

Abstract
The TAM (TYRO-3, AXL, MERTK) family receptor tyrosine kinases (RTKs) play an important role in promoting growth, survival, and metastatic spread of several tumor types. AXL and MERTK are overexpressed in head and neck squamous cell carcinoma (HNSCC), triple-negative breast cancer (TNBC), and non-small cell lung cancer (NSCLC), malignancies that are highly metastatic and lethal. AXL is the most well-characterized TAM receptor and mediates resistance to both conventional and targeted cancer therapies. Since AXL is highly expressed in aggressive tumor types, cancer patients are currently being enrolled in clinical trials testing AXL inhibitors. In the current study, we analyzed the effects of AXL inhibition using a small molecule AXL inhibitor, monoclonal antibody therapy, and siRNA in HNSCC, TNBC, and NSCLC preclinical models. Anti-AXL targeting strategies had limited efficacy across these different models which our data suggests could be attributed to upregulation of MERTK. MERTK expression was increased in cell lines and patient-derived xenografts treated with AXL inhibitors and inhibition of MERTK sensitized HNSCC, TNBC, and NSCLC preclinical models to AXL inhibition. Dual targeting of AXL and MERTK led to a more potent blockade of downstream signaling, synergistic inhibition of tumor cell expansion in culture, and reduced tumor growth in vivo. Furthermore, ectopic overexpression of MERTK in AXL inhibitor-sensitive models resulted in resistance to AXL-targeting strategies. These observations suggest that therapeutic strategies co-targeting both AXL and MERTK could be highly beneficial in a variety of tumor types where both receptors are expressed, leading to improved survival for patients with lethal malignancies.
Introduction:
The TAM family of receptor tyrosine kinases is comprised of three family members: TYRO3 (SKY), AXL (ARK or UFO), and MERTK. Cognate ligand binding to TAM receptors on the cell surface leads to receptor dimerization, kinase domain activation, and auto/trans-phosphorylation of tyrosine residues located on each receptor’s cytoplasmic tail (1). Activation of TAM receptors stimulates PI3K/AKT, RAS/RAF/MEK/ERK (MAPK), and other signaling cascades leading to increased tumor cell survival, proliferation, migration, invasion, and angiogenesis (1–4).
AXL and MERTK have been implicated in the development and progression of many malignancies including lung (3,5–9), breast (3,10–14), ovarian (15), colon (16), head and neck (17), thyroid (18), prostate (19), pancreatic (20), osteosarcoma (21), melanoma (22), leukemia (23), and Kaposi sarcoma (24). With the advancement of preclinical studies supporting the role of TAM family receptors in cancer, TAM family inhibitors are being clinically developed and will likely be beneficial in specific cancer settings. The most clinically advanced agent with AXL as a primary target is R428 (BGB324) (10), which is currently being tested in Phase Ib/II clinical trials for treatment of acute myeloid leukemia (AML), NSCLC, and melanoma (25). R428 has potent activity against AXL (IC50 = 14 nM) and fifty-fold selectivity for AXL relative to MERTK in cell-based assays (10). Similarly, UNC2025 is a MERTK and FLT3 tyrosine kinase inhibitor that has potent activity against MERTK (IC50 = 2.7 nM), forty-fold selectivity for MERTK relative to AXL in cell-based assays (26), and mediates therapeutic activity in preclinical leukemia and NSCLC xenograft models (26–28). Numerous other small molecule tyrosine kinase inhibitors that were developed to target other kinases have low nanomolar potency against AXL and/or MERTK, including agents that are FDA-approved (cabozantinb, crizotinib, sunitinib) or are being tested in clinical studies (merestinib, BMS-777607, S49076).
Resistance to single agent therapy is a major hurdle in cancer treatment. Cooperation of family members to overcome resistance is well documented (29–31). Here we describe an intrinsic and adaptive feedback mechanism of resistance to AXL-targeting agents mediated by upregulation of MERTK. Examination of a large panel of cell lines from different malignancies indicated that high expression of MERTK correlates with relative resistance to anti-AXL therapeutics. Targeting AXL with genetic silencing, a small molecule inhibitor, or an AXL-specific antibody increased MERTK protein expression in vitro and in vivo. Furthermore, combined targeting of MERTK and AXL inhibited tumor cell expansion in vitro and impacted tumor growth in vivo. Dual targeting of MERTK and AXL impacted several signaling pathways including AKT, STAT6, P70S6 kinase (P70S6K), S6 ribosomal protein (S6rp), and C-RAF. Finally, overexpression of MERTK in cells that were sensitive to AXL targeting led to activation of P70S6K, S6rp, and C-RAF and was sufficient to mediate resistance to AXL-targeting agents. Collectively, the data presented here indicate a critical role for MERTK in resistance to AXL-targeted therapy and provide rationale for clinical evaluation of strategies targeting both AXL and MERTK for treatment of cancer.
Materials and Methods:
Cell lines
All cell lines were obtained from indicated sources (Supplementary Materials and Methods). Cell line identities were confirmed using short tandem repeat (STR) analysis and publicly available databases. Validated STR profiles were not available for the H157 cell line, but the profile obtained did not match any other cell line.
TAM RTK Inhibitors
UNC2025 (MERTK inhibitor) was synthesized as previously described (26) or purchased from Selleck Chemicals or Apexbio. R428 (AXL inhibitor) was purchased from Selleck Chemicals or Apexbio. The monoclonal antibody targeting AXL (Mab173) was kindly provided by Dr. Parkash Gill, Department of Medicine and Pathology, University of Southern California, Los Angeles, CA (24). Sub-confluent cultures were treated with the indicated concentrations of Mab173, UNC2025, R428, or equivalent volumes of DMSO vehicle.
siRNA transfection
Non-targeting control siRNA (Cat# D-001810–01-05) and siRNAs targeting AXL (Cat#J-003104–10-0005, #J-003104–12-0005) were purchased from Thermo Scientific and utilized for transfection of NSCLC cells with final concentrations of 25nM siRNA and 1μL/mL DharmaFECT1 Transfection Reagent (Cat#T-2001–02). Non-targeting control pool siRNA (Cat#D-001810) and SMARTpool siRNA targeting AXL (Cat#L-003104) were purchased from Dharmacon, Inc and utilized for transfection of HNSCC and TNBC cell lines with a final concentration of 30nM siRNA with Lipofectamine RNAiMAX (Life Technologies).
Plasmids and transfection
pDONR223-MERTK was a gift from William Hahn & David Root (Addgene plasmid # 23900) and subcloned into the BamHI/NOTI restriction sites of the pcDNA6.0 expression vector (Life Technologies). pcDNA3.1-TYRO3 was purchased from GenScript USA Inc. (#OHu23055D, Piscataway, NJ, USA). Transfection was performed using Lipofectamine LTX and Opti-MEM I (Life Technology) according to the manufacturer’s instructions.
Immunoblotting
Cells were washed with PBS and scraped into lysis buffer (50mM HEPES pH 7.5, 150mM NaCl, 10mM EDTA, 10% glycerol, 1% Triton X-100, 1mM Na3VO4) supplemented with protease inhibitors (Complete Mini, Roche Molecular Biochemicals). Protein concentrations were determined and immunoblotting was performed as previously described (6) using antibodies shown in Supplemental Table 1. All antibodies were used as recommended by manufacturer’s instructions.
TAM kinase phosphorylation
Cells were cultured in medium with DMSO vehicle control or the indicated concentration of drug for 1 hour or 24 hours and then 120μM freshly-prepared pervanadate phosphatase inhibitor was added for an additional 3 minutes to stabilize phosphorylated proteins (26). Lysates were prepared, MERTK and AXL were immunoprecipitated with anti-MERTK (R&D Systems, MAB8912) or anti-AXL (R&D Systems, AF154) and Protein G beads (Invitrogen #10–1242) and phosphorylated and total MERTK and AXL proteins were detected by immunoblot as described above.
Cell expansion assays
CellTiter 96 AQueous One Solution reagent (Promega, Cat#G3580), Cell Counting Kit 8 (Dojindo Molecular Technologies, Cat#CK04), and crystal violet assay were used to determine relative numbers of viable cells 72 hours after transfection with siRNA targeting AXL (siAXL) or treatment with AXL and/or MERTK inhibitors. Alternatively, relative confluency was determined using the Incucyte® ZOOM live cell imaging system (Essen Biosciences). For studies using combined treatment with siAXL and UNC2025, cells were transfected and then 24 hours later were treated with UNC2025 for an additional 72 hours.
Clonogenic assay
H1299 NSCLC cells were cultured at low density in liquid medium containing R428, UNC2025, R428 and UNC2025, or vehicle. Colonies were stained with crystal violet after 10 days and counted using a GelCount automated counter (Oxford Optronix).
Xenograft Models
Female athymic nude mice (4–6 weeks old) were obtained from Envigo. Animal procedures and maintenance were conducted in accordance with institutional guidelines of University of Wisconsin. UW-SCC1 PDX tumors, UM-SCC1, and MDAMB231 cell lines were inoculated by subcutaneous injection into the dorsal flank of each mouse and tumor volume was measured using a caliper. When tumors attained a volume of 200 mm3 (for 28 day study) or 500 mm3 (for 4 day study), mice were randomized into groups and treatment was initiated. R428, UNC2025, R428+UNC2025, or an equivalent volume of vehicle (0.5% hydroxypropylmethylcellulose + 0.1% Tween 80) were administered twice daily by oral gavage for 5 days/week. Tumors were collected within 3 hours of the last treatment for analysis of biochemical markers (49).
Immunohistochemistry
Tumors were processed for immunohistochemistry (IHC) as previously described (50). Sections were heated in 10mmol/L citrate buffer (pH 6.0) or 10mmol/L Tris+1mmol/L EDTA buffer (pH 9.0) with a decloaking chamber and incubated overnight at 4ºC with pS6rpY235/236 (1:200), Ki67 (1:800), or MERTK (1:50) antibodies. Bound antibodies were detected using the VECTASTAIN Universal Kit/HRP (Vector Laboratories) and 3,3’-diaminobenzidine substrates. Tissues were counterstained with Mayer hematoxylin (ThermoFisher Scientific) and were examined using an Olympus BX51 microscope. Images are shown at a magnification of 20X.
Evaluation of therapeutic interactions
The fractional product method was used to evaluate therapeutic interactions between AXL inhibitors and UNC2025 in cell culture assays (51,52). This method utilizes relative cell density following treatment with each individual agent to calculate the expected (E) effect for an additive interaction as a product of the individual responses. The observed (O) effect is the relative cell density following dual treatment. The ratio of observed to expected (O:E) values was calculated and used to indicate synergy (O:E <1), additivity (O:E =1), or antagonism (O:E >1). Isobologram analysis was also performed as previously described (53). Experiments were repeated 3 times in triplicate.
Statistical analysis
Statistical analyses were performed using Prism 7 software (GraphPad Software, Inc.). Differences between multiple groups were evaluated using repeated measures ANOVA with Boneferroni’s post-test. Student’s t-test was used for comparisons between two groups. Differences were considered significant when p<0.05.
Results:
MERTK expression correlates with resistance to AXL-targeting agents
AXL has been identified as a novel molecular target in numerous human cancers including HNSCC, TNBC, and NSCLC (6,11,32–35), and anti-AXL targeting strategies are now in clinical development for these malignancies. Mechanisms of resistance to AXL inhibitors have not been evaluated but will likely play a role in limiting the clinical efficacy of these inhibitors. To examine potential resistance mechanisms, we assessed expression of TAM family receptors and sensitivity to the anti-AXL small molecule inhibitor R428 across a large panel of HNSCC (Fig 1A and B), TNBC (Fig 1C and D), and NSCLC (Fig 1E and F) cell lines. AXL, MERTK, and TYRO3 were differentially expressed in cancer cell lines and immunoblot analysis confirmed inhibition of AXL phosphorylation in response to treatment with R428 (Figure S1). R428 did not inhibit MERTK phosphorylation at the doses used (Figure S1C). Although all of the tumor cell lines expressed AXL to some degree, analysis of tumor cell expansion in culture revealed that only a subset of cell lines exhibited a statistically significant decrease in cell number in response to treatment with R428 and were scored as sensitive, while the majority were relatively resistant (Fig 1B, D and F). Interestingly, all five of the cell lines that expressed little or no MERTK were sensitive to AXL inhibition (HN30, UM-SCC38, SUM159, SUM149, H226) while eight of ten cell lines that expressed higher levels of MERTK had no significant response to single agent R428 treatment (UM-SCC1, UM-SCC6, UM-SCC47, UM-SCC104, SUM229, BT549, MDAMB231, H157), suggesting a role for MERTK in resistance to AXL-targeting agents.
Figure 1: Endogenous expression of AXL and MERTK predicts sensitivity to AXL inhibition.
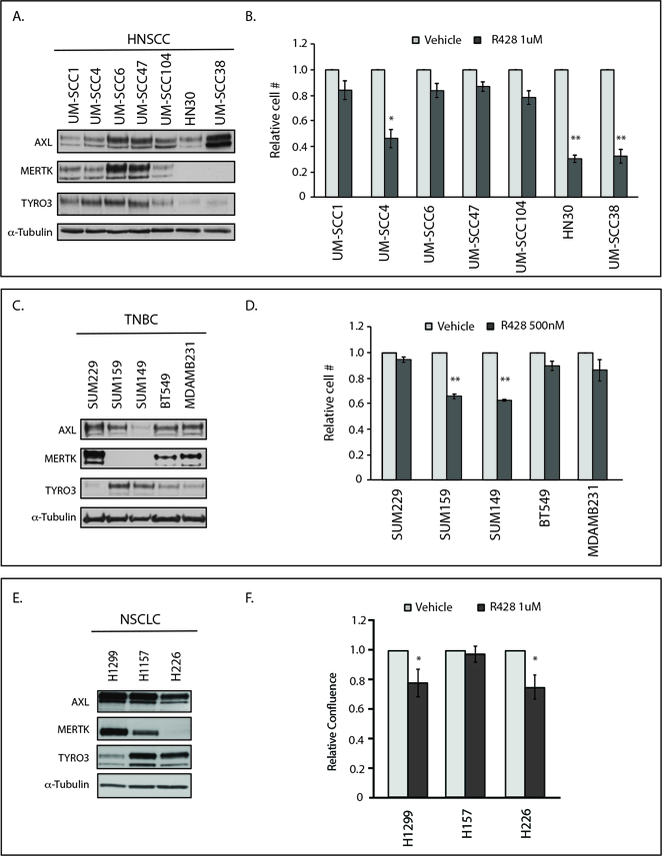
(A, C, E)Cell lysates were prepared from HNSCC, TNBC, and NSCLC cell lines and endogenous TAM kinase expression was determined by immunoblot. α-Tubulin is shown as loading control. (B, D, F) HNSCC, TNBC, and NSCLC cell lines were treated with vehicle or R428 for 72 hours and relative cell numbers were determined. Mean values, standard errors, and statistical analyses were derived from 3 (HNSCC and TNBC) or 7 (NSCLC) independent experiments. *p<0.05, **p<0.01.
Inhibition of AXL increases MERTK protein expression in vitro
Several studies have reported the cooperative nature of RTKs in cancer and resistance to RTK-targeting strategies. For example, the HER2 and HER3 receptors can be upregulated and mediate resistance to anti-epidermal growth factor receptor (EGFR) therapy (30,36,37). To determine whether alternative TAM family receptors are similarly upregulated in response to anti-AXL therapeutics, we analyzed the expression of MERTK and TYRO3 in HNSCC (UM-SCC47, HN30, UM-SCC4), TNBC (BT549, MDAMB231), NSCLC (H1299, H157), and esophageal cancer (KY270, KY410, SKGT4) cell lines after AXL inhibition mediated by genetic silencing (siAXL), small molecule inhibition (R428), or anti-AXL monoclonal antibody therapy (Mab173). First, cell lines were transfected with siAXL or a non-targeting siRNA control to examine the effects of genetic knockdown of AXL on MERTK and TYRO3 expression levels. While TYRO3 expression levels were unchanged in cells depleted of AXL, MERTK expression levels were increased by ~1.5 to 15-fold (Fig 2A, D, H and S2A). In contrast, expression of EGFR and cMET, RTKs outside the TAM family, did not change (Fig 2A and D). Similarly, MERTK expression was increased by ~1.5 to 15-fold in HNSCC, TNBC, NSCLC, and esophageal cancer cells treated with the AXL small molecule inhibitor R428 (Fig 2B, E, I and S2B) or the anti-AXL monoclonal antibody Mab173 (Fig 2F) compared to vehicle-treated controls. MERTK phosphorylation was also increased in select HNSCC, TNBC, NSCLC, and esophageal cell lines following treatment with R428 indicating increased MERTK activity coincident with MERTK upregulation (Fig 2C, G, J and S2C). EGFR and cMET protein levels were unchanged in response to AXL inhibition in HNSCC (Fig 2A, B) and TNBC (Fig 2D, E, F) cell lines. TYRO3 protein levels were only slightly increased in a subset of cell lines treated with R428 (Fig 2) and high levels of TYRO3 did not correlate with resistance to AXL inhibition (Fig 1, 2). Taken together, these data demonstrate selective upregulation of MERTK in tumor cell lines in response to AXL inhibition.
Figure 2: Inhibition of AXL increased MERTK protein expression and activity in vitro.
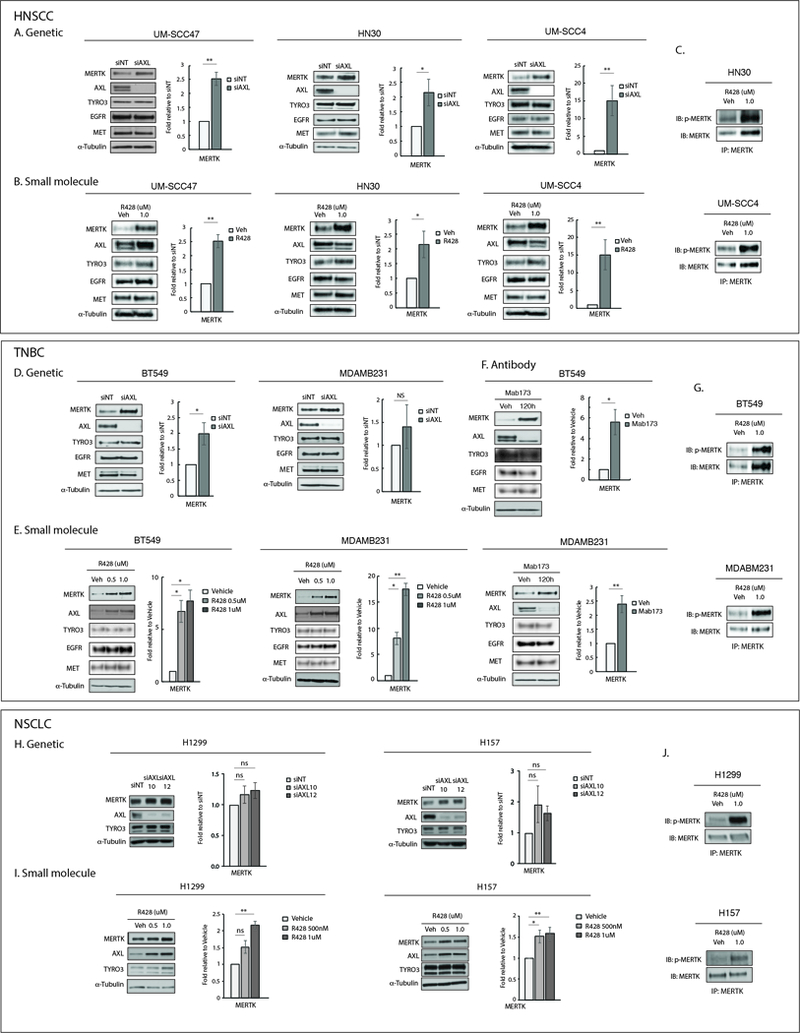
(A, D, H)Genetic inhibition of AXL in HNSCC, TNBC, and NSCLC cell lines. Cells were transfected with AXL siRNA (siAXL) or non-targeting siRNA (siNT). Cell lysates were prepared 72 hours later. (B, E, I) Small molecule inhibition of AXL in HNSCC, TNBC, and NSCLC cell lines. Cells were treated with vehicle or R428 for 72 hours prior to preparation of cell lysates. (F) Antibody inhibition of AXL in TNBC cell lines. Cells were treated with vehicle or Mab173 for 120 hours prior to preparation of cell lysates. Indicated proteins detected by immunoblot. α-Tubulin is shown as a loading control. MERTK expression was quantitated by densitometry using Image J software. Mean values and standard errors derived from 3 independent experiments are shown. *p<0.05, **p<0.01, ns=not significant. (C, G, J) The indicated cell lines were treated with vehicle or R428 for 24 hours and phosphorylated proteins were stabilized by treatment with pervanadate phosphatase inhibitor prior to preparation of cell lysates. MERTK was immunoprecipitated and phosphorylated (denoted by p-) and total MERTK proteins were detected by immunoblot. The data shown are representative of 2 or 3 independent experiments.
Combined inhibition of AXL and MERTK synergistically decreases tumor cell expansion and inhibits downstream signaling
Since expression of MERTK correlated with resistance to AXL inhibitors (Fig 1) and was upregulated in response to AXL suppression (Fig 2), we hypothesized that targeting MERTK would enhance the efficacy of AXL inhibitors. To test this hypothesis, HNSCC (UM-SCC1, UM-SCC4, UM-SCC6, UM-SCC47, UM-SCC104), TNBC (SUM229, BT549, MDAMB231), and NSCLC (H1299 and H157) cell lines that co-expressed both AXL and MERTK were either treated with a combination of R428 and UNC2025 (26) for 72 hours or transfected with siAXL and then treated with UNC2025 prior to determining relative cell numbers. Immunoblot analysis confirmed inhibition of MERTK phosphorylation in response to treatment with UNC2025 (Fig S3). AXL phosphorylation was not affected by treatment with UNC2025 at the doses used for these studies (Fig S3C). Combined treatment with AXL and MERTK kinase inhibitors significantly reduced tumor cell expansion relative to either agent alone in nearly all cell lines examined (Fig 3A, B, C, D, E, F). In addition, the interactions between R428 and UNC2025 or siAXL and UNC2025 were assessed in HNSCC, TNBC, and NSCLC cells using the fractional product method. The results demonstrated statistically significant differences between the expected relative cell number (E) assuming an additive interaction and the actual value observed (O) indicating synergy in 6 of 10 cell lines tested (Fig S4A). Synergistic interactions were also demonstrated in UM-SCC1 and MDAMB231 cell lines using isobologram analyses (Fig S4B). Similarly, reduced expansion was observed in esophageal cell lines expressing both AXL and MERTK after treatment with R428 and UNC2025 combination therapy (Fig S4C, D and E). Collectively, these data demonstrate combined inhibition of AXL and MERTK as a highly effective strategy to suppress expansion of tumor cells that co-express both receptors.
Figure 3: Dual targeting of AXL and MERTK can effectively inhibit cell proliferation in vitro.
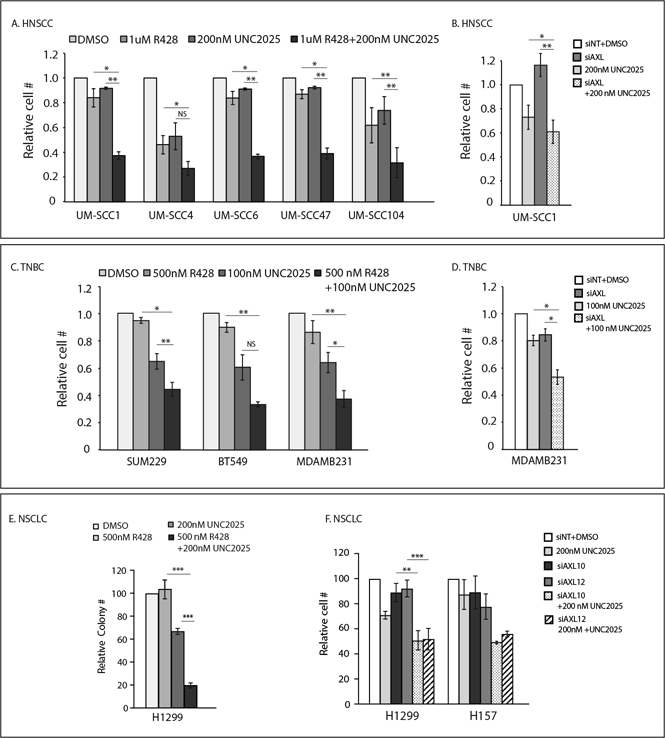
(A, B, C, D, F)HNSCC and TNBC cell lines were treated with vehicle (DMSO), R428, UNC2025, or R428+UNC2025 and relative cell numbers were determined after 72 hours. UM-SCC1, MDAMB231, and NSCLC cell lines were transfected with AXL siRNA (siAXL, siAXL10, siAXL12) or non-targeting siRNA (siNT) and then treated with vehicle or UNC2025 for 72 hours prior to determination of relative cell numbers. (E) NSCLC cell lines were cultured at low density and treated with vehicle (DMSO), R428, UNC2025, or R428+UNC2025 for 10 days. Colonies were stained and counted. *p<0.05, **p<0.01, ***p<0.001, NS=not significant.
To investigate potential mechanisms of synergistic anti-tumor activity mediated by the combination of R428 and UNC2025, we determined the status of downstream signaling pathways in the context of R428 and siAXL in combination with UNC2025. In UM-SCC1 and MDAMB231 cells treated with R428 and UNC2025, there was a substantial decrease in expression of phospho-STAT6, phospho-AKT, phospho-P70S6K, phospho-S6rp, and phospho-C-RAF (Fig 4A and B). In both cases, signaling was decreased to a greater degree in cultures treated with R428 and UNC2025 as compared to cultures treated with either single agent. Similarly, in H1299 NSCLC cells treated with siAXL and UNC2025, combined inhibition of AXL and MERTK led to more potent decreases in phosphorylation of STAT6 and AKT (Fig 4C). Collectively, these data demonstrate reduced downstream signaling in response to combined inhibition of AXL and MERTK and suggest potential biochemical mechanisms for the corresponding reduction in tumor cell expansion mediated by the combination treatment.
Figure 4: Dual inhibition of AXL and MERTK reduces downstream signaling through oncogenic pathways.

(A, B)UM-SCC1 and MDAMB231 cells were treated with vehicle (DMSO), R428, UNC2025, or R428+UNC2025 for 24 hours. (C) H1299 cells were transfected with AXL siRNA (siAXL10 or siAXL12) or non-targeting siRNA (siNT) and cells were treated with vehicle or UNC2025 for one hour. Expression of the indicated proteins was assessed in cell lysates by immunoblot. α-Tubulin is shown as a loading control. The data shown are representative of 3 independent experiments.
MERTK overexpression mediates resistance to AXL inhibition
To determine whether expression of MERTK is sufficient to mediate resistance to anti-AXL therapy, HNSCC and TNBC cell lines that are sensitive to anti-AXL targeting strategies (HN30 and SUM159) and do not express endogenous MERTK (Fig 1A-D) were manipulated to overexpress MERTK and then treated with AXL inhibitors R428 or Mab173 for 72 hours prior to assessment of relative cell numbers. While tumor cell expansion was inhibited by 45–65% in vector-expressing HN30 and SUM159 cells treated with R428 and Mab173, derivative cells with ectopic expression of MERTK were relatively resistant and cell numbers were only decreased by 3–23% (Fig 5A, C and D). In contrast, HN30 cells with ectopic expression of TYRO3 remained sensitive to R428 treatment (Fig S5). Consistent with the biochemical effects of MERTK inhibition, ectopic expression of MERTK increased downstream signaling through phospho-P70S6K, phospho-S6rp, and phospho-C-RAF (Fig 5B and E). Collectively, these results demonstrate a direct role for MERTK in resistance to AXL inhibitors.
Figure 5: Overexpression of MERTK confers resistance to AXL inhibition.
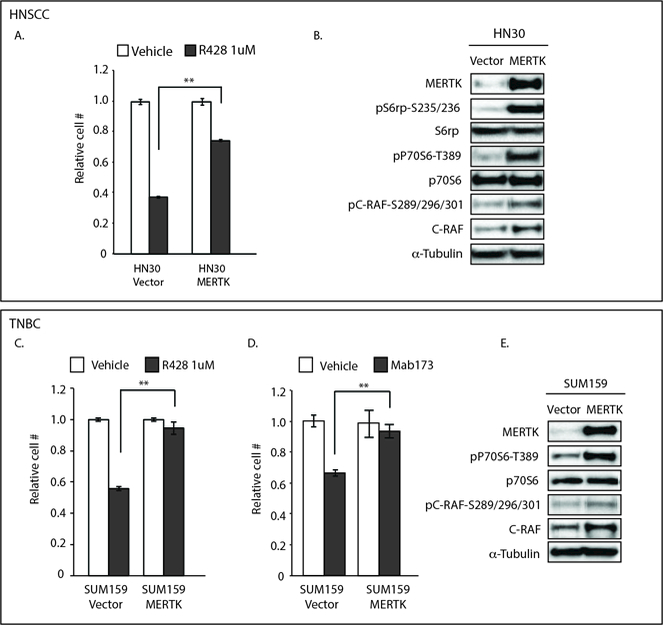
HN30 (A, B) and SUM159 (C, D, E) cells were transfected with pcDNA6.0-Vector (Vector) or pcDNA6.0-MERTK (MERTK) plasmids. (A, C, D) 24 hours after transfection, cells were treated with R428 (A, C) or Mab173 (D) for an additional 72 hours and then relative cell numbers were determined. Mean values, standard errors, and statistical analyses were derived from replicates within one experiment and are representative of 3 independent experiments. **p<0.01. (B, E) 72 hours after transfection, cells were harvested and lysates were prepared with the indicated proteins detected by immunoblot. α-Tubulin is shown as a loading control.
MERTK-mediated resistance to AXL inhibition can be overcome by dual targeting in vivo
The observed upregulation of MERTK in tumor cell lines in response to AXL inhibition (Fig 2) and the potent anti-tumor responses mediated by combined targeting of MERTK and AXL in vitro (Fig 3) implicate co-inhibition as an effective therapeutic strategy. To further investigate this potential, we determined whether a similar response could be observed in xenograft models. First, MDAMB231 (TNBC) xenografts were established in nude mice. Once tumors reached a volume of approximately 500 mm3, mice were treated with vehicle or R428 (50mg/kg/day) for 4 days (n=8 tumors per treatment group) and then tumors were harvested and analyzed for MERTK expression via immunoblotting and immunohistochemistry (IHC). In this study, MERTK expression was consistently increased in MDAMB231 tumors after AXL inhibition (Fig 6A). Similarly, a HNSCC patient-derived xenograft (PDX) was established in nude mice and treated with vehicle or R428 (50mg/kg/day) for 4 days or 28 days (n=8 tumors per treatment group). Again, MERTK was prominently upregulated in tumors treated with R428 and this increase persisted during the extended treatment period (Fig 6B). Consistent with this observation, tumor volume was not significantly different in mice treated with R428 relative to vehicle-treated mice. Thus, upregulation of MERTK was accompanied by relative resistance to AXL inhibition in this patient-derived xenograft model.
Figure 6: Inhibition of AXL increases MERTK protein expression and co-inhibition of AXL and MERTK suppresses tumor growth in vivo.
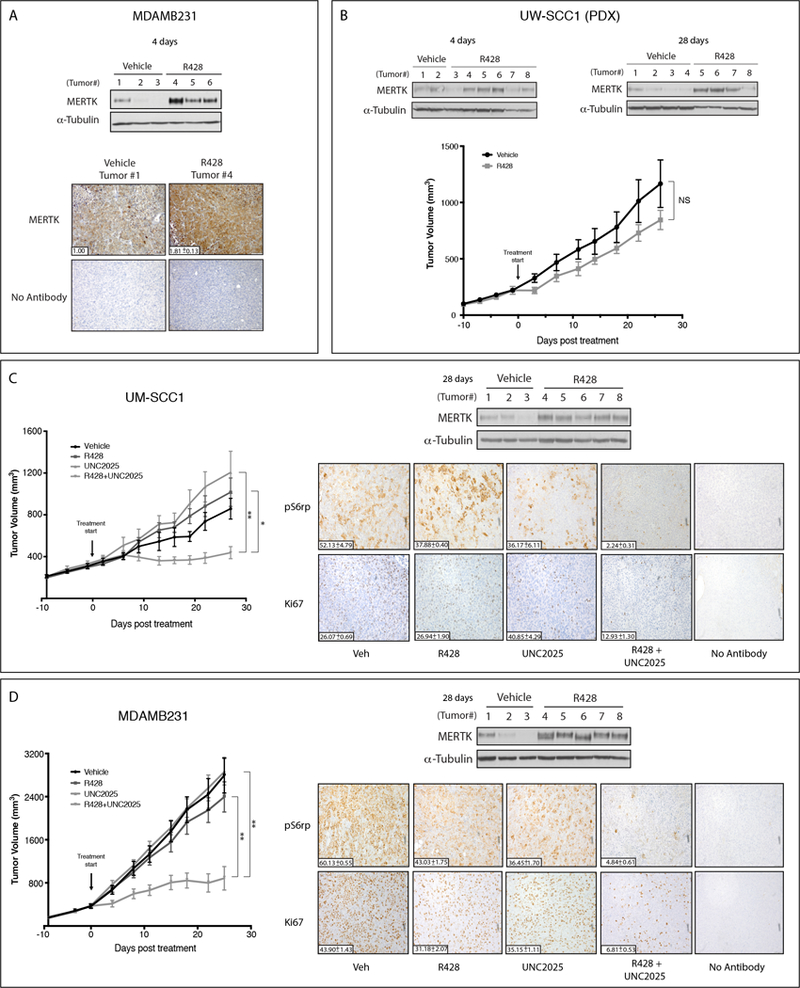
Subcutaneous xenografts were generated in nude mice using (A, D) the TNBC MDAMB231 cell line, (B) the HNSCC UW-SCC1 patient-derived xenograft (PDX), or (C) the HNSCC UM-SCC1 cell line. Tumor-bearing mice were treated with vehicle, 25mg/kg R428, 25 mg/kg UNC2025, or R428+UNC2025 administered twice daily via oral gavage. MERTK expression was assessed in tumor cell lysates by immunoblot. α-Tubulin was used as a loading control. (A) MERTK expression was also detected in MDAMB231 tumors using immunohistochemistry. (B, C, D) Tumor volume was measured (n=8 per group). (C, D) pS6rp and Ki67 were detected in tumor sections using IHC. Mean values and standard errors are shown. *p<0.05, **p<0.01, NS=not significant.
To test the therapeutic efficacy of co-targeting MERTK and AXL in vivo, subcutaneous xenografts of the HNSCC cell line UM-SCC1 and the TNBC cell line MDAMB231 were generated in nude mice. Once tumors reached approximately 200 mm3, mice were randomized into groups for treatment with vehicle, R428 (50mg/kg/day), UNC2025 (50mg/kg/day), or the combination of R428 and UNC2025 (n=16 tumors per treatment group). Therapies were administered twice daily by oral gavage. Combined treatment with R428 and UNC2025 significantly decreased tumor volume compared to treatment with either inhibitor alone in both models (Fig 6C and D). Furthermore, IHC analysis revealed robust decreases in phospho-S6rp and Ki67 in tumors treated with the combination therapy (~2 to 25-fold), indicating decreased tumor cell proliferation and downstream signaling consistent with the biochemical and functional effects of MERTK and AXL co-inhibition observed in vitro (Fig 4). MERTK expression was also upregulated in R428-treated UM-SCC1 and MDAMB231 tumors after 28 days of treatment (Fig 6C and D), consistent with our previous results in cell culture (Fig 2) and animal models (Fig 6A and B). Collectively, these data indicate that targeting MERTK can overcome resistance to AXL inhibition and enhance the therapeutic efficacy of AXL inhibitors in HNSCC and TNBC xenograft models.
Discussion:
AXL is overexpressed in numerous cancers and has been associated with resistance to both conventional and molecular-targeted therapies (1,3,5,9,14–21,24,32,33,35,38). Thus, AXL inhibition has emerged as a promising treatment strategy. While AXL-targeting strategies may have initial clinical benefit in tumor types that overexpress the receptor, intrinsic and acquired resistance to single agent kinase inhibitors is common and resistance to single agent AXL inhibitors will likely appear. Our previous studies utilizing shRNA demonstrated oncogenic roles for both MERTK and AXL in astrocytoma and NSCLC (6,39). Given this functional redundancy, we hypothesized a role for MERTK in resistance to AXL-targeted therapy. In the studies presented here, single agent anti-AXL therapy had limited efficacy and the TAM family receptor MERTK was upregulated in response to AXL suppression in HNSCC, TNBC, and NSCLC preclinical models. Moreover, ectopic expression of MERTK was sufficient to mediate resistance to AXL-targeting strategies and combined inhibition of both AXL and MERTK using a variety of different approaches provided potent anti-tumor activity in HNSCC, TNBC, and NSCLC cell culture and animal models. Collectively, these data demonstrate the importance of MERTK in resistance to AXL inhibitors in several tumor types and provide rationale for co-targeting AXL and MERTK in tumors that express both receptors.
Several preclinical studies have described similar adaptive responses to single agent RTK-targeting strategies. For example, HER2 and HER3 are upregulated in response to EGFR inhibition (29–31,36), and knockdown of the RTK RON results in upregulation of its close family member cMET. Co-targeting of RTK family members has been highly beneficial to overcome these adaptive responses in several tumor models (29–31,36,37,40,41). Additional studies have demonstrated activation of RTKs outside the immediate family of the targeted kinase, such as the induction of AXL expression in response to treatment with EGFR inhibitors (5,11,17,38). While genetic mechanisms such as mutations, polymorphisms, or copy number variations are likely to mediate or contribute to kinase inhibitor resistance in some cases, these findings illustrate the importance of bypass signaling as a mechanism of resistance and advocate the utility of therapeutic strategies targeting multiple receptors to prevent development of resistance, consistent with the data presented here.
Regulation of numerous signaling pathways known to play roles in tumorigenesis has been demonstrated downstream of TAM-family kinases, including RAF/MEK/ERK, JAK/STAT, PI3K/AKT, and mTOR/p70S6K (reviewed in (42)). Previous studies showed activation of AKT/mTOR via GAS6 stimulation of MERTK in NSCLC cells (6) and activation of PI3K/P70S6K via a chimeric MERTK receptor (43). Similarly, data presented here reveal activation of P70S6K and C-RAF in response to MERTK overexpression (Fig 5) and potent inhibition of P70S6K, C-RAF, and AKT in response to MERTK and AXL combination therapy (Fig 4). We have also recently found that AXL can mediate EGFR activation through C-RAF/ERK/C-JUN signaling (38). The therapeutic importance of some of these oncogenic pathways associated with TAM-family signaling has been investigated further. Recently Swick et al demonstrated that suppression of both PI3K/AKT/mTORC signaling and RAS/RAF/MAPK1 signaling resulted in dramatic growth suppression in HNSCC (44). Previous studies also associated P70S6K overexpression with increased risk of loco-regional recurrence in early breast cancer patients (45), and PF-4708671, a selective P70S6K inhibitor (46), has been utilized to reduce local relapse in breast cancer models (47) and inhibit NSCLC tumorigenesis (48). Thus, our observation that combined targeting of AXL and MERTK can synergistically inhibit these oncogenic signaling pathways suggests possible mechanisms for the synergistic anti-tumor activity described here. Given the therapeutic impact of these signaling pathways, inhibition of these pathways via combined targeting of AXL and MERTK underscores the clinical benefit that this treatment strategy may provide.
Small molecule inhibitors, monoclonal antibodies, and soluble decoy receptors targeting AXL are currently being tested in early phase clinical trials for treatment of several different malignancies (clinicaltrails.gov). R428 (BGB324) is in phase I/II clinical trials enrolling patients with NSCLC, TNBC, AML, myelodysplastic syndromes, and melanoma. Our data suggest that MERTK upregulation is a common occurrence in at least the broad group of epithelial malignancies tested here. Whether increased by AXL inhibition or present constitutively, MERTK mediated resistance to AXL-targeting agents in functional assays. In addition, cell lines that expressed very little or no MERTK protein were invariably sensitive to AXL inhibition and 80% of cell lines that expressed higher levels of MERTK were resistant to AXL inhibition (Fig 1). While MERTK expression levels alone did not always predict resistance to AXL inhibition (e.g. H1299, UM-SCC4), these data suggest that in many cases MERTK expression could be useful as a predictive biomarker to identify tumors that are less likely to respond to single agent AXL therapy. Targeting of a non-mutant RTK alone has not been very successful; however, we are learning through multiple examples (e.g mutant EGFR and ALK) that patients can benefit from second and third generation inhibitors and combinations of RTK inhibitors that reduce tumor cell survival signaling and other oncogenic pathways. Indeed, inhibition of MERTK re-sensitized tumors to AXL inhibition mediated by R428 in preclinical models, providing strong rationale for clinical development of therapeutic strategies targeting both AXL and MERTK.
Supplementary Material
Acknowledgements:
We thank Parkash Gill at the University of Southern California for use of the AXL antibody (Mab173), the Daniel Horyn Foundation for the gift of esophageal cell lines. The authors also thank Masami Glines for expert technical assistance, and the University of Wisconsin Carbone Cancer Center Support Grant (P30 CA014520).
Financial Support: Research reported in this publication was supported by the Wisconsin Head & Neck Cancer SPORE (DLW, RJK, and PMH P50 DE026787), Vilas Faculty Early Career Investigator Award (DLW, MSN189325), an NIH SPORE in Lung Cancer grant (KDD, P50CA058187), the Lung Cancer Colorado Fund (KDD) and an NIH grant from Cellular and Molecular Pathology Graduate Training Program (NKM, 5 T32 GM81061–7). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest: DKG, SVF, HSE, XW, and DD have ownership in Meryx, Inc. SVF, HSE and DKG have leadership positions in Meryx. Inc. No potential conflicts of interest were disclosed for others.
References:
- 1.Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Advances in cancer research 2008;100:35–83 doi 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verma A, Warner SL, Vankayalapati H, Bearss DJ, Sharma S. Targeting Axl and Mer kinases in cancer. Mol Cancer Ther 2011;10(10):1763–73 doi 10.1158/1535-7163.MCT-11-0116. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J, et al. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009;28(39):3442–55 doi 10.1038/onc.2009.212. [DOI] [PubMed] [Google Scholar]
- 4.Linger RM, Keating AK, Earp HS, Graham DK. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert opinion on therapeutic targets 2010;14(10):1073–90 doi 10.1517/14728222.2010.515980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nature genetics 2012;44(8):852–60 doi 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linger RM, Cohen RA, Cummings CT, Sather S, Migdall-Wilson J, Middleton DH, et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013;32(29):3420–31 doi 10.1038/onc.2012.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013;19(1):279–90 doi 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye X, Li Y, Stawicki S, Couto S, Eastham-Anderson J, Kallop D, et al. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010;29(38):5254–64 doi 10.1038/onc.2010.268. [DOI] [PubMed] [Google Scholar]
- 9.Shieh YS, Lai CY, Kao YR, Shiah SG, Chu YW, Lee HS, et al. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005;7(12):1058–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res 2010;70(4):1544–54 doi 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]
- 11.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal 2013;6(287):ra66 doi 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang YX, Knyazev PG, Cheburkin YV, Sharma K, Knyazev YP, Orfi L, et al. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res 2008;68(6):1905–15 doi 10.1158/0008-5472.CAN-07-2661. [DOI] [PubMed] [Google Scholar]
- 13.Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014;33(10):1316–24 doi 10.1038/onc.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Alfonso TM, Hannah J, Chen Z, Liu Y, Zhou P, Shin SJ. Axl receptor tyrosine kinase expression in breast cancer. Journal of clinical pathology 2014;67(8):690–6 doi 10.1136/jclinpath-2013-202161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rankin EB, Fuh KC, Taylor TE, Krieg AJ, Musser M, Yuan J, et al. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res 2010;70(19):7570–9 doi 10.1158/0008-5472.CAN-10-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunne PD, McArt DG, Blayney JK, Kalimutho M, Greer S, Wang T, et al. AXL Is a Key Regulator of Inherent and Chemotherapy-Induced Invasion and Predicts a Poor Clinical Outcome in Early-Stage Colon Cancer. Clin Cancer Res 2014;20(1):164–75 doi 10.1158/1078-0432.CCR-13-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giles KM, Kalinowski FC, Candy PA, Epis MR, Zhang PM, Redfern AD, et al. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol Cancer Ther 2013;12(11):2541–58 doi 10.1158/1535-7163.MCT-13-0170. [DOI] [PubMed] [Google Scholar]
- 18.Avilla E, Guarino V, Visciano C, Liotti F, Svelto M, Krishnamoorthy G, et al. Activation of TYRO3/AXL tyrosine kinase receptors in thyroid cancer. Cancer Res 2011;71(5):1792–804 doi 10.1158/0008-5472.CAN-10-2186. [DOI] [PubMed] [Google Scholar]
- 19.Paccez JD, Vasques GJ, Correa RG, Vasconcellos JF, Duncan K, Gu X, et al. The receptor tyrosine kinase Axl is an essential regulator of prostate cancer proliferation and tumor growth and represents a new therapeutic target. Oncogene 2013;32(6):689–98 doi 10.1038/onc.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song X, Wang H, Logsdon CD, Rashid A, Fleming JB, Abbruzzese JL, et al. Overexpression of receptor tyrosine kinase Axl promotes tumor cell invasion and survival in pancreatic ductal adenocarcinoma. Cancer 2011;117(4):734–43 doi 10.1002/cncr.25483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han J, Tian R, Yong B, Luo C, Tan P, Shen J, et al. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochem Biophys Res Commun 2013;435(3):493–500 doi 10.1016/j.bbrc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 22.Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014;5:5712 doi 10.1038/ncomms6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huey MG, Minson KA, Earp HS, DeRyckere D, Graham DK. Targeting the TAM Receptors in Leukemia. Cancers (Basel) 2016;8(11) doi 10.3390/cancers8110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu R, Gong M, Li X, Zhou Y, Gao W, Tulpule A, et al. Induction, regulation, and biologic function of Axl receptor tyrosine kinase in Kaposi sarcoma. Blood 2010;116(2):297–305 doi 10.1182/blood-2009-12-257154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.BerGenBio. 2017 21 Jun. Pipeline: BGB324. http://www.bergenbio.com/pipeline/bgb324. Accessed 2017 21 Jun.
- 26.Zhang W, DeRyckere D, Hunter D, Liu J, Stashko MA, Minson KA, et al. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J Med Chem 2014;57(16):7031–41 doi 10.1021/jm500749d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeRyckere D, Lee-Sherick AB, Huey MG, Hill AA, Tyner JW, Jacobsen KM, et al. UNC2025, a MERTK Small-Molecule Inhibitor, Is Therapeutically Effective Alone and in Combination with Methotrexate in Leukemia Models. Clin Cancer Res 2017;23(6):1481–92 doi 10.1158/1078-0432.CCR-16-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cummings CT, Zhang W, Davies KD, Kirkpatrick GD, Zhang D, DeRyckere D, et al. Small Molecule Inhibition of MERTK Is Efficacious in Non-Small Cell Lung Cancer Models Independent of Driver Oncogene Status. Mol Cancer Ther 2015;14(9):2014–22 doi 10.1158/1535-7163.MCT-15-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen FL, Xia W, Spector NL. Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res 2008;14(21):6730–4 doi 10.1158/1078-0432.CCR-08-0581. [DOI] [PubMed] [Google Scholar]
- 30.Mancini M, Gaborit N, Lindzen M, Salame TM, Dall’Ora M, Sevilla-Sharon M, et al. Combining three antibodies nullifies feedback-mediated resistance to erlotinib in lung cancer. Sci Signal 2015;8(379):ra53 doi 10.1126/scisignal.aaa0725. [DOI] [PubMed] [Google Scholar]
- 31.Yamaguchi H, Chang SS, Hsu JL, Hung MC. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene 2014;33(9):1073–81 doi 10.1038/onc.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Massenhausen A, Bragelmann J, Billig H, Thewes B, Queisser A, Vogel W, et al. Implication of the Receptor Tyrosine Kinase AXL in Head and Neck Cancer Progression. Int J Mol Sci 2016;18(1) doi 10.3390/ijms18010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Coan JP, et al. AXL Is a Logical Molecular Target in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res 2015;21(11):2601–12 doi 10.1158/1078-0432.CCR-14-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leconet W, Chentouf M, du Manoir S, Chevalier C, Sirvent A, Ait-Arsa I, et al. Therapeutic Activity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin Cancer Res 2017;23(11):2806–16 doi 10.1158/1078-0432.CCR-16-1316. [DOI] [PubMed] [Google Scholar]
- 35.Wu X, Ma W, Zhou Q, Yan H, Lim ZF, Huang M, et al. AXL-GAS6 expression can predict for adverse prognosis in non-small cell lung cancer with brain metastases. J Cancer Res Clin Oncol 2017. doi 10.1007/s00432-017-2408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008;27(28):3944–56 doi onc200819 [pii] 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iida M, Bahrar H, Brand TM, Pearson HE, Coan JP, Orbuch RA, et al. Targeting the HER Family with Pan-HER Effectively Overcomes Resistance to Cetuximab. Mol Cancer Ther 2016;15(9):2175–86 doi 10.1158/1535-7163.MCT-16-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Luthar N, et al. AXL Mediates Resistance to Cetuximab Therapy. Cancer Res 2014;74(18):5152–64 doi 10.1158/0008-5472.CAN-14-0294. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 39.Keating AK, Kim GK, Jones AE, Donson AM, Ware K, Mulcahy JM, et al. Inhibition of Mer and Axl receptor tyrosine kinases in astrocytoma cells leads to increased apoptosis and improved chemosensitivity. Mol Cancer Ther 2010;9(5):1298–307 doi 10.1158/1535-7163.MCT-09-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iida M, Brand TM, Starr MM, Huppert EJ, Luthar N, Bahrar H, et al. Overcoming acquired resistance to cetuximab by dual targeting HER family receptors with antibody-based therapy. Mol Cancer 2014;13:242 doi 10.1186/1476-4598-13-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawada I, Hasina R, Arif Q, Mueller J, Smithberger E, Husain AN, et al. Dramatic antitumor effects of the dual MET/RON small-molecule inhibitor LY2801653 in non-small cell lung cancer. Cancer Res 2014;74(3):884–95 doi 10.1158/0008-5472.CAN-12-3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer 2014;14(12):769–85 doi 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 43.Ling L, Kung HJ. Mitogenic signals and transforming potential of Nyk, a newly identified neural cell adhesion molecule-related receptor tyrosine kinase. Mol Cell Biol 1995;15(12):6582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swick AD, Prabakaran PJ, Miller MC, Javaid AM, Fisher MM, Sampene E, et al. Cotargeting mTORC and EGFR Signaling as a Therapeutic Strategy in HNSCC. Mol Cancer Ther 2017;16(7):1257–68 doi 10.1158/1535-7163.MCT-17-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Hage JA, van den Broek LJ, Legrand C, Clahsen PC, Bosch CJ, Robanus-Maandag EC, et al. Overexpression of P70 S6 kinase protein is associated with increased risk of locoregional recurrence in node-negative premenopausal early breast cancer patients. Br J Cancer 2004;90(8):1543–50 doi 10.1038/sj.bjc.6601741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, et al. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem J 2010;431(2):245–55 doi 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- 47.Segatto I, Berton S, Sonego M, Massarut S, D’Andrea S, Perin T, et al. Inhibition of breast cancer local relapse by targeting p70S6 kinase activity. J Mol Cell Biol 2013;5(6):428–31 doi 10.1093/jmcb/mjt027. [DOI] [PubMed] [Google Scholar]
- 48.Qiu ZX, Sun RF, Mo XM, Li WM. The p70S6K Specific Inhibitor PF-4708671 Impedes Non-Small Cell Lung Cancer Growth. PLoS One 2016;11(1):e0147185 doi 10.1371/journal.pone.0147185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iida M, Brand TM, Starr MM, Li C, Huppert EJ, Luthar N, et al. Sym004, a Novel EGFR Antibody Mixture, Can Overcome Acquired Resistance to Cetuximab. Neoplasia 2013;15(10):1196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li C, Brand TM, Iida M, Huang S, Armstrong EA, van der Kogel A, et al. Human epidermal growth factor receptor 3 (HER3) blockade with U3–1287/AMG888 enhances the efficacy of radiation therapy in lung and head and neck carcinoma. Discov Med 2013;16(87):79–92. [PMC free article] [PubMed] [Google Scholar]
- 51.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70(2):440–6 doi 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 52.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- 53.Irwin ME, Mueller KL, Bohin N, Ge Y, Boerner JL. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol 2011;226(9):2316–28 doi 10.1002/jcp.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.