

Abstract
Neuroblastoma is the most common extracranial malignancy in the pediatric population, accounting for over 9% of all cancer-related deaths in children. Autophagy is a cell self-protective mechanism that promotes tumor cell growth and survival, making it an attractive target for treating cancer. However, the role of autophagy in neuroblastoma tumor growth and metastasis is largely undefined. Here we demonstrate that targeted inhibition of an essential autophagy kinase, unc-51 like autophagy kinase 1 (ULK1), with a recently developed small molecular inhibitor of ULK1, SBI-0206965, significantly reduces cell growth and promotes apoptosis in SK-N-AS, SH-SY5Y, and SK-N-DZ neuroblastoma cell lines. Furthermore, inhibition of ULK1 by a dominant-negative mutant of ULK1 (dnULK1K46N) significantly reduces growth and metastatic disease and prolongs survival of mice bearing SK-N-AS xenograft tumors. We also show that SBI-0206965 sensitizes SK-N-AS cells to TRAIL treatment, but not to mTOR inhibitors (INK128, Torin1) or topoisomerase inhibitors (doxorubicin, topotecan). Collectively, these findings demonstrate that ULK1 is a viable drug target and suggest that inhibitors of ULK1 may provide a novel therapeutic option for the treatment of neuroblastoma.
INTRODUCTION
Neuroblastoma is a cancer of the primordial neural crest cells, which gives rise to the sympathetic nervous system and generally occurs in infants and young children. Neuroblastoma is the most common extracranial solid tumor in the pediatric population, accounting for 7 to 10% of all pediatric cancers and over 9% of all cancer-related deaths in children (1). Clinically, tumors can occur anywhere along the sympathetic nervous system and symptoms can develop from compression of vital structures, including the spinal column, from tumors that arise in the parasympathetic ganglia. However, the majority of tumors arise in the adrenal gland, which can progress to high-stage tumors that infiltrate local organ structures and metastasize to lymph nodes, bone marrow, and the liver. The behavior of neuroblastoma varies widely from spontaneous remission to aggressive metastatic disease. Several prognostic factors are used to stratify neuroblastoma into very low-risk, low-risk, intermediate-risk, and high-risk classes (1,2). These factors include the age at diagnosis, DNA index (ploidy), MYCN amplification, and tumor stage, which is based upon the surgical-pathologic International Neuroblastoma Staging System (INSS) (1–3). Distant metastases are detected in approximately 50% of patients at diagnosis, and most commonly occur in the bone, bone marrow, lymph nodes, and liver (1). Therapeutic strategies for high risk neuroblastoma are aggressive, consisting of multi-agent chemotherapy, surgery, radiation, and myeloablative chemotherapy regiments with subsequent autologous bone marrow transplant (2). However, despite these treatments, over half of patients with high risk neuroblastoma will relapse (4), and the 5-year overall survival rate is only 50% (5). Thus, there is an urgent need for new therapeutic options, particularly for patients with high-risk neuroblastoma and those with recurrent or relapsed neuroblastoma.
Macro-autophagy (hereafter referred to as autophagy) is an evolutionarily-conserved degradation process that maintains cellular homeostasis through regular turnover of dysfunctional proteins and organelles (6,7). Autophagy is upregulated by cellular stressors such as nutrient deprivation and provides an important cell survival mechanism by facilitating the recycling of essential nutrients, preventing the accumulation of misfolded proteins and reactive oxygen species (ROS), maintaining organelle function, and regulating intracellular signaling pathways (7). Importantly, autophagy can support established tumors by generating metabolic fuel and reducing oxidative stress (7,8), and inhibition of autophagy can reduce growth and metastasis in certain tumor types (9), such as in pancreatic and lung cancer (10–13). This role of autophagy in cancer survival has made it a popular therapeutic target during the development of new anti-cancer agents (14,15). The limited research available indicates that autophagy promotes resistance to several chemotherapeutic agents used to treat neuroblastoma, such as vincristine and doxorubicin, amongst others (16–18). However, the role of autophagy in neuroblastoma tumor growth and metastasis is still unknown.
A major challenge for targeting autophagy in any cancer is the lack of potent and selective pharmaceutical autophagy inhibitors. The efforts to pharmacologically target autophagy have relied heavily on bafilomycin A1, chloroquine, and hydroxychloroquine, which target not only autophagy flux but also the endolysosomal pathway by inhibiting late endosome and lysosome function. Recently, a small molecule kinase inhibitor called SBI-0206965 has been developed that targets an essential autophagy kinase, unc-51 like autophagy kinase 1 (ULK1) (19). The autophagy process is regulated by more than 30 autophagy-related genes (ATGs) through four discrete steps: (1) the initiation of autophagosome biogenesis, (2) nucleation of the phagophore, (3) the expansion of the phagophore to a mature autophagosome, and (4) the fusion of the autophagosome to the lysosome (6,7). Importantly, initiation of autophagy is predominantly mediated by ULK1, a mammalian homolog of yeast Atg1 that is the only serine/threonine kinase among the ATG proteins (20). ULK1 forms a complex with multiple regulatory subunits including ATG13 and FIP200, a mammalian counterpart of yeast Atg17. This complex is negatively regulated by the mechanistic target of rapamycin complex 1 (mTORC1) through hyper-phosphorylation of ULK1 in nutrient-rich environments, while AMP-activated protein kinase (AMPK) binds and phosphorylates ULK1 to activate autophagy under conditions of energy scarcity (21,22). Upon activation, ULK1 induces the next step of autophagy, nucleation of the immature autophagosome, by phosphorylating the downstream BECN1 complex. Thus, ULK1 is an upstream kinase that regulates the initiation of autophagy, making it an excellent drug target to inhibit autophagy.
SBI-0206965 has been previously characterized as a potent and selective inhibitor of ULK1, exhibiting an IC50 of ULK1 kinase activity at 108nM (19). Importantly, SBI-0206965 was demonstrated to synergize with nutrient deprivation and mTOR inhibitors to induce apoptosis in several cell types (19). Although still in the early stages of development, SBI-0206965 provides new opportunity for pharmacologic inhibition of autophagy that can potentially translate to the clinic. In this study, we establish the important role of ULK1 and autophagy in neuroblastoma growth and response to chemotherapy. We determine that SBI-0206965 exhibits cytotoxicity across multiple neuroblastoma cell lines and that genetic inhibition of ULK1 significantly reduces neuroblastoma tumor growth and metastasis in vivo. These results demonstrate that ULK1-mediated autophagy plays a key role in neuroblastoma tumor growth and progression and that ULK1 is a viable drug target for the treatment of neuroblastoma.
MATERIALS AND METHODS
Cell Culture
Human cell lines SK-N-AS (ATCC CRL-2137), SK-N-DZ (ATCC CRL-2149), SH-SY5Y (ATCC CRL-2266), and A549 (ATCC CCL-185) were purchased from American Type Culture Collection (ATCC). Cells were cultured in Dulbecco’s Modification of Eagle’s Medium (DMEM)/Ham’s F-12 50/50 Mix containing L-glutamine and 15mM HEPES supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) and 1% antibiotic and antimycotic solution at 37°C in a humidified incubator with 5% CO2. All cell lines were passaged in our laboratory for fewer than 6 months before use and periodically authenticated by morphologic inspection and mycoplasma testing. Where indicated, cells were starved in amino acid- and FBS-deficient medium (Wako, cat#048–33575).
Chemicals
The ULK1 inhibitor SBI-0206965 was purchased from Xcessbio Biosciences (cat# M60268–2) and was dissolved in DMSO. Doxorubicin was obtained from Selleck Chemicals (catalog# S1208), Topotecan from Alexis Biochemicals (cat# 350–133-M001), INK128 from Active Biochem (cat# A-1023), Torin1 from Fisher Scientific (cat# NC0418592), and TRAIL from vendor VWR (cat# 10787–196). Subsequent dilutions of stock solutions of compounds were made in culture media just before use. In all experiments, the final concentration of DMSO did not exceed 0.1% (v/v), a concentration that is nontoxic to the cells.
Xenograft Mouse Model of Neuroblastoma
All animal studies were performed according to the guidelines established by the Institutional Animal Care and Use Committee (IACUC) at the Penn State College of Medicine. A xenograft neuroblastoma model was generated by injecting 4.0×106 SK-N-AS cells into the subcutaneous tissue in the flank of NOD SCID Gamma (NSG; Jackson Laboratory, cat# 005557) male and female mice, aged 6–8 weeks. Cells stably expressing a firefly luciferase gene (luc2) were injected in a 50:50 mixture of PBS and matrigel basement membrane matrix (Fisher Scientific, cat# CB-40234). Mice were imaged for luciferase expression on a weekly basis for 5 weeks via a Xenogen IVIS bioluminescent imager. Mice were injected with 5ul/gram body weight of 30mg/ml Luciferin-D (Gold Biotechnology, cat# LUCK-1G) in PBS, 5 minutes prior to imaging. Photon flux was calculated using region of interest (ROI) measurements of either the primary tumor site, or the ventral thoracic area for lung metastasis. Tumor volume was also measured using calipers and calculated as (length2 x width)/2. At the experimental end point, mice were euthanized and tumors were harvested for ex vivo analysis and subsequent histology.
Tail Vein Injection Metastasis Assay
A mouse model of neuroblastoma metastasis was created by injecting 5.0×105 SK-N-AS cells stably expressing the luc2 gene into the tail vein of 6- to 8-week old male NSG mice. Mice were imaged on the ventral side for luciferase expression on a weekly basis for a total of 8 weeks via Xenogen IVIS imaging, as described above. At the experimental end point, mice were euthanized and tumor, bone, and liver tissues were harvested for ex vivo examination and subsequent histologic analysis. Lung, abdominal lymph nodes, and other abdominal organs were examined for metastatic lesions, but none were found.
Immunoblotting
Treated and untreated cells were lysed in RIPA lysis buffer containing protease and phosphatase inhibitors and subjected to immunoblotting with primary antibodies: Flag (Sigma-Aldrich, cat# F1804); p62 (American Research Products, cat# 03-GP62-C); β-actin (Sigma-Aldrich, cat# A5441); LC3 (Novus Biologicals, NB100–2220), PARP (Cell Signaling, cat# 9542), Cleaved Caspase-3 (Cell Signaling, cat# 9661), Phospho-AKT (Ser473) (Cell Signaling, cat# 4058), Phospho-4EBP1 (Thr37/46) (Cell Signaling, cat# 2855), 4EBP1 (Cell Signaling, cat# 9644), AKT (Cell Signaling, cat# 4691), ULK1 (Cell Signaling, cat# 8054S) followed by fluorophore-conjugated secondary antibodies and detection with a Li-Cor Odyssey CLx Imager.
Immunohistochemistry
Tumor and liver tissues were harvested and fixed in 10% formalin and paraffin embedded. Sections were deparaffinized, hydrated, and boiled in citrate buffer for antigen retrieval. VECTASTAIN elite ABC kit (cat# PK-6101) was used for Ki67 (Novus Biologicals, cat# NB500–170) and Cleaved Caspase-3 (Cell Signaling, cat# 9661) staining. Ki67 scoring was performed blind by two independent observers. Liver sections were stained with hematoxylin and eosin.
Viability and Apoptosis Assays
Cell viability was measured by PrestoBlue Cell Viability Reagent (Invitrogen, cat# A-13262) per the manufacturer’s instructions. Caspase-8 and Caspase-3/7 activities were measured using Caspase-Glo 8 (Promega, cat# G8200) and Caspase-Glo 3/7 (Promega, cat# G8090) Assay Systems, respectively. For flow cytometry cell death analyses, treated cells were washed once with PBS and stained with 5% APC Annexin-V and 7-AAD (BioLegend, cat# 640941) for 15 min before flow cytometry analyses. All data were normalized to their non-treated controls.
Statistics
Data were analyzed using Graph Pad Prism and statistical software SAS version 9.4 (SAS Institute, Cary, NC, USA). Student t-test (two-tailed) was used for single comparisons. Group differences were evaluated using ANOVA or repeated-measure ANOVA models. Data were considered statistically significant when p<0.05.
RESULTS
SBI-0206965 reduces cell growth and promotes apoptosis in neuroblastoma cell lines
To validate that the ULK1 inhibitor, SBI-0206965 (Figure 1A), suppresses autophagy in neuroblastoma, we assessed autophagic flux in SK-N-AS cells treated with SBI-0206965 compared to DMSO control. Total ULK1 protein level was reduced in SK-N-AS cells treated with SBI-0206965 compared to control cells treated with DMSO, particularly in starvation conditions, as previously reported (19). Furthermore, as expected, treatment with SBI-0206965 resulted in an accumulation of p62, an autophagy substrate that is widely used as a reporter of autophagic degradation (23), in both complete and starvation medium (Figure 1B). SBI-0206965-treated SK-N-AS cells also displayed a decrease in LC3-II accumulation in the presence of bafilomycin A1, which blocks lysosomal degradation and the autophagosome-lysosome fusion, under starvation conditions (Figure 1B). Together, these results demonstrate that SBI-0206965 reduces LC3 lipidation and autophagic flux in SK-N-AS cells. To determine if pharmacologic inhibition of ULK1 exhibits cytotoxic effects in neuroblastoma, we treated three different neuroblastoma cell lines (SK-N-AS, SH-SY5Y, and SK-N-DZ) with SBI-0206965 (Figure 1C). Western blot analysis of SBI-0206965-treated neuroblastoma cells revealed an accumulation of p62, verifying that SBI-0206965 inhibited autophagy in all three cell lines (Figure 1C). Importantly, SBI-0206965 treatment increased PARP and caspase-3 cleavage in all neuroblastoma cell lines, indicating that SBI-0206965 promoted apoptosis (Figure 1C). The influence of SBI-0206965 treatment on cell growth compared to DMSO control was measured over a 72-hour time course. In normal cell culture conditions, treatment with SBI-0206965 significantly reduced cell growth (Figure 1D-F). As autophagy is known to promote survival under starvation conditions, the neuroblastoma cell lines were also treated with SBI-0206965 in nutrient-deprived culture medium, which further enhanced the cytotoxicity of SBI-0206965 (Figure 1G-I). Furthermore, flow cytometry analysis of SBI-0206965-treated SK-N-AS cells displayed an increase in annexin-V staining compared to cells treated with DMSO control (Figure 1J-K). This effect was further increased under nutrient deprivation, where SBI-0206965 treatment significantly enhanced cell death beyond starvation-induced apoptosis, as previously reported (19) (Figure 1J-K). Similarly, both SH-SY5Y and SK-N-DZ cells also displayed increased annexin-V staining upon SBI-0206965 treatment compared to DMSO control (Figure 1L). Together, these data indicate that SBI-0206965 inhibits autophagy and promotes apoptosis in multiple neuroblastoma cell lines.
Figure 1. SBI-0206965 reduces cell growth and promotes apoptosis in neuroblastoma cell lines.
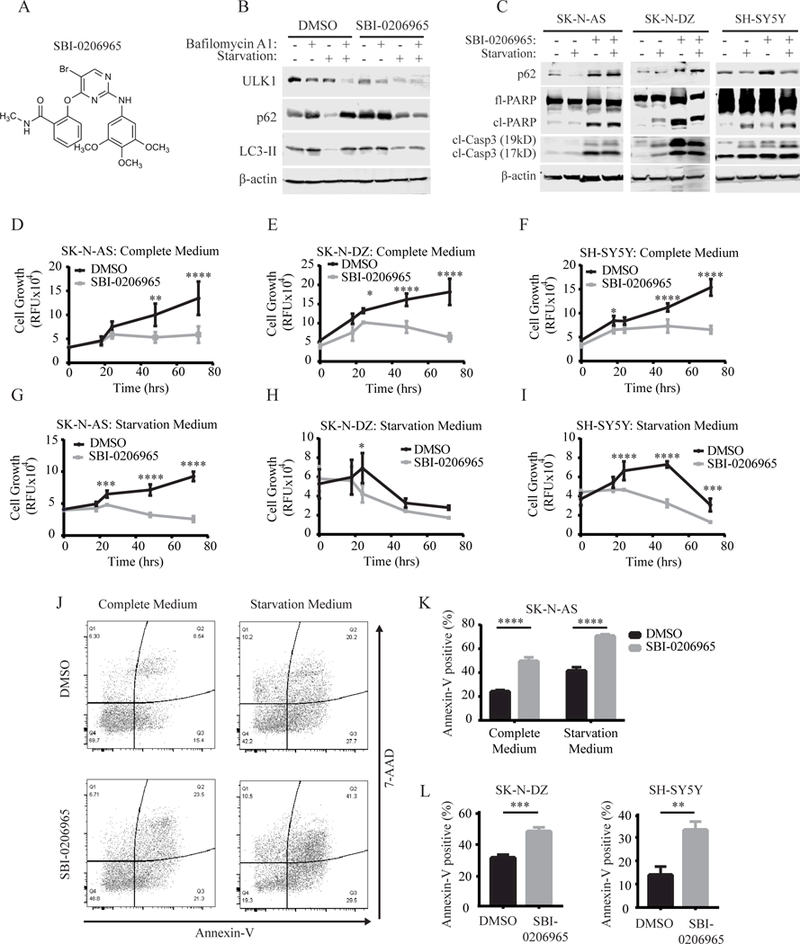
(A) Chemical structure of SBI-0206965. (B) Autophagic flux assay assessing the functional effects of SBI-0206965 on autophagy and ULK1 protein levels. (C)Western blot analysis of autophagy (p62) and apoptosis (cleaved-PARP and cleaved-caspase 3) markers after treatment with 10¼M SBI-0206965 for 48 hours in SK-N-AS and 24 hours in SK-N-DZ and SH-SY5Y cells. (D-I) Cell growth as assessed by Prestoblue in the indicated neuroblastoma cell lines treated with either DMSO or 10¼M SBI-0206965 over 72 hours in complete medium (D-F) or starvation medium (G-I). (J-K) Annexin-V/7-AAD flow cytometry analysis of SK-N-AS cells treated with 10¼M SBI-0206965 or DMSO for 48 hours in complete medium or starvation medium. (L) Annexin-V/7-AAD flow cytometry analysis of SK-N-DZ and SH-SY5Y cells treated with DMSO or 10¼M SBI-0206965 for 24 hours in starvation medium. *p<0.05, **p<0.01, ***p<0.001, p<0.0001.
Inhibition of ULK1 by dnULK1 promotes apoptosis in SK-N-AS cells
To further validate ULK1 as a drug target for neuroblastoma, we generated SK-N-AS cells that express a kinase-dead dominant-negative ULK1K46N gene (dnULK1), as well as control SK-N-AS cells expressing an empty-vector (empty). Expression of dnULK1 resulted in an accumulation of p62, indicating inhibition of autophagy (Figure 2A). Although SBI-0206965-treated cells exhibited higher levels of apoptosis in complete medium conditions, both SBI-0206965 and dnULK1 promoted apoptosis compared to control, as evident by increased cleaved-caspase-3 and cleaved-PARP (Figure 2A). In agreement, the enzymatic activity of caspase-3/7 and caspase-8 increased in both dnULK1-expressing cells and SBI-0206965-treated cells compared to control, which was particularly evident in starvation conditions (Figure 2B-C). Furthermore, annexin-V staining was increased in dnULK1-expressing SK-N-AS cells compared to empty-vector control (Figure 2D-E). Together, these data demonstrate that inhibition of ULK1 by either dnULK1 or SBI-0206965 promotes apoptosis in SK-N-AS cells, validating ULK1 as an attractive target for neuroblastoma treatment.
Figure 2. Inhibition of ULK1 by dnULK1 promotes apoptosis in SK-N-AS cells.
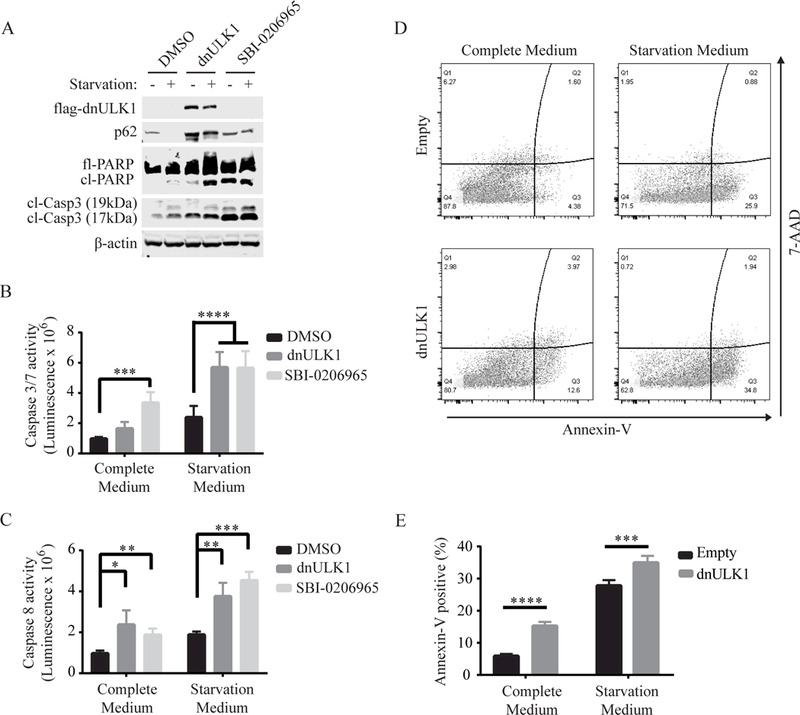
(A) Western blot analysis of SK-N-AS cells expressing dnULK1K46N or treated with 10¼M SBI-0206965 or DMSO for 48 hours in complete or starvation medium. (B-C) Assessment of caspase-3/7 activity (B) or caspaspe-8 activity (C) of SK-N-AS cells expressing dnULK1K46N construct or treated with 10¼M SBI-0206965 or DMSO for 24 hours in complete or starvation medium. (D-E) Annexin-V/7-AAD flow cytometry assay of SK-N-AS cells expressing dnULK1K46N or empty-vector control in complete medium or starvation medium for 48 hours. *p<0.05, **p<0.01, ***p<0.001, p<0.0001.
Inhibition of ULK1 by dnULK1 reduces SK-N-AS xenograft tumor growth and promotes apoptosis
The pharmacological properties of SBI-0206965 in animal models are currently unclear and require further development. Thus, to determine if ULK1 is a viable drug target for neuroblastoma and to determine the effects of ULK1 kinase on neuroblastoma tumor growth, we utilized a genetic approach to inhibit ULK1 in neuroblastoma xenograft mouse models. Specifically, SK-N-AS cells stably expressing dnULK1 or empty-vector were injected into the subcutaneous tissue of NSG mice. A luciferase reporter gene (luc2) was also introduced into the cells to allow for noninvasive monitoring of tumor growth. Weekly measurements of the primary tumor site in the flank revealed that the dnULK1 xenografts grew significantly slower compared to the control empty-vector xenografts (Figure 3A-C). Consistently, ex vivo assessment of both tumor weight and volume showed a significant reduction in the dnULK1-expressing group after 4 weeks of growth (Figure 3D-E). Importantly, primary tumors expressing dnULK1 contained higher levels of p62, indicating that autophagy was suppressed in vivo (Figure 3F). Furthermore, dnULK1-expressing tumors displayed an increased cleavage of PARP and caspase-3, indicating that dnULK1 promoted cell death in vivo (Figure 3F). An increase in cleaved caspase-3 was also detected by immunohistochemistry (IHC) in dnULK1 tumors compared to comparable areas in control tumors (Figure 3G). Interestingly, for reasons that are not currently clear, only an increase in the 19kDa fragment of caspase-3 was detected in tumor lysate by western blot, whereas the level of the 17kDa fragment remained similar between groups. Similarly, although cl-PARP 89kD is slightly increased in dnULK1 SK-N-AS xenografts, we observed significant increases in the 55kD and 42kD PARP cleavage fragments. These fragments are generated from PARP-cleavage by lysosomal proteases, such as cathepsin B and D, and are indicative of necrosis (24,25). Thus, loss of ULK1 function may increase both apoptosis and tumor necrosis in vivo. Additionally, dnULK1-expressing tumors demonstrated a significant reduction in Ki67-positive cells, indicating reduced cell proliferation in vivo. Together, these data indicate that the loss of ULK1 function promotes tumor cell death and reduces tumor growth.
Figure 3. Inhibition of ULK1 by dnULK1 reduces SK-N-AS xenograft tumor growth and promotes apoptosis.
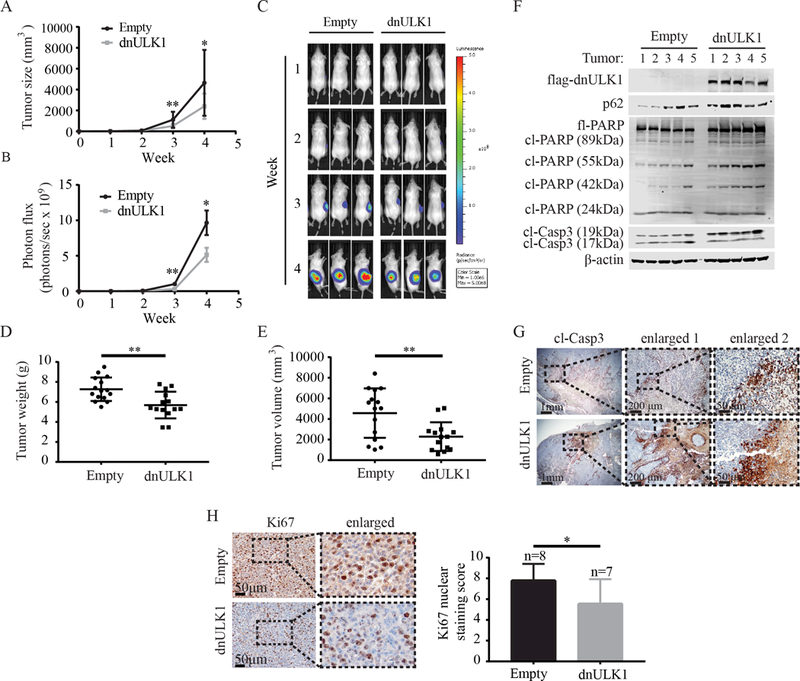
(A) Tumor volume of SK-N-AS xenografts expressing either dnULK1 or empty-vector over 4 weeks (mean ± SD, n=16 mice per group). (B) Quantification of luciferase intensity (photons/sec) in dnULK1 and empty-vector tumors (mean ± SEM, n=16 mice per group). (C) Representative images of luciferase expression from primary xenograft tumors over a 4-week time period. (D) Ex vivo analysis of xenograft tumor weight (mean ± SD). (E) Ex vivo analysis of xenograft tumor volume (mean ± SD). (F) Western blot analysis of xenograft tumors (n=5 tumors per group). (G) Representative image of immunohistochemical staining for cleaved caspase-3 of primary xenograft tumors. (H) Quantification and representative image of immunohistochemical staining for Ki67 of primary xenograft tumors (mean ± SD). *p<0.05, **p<0.01
Inhibition of ULK1 by dnULK1 reduces metastatic tumor growth and improves overall survival in an experimental metastasis mouse model of neuroblastoma
As metastasis is detected in approximately 50% of patients at diagnosis and is generally associated with worse prognosis (1), we examined the effects of ULK1 inhibition in a mouse model of neuroblastoma metastasis. Here, SK-N-AS cells expressing luc2 and dnULK1 or empty-vector were injected into the bloodstream of mice via the tail-vein and metastatic burden, which occurred primarily in the liver, was monitored by luciferase expression over 8 weeks. Importantly, mice receiving dnULK1-expressing SK-N-AS cells survived significantly longer compared to control (Figure 4A) and exhibited a significant reduction in growth at the metastatic site (i.e. liver) (Figure 4B-C). Interestingly, the pattern of tumor growth in the liver was drastically different between the two groups (Figure 4D). At the time of sacrifice, the livers in the empty-vector group were enlarged, displayed large fluid-filled cysts, and lacked defined solid tumor masses (Figure 4D-E). Comparatively, livers in the dnULK1-expressing SK-N-AS group were smaller with solid tumor masses and contained significantly fewer cysts (Figure 4E-F). At this point, it is unclear how engrafted empty-vector SK-N-AS cells cause the cystic liver phenotype and why inhibition of ULK1 kinase significantly reduces this effect.
Figure 4. Inhibition of ULK1 by dnULK1 promotes survival and reduces metastatic tumor growth.
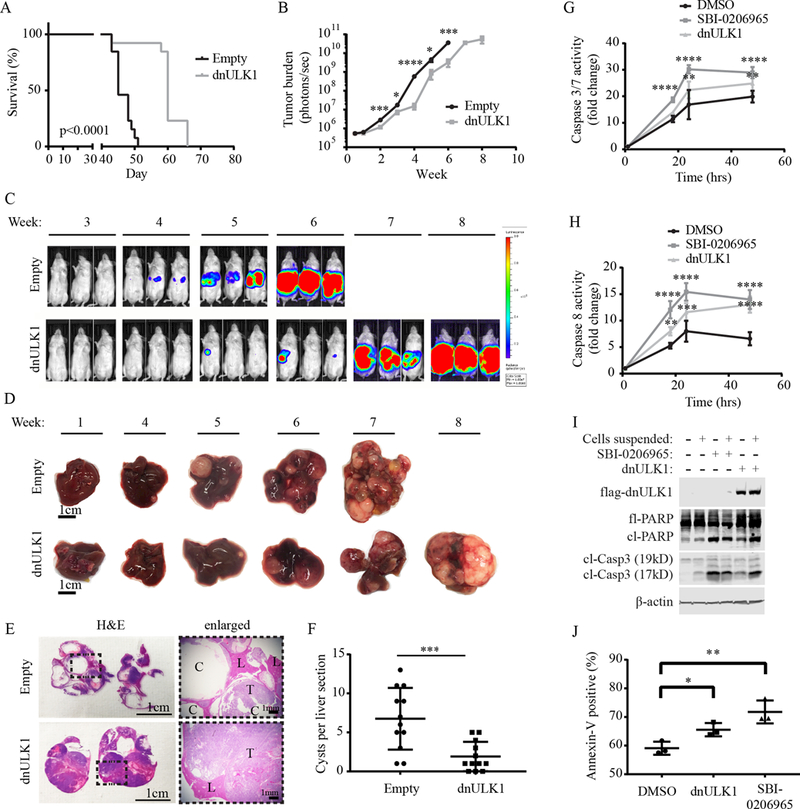
(A) Kaplan-Meier survival curve of mice injected via the tail vein with dnULK1-expressing SK-N-AS cells compared to empty-vector control. (B) Quantification of metastatic tumor growth via luciferase expression (mean ± SEM, n = 13). (C) Representative images of luciferase expression in SK-N-AS xenografts. (D) Representative images of tumor-bearing livers over an 8-week time period. (E) Representative images of H&E staining of tumor-bearing liver sections from empty-vector mice at week 7 and dnULK1 mice at week 8. (C=cyst, L=liver tissue, T=tumor tissue). (F) Quantification of cysts in empty-vector and dnULK1 liver sections. (G) Caspase 3/7 activity in suspended SK-N-AS cells expressing dnULK1 or treated with 10¼M SBI-0206965 or DMSO over 48 hours. (H) Caspase 8 activity in suspended SK-N-AS cells expressing dnULK1 or treated with 10¼M SBI-0206965 or DMSO over 48 hours. (I) Western blot analysis of adherent and suspended SK-N-AS cells expressing dnULK1 or treated with 10¼M SBI-0206965 or DMSO for 48 hours. (J) Assessment of annexin-V staining by flow cytometry in suspended SK-N-AS cells expressing dnULK1 or treated with 10¼M SBI-0206965 or DMSO for 48 hours. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Evasion of anoikis, a form of apoptotic cell death that occurs when cells experience prolonged detachment from the extracellular matrix (26), contributes to circulating tumor cell (CTC) survival and metastasis (9,27). As autophagy is known to promote anoikis -resistance, we tested whether inhibition of ULK1 sensitizes neuroblastoma cells to anoikis. Indeed, both dnULK1 and SBI-0206965 treatment enhanced caspase-3/7 and caspase-8 activity in suspended SK-N-AS cells over 48 hours (Figure 4G-H). In agreement, inhibition of ULK1 by dnULK1 or SBI-0206965 in suspended SK-N-AS cells increased cleaved-PARP and cleaved-caspase-3 protein levels (Figure 4I). Moreover, inhibition of ULK1 resulted in an increase in annexin-V positive cells (Figure 4J). Together, these data suggest that targeted inhibition of ULK1 kinase promotes anoikis, which may contribute to metastasis suppression by reducing CTC survival.
SBI-0206965 sensitizes SK-N-AS cells to TRAIL but not mTOR inhibitors or topoisomerase inhibitors
Finally, we aimed to determine if SBI-0206965 synergizes with other chemotherapeutic agents, as SBI-0206965 was previously reported to synergize with mTOR inhibitors to target lung cancer cells, and because inhibition of autophagy enhances the efficacy of conventional therapeutic modalities (14,19). First, we tested if SBI-0206965 sensitizes SK-N-AS cells to two FDA-approved topoisomerase inhibitors (doxorubicin and topotecan) that are used clinically to treat neuroblastoma. However, no significant increase in cell death was observed in combination treatments (Figure 5A-B). Similarly, when SBI-0206965 was combined with the mTOR inhibitors INK128 or Torin1 (28,29), no significant increase in cytotoxicity was observed (Figure 5C-D). In agreement with cell viability, co-treatment of SBI-0206965 with INK128 or Torin1 did not significantly increase PARP cleavage or annexin-V staining (Figure 5E-F). As these findings are in opposition to those previously reported (19), we validated that mTORC1/2 signaling was inhibited by INK128 and Torin1 in SK-N-AS cells, as evident by reduced phospho-4eBP1(T37/46) and phospho-AKT(S473) levels (Figure 5F). We then tested SH-SY5Y and SK-N-DZ neuroblastoma cells, as well as A549 lung cancer cells, which were previously reported to exhibit cytotoxic synergism to co-treatments of SBI-0206965 and mTOR inhibitors (19). As expected, A549 cells displayed a significant increase in annexin-V staining and PARP cleavage when co-treated with SBI-0206965 and INK128 or Torin 1(Figure 5G-H). However, as with SK-N-AS cells, both SH-SY5Y and SK-N-DZ exhibited limited additive effect to co-treatments of SBI-0206965 and INK128 or Torin 1 (Figures 5G-H), further demonstrating that SBI-0206965 does not exhibit synergism with mTORC1/2 inhibitors in neuroblastoma. Interestingly, A549 cells display higher levels of phospho-AKT(S473) as compared to SK-N-AS, SH-SY5Y and SK-N-DZ cells, and SBI-0206965 reduces phospho-AKT(S473), particularly in neuroblastoma cells (Figure 5I). The downstream target of mTORC1, phospho-4eBP1(T37/46), remained unaffected (Figure 5I). Thus, the lack of combined effect of SBI-0206965 and mTOR inhibition in neuroblastoma may be due to reduced activity and dependence on AKT signaling in neuroblastoma cell lines, as well as due to off-target effects of SBI-0206965 that reduce AKT activity through an undefined mechanism. In support of this, expression of dnULK1 in SK-N-AS cells did not reduce phospho-AKT(S473) (Figure 5J), indicating that SBI-0206965-mediated reduction of phospho-AKT(S473) is likely independent of ULK1 function. Furthermore, treatment of dnULK1-expressing SK-N-AS cells with mTOR inhibitors promoted apoptosis at levels similar to that of SBI-0206965-treated SK-N-AS cells (Figure 5K). These results indicate that SBI-0206965 has increased cytotoxicity compared to dnULK1-mediated inhibition of ULK1 in neuroblastoma due to off-target inhibition of AKT, and that the use of a more specific inhibitor of ULK1 can sensitize neuroblastoma cells to mTORC1/2 inhibitors.
Figure 5. SBI-0206965 does not affect the cytotoxicity of topoisomerase or mTOR inhibitors in neuroblastoma cells.
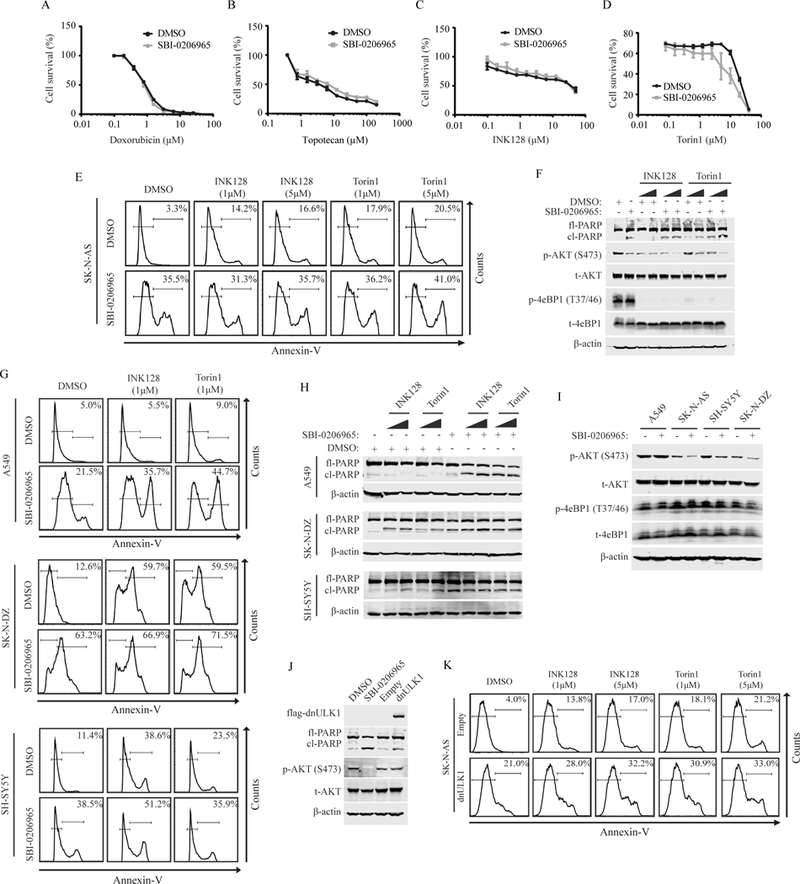
(A-D) Survival curve measured by Prestoblue of SK-N-AS cells co-treated with increasing concentrations of doxorubicin (A), topotecan (B), INK128 (C), or Torin1 (D) and 10μM SBI-0206965 or DMSO vehicle for 24 hours. (E) Annexin-V flow cytometry analysis of SK-N-AS cells treated with 10μM SBI-0206965 and 5¼M INK128 or Torin1 alone or in combination for 48 hours. (F) Western blot analysis of SK-N-AS cells treated with 10μM SBI-0206965 and INK128 or Torin1 at 1¼M and 5¼M alone or in combination for 48 hours. (G) Annexin-V flow cytometry analysis of A549, SH-SY5Y and SK-N-DZ cells treated with 10μM SBI-0206965 and 1¼M INK128 or Torin1 alone or in combination. A549 cell were treated for 48 hours. SH-SY5Y and SK-N-DZ cells were treated for 24 hours. (H) Western blot analysis of A549, SH-SY5Y and SK-N-DZ cells treated with 10μM SBI-0206965 and INK128 or Torin1 at 1¼M and 5¼M alone or in combination. A549 cell were treated for 48 hours. SH-SY5Y and SK-N-DZ cells were treated for 24 hours. (I) Western blot analysis assessing mTORC1/2 downstream signaling in A549, SK-N-AS, SH-SY5Y and SK-N-DZ treated with SBI-0206965 or DMSO control for 24 hours. (J) Western blot analysis comparing phosphorylated AKT(S473) levels in SK-N-AS cells expressing dnULK1K46N or treated with SBI-0206965 for 48 hours and empty-vector or DMSO control. (K) Annexin-V flow cytometry analysis of SK-N-AS expressing dnULK1 or empty-vector treated with INK128 or Torin1 at 1¼M and 5¼M for 48 hours.
As tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a potent apoptosis inducer that has been proposed for the treatment of pediatric malignancies (30), we tested whether SBI-0206965 synergizes with TRAIL to kill neuroblastoma cells. When TRAIL was combined with SBI-0206965 in SK-N-AS cells, a significant increase in apoptosis was observed compared to treatment with SBI-0206965 or TRAIL alone, as measured by annexin-V staining (Figure 6A-B). Moreover, TRAIL sensitized SK-N-AS cells to SBI-0206965 at concentrations as low as 10ng/mL (Figure 6C). In agreement, expression of dnULK1 also increased TRAIL-induced apoptosis in SK-N-AS cells, which was further enhanced in starvation conditions (Figure 6D). Furthermore, TRAIL treatment induced autophagic flux in SK-N-AS cells (Figure 6E), indicating that autophagy may be upregulated to suppress TRAIL-induced apoptosis, as inhibition of ULK1 sensitizes SK-N-AS cells to TRAIL treatment.
Figure 6. SBI-0206965 sensitizes SK-N-AS cells to TRAIL treatment.
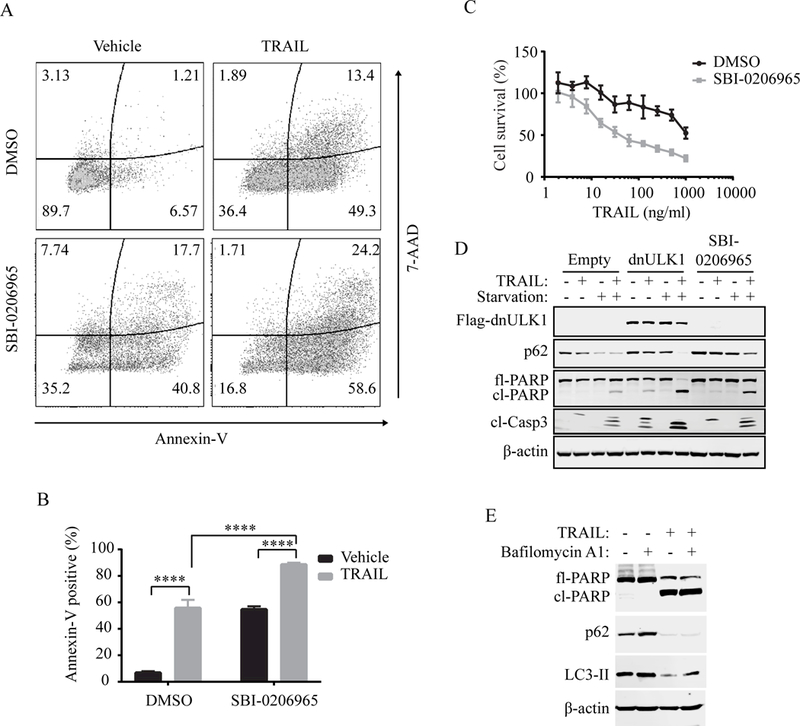
(A-B) Annexin-V/7-AAD flow cytometry analysis of SK-N-AS cells. Cells were treated with 10¼M SBI-0206965 or DMSO alone for 44 hours, followed by the addition of 100ng/ml TRAIL or vehicle for 4 hours, for a total treatment time of 48 hours. (C) Survival curve measured by Prestoblue of SK-N-AS cells co-treated with increasing concentrations of TRAIL and 10¼M SBI-0206965 or DMSO for 24 hours. (D) Western blot analysis of SK-N-AS cells expressing empty-vector or dnULK1 or treated with 10¼M SBI-0206965 and 100ng/ml TRAIL alone or in combination for 4 hours in normal or starvation medium. ****p<0.0001. (E) Western blot analysis of autophagy markers in SK-N-AS cells treated with TRAIL or vehicle control for 6 hours.
DISCUSSION
Neuroblastoma is a type of cancer that develops in early nerve cells of the sympathetic nervous system and most often affects infants and children younger than 10 years of age. There are about 700 new cases of neuroblastoma each year in the United States that account for about 7–10% of all cancers in children and more than 9% of all pediatric cancer-related deaths (1–3). There is an urgent need for the development of more effective therapies for neuroblastoma. Increasing evidence suggests that dysregulation of autophagy contributes to the pathogenesis and drug resistance of cancer (8,9,14,15). Here, we demonstrate that targeted inhibition of an essential autophagy kinase, ULK1, promotes tumor cell death as well as reduced proliferation in two mouse models of neuroblastoma, indicating that the loss of ULK1 is both cytotoxic and cytostatic. Although markers of apoptosis were enriched by dnULK1 expression in vitro, expression of dnULK1 in animal models enhanced both apoptosis and necrosis markers, indicating that the mechanism of cell death may be altered and multifaceted in an in vivo setting. Nevertheless, inhibition of ULK1 function reduced neuroblastoma xenograft tumor growth and metastatic burden in the liver, as well as significantly increased survival in a mouse model of neuroblastoma metastasis. This decrease in metastatic disease, in particular, demonstrates that inhibition of ULK1 may have important clinical implications in neuroblastoma, as metastasis is detected in many neuroblastoma patients at diagnosis and predicts worse prognosis (1–3). Thus, targeting ULK1 kinase has the potential to provide a novel therapeutic approach for the treatment of neuroblastoma. It is encouraging that the recently-developed small molecule inhibitor of ULK1, SBI-0206965, significantly reduces cell growth and promotes apoptosis in several neuroblastoma cell lines. Future work characterizing and optimizing the pharmacological properties and on-target efficacy of SBI-0206965 in animal models will be an important next step in determining whether SBI-0206965 is a viable pharmaceutical that can translate to the clinic.
Inhibition of autophagy has been shown to enhance the efficacy of conventional therapeutic modalities in patients with advanced solid malignancies (31). Moreover, SBI-0206965 has been previously demonstrated to synergize with mTOR inhibitors against lung cancer cells (19). In this study, SBI-0206965 has limited additive effect on the cytotoxicity of either mTOR inhibitors (INK128 and Torin1) or conventional chemotherapies (doxorubicin and topotecan) in neuroblastoma cell lines. However, SBI-0206965 was observed to reduce AKT(S473) phosphorylation, a predictor of poor outcome in neuroblastoma (32), indicating that SBI-0206965 reduces key oncogenic pathways in neuroblastoma. Moreover, the influence of SBI-0206965 on AKT activity may explain why we fail to see synergistic effects from co-treatments of SBI-0206965 and mTOR inhibitors. In support of this, expression of dnULK1 did not reduce AKT(S473) phosphorylation and sensitized SK-N-AS cells to mTORC1/2 inhibition, resulting in levels of apoptosis similar to that of SBI-0206965 treatment. Although it is possible that the reduction in AKT phosphorylation is directly due to off-target effects of SBI-0206965, previous profiling of SBI-0206965 kinase selectivity did not report AKT as a target (19), suggesting that AKT may be down-regulated through off-target inhibition of other kinases, such as SRC. Thus, further investigation into the specificity of SBI-0206965 in neuroblastoma is warranted. However, this off-target effect of SBI-0206965 provides duel inhibition of both ULK1 and AKT activity, which may be advantageous for treating neuroblastoma.
The tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has been implicated as a potential anticancer therapeutic agent for its ability to strongly trigger the extrinsic pathway of apoptosis by binding to its receptors (TRAIL-R1/2) to promote cancer cell death without causing significant toxicity in nonmalignant cells (30,33–35). Moreover, TRAIL receptor agonists (TRAs) are well-tolerated and demonstrate some therapeutic efficacy in a phase 1 clinical trial of pediatric patients with solid tumors (36). However, the majority of clinical trials using TRAs have provided limited efficacy due to tumor cell resistance to apoptosis induction (35,37–41). To translate the promising preclinical data of TRAs to the clinic, current research is focused on the development of more effective TRAs (42) and the elucidation of the mechanisms of TRAIL-resistance in order to develop potent TRAIL-sensitizers (33,35). Here, we demonstrate that SBI-0206965 greatly sensitizes SK-N-AS neuroblastoma cells to TRAIL treatment, suggesting an opportunity for an effective co-therapy development. Reversal of TRAIL resistance by autophagy inhibition has been previously demonstrated in many other cancer types including T-cell leukemia (43), colon carcinoma (43,44), breast cancer (44,45), lung cancer (46), cervical cancer (47), pancreatic cancer (48), and thyroid cancer (49). Moreover, increased autophagic activity promotes TRAIL-resistance in breast cancer (45), and autophagy is stimulated by TRAIL-treatment to promote survival in lung cancer cells (46). Thus, our results are in agreement with previous reports that autophagy inhibition promotes TRAIL-sensitization. The mechanisms through which SBI-0206965 sensitizes neuroblastoma to TRAIL are currently unclear. Further investigation into these mechanisms will advance our understanding of both TRAIL-resistance and the crosstalk between ULK1 and apoptosis signaling.
In summary, we establish that inhibition of ULK1 kinase in neuroblastoma promotes apoptosis and reduces tumor growth and metastatic disease. To our best knowledge, this work is the first to demonstrate the antitumor effects of targeting an essential autophagy gene in neuroblastoma mouse models. These data suggest that inhibition of ULK1 kinase is a viable therapeutic option for the treatment of neuroblastoma and that further development and characterization of ULK1 inhibitors such as SBI-0206965 is warranted. Future research characterizing the bioavailability, specificity, toxicity, and efficacy of SBI-026965 in neuroblastoma mouse models will be an essential component of preclinical development. Additionally, it will be interesting to determine whether the observed reduction in neuroblastoma growth and induction of apoptosis are simply due to autophagy inhibition or due to non-canonical functions of ULK1 (50). Regardless, inhibition of ULK1 kinase significantly suppresses tumor growth and metastasis and promotes overall survival in animal models of neuroblastoma, making it a promising drug target for the treatment of neuroblastoma.
ACKNOWLEDGEMENTS
We thank the members of the Wang lab who gave insight and technical help. We are grateful to the Penn State College of Medicine’s Flow Cytometry Core and the Department of Comparative Medicine. This work was supported in part by National Institutes of Health grants CA171501 (to H.G. Wang) and GM117014 (to B.A. Miller) and the Lois High Berstler Research Endowment Fund and the Four Diamonds Fund of the Pennsylvania State University.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
REFERENCES
- 1.Irwin MS, Park JR. Neuroblastoma: paradigm for precision medicine. Pediatr Clin North Am. 2015;62:225–56. [DOI] [PubMed] [Google Scholar]
- 2.Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primer. 2016;2:16078. [DOI] [PubMed] [Google Scholar]
- 3.Vo KT, Matthay KK, Neuhaus J, London WB, Hero B, Ambros PF, et al. Clinical, Biologic, and Prognostic Differences on the Basis of Primary Tumor Site in Neuroblastoma: A Report From the International Neuroblastoma Risk Group Project. J Clin Oncol. 2014;32:3169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341:1165–73. [DOI] [PubMed] [Google Scholar]
- 5.Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33:3008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–72. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimmelman AC, White E. Autophagy and Tumor Metabolism. Cell Metab. 2017;25:1037–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dower CM, Wills CA, Frisch SM, Wang H-G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy. 2018;1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim M-J, Woo S-J, Yoon C-H, Lee J-S, An S, Choi Y-H, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011;286:12924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4:466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy sustains pancreatic cancer growth through both cell autonomous and non-autonomous mechanisms. Cancer Discov. 2018;CD-17–0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belounis A, Nyalendo C, Le Gall R, Imbriglio TV, Mahma M, Teira P, et al. Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer. 2016;16:891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aveic S, Pantile M, Seydel A, Esposito MR, Zanon C, Li G, et al. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget. 2016;7:5646–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radogna F, Cerella C, Gaigneaux A, Christov C, Dicato M, Diederich M. Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene. 2016;35:3839. [DOI] [PubMed] [Google Scholar]
- 19.Egan DF, Chun MGH, Vamos M, Zou H, Rong J, Miller CJ, et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59:285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest. 2015;125:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egan DF, Kim J, Shaw RJ, Guan K-L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:645–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JW, Park S, Takahashi Y, Wang H-G. The association of AMPK with ULK1 regulates autophagy. PloS One. 2010;5:e15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gobeil S, Boucher CC, Nadeau D, Poirier GG. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 2001;8:588–94. [DOI] [PubMed] [Google Scholar]
- 25.Chaitanya GV, Alexander JS, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal CCS. 2010;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strilic B, Offermanns S. Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell. 2017;32:282–93. [DOI] [PubMed] [Google Scholar]
- 28.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective Mammalian Target of Rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem. 2010;53:7146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gasparini C, Brumatti LV, Zauli LM and G. TRAIL-Based Therapeutic Approaches for the Treatment of Pediatric Malignancies. Curr. Med. Chem. 2013;20:2254–71. [DOI] [PubMed] [Google Scholar]
- 31.Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Opel D, Poremba C, Simon T, Debatin K-M, Fulda S. Activation of Akt Predicts Poor Outcome in Neuroblastoma. Cancer Res. 2007;67:735–45. [DOI] [PubMed] [Google Scholar]
- 33.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor–related apoptosis–inducing ligand in vivo. Nat Med. 1999;5:157. [DOI] [PubMed] [Google Scholar]
- 34.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Miguel D, Lemke J, Anel A, Walczak H, Martinez-Lostao L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016;23:733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merchant MS, Geller JI, Baird K, Chou AJ, Galli S, Charles A, et al. Phase I Trial and Pharmacokinetic Study of Lexatumumab in Pediatric Patients With Solid Tumors. J Clin Oncol. 2012;30:4141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O’Dwyer PJ, Gordon MS, et al. Phase I Dose-Escalation Study of Recombinant Human Apo2L/TRAIL, a Dual Proapoptotic Receptor Agonist, in Patients With Advanced Cancer. J Clin Oncol. 2010;28:2839–46. [DOI] [PubMed] [Google Scholar]
- 38.Soria J-C, Smit E, Khayat D, Besse B, Yang X, Hsu C-P, et al. Phase 1b Study of Dulanermin (recombinant human Apo2L/TRAIL) in Combination With Paclitaxel, Carboplatin, and Bevacizumab in Patients With Advanced Non-Squamous Non–Small-Cell Lung Cancer. J Clin Oncol. 2010;28:1527–33. [DOI] [PubMed] [Google Scholar]
- 39.Cheah CY, Belada D, Fanale MA, Janikova A, Czucman MS, Flinn IW, et al. Dulanermin with rituximab in patients with relapsed indolent B-cell lymphoma: an open-label phase 1b/2 randomised study. Lancet Haematol. 2015;2:e166–174. [DOI] [PubMed] [Google Scholar]
- 40.Soria J-C, Márk Z, Zatloukal P, Szima B, Albert I, Juhász E, et al. Randomized Phase II Study of Dulanermin in Combination With Paclitaxel, Carboplatin, and Bevacizumab in Advanced Non–Small-Cell Lung Cancer. J Clin Oncol. 2011;29:4442–51. [DOI] [PubMed] [Google Scholar]
- 41.Lemke J, Karstedt S von, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21:1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holland PM. Death receptor agonist therapies for cancer, which is the right TRAIL? Cytokine Growth Factor Rev. 2014;25:185–93. [DOI] [PubMed] [Google Scholar]
- 43.Han J, Hou W, Goldstein LA, Lu C, Stolz DB, Yin X-M, et al. Involvement of Protective Autophagy in TRAIL Resistance of Apoptosis-defective Tumor Cells. J Biol Chem. 2008;283:19665–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Enhancement of tumor-TRAIL susceptibility by modulation of autophagy. Autophagy. 2008;4:940–3. [DOI] [PubMed] [Google Scholar]
- 45.Lv S, Wang X, Zhang N, Sun M, Qi W, Li Y, et al. Autophagy facilitates the development of resistance to the tumor necrosis factor superfamily member TRAIL in breast cancer. Int J Oncol. 2015;46:1286–94. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Zhou X, Qiao J, Bao A. Autophagy is a regulator of TRAIL-induced apoptosis in NSCLC A549 cells. J Cell Commun Signal. 2017;11:219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, et al. Autophagy Controls the Kinetics and Extent of Mitochondrial Apoptosis by Regulating PUMA Levels. Cell Rep. 2014;7:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monma H, Iida Y, Moritani T, Okimoto T, Tanino R, Tajima Y, et al. Chloroquine augments TRAIL-induced apoptosis and induces G2/M phase arrest in human pancreatic cancer cells. PloS One. 2018;13:e0193990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin S-M, Jang HW, Sohn SY, Kim NK, Joung JY, Cho YY, et al. Role of autophagy in the resistance to tumour necrosis factor-related apoptosis-inducing ligand-induced apoptosis in papillary and anaplastic thyroid cancer cells. Endocrine. 2014;45:256–62. [DOI] [PubMed] [Google Scholar]
- 50.Lindqvist LM, Simon AK, Baehrecke EH. Current questions and possible controversies in autophagy. Cell Death Discov. 2015;1. [DOI] [PMC free article] [PubMed] [Google Scholar]