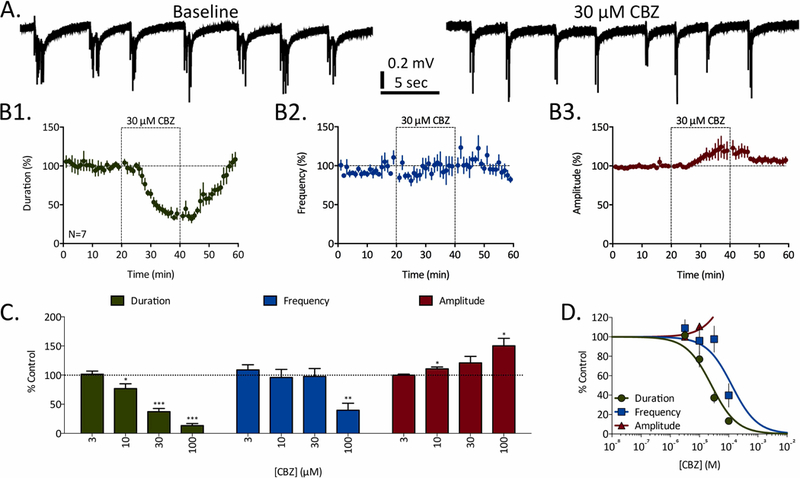
Concentration-dependent inhibition of REDs by carbamazepine (CBZ). A) Representative traces of REDs recorded before (baseline) and in the presence of 30 µM CBZ. B) Time courses for 30 µM CBZ’s effects of RED duration (B1, green), frequency (B2, blue), and amplitude (B3, red). Dashed-line boxes from 20 to 40 minutes represent the duration of CBZ exposure. Data points represent mean ± SEM for RED parameters in 1-minute bins. C) Concentration-dependent effects of CBZ on RED duration (green), frequency (blue), and amplitude (red). Bars represent mean ± SEM of N slices (see Table 1) and * = p<0.05, ** = p<0.01, *** = p<0.001. D) Concentration-response curves fit to data from panel C for CBZ’s effects on RED duration (green), frequency (blue), and amplitude (red).
Although not significant at 30 µM, but significant at 10 µM and 100 µM, CBZ also increased the average amplitudes of high-frequency spikes recorded during REDs (Fig. 2B3). Similar significant RED amplitude increases were observed with other ASDs and were nearly always correlated with significant attenuations of RED duration (11 of 16 instances, Table 1). In the cases of eslicarbazepine and lamotrigine, these amplitude increases reversed to significantly attenuated RED amplitudes at higher concentrations where duration and frequency were also more dramatically attenuated. Accordingly, significant amplitude increases are interpreted as transient temporal summations of compound action potentials as RED durations become briefer at intermediate effective concentrations.
Several ASDs demonstrated minimal or no significant effects on RED parameters at the concentrations tested in these experiments (e.g. felbamate, gabapentin, levetiracetam, and valproate). Gabapentin (GBP), in particular, demonstrated no significant effects on any RED parameters at all concentrations tested (Fig. 3). Representative traces observed after a 20-minute exposure to 1 mM GBP, the highest concentration tested here, were unremarkable compared to baseline control traces (Fig. 3A). Time course data confirmed these observations; no significant deviations from baseline RED parameters were measured in the presence of 1 mM GBP (Fig. 3B). Concentration-response and significance data revealed no significant effects on any RED parameters at any concentrations tested; accordingly, no EC50 quantifications were determined (Fig. 3C-D).
Figure 3:
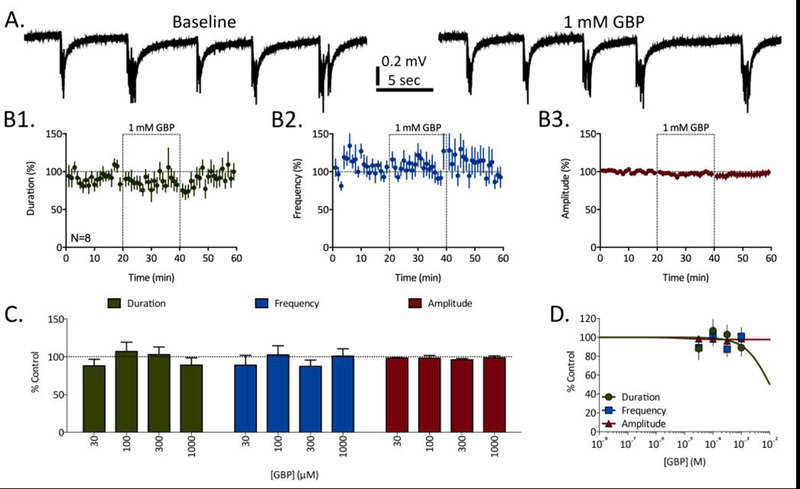
Gabapentin (GBP) fails to affect REDs at any concentrations tested. A) Representative traces of REDs recorded before (baseline) and in the presence of 1 mM GBP. B) Time courses for 1 mM GBP’s effects of RED duration (B1, green), frequency (B2, blue), and amplitude (B3, red). Dashed-line boxes from 20 to 40 minutes represent the duration of GBP exposure. Data points represent mean ± SEM for RED parameters in 1-minute bins. C) Concentration-dependent effects of GBP on RED duration (green), frequency (blue), and amplitude (red). Bars represent mean ± SEM of N slices (see Table 1) and * = p<0.05, ** = p<0.01, *** = p<0.001. D) Concentration-response curves fit to data from panel C for GBP’s effects on RED duration (green), frequency (blue), and amplitude (red).
As a final example of the different qualitative outcomes observed in these experiments, several ASDs significantly increased RED duration in a concentration-dependent manner (e.g. clobazam, ethosuximide, stiripentol, topiramate, and vigabatrin). For example, topiramate (TPM) produced a concentration-dependent increase in RED duration that was significant at three of the four concentrations tested (Fig. 4). The increase in RED duration produced by 100 µM TPM was clearly visible in representative traces (Fig. 4A) as well as time course data (Fig. 4B1). In addition to the significant and progressive increases in RED duration at 30–300 µM, a relatively small but significant attenuation in RED frequency was also observed at 100–300 µM (Fig. 4C). When fit with a sigmoidal concentration-response curve (Fig. 4D), the EC50 for TPM’s effect on RED duration was 38.5 µM, and the predicted effect plateau was 151.0% of baseline duration. Additionally, the EC50 for TPM’s effect on RED frequency was 26.9 µM, and the predicted plateau was 73.0% of baseline frequency.
Figure 4:
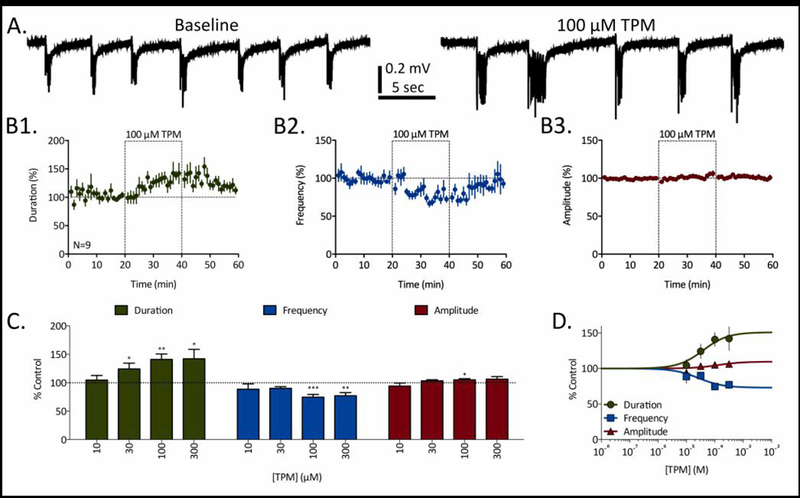
Topiramate (TPM) prolongs RED duration in a concentration-dependent manner. A) Representative traces of REDs recorded before (baseline) and in the presence of 100 µM TPM. B) Time courses for 100 µM TPM’s effects of RED duration (B1, green), frequency (B2, blue), and amplitude (B3, red). Dashed-line boxes from 20 to 40 minutes represent the duration of TPM exposure. Data points represent mean ± SEM for RED parameters in 1-minute bins. C) Concentration-dependent effects of TPM on RED duration (green), frequency (blue), and amplitude (red). Bars represent mean ± SEM of N slices (see Table 1) and * = p<0.05, ** = p<0.01, *** = p<0.001. D) Concentration-response curves fit to data from panel C for TPM’s effects on RED duration (green), frequency (blue), and amplitude (red).
A summary of concentration-response analyses, expressed as EC50 values when data were successfully fit by concentration-response curves, is presented in Table 2. Additionally, concentration-response curve plateaus were used to predict maximum parameter effects at concentrations above those directly tested in these experiments. Furthermore, EC50 values given for the most potent RED parameter effects are compared to reported average plasma concentrations in humans when these ASDs are taken at therapeutic doses; this analysis was conducted in order to facilitate informed judgements on the translational significance of a given ASD’s effects on REDs in this model. Based on these data, ASDs are grouped according to mechanistic target classes (VGSCs and potassium channels, GABA modulators, and other/mixed targets) and then rank ordered according to EC50 / Ave. Human Plasma Concentration ratios. These data reveal that a) ASDs that target VGSCs and potassium channels robustly attenuate REDs at concentrations approximately 1–3x their average plasma concentrations, b) GABA modulators generally have weaker attenuating effects at concentrations approximately 3–40x their average plasma concentrations, and often increase RED duration, and c) all other ASDs increase RED duration, incompletely attenuate RED frequency, or have no effects on any RED parameters.
Table 2.
Concentration-Response Summary of ASD effects on RED duration, frequency, and amplitude in layer II of the mEC.
| Target Class | ASD | Duration EC50 (µM) Predicted Max (%) |
Frequency EC50 (µM) Predicted Max (%) |
Amplitude EC50 (µM) Predicted Max (%) |
EC50 / Ave. human plasma conc. |
|---|---|---|---|---|---|
| Na+ | Carbamazepine | 25.8 0% |
128.7 0% |
153.6 227.1% |
0.81 |
| Na+ | Eslicarbazepine | 299.2 0% |
91.9 0% |
631.9 0% |
1.43 |
| Na+ | Rufinamide | - | 276.0 31.1% |
- | 1.88 |
| Na+ | Phenytoin | 356.7 0% |
127.8 0% |
- | 2.15 |
| Na+ / h-current | Lamotrigine | 79.3 0% |
84.0 0% |
787.5 0% |
2.32 |
| Na+ / slow inactivation | Lacosamide | 318.1 8.9% |
78.6 35.3% |
- | 2.62 |
| K+ | Ezogabine | 2.8 0% |
11.8 0% |
21.4 0% |
2.80 |
| GABA / reuptake | Tiagabine | 0.4 42.5% |
472.7 0% |
- | 2.50 |
| GABA / barbiturate | Phenobarbital | 477.5 0% |
270.1 0% |
- | 3.58 |
| GABA-T inhibitor | Vigabatrin | 2767.0 289.1% |
1909.0 0% |
- | 13.40 |
| GABA / 1,5- benzodiazepine |
Clobazam | - | 79.1 42.2% |
- | 39.55 |
| GABA / 1,4- benzodiazepine |
Midazolam | 21.3 165% |
112.3 0% |
- | 42.60 |
| GABA / mixed | Stiripentol | - | - | - | - |
| NKCC1 | Bumetanide | - | 0.9 82.0% |
- | 0.15 |
| Ca2+ | Ethosuximide | 1496.0 180.0% |
290.2 88.9% |
- | 0.59 |
| Mixed | Topiramate | 38.5 151.0% |
26.9 73.0% |
- | 0.73 |
| SV2A | Levetiracetam | - | - | - | - |
| Mixed | Valproate | - | - | - | - |
| Mixed | Felbamate | - | - | - | - |
| Ca2+ | Gabapentin | - | - | - | - |
“-” = Not Calculable. EC50: Half-maximum Effective Concentration. Predicted Max (%): computationally derived concentration-response curve plateaus. Average (Ave.) plasma concentrations were adapted from Bialer, M., Twyman, R. E. & White, H. S. Epilepsy Behav 5, 866–872 (2004)37, Löscher, W. Epilepsia 48, 1245–1258 (2007),38 Krasowski, M. D. Pharmaceuticals 3, 1909–1935 (2010)39, Rey, E., Tréluyer, J. M. & Pons, G. Clin Pharmacokinet 36, 409–424 (1999)40, and Gunthorpe, M. J., Large, C. H. & Sankar, R. Epilepsia 53, 412–424 (2012)26.
4. Discussion
Brain slices containing hippocampus and mEC have long been used to study the dynamics and pharmacology of spontaneous seizure-like activity.22 Many of these studies reported differential resistances of epileptiform discharges to ASDs and have suggested that these data demonstrate the utility of brain slice preparations to model pharmacoresistance. However, differences in the specific methods used complicates the cross-comparison of the efficacy of ASDs in these models.23 The present study seeks to facilitate such cross-comparisons by examining the concentration-dependent effects of 20 ASDs on spontaneous epileptiform discharges in a model predicated on brain slices derived from a translationally-relevant epilepsy substrate, the KA-induced SE model of TLE. These data illustrate a spectrum of ASD-mediated effects on REDs and are expected to serve as a foundation upon which future therapeutics may be identified, differentiated, and assessed for potentially translatable efficacy in refractory epilepsy patients.
4.1. Glutamate receptor antagonists differentially affect REDs
The brain slices used in these experiments were perfused with a low Mg2+ / elevated K+-based ACSF in order to facilitate the occurrence of REDs.11,19 While electrically evoked fEPSPs are largely mediated by AMPA receptors24, epileptiform discharges observed under similar conditions are more susceptible to inhibition by NMDA receptor antagonists.8,25 However, data presented here demonstrates that neither CNQX nor APV were able to fully inhibit REDs. These data can be contrasted with findings from the late Uwe Heinemann’s laboratory that used similar conditions (low Mg2+) but obtained tissues from naïve rodents.8 In this seminal work, 30 µM APV fully inhibited LRDs (equivalent to REDs). In contrast, 100 µM APV failed to fully block REDs under our conditions. This enhanced resistance to NMDA receptor antagonists may reflect molecular and/or network-level changes that are present in the mEC of KA-rats. Additionally, Zhang et al. showed 60 µM CNQX had no effect on LRDs in naïve rat slices; in contrast, 10 µM CNQX significantly reduced RED frequency and, interestingly, prolonged RED duration. These prolonged duration REDs, like those observed in the presence of some GABAergic synaptic transmission modulators, may resemble the more pharmacosensitive SLEs described by the Heinemann lab.8,9
4.2. ASDs that target voltage-gated sodium or potassium channels
Data presented here demonstrate that ASDs targeting VGSCs and potassium channels attenuate REDs at concentrations ~1–3x their average plasma concentrations. Ezogabine, which targets KCNQ2–5 potassium channels, was the only ASD to experimentally achieve complete block of REDs. Ezogabine’s EC50 for reducing RED duration (2.8 µM) was consistent with its EC50 for modulating KCNQ channels (0.6 – 6.4 µM) and the therapeutic plasma concentrations necessary to achieve 50% seizure reduction (~1 µM).26 Ezogabine’s EC50 for reducing RED frequency (11.8 µM) was also in agreement with reports of its effects on RED-like frequencies in a low-Mg2+ slice model from naïve rodents (EC50 ~10µM).27 All other ASDs tested in this mechanistic category are known to inhibit VGSCs through a variety of mechanisms. When comparing our data to similar studies, some differences present themselves. Unlike data reported here, both carbamazepine (50 µM) and phenytoin (50 µM) were reported to increase RED-like frequency in both low Mg2+- and 4-AP-based models in tissues from naïve rodents.7,28 To our knowledge, this is the first report for the effects of eslicarbazepine, lamotrigine, lacosamide, and rufinamide on REDs recorded from the mEC in slices from either naïve or post-SE models.
4.3. ASDs that target GABAergic synaptic transmission
ASDs that target GABAergic synaptic transmission shared some unique effects on REDs in this model. With the exceptions of tiagabine and phenobarbital, prolongation of burst duration was a common feature amongst these ASDs. Concentration-response curve plateaus predicted increased RED durations above baseline for vigabatrin and midazolam. This increase in RED duration was similar to that reported for muscimol in a low-Mg2+-based model; in that study, muscimol changed LRDs (high frequency / short duration / pharmacoresistant) into SLEs (low frequency / long duration / pharmacosensitive).29 The pharmacosensitivity of the prolonged-duration REDs observed here remains to be determined. However, as was observed with clobabzam (and to a lesser degree with phenobarbital), prolongation of RED duration reversed to significantly shorter durations at higher concentrations. This reversal was coincident with significant attenuations of RED frequency.
Another distinguishing characteristic of these GABAergic targeted ASDs was the elevated concentrations over which they attenuated REDs; with the exception of tiagabine, EC50s were approximately 4–40x their average plasma concentrations. As for tiagabine, the maximum effect on RED duration was plateaued at 42.5% of baseline. Taken together, these data suggest that REDs in this model have a pharmacoresistant-like decreased sensitivity to GABAergic targeted ASDs at therapeutically relevant concentrations.
Some differences become apparent when comparing our data in tissues taken from KA-rats to similar studies in tissues from naïve rats. Tiagabine was reported to have no effects on LRD frequency at 50–250 µM in a low-Mg2+ model29, whereas we report a significant attenuation of RED frequency at 100 µM. Also contrasting with our data, phenobarbital and midazolam were reported to have no effect on LRDs at 150 µM and 50 µM respectively.7 However, vigabatrin (1mM) similarly reduces LRD frequency (and increases LRD duration) in a low-Mg2+ model.30 To our knowledge, no data has been reported for clobabzam or stiripentol in similar slice models of epileptiform activity in the mEC.
4.4. ASDs with other, mixed, or poorly defined mechanisms of action
ASDs with other mechanisms of action were also shown here to weakly or incompletely inhibit RED frequency (bumetanide, ethosuximide, and topiramate), increase RED duration (ethosuximide and topiramate), or have no significant effects that were consistently measureable (levetiracetam, valproate, felbamate, and gabapentin). In naïve rodent slices, ethosuximide reportedly shortened the duration and increased the frequency of LRDs7, while valproate had no effects.9,31 While we observed similar results with valproate, our results with ethosuximide showed a significant enhancement of RED duration. In 4-AP-based models, both topiramate and valproate were previously shown to have no effects on epileptiform discharges.32 Again, while these results are similar for valproate, our data using topiramate demonstrate an increase in RED duration and a decrease in frequency. Although topiramate is known to possess several mechanisms of action33, these concentrations positively modulate GABA-A receptors.34,35 This mechanism may explain topiramate’s qualitatively similar effects with other GABA-modulating ASDs on RED duration. Lastly, this report is the first to describe the effects of bumetanide, levetiracetam, felbamate, and gabapentin in any slice model of epileptiform activity measured in the mEC.
4.5. Conclusions
Taken together, these data suggest that epileptiform activity recorded from the superficial layers of the mEC in slices obtained from KA-rats are differentially sensitive to existing ASDs. In the context of previously reported data using tissues from naïve rodents, the different sensitivities of REDs to these ASDs may reflect persistent molecular and/or network changes resulting from disease. Indeed, differential sensitivities to pharmacological agents have been described in slices when tissues derived from epilepsy models were compared to control tissues.10–14,18,19 Therefore, we propose that in-vitro models of spontaneous epileptiform activity that employ tissues from translationally relevant epilepsy models may better reflect the underlying molecular and/or network substrate and, therefore, better predict the efficacy of investigational therapeutics, particularly those with novel mechanisms of action. Accordingly, this model is used, along with other in-vivo models of acute seizures and epilepsy, to evaluate investigational pharmacoresistant epilepsy therapies in the ETSP.36
Supplementary Material
Key Point Box [3–5 bullets, each <140 characters each].
The present study describes the effects of 20 ASDs on recurrent epileptiform discharges (REDs) in slices from kainate-treated rats.
ASDs targeting sodium and potassium channels attenuate REDs at therapeutic concentrations.
GABAergic synaptic transmission modulating ASDs attenuate REDs only at higher concentrations and often increase RED duration.
ASDs with other mechanisms of action weakly inhibit RED frequency, increase RED duration, or have no significant effects.
Thus, REDs recorded from the mEC in slices obtained from kainate-treated rats are differentially sensitive to existing ASDs.
Acknowledgements
The authors would like to acknowledge and thank all the members of the Epilepsy Therapy Screening Program (ETSP) contract site at the University of Utah (formerly known as the Anticonvulsant Drug Development, ADD, Program) for scientific, technical, and logistical support. In particular, we would like to thank Kristina Johnson, Elisa Koehler, Kyle Thomson, and Sharon Edwards. The authors would also like to thank the ETSP at the National Institutes of Neurological Diseases and Stroke (NINDS) for their review and comments on this manuscript. Finally, we would like to acknowledge and thank the external consultation board members for the ETSP (Dr. Amy Brooks Kayal, Dr. Henrik Klitgaard, Dr. Wolfgang Loscher, and Dr. Steve Perrin) for their continuing helpful guidance. This project was supported by Federal funds from the NINDS, ETSP, National Institutes of Health, Department of Health and Human Services, under NINDS Contract No. HHSN271201100029C (KSW).
Abbreviations:
- ASD
antiseizure drug
- ACSF
artificial cerebral spinal fluid
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- APV
(2R)-amino-5-phosphonovaleric acid
- CBZ
carbamazepine
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- CNS
central nervous system
- DMSO
dimethyl sulfoxide
- EC50
half-maximal effective concentration
- EZG
ezogabine
- fEPSPs
field excitatory postsynaptic potentials
- GBP
gabapentin
- KA
kainic acid
- KA-rats
rats from the multiple low-dose KA model of TLE
- LRDs
late recurrent discharges
- mEC
medial entorhinal cortex
- NMDA
N-Methyl-D-aspartic acid
- REDs
recurrent epileptiform discharges
- SE
status epilepticus
- SLEs
seizure-like events
- TLE
temporal lobe epilepsy
- TPM
topiramate
- VGSCs
voltage-gated sodium channels
Footnotes
Disclosure of conflicts of interest
KSW is a scientific advisor and receives stock options for Blackfynn, Inc. CSM is a scientific advisor and stockholder in Sea Pharmaceuticals, LLC. HSW has served as a consultant to Greenwich Pharmaceuticals and Bial Pharmaceuticals in the previous 12 months. HSW is also a scientific co-founder of NeuroAdjuvants, Inc., SLC, UT. None of the other authors have any conflict of interest to disclose.
Ethical publication statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Hesdorffer DC, Logroscino G, Benn EKT, et al. Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology 2011; 76:23–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt D, Sillanpää M. Evidence-based review on the natural history of the epilepsies. Curr Opin Neurol 2012; 25:159–63. [DOI] [PubMed] [Google Scholar]
- 3.Wilcox KS, Dixon-Salazar T, Sills GJ, et al. Issues related to development of new antiseizure treatments. Epilepsia 2013; 54 Suppl 4:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010; 51:1069–77. [DOI] [PubMed] [Google Scholar]
- 5.Tang F, Hartz AMS, Bauer B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front Neurol 2017; 8:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Löscher W Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res 2016; 126:157–84. [DOI] [PubMed] [Google Scholar]
- 7.Zhang CL, Dreier JP, Heinemann U. Paroxysmal epileptiform discharges in temporal lobe slices after prolonged exposure to low magnesium are resistant to clinically used anticonvulsants. Epilepsy Res 1995; 20:105–11. [DOI] [PubMed] [Google Scholar]
- 8.Zhang CL, Gloveli T, Heinemann U. Effects of NMDA- and AMPA-receptor antagonists on different forms of epileptiform activity in rat temporal cortex slices. Epilepsia 1994; 35 Suppl 5:S68–73. [DOI] [PubMed] [Google Scholar]
- 9.Dreier JP, Heinemann U. Late low magnesium-induced epileptiform activity in rat entorhinal cortex slices becomes insensitive to the anticonvulsant valproic acid. Neurosci Lett 1990; 119:68–70. [DOI] [PubMed] [Google Scholar]
- 10.Carter DS, Deshpande LS, Rafiq A, et al. Characterization of spontaneous recurrent epileptiform discharges in hippocampal-entorhinal cortical slices prepared from chronic epileptic animals. Seizure 2011; 20:218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith MD, Adams AC, Saunders GW, et al. Phenytoin- and carbamazepine-resistant spontaneous bursting in rat entorhinal cortex is blocked by retigabine in vitro. Epilepsy Res 2007; 74:97–106. [DOI] [PubMed] [Google Scholar]
- 12.Shiri Z, Herrington R, Lévesque M, et al. Neurosteroidal modulation of in vitro epileptiform activity is enhanced in pilocarpine-treated epileptic rats. Neurobiology of Disease 2015; 78:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maslarova A, Salar S, Lapilover E, et al. Increased susceptibility to acetylcholine in the entorhinal cortex of pilocarpine-treated rats involves alterations in KCNQ channels. Neurobiology of Disease 2013; 56:14–24. [DOI] [PubMed] [Google Scholar]
- 14.Klaft Z-J, Schulz SB, Maslarova A, et al. Extracellular ATP differentially affects epileptiform activity via purinergic P2X7 and adenosine A1 receptors in naive and chronic epileptic rats. Epilepsia 2012; 53:1978–86. [DOI] [PubMed] [Google Scholar]
- 15.Zahn RK, Liotta A, Kim S, et al. Reduced ictogenic potential of 4-aminopyridine in the hippocampal region in the pilocarpine model of epilepsy. Neurosci Lett 2012; 513:124–8. [DOI] [PubMed] [Google Scholar]
- 16.Sandow N, Zahn RK, Gabriel S, et al. Glutamine induces epileptiform discharges in superficial layers of the medial entorhinal cortex from pilocarpine-treated chronic epileptic rats in vitro. Epilepsia 2009; 50:849–58. [DOI] [PubMed] [Google Scholar]
- 17.Zimmerman G, Njunting M, Ivens S, et al. Acetylcholine-induced seizure-like activity and modified cholinergic gene expression in chronically epileptic rats. Eur J Neurosci 2008; 27:965–75. [DOI] [PubMed] [Google Scholar]
- 18.Zahn RK, Tolner EA, Derst C, et al. Reduced ictogenic potential of 4-aminopyridine in the perirhinal and entorhinal cortex of kainate-treated chronic epileptic rats. Neurobiology of Disease 2008; 29:186–200. [DOI] [PubMed] [Google Scholar]
- 19.Smith MD, Saunders GW, Clausen RP, et al. Inhibition of the betaine-GABA transporter (mGAT2/BGT-1) modulates spontaneous electrographic bursting in the medial entorhinal cortex (mEC). Epilepsy Res 2008; 79:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972; 32:281–94. [DOI] [PubMed] [Google Scholar]
- 21.Thomson KE, Modi AC, Glauser TA, et al. The impact of nonadherence to antiseizure drugs on seizure outcomes in an animal model of epilepsy. Epilepsia 2017; 58:1054–62. [DOI] [PubMed] [Google Scholar]
- 22.Avoli M, D’Antuono M, Louvel J, et al. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol 2002; 68:167–207. [DOI] [PubMed] [Google Scholar]
- 23.Raimondo JV, Heinemann U, de Curtis M, et al. Methodological standards for in vitro models of epilepsy and epileptic seizures. A TASK1-WG4 report of the AES/ILAE Translational Task Force of the ILAE. Epilepsia 2017; 58 Suppl 4:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neagu B, Strominger NL, Carpenter DO. Contribution of NMDA receptor-mediated component to the EPSP in mouse Schaffer collateral synapses under single pulse stimulation protocol. Brain Res 2008; 1240:54–61. [DOI] [PubMed] [Google Scholar]
- 25.Lopantsev V, Avoli M. Laminar organization of epileptiform discharges in the rat entorhinal cortex in vitro. J Physiol (Lond) 1998; 509 ( Pt 3):785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 2012; 53:412–24. [DOI] [PubMed] [Google Scholar]
- 27.Armand V, Rundfeldt C, Heinemann U. Effects of retigabine (D-23129) on different patterns of epileptiform activity induced by low magnesium in rat entorhinal cortex hippocampal slices. Epilepsia 2000; 41:28–33. [DOI] [PubMed] [Google Scholar]
- 28.Brückner C, Heinemann U. Effects of standard anticonvulsant drugs on different patterns of epileptiform discharges induced by 4-aminopyridine in combined entorhinal cortex-hippocampal slices. Brain Res 2000; 859:15–20. [DOI] [PubMed] [Google Scholar]
- 29.Pfeiffer M, Draguhn A, Meierkord H, et al. Effects of gamma-aminobutyric acid (GABA) agonists and GABA uptake inhibitors on pharmacosensitive and pharmacoresistant epileptiform activity in vitro. Br J Pharmacol 1996; 119:569–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engel D, Endermann U, Frahm C, et al. Acute effects of gamma-vinyl-GABA on low-magnesium evoked epileptiform activity in vitro. Epilepsy Res 2000; 40:99–107. [DOI] [PubMed] [Google Scholar]
- 31.Sokolova S, Schmitz D, Zhang CL, et al. Comparison of effects of valproate and trans-2-en-valproate on different forms of epileptiform activity in rat hippocampal and temporal cortex slices. Epilepsia 1998; 39:251–8. [DOI] [PubMed] [Google Scholar]
- 32.D’Antuono M, Köhling R, Ricalzone S, et al. Antiepileptic drugs abolish ictal but not interictal epileptiform discharges in vitro. Epilepsia 2010; 51:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White HS, Rho JM. Mechanisms of Action of Antiepileptic Drugs Professional Communications; 2010. [Google Scholar]
- 34.Simeone TA, Wilcox KS, White HS. Subunit selectivity of topiramate modulation of heteromeric GABA(A) receptors. Neuropharmacology 2006; 50:845–57. [DOI] [PubMed] [Google Scholar]
- 35.White HS, Brown SD, Woodhead JH, et al. Topiramate modulates GABA-evoked currents in murine cortical neurons by a nonbenzodiazepine mechanism. Epilepsia 2000; 41 Suppl 1:S17–20. [PubMed] [Google Scholar]
- 36.Barker-Haliski ML, Johnson K, Billingsley P, et al. Validation of a Preclinical Drug Screening Platform for Pharmacoresistant Epilepsy. Neurochem Res 6 ed. 2017; 161:695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bialer M, Twyman RE, White HS. Correlation analysis between anticonvulsant ED50 values of antiepileptic drugs in mice and rats and their therapeutic doses and plasma levels. Epilepsy Behav 2004; 5:866–72. [DOI] [PubMed] [Google Scholar]
- 38.Löscher W The Pharmacokinetics of Antiepileptic Drugs in Rats: Consequences for Maintaining Effective Drug Levels during Prolonged Drug Administration in Rat Models of Epilepsy. Epilepsia 5 ed. 2007; 48:1245–58. [DOI] [PubMed] [Google Scholar]
- 39.Krasowski MD. Therapeutic Drug Monitoring of the Newer Anti-Epilepsy Medications. Pharmaceuticals 2010; 3:1909–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rey E, Tréluyer JM, Pons G. Pharmacokinetic optimization of benzodiazepine therapy for acute seizures. Focus on delivery routes. Clin Pharmacokinet 1999; 36:409–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.