

Abstract
We describe a multi-purpose technology platform, termed μSCALE (Microcapillary Single Cell Analysis and Laser Extraction), that enables massively parallel, quantitative biochemical and biophysical measurements on millions of protein variants expressed from yeast or bacteria. μSCALE spatially segregates single cells within a microcapillary array, enabling repeated imaging, cell growth, and protein expression. We performed high-throughput analysis of cells and their protein products using a range of fluorescent assays, including binding affinity measurements and dynamic enzymatic assays. A precise laser-based extraction method allows rapid recovery of live clones and their genetic material from microcapillaries for further study. With μSCALE, we discovered a new antibody against a clinical cancer target, evolved an orange fluorescent protein biosensor, and engineered an enzyme with reduced sensitivity to its inhibitor. These three distinct protein analysis and engineering applications, each with unique assay requirements and different host organisms, highlight the flexibility and technical capabilities of our platform.
Introduction:
Over the past decade, high-throughput technologies have allowed researchers to gain unprecedented insights into intrinsically complex and interconnected biological systems. As examples, whole-genome sequencing has enabled the identification of crucial genes and mutations underlying disease pathophysiology,1,2 DNA microarrays have been used to elucidate transcription patterns involved in healthy and diseased states,3 and large-scale proteomics methods have helped map the connectivity of cell signaling networks that orchestrate responses to growth factors and other external stimuli.4 In contrast, analogously powerful approaches for rapidly and deeply interrogating the sequence-structure-activity relationship of proteins, with functional read-outs that span a range of biophysical and biochemical measurements, have lagged because of technical challenges. Here, we describe the development of a new technology platform that addresses this need and showcase its capabilities and breadth through applications on three distinct protein classes: antibody therapeutics, fluorescent protein biosensors, and enzymes.
Protein engineers rely heavily on directed evolution, a powerful method that iterates rounds of library mutagenesis and screening.5 In a directed evolution experiment, randomly generated protein libraries are mined for variants with desirable characteristics, such as high affinity binding to a target of interest,6 stability,7 fluorescence,8 or enzymatic activity.9 Maintaining a genotype-to-phenotype linkage is a fundamental requirement for any directed evolution effort, allowing for the identification of protein variants through their corresponding DNA sequence following a screen. Genotype-to-phenotype linkages are most easily preserved in screens that probe for protein binding partners. As examples, genetic fusion of protein variants to the exterior surface components of microbes or phage, or to protein translation machinery, enable rapid identification of target binders from large protein libraries (107–1014 variants) using fluorescence-activated cell sorting (FACS) or panning methods.10,11
For protein engineering applications that extend beyond binding interactions, spatial segregation establishes the genotype-to-phenotype linkage. For this purpose, researchers express and assay individual protein variants within separate wells of microtiter plates or through phenotypic screens based on colony selection on petri dishes. While liquid handling robots have eased labor, these engineering endeavors are generally limited in throughput to 103–105 variants in a typical screen.12 Such relatively small library sizes are restrictive relative to the vast amino acid search space available to a typical protein.13 Oil-water emulsion droplets generated in bulk or in microfluidic chips have enabled high-throughput enzyme engineering applications with libraries of 107–108 variants.14–18 Additionally, two past efforts use miniaturized microwell arrays to increase throughput for enzyme engineering.19,20 Notably, one effort describes an enzyme screening platform centered on reusable plates with 106 wells, achieving throughputs of up to 107 assays per day.19 Improvements in handling or cell retrieval methods would facilitate widespread application of these technologies to protein characterization and engineering efforts.
Here, we describe μSCALE (Microcapillary Single Cell Analysis and Laser Extraction), a multi-purpose platform technology capable of interrogating a dense array of millions of spatially segregated single cells or their protein products within a time frame of minutes. A key feature of μSCALE is the ability to isolate target cells post analysis from the microcapillary array using a precise laser-based extraction technique. Previous studies demonstrate the potential utility of microchamber, microcapillary, or microwell arrays for single enzyme characterization,21,22 digital ELISA,23 selection of antibody-producing cells,24,25 and isolation of mammalian cells26 or circulating tumor cells.27 While these applications have mostly focused on either time-resolved kinetic analysis or cell sorting, we used μSCALE to achieve both capabilities simultaneously. To highlight the flexibility and technical capabilities of our technology, we describe three distinct protein analysis and engineering applications, using libraries expressed in yeast or bacteria.
Results:
Microcapillary Single Cell Analysis and Laser Extraction
We describe the overall concept and workflow of the μSCALE platform in Figure 1a, b and Supplementary Results, Supplementary Figure 1. One core component of the technology is a dense glass-substrate array of millions of spatially-segregated, high-aspect ratio microcapillaries (1 mm thick, 10 μm or 20 μm in diameter) (Fig. 1c). We apply a cell suspension that is mixed with magnetic microparticles to the array by pipetting. While the cells randomly distribute into the array following a Poisson distribution, the observed and expected means differ by 2–3 fold due to the high aspect ratio of the microcapillaries (Supplementary Fig. 2). The microcapillary array is unsealed on the bottom, yet the liquid sample is held in place by surface tension. The passive nature of the filling process results in a uniform meniscus level across the entire array. This uniformity, coupled with gravitational sedimentation of the loaded cells, simplifies establishment of the imaging focus plane without the need for autofocus.
Figure 1.
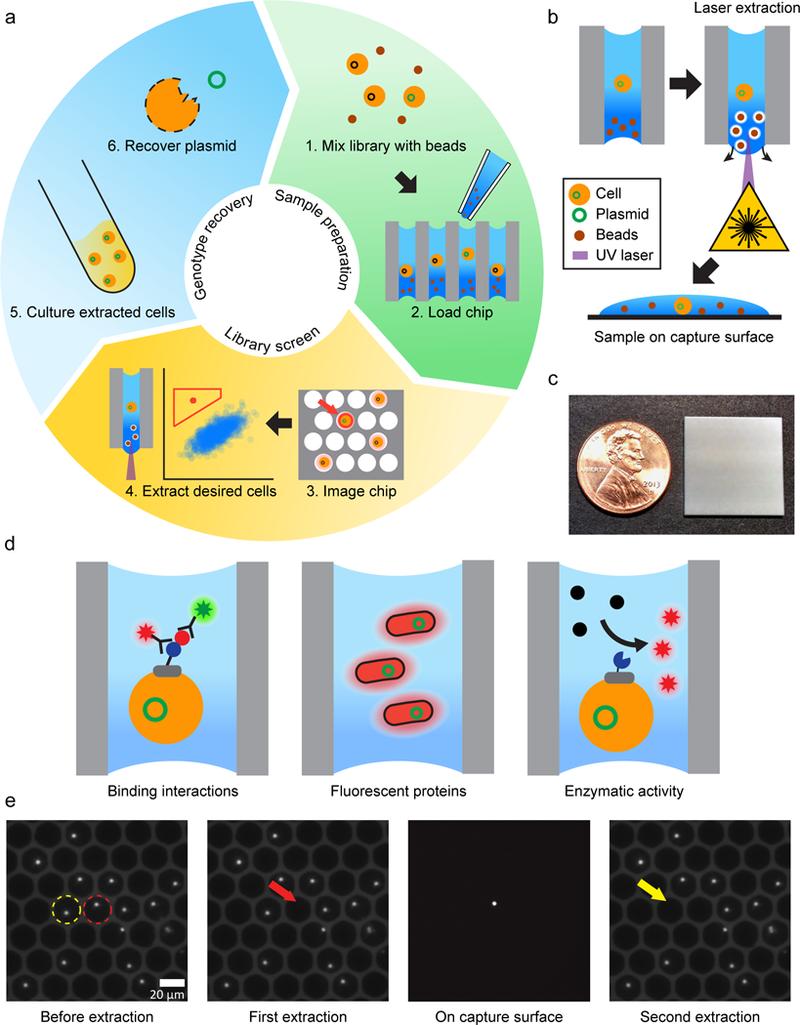
μSCALE overview. (a) Platform workflow. 1) A library of protein variants is mixed with opaque microbeads. 2) The mixture is pipetted into the array at a concentration that results in a mean single-cell occupancy. A biochemical assay is performed using fluorescence as a read-out. 3) The array is imaged via fluorescent microscopy. 4) The fluorescent intensity of each microcapillary is quantified, and desired clones are isolated with a laser-based extraction method. 5) The extracted cells are cultured. 6) Cells are lysed and the plasmid is recovered. (b) Laser-based extraction method. A UV laser is positioned and pulsed, disrupting the surface tension of the microcapillary and ejecting its contents onto a capture surface below the array. (c) Image of microcapillary array with penny for scale. (d) Three assays performed using the μSCALE platform. Left, Binding interactions of a yeast surface-displayed scFv library pre-stained with fluorescent antibodies. Middle, Fluorescent protein variants expressed in E. coli clones cultured within the microcapillaries. Right, Real-time kinetic measurements on a library of yeast surface-displayed enzyme variants that convert a substrate to a fluorescent product. (e) Two extractions of neighboring microcapillaries loaded with fluorescent particles. In the left panel, the first and second capillaries to be extracted are outlined with a red or yellow circle respectively. The second panel demonstrates successful extraction without disturbing neighboring microcapillaries. The third panel shows the isolated particle on the capture surface below the array. The final panel demonstrates the second successful microcapillary extraction.
We demonstrated a variety of biochemical assays using the μSCALE platform (Fig. 1d). In each case, we first load bacteria or yeast cells expressing a protein of interest into a microcapillary array. The humid environment of the array permits time-resolved experimental measurements or cell growth over a period of days, if desired. We then image the microcapillaries using a high numerical aperture microscope objective illuminated using a band-pass filtered high intensity arc lamp in an epi-illumination configuration. A high-sensitivity, cooled CCD camera detects the fluorescent emission through a bandpass filter. Following established guidelines for quantitative wide-field microscopy, we measured the inter-capillary variability in fluorescence signals detected from the array and found it to be comparable to other high-throughput methods (Supplementary Fig. 3).28 We routinely image the entire array at multiple positions and at multiple wavelengths at a rate of approximately 10,000 microcapillaries per second, followed by analysis using standard image processing functions in MATLAB. The high resolution of the images allowed us to distinguish promising candidates from cell debris and other false fluorescence signals via quantified metrics (size, roundness, and fluorescence intensity) or by visual inspection.
Following analysis, we recover the contents of individual microcapillaries of interest by laser extraction. Alternatively, we can designate multiple microcapillaries to be extracted (termed pooled extraction) at a rate of approximately 2 microcapillaries per second. A pulsed UV laser interacts with the magnetic particles to disrupt the surface tension within a single microcapillary, extracting its contents without affecting its neighbors (Fig. 1b and e).
We eject the contents of the microcapillary onto a capture surface below the array; this allows us to perform imaging to confirm extraction and carry out additional morphological studies. To refine this technique, we varied the laser intensity and titrated the magnetic bead concentrations to establish a range of conditions that achieved complete and specific extraction (Supplementary Fig. 4).
Analysis and screening of protein binding interactions
As an initial demonstration, we used μSCALE to measure known protein-protein binding interactions (Fig. 2a, b), and subsequently to rapidly screen and identify antibody binders against a clinical target (Fig. 2c, d). Growth arrest specific 6 (Gas6) binds to the Axl receptor tyrosine kinase and mediates immune function, blood coagulation, and tumor cell invasion and migration.29,30 We first evaluated the ability to distinguish a wild-type (WT) Gas6 binding variant of the Axl receptor tyrosine kinase and a non-binding variant on the yeast surface. Two-color fluorescence imaging was used to quantify yeast expression levels (measured by primary and secondary antibodies against a C-terminal c-myc epitope tag), and ligand binding (measured using an antibody against a hexahistidine tag on the ligand). We observed a clear distinction between the WT and non-binding Axl variant yeast populations (Fig. 2a, b). Next, to characterize our ability to isolate rare clones of interest, we screened mock libraries of yeast–displayed proteins in ratios spanning 1:10 to 1:100,000 ligand binding:non-binding clones (Fig. 2b, Supplementary Fig. 5, and Supplementary Table 1). Individual yeast cells were extracted from the array, cultured, and evaluated by DNA recovery and sequencing. Across the six mock libraries, we correctly identified 37 out of 39 extracted clones as the binding variant, achieving an enrichment ratio of 100,001 in the most stringent condition. Additionally, we confirmed that all 19 extracted clones selected as non-binders were indeed non-binders. These mock library screens validated the ability of μSCALE to differentiate rare functional clones within a large population of non-functional variants.
Figure 2.
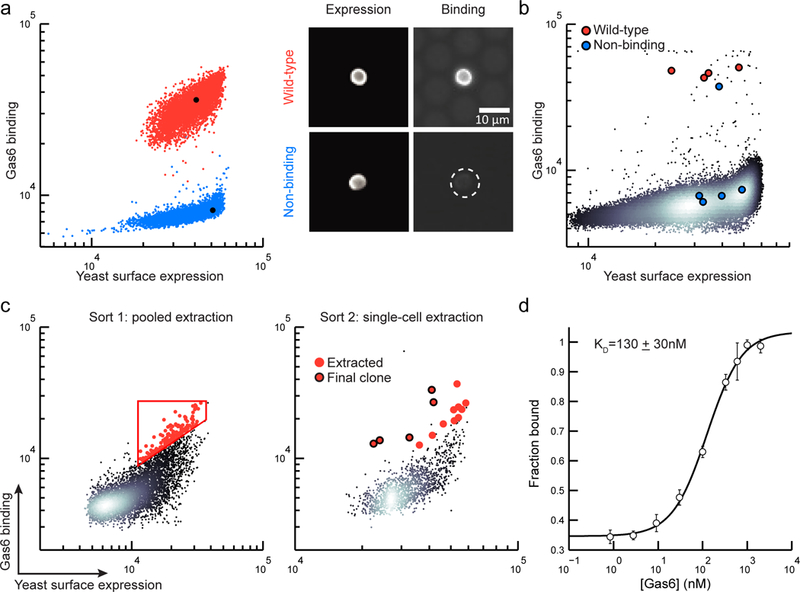
High-throughput screening of binding proteins using μSCALE. (a) Two-parameter scatter plots (left), where each dot represents a microcapillary (n = 5000 microcapillaries per condition), and microscope images (right) of the WT Axl (red) and the non-binding Axl variant (blue). (b) A scatter plot from a representative mock library screen (ratio of WT:non-binding variant = 1:10,000, n=147,849 microcapillaries). Extracted clones identified as WT Axl (red dots) and non-binding Axl variants (blue dots). (c) Two rounds of μSCALE screening of a yeast surface-displayed naïve scFv library for Gas6 binders. Sort 1: automated pooled extraction of 143 capillaries with a user-drawn gate (left), Sort 2: manual single-cell extraction of 15 capillaries (right). Red dots, extracted clones; Red dots with black outline, clones with highest binding to Gas6. (d) Gas6 binding curve of the yeast-displayed scFv identified by μSCALE, represented as the fraction of Gas6 bound versus the concentration added. Error bars correspond to the standard deviation of three technical replicates.
We next applied μSCALE to screen a large, naïve library of yeast-displayed single-chain variable fragments (scFvs) to identify a variant that binds to Gas6 with high affinity. We first cleared the library of scFv binders against screening reagents using magnetic-activated cell sorting (MACS), followed by a round of MACS using Gas6-coated magnetic beads; this approach reduced the theoretical library size to 2.8 × 106 clones. We then enriched the resulting pool of scFv variants for binders to Gas6 using two rounds of screening on the μSCALE platform (Fig. 2c). In the first round, we added a relatively low concentration of Gas6 (33 nM) to the scFv library and allowed it to reach binding equilibrium. Using pooled extraction, we isolated approximately 150 microcapillaries containing yeast with the highest Gas6 binding levels, normalized to the amount of scFv expression (Fig 2c, left). We cultured this extracted yeast pool and induced scFv expression, and carried out a second μSCALE screen under more stringent conditions (10 nM Gas6). In the second screen, we isolated 15 individual microcapillaries and sequenced the extracted contents (Fig. 2c, right and Supplementary Fig. 6e). Flow cytometry validated the enrichment achieved with each screening step (Supplementary Fig. 6). Retrospective examination of the μSCALE screen revealed that the 5 microcapillaries with the highest observed Gas6 binding levels (normalized to scFv expression) contained the same clone (Fig. 2c, right). This resulting scFv variant bound to Gas6 with a KD of 130 ± 30 nM (Fig. 2d), comparable to other binders mined from naïve scFv libraries.31
Cell growth and screening of fluorescent proteins
Next, we demonstrated the use of μSCALE to engineer a new fluorescent protein variant, expanding the color palette of available biosensors and highlighting the compatibility of the platform with E. coli libraries. Traditionally, fluorescent proteins have been engineered by screening individual E. coli colonies grown on agar dishes to identify desired color hues and/or fluorescence brightness.32 While straightforward, this low-throughput method relies on basic optics and laborious colony picking, which is time consuming, subjective, and hampers recovery of the most desirable variants. We first established the ability to grow spatially segregated cultures from individual E. coli cells in the microcapillary array. We loaded E. coli suspensions expressing green fluorescent protein into the array at a concentration approximating a single cell per capillary on average, incubated the array at 37 °C, and imaged at regular time intervals. The resulting time course shows robust clonal expansion and fluorescent protein production, with E. coli growth confined to create a dense array of segregated cultures (Fig. 3a).
Figure 3.
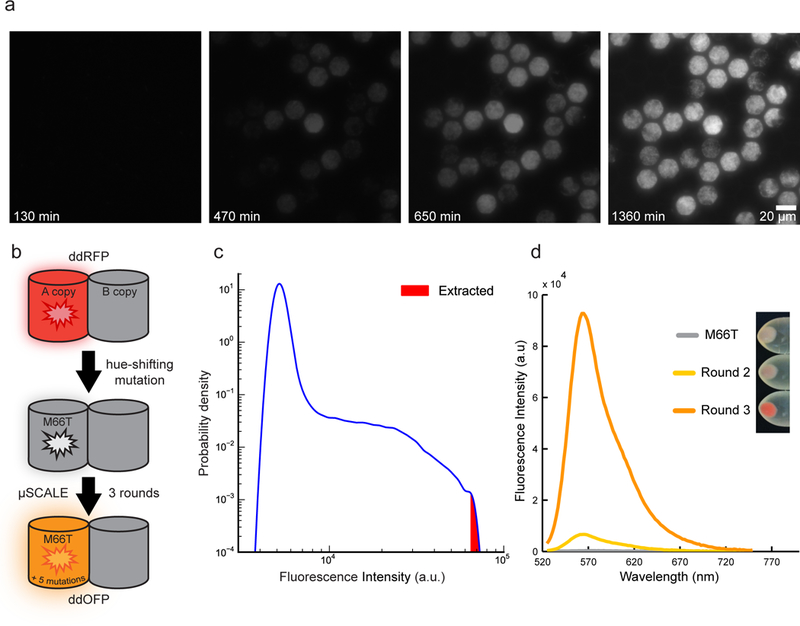
Engineering an orange-hued fluorescent protein using μSCALE. (a) Growth of spatially segregated E. coli cultures expressing GFP. Images depict a small portion of a 20 μm array, which was loaded with, on average, a single bacterial cell per microcapillary, cultured at 37 °C for the indicated times, and serially imaged. (b) Engineering strategy used to generate ddOFP from the ddRFP template. A M66T mutation was introduced into the A copy of ddRFP (starburst), generating a largely non-fluorescent hue-shifted variant. Three rounds of mutagenesis and μSCALE screening of protein libraries expressed in E. coli resulted in ddOFP, which has 5 mutations in addition to M66T. (c) Quantification of μSCALE screen of the third E.coli library. The distribution of observed fluorescence intensities of the library variants is shown in blue (n = 305,279 microcapillaries). The 10 brightest cells at the upper tail of this distribution (red) were extracted from the array. (d) Iterative directed evolution results in variants with increased fluorescence intensity. Plot depicts fluorescence emission spectra of E. coli crude lysates expressing M66T, a variant from round 2, and ddOFP from round 3. Inset depicts the increase in color hue of the cell pellets of E. coli expressing FP variants described above.
We then applied μSCALE to generate a hue-shifted, new color variant of a dimerization-dependent fluorescent protein (termed ddFP) (Supplementary Fig. 7).We used a red dimerization-dependent fluorescent protein (ddRFP) as a template and introduced a blue-shifting and chromophore-disrupting mutation (M66T), which eliminated fluorescence (Fig. 3b).8,33 To rescue and enhance fluorescence, we performed three sequential rounds of directed evolution, using μSCALE to screen libraries created by error-prone PCR mutagenesis and expressed in E. coli. For directed evolution rounds 1 and 2, we isolated approximately a dozen clones demonstrating weak fluorescence and used them to generate a gene template mix for a subsequent round of mutagenesis. By round 3, a fraction of the library exhibited fluorescence levels substantially higher than the library mean. We extracted, cultured, and characterized E. coli from the 10 microcapillaries with the brightest fluorescence (Fig. 3c, red region). The brightest fluorescent protein variant from round 3, designated dimerization-dependent orange fluorescent protein (ddOFP), acquired 5 mutations in addition to the original M66T mutation (Supplementary Fig. 8a) that conferred a dramatic increase in fluorescence intensity relative to the best mutant from round 2 and the ‘parent’ M66T clone (Fig. 3d). ddOFP has absorption and emission maxima at 535 nm and 565 nm, respectively (Supplementary Fig. 8b), filling the spectral gap between ddRFP and ddYFP variants (Supplementary Fig. 9). In addition, ddOFP exhibits comparable brightness and fluorogenic properties to other ddFPs (Supplementary Table 2),8,34 making it a suitable module for biosensing applications such as fluorescent protein exchange.35
Analysis and screening for improved enzyme catalysts
As a final demonstration of its capabilities and versatility, we used μSCALE to measure enzyme kinetics and to identify an improved biocatalyst. Decades of published biochemical insights for alkaline phosphatase qualified this enzyme as a model system for these studies.36 We measured catalytic activity using the substrate 9H-(1,3-dichloro-9,9-dimethylacridin-2-one-7-yl) phosphate (DDAOP), which yields a fluorescent product upon hydrolysis. The Michaelis-Menten kinetics and phosphate inhibition profiles of yeast-displayed WT alkaline phosphatase were within 2-fold of values measured from purified enzyme harvested from E. coli (Supplementary Fig. 13). Moreover, yeast-displayed constructs preserved the kinetic differences between WT alkaline phosphatase and a previously characterized R166S mutant, which exhibits a significant reduction in the apparent second-order rate constant (kcat/KM) for the DDAOP substrate37 (Supplementary Table 4).
First, we used μSCALE to measure single-cell reaction time courses for yeast-displayed WT alkaline phosphatase and the R166S mutant, demonstrating that the platform can provide time-resolved, kinetic information on the activity of enzyme variants. The microcapillaries maintained robust spatial segregation of yeast-displayed enzymes and accumulating product 7-hydroxy-9H-(1,3-dichloro-9,9-dimethylacridin-2-one (DDAO) throughout each reaction time course in the arrays, enabling facile and quantitative kinetic discrimination of microcapillaries containing highly active yeast-displayed enzymes from less active neighbors or empty microcapillaries (Fig. 4a and Supplementary Fig. 14). Over 20-min or 3-hr reactions (for WT and R166S, respectively) in the arrays, we quantified approximately 5,000 single-cell traces for WT alkaline phosphatase and the R166S variant. Histograms of the time course slopes are shown in Figure 4b and define the activity distribution of each mutant in the microcapillary array. Despite cell-to-cell variability in enzymatic activity within each clonal population, likely due to heterogeneity in protein expression levels (Supplementary Fig. 17), the μSCALE platform clearly delineates the R166S mutant from the WT enzyme on the basis of reaction kinetics.
Figure 4:
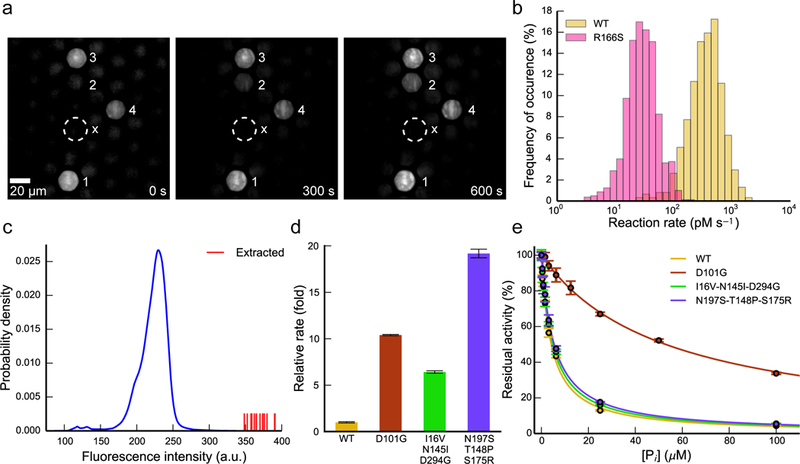
High-throughput kinetic measurements and enzyme library screening using μSCALE. (a) Representative images from a time-resolved enzymatic assay. Four microcapillaries harboring yeast cells displaying active enzyme are denoted in white numbering, and a nearby empty capillary is marked with an “x” for comparison. Image contrast artificially enhanced for clarity. (b) Single-cell kinetic profiles of yeast-displayed WT alkaline phosphatase and an R166S point mutant with reduced catalytic efficiency. Reactions were carried out with 10 μM DDAOP substrate and were recorded for up to 3 hr. Slopes of the linear portion of each time course were binned to yield the distribution of rates. (WT, n = 618 traces; R166S, n = 812 traces) (c) Quantification of μSCALE screen of randomly mutated alkaline phosphatase library. The distribution of observed enzymatic activity during the screen is shown in blue (n = 309,895 microcapillaries). The fifteen most active cells (shown as red vertical lines) were extracted. (d) Relative rates of WT alkaline phosphatase and the three most active isolated variants. Rates were measured with yeast-displayed constructs in bulk biochemical assays under identical conditions to those used in the screen and were normalized to the WT value. (e) Phosphate inhibition curves for WT enzyme and the three isolated variants. Reactions were carried out with substrate concentrations at least 10-fold lower than the apparent KM of each mutant. Fits to a competitive inhibition model are overlaid above the data as solid lines. In above experiments, error bars correspond to the standard deviation of three technical replicates.
We next demonstrated the ability of μSCALE to identify improved enzyme variants. An alkaline phosphatase library was created by error prone PCR mutagenesis and screened using 1 μM DDAOP substrate (4-fold lower than the apparent KM for WT alkaline phosphatase) in the presence of 15 μM inorganic phosphate (4-fold higher than the measured KI for WT alkaline phosphatase) (see Methods and Supplementary Tables 3 and 4). These conditions enrich for enzyme variants with increased tolerance to inhibitor, a common objective in many diverse subfields of enzyme engineering.38–40 Using μSCALE, we rapidly analyzed the catalytic activity of 3 × 105 enzyme variants. By single-cell extraction, we isolated fifteen putative hits with enhanced activity relative to WT alkaline phosphatase under the screening conditions tested, then cultured and rescreened them in bulk under the same conditions described above (Fig. 4c). We selected three variants for sequencing and additional characterization based on their significant rate accelerations relative to WT alkaline phosphatase in bulk biochemical assays (Fig. 4d). One variant, which contained a single point mutation D101G, exhibited significantly reduced sensitivity to inorganic phosphate, with a measured KI that was 10-fold higher than that of the WT enzyme (Fig. 4e and Supplementary Fig. 15). The improved performance of this D101G mutant relative to the WT enzyme remained upon soluble expression and purification from E. coli (Supplementary Table 5). The higher basal levels of activity of the other two yeast-displayed variants compared to WT alkaline phosphatase (Supplementary Fig. 16) can be in part explained by their increased yeast surface expression levels (Supplementary Fig. 17). However, both variants were inactive when purified from E. coli (Supplementary Tables 5 and 6), suggesting that the beneficial mutations acquired by these clones are specific to the yeast surface environment and incompatible with recombinant expression in bacteria.
Discussion
In this work we demonstrate three disparate protein engineering applications using μSCALE: isolation of a novel protein binder from a naïve antibody library, creation of a new fluorescent protein biosensor, and identification of an improved enzyme variant. In all three examples, the μSCALE platform afforded robust spatial segregation of microcapillary contents and the ability to recover single cells using a laser-based extraction method.
μSCALE offers clear advantages over microtiter plates and petri dishes traditionally used to analyze the functional activity of protein variants within a library screen. In addition to greater sample throughput, the microcapillary arrays can contain up to four million E.coli cultures per square inch, five orders of magnitude higher than in a standard petri dish or microtiter plate. As we demonstrated in our fluorescent protein engineering example, clonal expansion of E.coli from a single cell into a colony provides signal amplification and the ability to measure the population mean, thereby reducing variability. Additionally, the μSCALE technology platform enables real-time kinetic measurements as demonstrated in our example of alkaline phosphatase engineering. This capability for repeat imaging helps separate enzyme variants by their reaction kinetics rather than by fluorescence intensity at a single time point, potentially reducing false positives. However, the requirement for a fluorescent readout in μSCALE screens necessitates the use of a surrogate fluorogenic substrate (such as DDAOP) in enzymatic assays, which may poorly approximate the desired substrate for downstream engineering applications. In contrast, microtiter plate-based assays support a broader range of possible surrogate substrates due to the choice of absorbance, fluorescence, or chemiluminescence readouts. Direct chromatographic detection of the desired product is also possible from a microtiter plate-based screen, albeit with greatly reduced throughput.41
μSCALE affords several additional distinctions compared to FACS-based library screening methods, including the capability for direct cell imaging and temporal decoupling of cell analysis and sorting. Before the user sets the sorting parameters, μSCALE allows for imaging and analyzing of the entire population of microcapillaries. In addition, μSCALE enables robust isolation of rare clones with high precision (enrichment ratios up to 100,001; Supplementary Table 1), in contrast to the modest enrichment ratios supported by FACS.6 As such, efficient library screening and clone recovery is achievable in only 1 or 2 rounds of μSCALE screening, as evidenced by our rapid isolation of an antibody fragment against the Gas6 ligand.
While other microchamber technologies primarily rely on micromanipulators,19,20, air,25 or optical tweezers26 to recover samples, we present an alternative laser-based sample recovery mechanism. Laser-induced cavitation is a well-studied phenomenon frequently used by researchers to disrupt mammalian cell membranes for delivery or specific cell targeting.42,43 Based on these studies, in our application it is likely that the laser energy rapidly heats the surface of the magnetic microparticles, forming a cavitation bubble that disrupts the microcapillary meniscus and empties the contents onto the extraction surface. Because most of the laser energy is absorbed by the magnetic beads rather than the cells, we observe high viability in commonly used protein engineering cell lines (up to 80% and 100% survival for extracted yeast (S. cerevisiae) and bacteria (E.coli and B.subtilis), respectively; Supplementary Table 7). Given that optical cavitation has been used for membrane disruption and targeted cell killing, the extension of this cell recovery mechanism to more delicate cell types, such as mammalian cells, requires additional study.
In summary, μSCALE provides a new approach for quantifying the functional activity of millions of protein variants and isolating desirable mutants from library screens. In addition to protein characterization and engineering, μSCALE has broad applicability to high-throughput analysis of single cells and cell cultures. For example, μSCALE has potential uses for probing cell-to-cell interactions, single-cell growth and signaling dynamics, gene expression differences, recombinant host cell expression levels, or other applications requiring massively parallel biological measurements.
Methods
μSCALE instrumentation and software.
μSCALE experiments were performed with a Veritas laser capture microdissection (LCM) system (Arcturus), adapted with hardware and software modifications to enable microcapillary screening applications. The system contains an inverted fluorescence microscope with motorized stage and the Triton UV laser (diode-pumped Q-switched Nd:YLF laser, Spectra-Physics) (Supplementary Fig. 1). The laser system uses a wavelength of 349 nm, a focused beam diameter of 5 μm, and a pulse duration of 20 ns. For fluorescence imaging, an X-cite 120 illumination system (EXFO Photonic Solutions Inc.) was used along with the XF410 QMAX FITC and the XF406 QMAX red filter set (Omega Optical). All images were acquired with a 20x Plan Fluorite objective (numerical aperture: 0.45, CFI, WD: 7.4, Nikon) by an ORCA-ER cooled CCD camera (Hamamatsu).
Custom software, written in MATLAB code, was used to control the LCM microscope and laser. The software suite was used to automatically move the stage and objective to acquire multi-color fluorescent and bright-field images of every microcapillary. To quantify each microcapillary, image segmentation was performed using Otsu’s method to threshold the chosen color image (frequently the bright-field image), creating a binary mask.44 This binary mask was used to quantify the fluorescence intensity at each region of interest. The quantified data was then filtered by size, roundness (eccentricity), and minimal fluorescence intensity with user-defined parameters to minimize false positives, such as cell debris or other fluorescent debris. These image analysis steps were performed with standard functions in the MATLAB Image Processing Toolbox (available upon request). The software was also used to retrieve cells from desired microcapillaries in two different modalities: single-capillary extraction and pooled extraction. In single-capillary extraction, the software was used to move the stage to center the selected microcapillary, allowing further examination and extraction of the desired individual cell onto a sterile capture surface. In pooled extraction, sort gates were established and the software was used to sequentially move the stage and fire the laser to extract the contents of all specified microcapillaries onto a single capture surface, resulting in a population of enriched cells.
Microcapillary array preparation, loading, and extraction.
Microcapillary arrays (10 and 20 μm diameter, 1mm thick; INCOM, Inc.) were sterilized in ethanol and dried. The loading side of the arrays was treated using a corona wand (BD-20AC Electro-Technic Products) to generate a hydrophilic surface, which facilitates loading. To achieve the maximum fraction of wells containing single cells, cell suspensions were diluted prior to loading according to Poisson statistics (~3,200 cells/μl in 20 μm arrays and ~12,800 cells/μl in 10 μm arrays). Specific loading conditions for each protein engineering application are described in subsequent sections. Cell suspensions were mixed with magnetic microbeads (Life Technologies, 37002D) to a final bead concentration of 10 mg/ml and pipetted into the arrays. The magnetic microparticles are opaque and impart a brownish color to the cell suspensions (Supplementary Fig. 1 inset). The addition of these microparticles uniformly occludes the fluorescence signal to a limited degree and prevents bright-field imaging of the contents of the microcapillary (Supplementary Fig. 18). A 2 mm slab of 1% weight/volume agarose was overlaid on the array to help prevent evaporation.
The Triton UV laser in the LCM system was used to extract the contents of desired capillaries. As described in Supplementary Figure 4, the laser power was adjusted so that the extraction efficiency is close to 100%. With our optimized extraction parameters, the laser operates for 18 ± 2 ms (n = 5 measurements), delivering a train of pulses at 2.5 kHz with a total energy of approximately 100 μJ. The microcapillary contents were extracted onto a glass coverslip, which was then placed in yeast or bacterial growth media (liquid medium or agar plates) to propagate the extracted cells. After each experiment, the arrays were cleaned by removing their contents with a strong stream of distilled water, followed by brief sonication in 1 M NaOH, and storage in 100% ethanol prior to reuse. In our hands, the microcapillary arrays can be reused at least 10 times without observable deterioration.
Preparation of Axl protein constructs and libraries.
DNA encoding human Axl Ig1 (amino acids Ala19-Pro131) and a non-binding Axl variant (E59R, T77R) were cloned into the pCT yeast display plasmid between NheI and BamHI restriction sites.6 Plasmid DNA was transformed into the S. cerevisiae strain EBY100 by electroporation for yeast surface display studies. Soluble Gas6 was recombinantly expressed in human embryonic kidney (HEK) cells using FreeStyle Max 293 Expression System (Invitrogen) and purified as previously described.6 The yeast surface-displayed naïve scFv library (~7 × 108 variants) was generously provided by James Van Deventer and K. Dane Wittrup (MIT)45 and Sachdev Sidhu (U. Toronto). All yeast cells harboring the pCT display plasmid were grown in selective media and induced as previously described.46
Axl Ig1 mock library screening.
For the Axl Ig1 mock libraries, induced yeast cells displaying wild-type (WT) and the non-binding Axl variant were mixed at defined cell ratios of WT: non-binding of 1:10, 1:100, 1:1,000, 1:10,000, and 1:100,000. Approximately 3–6 × 106 yeast cells (depending on the mock library) were incubated at room temperature for 6 hr with 1 nM purified Gas66 in phosphate-buffered saline with 1 mg/ml BSA (PBSA). Yeast were then washed and resuspended in PBSA containing a 1:250 dilution of chicken anti-c-Myc antibody (Life Technologies, A21281) for 45 min at 4 °C. To detect Gas6 binding, yeast were washed and resuspended in PBSA containing a 1:100 dilution of mouse anti-His IgG Hilyte Fluor 555 (Anaspec, 61250-H555) for 45 min at 4 °C. To enhance the binding signal, yeast were washed and resuspended in PBSA, and a 1:100 dilution of goat anti-mouse IgG Hilyte Fluor 555 (Anaspec, AS-28175–05-H555) was subsequently added for 30 min at 4 °C. Finally, to detect c-Myc tag expression, yeast were washed and resuspended in PBSA containing a 1:100 dilution of goat anti-chicken IgG Alexa Fluor 488 (Life Technologies, A11039) for 30 min at 4 °C. Labeled yeast were diluted to ~12,800 cells/μL, loaded on a 10 μm array, and analyzed for Gas6 binding with 475/40 nm excitation/510 nm long pass emission filters, and for c-Myc cell surface expression with 525/45 nm excitation/565 nm long pass emission filters. Yeast cells predicted to express WT or the non-binding Axl variant were extracted via single-cell sorting and cultured on agar plates containing selective yeast media. To identify the extracted cells, plasmid DNA was recovered using a Zymoprep kit (Zymo Research Corporation), amplified by PCR, and sequenced using the Sanger method (Sequetech).
scFv naïve library screening.
The yeast displayed scFv library was grown and induced for expression as previously described.45 The library was subjected to two rounds of magnetic-activated cell sorting (MACS) to reduce the diversity prior to μSCALE screening: a negative selection for cells that did not bind His-tag Isolation beads (Dynabeads, 10103D, Life Technologies) and a positive selection for cells that bind Gas6-coated magnetic heads. Both sorts were performed according to manufacturer’s instructions (Dynabeads® His-Tag Isolation & Pulldown protocol) and modeled after sorts previously described.46 Gas6-coated magnetic beads (4 mg) were prepared by incubating Dynabeads with a saturating amount of His-tagged Gas6 on a rotator for 2 hr at 4 °C. These beads were washed using a magnetic holder (MPC-S, Dynal) with Binding/Wash Buffer (50 mM sodium phosphate pH 8.0, 300 mM NaCl, and 0.01% Tween-20) to removed unconjugated Gas6. For sort 1, 8 × 109 yeast cells were washed with Pull-down Buffer (3.25 mM sodium phosphate pH 7.4, 70 mM NaCl, and 0.01% Tween-20), and incubated with non-coated His-tag Dynabeads in Pull-down Buffer for 2 hr at 4 °C. After incubation, cells and beads were placed in a magnetic holder and unbound cells were collected; these non-binding yeast were transferred to fresh tubes and incubated with the prepared Gas6-coated beads for 2 hr at 4 °C. Using the magnetic holder, the yeast and beads were washed several times with Binding/Wash Buffer. Gas6-bound yeast cells were eluted from the magnetic beads with His-Elution Buffer (150 mM imidazole, 25 mM sodium phosphate pH 8.0, 150 mM NaCl, and 0.005% Tween-20) and grown in selective yeast media. Using this protocol, MACS reduced the library to ~2 × 106 variants, which corresponds to 0.4% of the original library size.
μSCALE was used for two sequential screening rounds of the MACS-reduced scFv library. Yeast were incubated at room temperature for 2 hr in PBSA containing 33 nM Gas6 (sort 1) or 10 nM Gas6 (sort 2). Following incubation with Gas6, cells were treated with PBSA containing a 1:250 dilution of chicken anti-c-Myc antibody (Life Technologies, A21281) for 30 min at 4 °C. Cells were then incubated with PBSA containing a 1:100 dilution of mouse anti-His Tag IgG Hilyte Fluor 488 (Anaspec, 61250-H488) for 30 min at 4 °C. Finally, cells were incubated in PBSA containing a 1:100 dilution of both goat anti-mouse IgG Alexa Fluor 488 (Life Technologies, A11001) and goat anti-chicken IgG Alexa Fluor 555 (Life Technologies, A21437) for 30 min at 4 °C. Labeled yeast were diluted to ~12,800 cells/μl and loaded on a 10 μm array and analyzed for Gas6 binding using excitation/emission parameters described above. For μSCALE round 1, 143 capillaries with the highest Gas6 binding/c-Myc expression ratio were auto-extracted within a single pool, resulting in a population enriched for binding affinity to Gas6. In μSCALE round 2, the top 15 capillaries were extracted via single-cell sorting. After round 2, plasmid DNA was recovered using a Zymoprep kit (Zymo Research Corporation), PCR amplified, and sequenced (Sequetech).
The Gas6 binding affinity of the best scFv clone was measured by incubating 105 induced yeast cells with varying concentrations of Gas6 in PBSA200 (0.1% BSA + 200 mM NaCl in PBS) for 2 hr at room temperature. Cells were incubated with PBSA200 containing a 1:250 dilution of chicken anti-c-Myc antibody (Life Technologies, A21281) for 15 min at 4 °C. Secondary antibody labeling was carried out in PBSA200 containing a 1:100 dilution of mouse anti-His Tag IgG Hilyte Fluor 488 (Anaspec, 61250-H488) and goat anti-chicken IgY PE (Santa Cruz Biotechnology, sc-3730) for 30 min at 4 °C. Binding and expression signals of the labeled cells were measured by flow cytometry (FACS Calibur, BD Biosciences). The error for the reported KD corresponds to a 1-σ (68%) confidence interval for the estimated binding affinity, and was calculated with the parametric bootstrap method using data from three technical replicates of each fit point.47
Fluorescent protein library construction and screening.
ddFP technology involves the reversible association of two dark (i.e., non-fluorescent) FP monomers to form a fluorescent heterodimer (Supplementary Fig. 7). The convention for naming the constituent FP monomers is ddFP-A copy and ddFP-B copy. The A copy monomer possesses the quenched chromophore, whereas the B copy monomer lacks a chromophore. We designate the non-covalent complex as simply as ddFP (e.g., ddOFP) and the genetically fused tandem dimer as tdFP. tdRFP was cloned into the pBAD expression plasmid between XhoI and EcoRI restriction sites (where ddRFP-B was the 5’ partner in the tandem gene fusion) and used as a template for protein engineering experiments. First, M66T was introduced into the ddRFP-A copy by site-directed mutagenesis. A library was then created by randomly mutagenizing the ddRFP-A copy (M66T) gene using the following error-prone PCR conditions: 0.15 or 0.075 mM MnCl2, 10 ng template, 200 μM dATP, 200 μM dGTP, 1 mM dTTP, 1 mM dCTP, and 4 mM MgCl2. Forty cycles of PCR amplification were carried out for each reaction using Taq polymerase with an extension temperature of 68 °C. Mutant gene libraries were cloned 3’ to the ddRFP-B copy, to generate a library of tandem ddFP variants, and transformed into DH10B electrocompetent E. coli for growth and screening within the μSCALE platform.
To create a highly fluorescent ddOFP, μSCALE was used to screen three successive E. coli libraries, with error-prone PCR performed on the isolated DNA between each round of screening. In each library cycle, an overnight outgrowth of cells in LB media at 37 °C with shaking was performed after transformation to allow E. coli to recover. Cultures were then diluted to ~12,800 cells/μl in expression media (LB supplemented with ampicillin (100 μg/ml) and arabinose (0.1%)), loaded on a 10 μm array, and incubated overnight at 37 °C in a sealed Petri dish with a moistened Kimwipe. After overnight growth, arrays containing 3 × 105 clones each were analyzed for fluorescence intensity with 525/45 nm excitation/565 nm long pass emission filters. For μSCALE round 1, 10 capillaries were auto-extracted as a bulk population, which was used to create the next library via error-prone PCR as described above. For μSCALE round 2, 12 capillaries were extracted via single-cell sorting. The four clones with the highest fluorescence signal were used to create the third generation error-prone library with the same conditions above. For the third and final μSCALE round, the top 10 capillaries were extracted via single-cell sorting. To evaluate protein characteristics after each library screen, extracted cells were cultured overnight at 37 °C with shaking in expression media. Fluorescent proteins were extracted using B-Per (Pierce), and spectra were collected using a Biotek Synergy H4 plate reader.
ddOFP spectral properties.
Prior to spectral analysis, all proteins were purified using metal chelating Ni-NTA chromatography and dialyzed into PBS. Emission spectra were recorded with a Biotek Synergy H4 plate reader. Absorbance measurements were made on a Varian Cary 50 UV/Vis Spectrophotometer using a 1 cm quartz microcell cuvette. The alkaline chromophore denaturation method was used to determine ε values. mCitrine (Φ = 0.76) was used as the reference for quantum yield determination for ddOFP. pH titrations were performed by incubating purified proteins in buffers of desired pH and acquiring emission spectra with a 96-well Biotek Synergy H4 plate reader. A 1 μM solution of fluorescent protein was prepared in PBS and diluted 1:10 with a universal buffer of desired pH. This universal buffer solution was prepared by mixing equal volumes of 0.04 M H3BO3, 0.04 M CH3COOH, and 0.04 M H3PO4. The pH was adjusted to the desired value by adding 1M NaOH to the prepared stock solution. The pKa was determined by fitting the experimental data to the equation: , where F is fluorescence, A and B are variables that define the baselines, and nH is the Hill coefficient. Mean integrated emission peak intensities were normalized and plotted as a function of pH.
Preparation of alkaline phosphatase protein constructs.
The E. coli WT alkaline phosphatase gene and R166S variant used for kinetic comparisons were cloned into the pCT yeast display plasmid between NheI and BamHI restriction sites. Plasmid DNA was subsequently transformed into S. cerevisiae EBY100 by electroporation for yeast surface display studies. Soluble versions of WT alkaline phosphatase and the R166S mutant were also expressed and purified from E. coli SM547λDE3 cells as described previously.48 Yeast cells transformed with pCT display plasmid were grown in selective media and induced as described above. The yeast induction media was supplemented with 500 μM ZnCl2 and 500 μM MgCl2 to provide the required metal ions for catalysis. To quantify the level of yeast surface-displayed enzyme, antibody labeling of a C-terminal c-Myc tag fused to each enzyme was performed. Approximately 8 × 104 induced yeast cells were washed and resuspended in PBSA containing a 1:250 dilution of chicken anti-c-Myc antibody (Life Technologies, A21281) for 45 min at room temperature. After incubation, yeast were washed and resuspended in PBSA containing a 1:100 dilution of goat anti-chicken IgG Alexa Fluor 488 (Life Technologies, A11039) for 30 min at room temperature. c-Myc expression levels were then measured using a Guava easyCyte flow cytometer (Millipore).
Measurement of DDAO product pKa.
pH titrations were performed on 7-hydroxy-9H-(1,3-dichloro-9,9-dimethylacridin-2-one (DDAO) to determine the pH-dependence of product fluorescence (Supplementary Fig. 10). A 1 μM stock solution of DDAO was prepared in Milli-Q water and titrated with 1M HCl and 1M NaOH to adjust the solution pH. Using a Biotek Synergy H4 plate reader, fluorescence measurements at an excitation/emission of 590/660 nm were performed on aliquots from each titration step. A fit of the data to the Henderson-Hasselbalch equation yielded a pKa of 5.07 for the DDAO product. Thus, subsequent enzymatic assays were conducted at pH 8.0 to eliminate fluorescence quenching arising from protonation of the DDAO nitrogen.
Analysis of alkaline phosphate activity
Reaction conditions and calibration curves were established for the substrate 9H-(1,3-dichloro-9,9-dimethylacridin-2-one-7-yl) phosphate (DDAOP) and its product DDAO (Supplementary Fig. 11). Induced yeast cells were diluted to a final concentration of ~3200 cells/μl, washed twice in AP Reaction Buffer (100 mM sodium MOPS pH 8.0, 0.5 M NaCl, 1 mM MgCl2, 100 μM ZnSO4, 0.2% BSA, 1 mM mannose)48 and resuspended in AP Reaction Buffer containing variable concentrations of DDAOP and inorganic phosphate immediately prior to analysis. Reaction mixtures were then loaded onto either 96-well microtiter plates or μSCALE arrays for bulk and single-cell experiments, respectively. Bulk kinetic assays were conducted by measuring fluorescence time courses at an excitation/emission of 590/660 nm on a Biotek Synergy H4 plate reader spectrophotometer, which allowed linear correlation of the fluorescent signal with DDAO concentration over a range from 0–3 μM (Supplementary Fig. 11a). For kinetic measurements on the μSCALE platform, a series of images were acquired at set intervals with 600/60 nm excitation/655 nm long pass emission filters to generate single-cell reaction time courses for each capillary in the array. The μSCALE filter set and camera detected a linear correlation of product formation with fluorescence over a comparable dynamic range to the plate reader (Supplementary Fig. 11b). In both cases, the linear regime of each kinetic time series was used to estimate reaction rates. An effective enzyme concentration of 50 pM (assuming ~ 104 displayed proteins per cell14,46) was used for back-calculating rate constants from kinetic data.
Measurement of inorganic phosphate contamination in alkaline phosphatase reactions.
Inorganic phosphate, which affects measurement of KM and KI values, was measured in buffer components and reagents using fluorescently labeled phosphate-binding protein (PBP) from E. coli (Life Technologies, PV4406) (Supplementary Fig. 12 and Supplementary Table 3) as described previously.49 Briefly, 100 μl of each sample was mixed with 100 μl of 1 μM PBP in Phosphate Sensor Detection Buffer (20 mM TrisHCl, pH 7.6, 0.05% Triton X-100), and fluorescence of the mixture was immediately measured at an excitation/emission of 430/450 nm on a Biotek Synergy H4 plate reader spectrophotometer. When quantifying phosphate content in the AP Reaction Buffer, the buffer was diluted 1:20 in water prior to incubation with the sensor in order to minimize interference from the concentrated MOPS and salt solutions on the PBP binding thermodynamics. PBP stocks were stored as 10 μM aliquots in Phosphate Sensor Storage Buffer (10 mM TrisHCl, pH 7.6, 50 mM NaCl) at –80 °C and individually thawed prior to use. Fluorescence measurements were converted to phosphate concentrations using a standard curve (Supplementary Fig. 12), which was fit to a two-state binding isotherm of the form F = Fmin + (Fmax – Fmin) × (1 + KD / [Pi])–1. The binding parameters were determined to be: Fmin = 14200 ± 500 RFU, Fmax = 85000 ± 1000 RFU, and KD = 0.94 ± 0.05 μM. The presence of phosphate contamination in the AP Reaction Buffer (Supplementary Table 3) accounts for the discrepancy between kinetic parameters measured for WT alkaline phosphatase in this work and values previously reported in the literature.37
Alkaline phosphatase library construction and screening.
Error-prone PCR was used to randomly mutate the WT alkaline phosphatase gene using the following reaction conditions: 0.1 mM MnCl2, 10 ng WT template, 200 μM dATP, 200 μM dGTP, 1 mM dTTP, 1 mM dCTP, and 4 mM MgCl2. PCR amplification was performed as described above. The mutated alkaline phosphatase insert DNA and NheI/BamHI-digested pCT plasmid were electroporated in a 5:1 weight ratio into EBY100 yeast and assembled in vivo via homologous recombination.46 Library size was estimated to be ~107 by dilution plating.
μSCALE was used to screen 3 × 105 variants from the alkaline phosphatase library to identify variants with improved activity under the assay conditions. Induced yeast cells were washed twice in AP Reaction Buffer, resuspended at ~3,200 cells/μl in AP Reaction Buffer containing 1 μM DDAOP and 15 μM inorganic phosphate, and loaded into the array. After 30 min of reaction time, the fluorescent intensity of each capillary was analyzed as described in the alkaline phosphatase kinetic studies. The 15 capillaries with the highest fluorescent intensity were individually extracted and grown on agar plates containing selective yeast media. Kinetic parameters of yeast-displayed alkaline phosphatase variants from the extracted cells were measured as described above. Plasmid DNA was recovered using a Zymoprep kit (Zymo Research Corp.), transformed into DH10B electrocompetent E. coli, isolated using a plasmid miniprep kit (Thermo Scientific), and sequenced.
Assessment of metal binding in purified alkaline phosphatase constructs.
Metal content in alkaline phosphatase variants purified from E. coli was determined by equilibrium dialysis followed by atomic emission spectroscopy, as described previously.50 Briefly, purified alkaline phosphatase mutants were incubated in AP Storage Buffer (10 mM Tris pH 7.5, 50 mM NaCl, 100 μM ZnCl2, 1 mM MgCl2) for 4 d at room temperature. Enzyme aliquots at 10 μM concentration were then dialyzed in storage buffer lacking zinc or magnesium using Amicon Ultra-15 centrifugal filter units (10 kDa MWCO). Dialyzed samples were diluted in 1 M sodium acetate pH 5.5 and analyzed by atomic emission spectroscopy, using the following wavelengths for detection: 213.8 nm for Zn, 280.2 nm for Mg, 213.6 nm for P, and 180.7 nm for S. A standard curve containing control samples of dialysis buffer with known concentrations of sulfur, phosphorous, zinc, and magnesium (prepared as stock solutions in 2% nitric acid) was measured in the same manner and used to correct for background. Measured levels of phosphorous, zinc, and magnesium in each enzyme sample were normalized to protein levels (determined from sulfur content using a conversion of 12 sulfur atoms per alkaline phosphatase monomer) in order to determine the active site stoichiometry of each cofactor.
Supplementary Material
Acknowledgements:
The authors thank K. Dane Wittrup and James Van Deventer (MIT) and Sachdev Sidhu (U. Toronto) for providing the yeast-displayed scFv library. This project was funded in part by the Stanford-Wallace H. Coulter Translational Partnership Award Program, the Siebel Stem Cell Institute and the Thomas and Stacey Siebel Foundation (to I.K.D.), the Stanford Photonics Research Center, a Hitachi America Faculty Scholar Award (to J.R.C) and the NIH (GM49243 to D.H.). We acknowledge support from the National Science Foundation Graduate Fellowship Program (B.C., A.K.), Howard Hughes Medical Institute International Student Research Program (S.L.), Fannie and John Hertz Foundation Graduate Fellowship (A.K.), Stanford Bio-X Fellowship Program (S.L.), Stanford Graduate Fellowship Program (B.C.), and Stanford Dean’s Fellowship Program (S.C.A.).
Footnotes
Competing financial interests
The authors are listed as inventors on pending patent applications (PCT/US2013/047792) related to technology described in this work.
References
- 1.Lander ES Initial impact of the sequencing of the human genome. Nature 470, 187–197 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Rahman N Realizing the promise of cancer predisposition genes. Nature 505, 302–308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhodes DR & Chinnaiyan AM Integrative analysis of the cancer transcriptome. Nat. Genet~. 37 Suppl, S31–S37 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y et al. Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol. Cell. Proteomics 4, 1240–1250 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Romero PA & Arnold FH Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kariolis MS et al. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 10, 977–983 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu I & Arnold FH Engineered thermostable fungal Cel6A and Cel7A cellobiohydrolases hydrolyze cellulose efficiently at elevated temperatures. Biotechnol. Bioeng. 110, 1874–1883 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Alford SC, Ding Y, Simmen T & Campbell RE Dimerization-dependent green and yellow fluorescent proteins. ACS Synth. Biol. 1, 569–575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen I, Dorr BM & Liu DR A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U. S. A. 108, 11399–11404 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gai SA & Wittrup KD Yeast surface display for protein engineering and characterization. Curr. Opin. Struct. Biol. 17, 467–473 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CV et al. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J. Mol. Biol. 340, 1073–1093 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Martis E High-Throughput Screening: The Hits and Leads of Drug Discovery. J. Appl. Pharm. Sci. 01, 02–10 (2011). [Google Scholar]
- 13.Povolotskaya IS & Kondrashov FA Sequence space and the ongoing expansion of the protein universe. Nature 465, 922–926 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Agresti JJ et al. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl. Acad. Sci. U. S. A. 107, 4004–4009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ostafe R, Prodanovic R, Nazor J & Fischer R Ultra-high-throughput screening method for the directed evolution of glucose oxidase. Chem. Biol. 21, 414–421 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Zinchenko A et al. One in a million: flow cytometric sorting of single cell-lysate assays in monodisperse picolitre double emulsion droplets for directed evolution. Anal. Chem. 86, 2526–2533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischlechner M et al. Evolution of enzyme catalysts caged in biomimetic gel-shell beads. Nat. Chem. 6, 791–796 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Romero P. a., Tran TM & Abate AR Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. 112, 201422285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lafferty M & Dycaico MJ GigaMatrix: An ultra high-throughput tool for accessing biodiversity. JALA - J. Assoc. Lab. Autom. 9, 200–208 (2004). [Google Scholar]
- 20.Fukuda T, Shiraga S, Kato M & Yamamura S Construction of novel single-cell screening system using a yeast cell chip for nano-sized modified-protein-displaying libraries. NanoBiotechnology 1, 105–111 (2005). [Google Scholar]
- 21.Gorris HH & Walt DR Mechanistic aspects of horseradish peroxidase elucidated through single-molecule studies. J. Am. Chem. Soc. 131, 6277–6282 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Liebherr RB, Renner M & Gorris HH A single molecule perspective on the functional diversity of in vitro evolved β-glucuronidase. J. Am. Chem. Soc. (2014). [DOI] [PubMed] [Google Scholar]
- 23.Rissin DM et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 28, 595–599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love JC, Ronan JL, Grotenbreg GM, van der Veen AG & Ploegh HL A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat. Biotechnol. 24, 703–707 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Fitzgerald V et al. Exploiting Highly Ordered Subnanoliter Volume Microcapillaries as Microtools for the Analysis of Antibody Producing Cells. Anal Chem 87, 997–103 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Kovac JR & Voldman J Intuitive, image-based cell sorting using optofluidic cell sorting. Anal. Chem. 79, 9321–9330 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gach PC, Attayek PJ, Whittlesey RL, Yeh JJ & Allbritton NL Micropallet arrays for the capture, isolation and culture of circulating tumor cells from whole blood of mice engrafted with primary human pancreatic adenocarcinoma. Biosens. Bioelectron. 54, 476–483 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waters JC Accuracy and precision in quantitative fluorescence microscopy. J. Cell Biol. 185, 1135–1148 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemke G & Rothlin CV Immunobiology of the TAM receptors. Nat. Rev. Immunol. 8, 327–336 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graham DK, DeRyckere D, Davies KD & Earp HS The TAM family: phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 14, 769–785 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Sidhu SS et al. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J. Mol. Biol. 338, 299–310 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Ai H-W, Baird MA, Shen Y, Davidson MW & Campbell RE Engineering and characterizing monomeric fluorescent proteins for live-cell imaging applications. Nat. Protoc. 9, 910–928 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Shaner NC et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Alford SC, Abdelfattah AS, Ding Y & Campbell RE A fluorogenic red fluorescent protein heterodimer. Chem. Biol. 19, 353–360 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding Y et al. Ratiometric biosensors based on dimerization-dependent fluorescent protein exchange. Nat. Methods 12, 195–198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coleman J Structure and mechanism of alkaline phosphatase. Annu. Rev. Biophys. Biomol. Struct. 21, 441–483 (1992). [DOI] [PubMed] [Google Scholar]
- 37.O’Brien PJ & Herschlag D Functional interrelationships in the alkaline phosphatase superfamily: Phosphodiesterase activity of Escherichia coli alkaline phosphatase. Biochemistry 40, 5691–5699 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Alberstein M, Eisenstein M & Abeliovich H Removing allosteric feedback inhibition of tomato 4-coumarate:CoA ligase by directed evolution. Plant J. 69, 57–69 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Yang J-S, Seo SW, Jang S, Jung GY & Kim S Rational engineering of enzyme allosteric regulation through sequence evolution analysis. PLoS Comput. Biol. 8, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu X, Robin S, O’Connell S, Walsh G & Wall JG Engineering of a fungal beta-galactosidase to remove product inhibition by galactose. Appl. Microbiol. Biotechnol. 87, 1773–1782 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Coelho PS, Brustad EM, Kannan A & Arnold FH Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 339, 307–10 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Pitsillides CM, Joe EK, Wei X, Anderson RR & Lin CP Selective cell targeting with light-absorbing microparticles and nanoparticles. Biophys. J. 84, 4023–4032 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Y-C et al. Massively parallel delivery of large cargo into mammalian cells with light pulses. Nat. Methods 12, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otsu N A threshold selection method from gray-level histograms. IEEE Transactions on Systems, Man, and Cybernetics 9, 62–66 (1979). [Google Scholar]
- 45.Deventer J. A. Van & Wittrup KD Yeast Surface Display for Antibody Isolation: Library Construction, Library Screening, and Affi nity Maturation. 1131, (Humana Press, 2014). [DOI] [PubMed] [Google Scholar]
- 46.Chao G et al. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Benton D & Krishnamoorthy K Performance of the parametric bootstrap method in small sample interval estimates. Adv. Appl. Stat. 2, 269–285 (2002). [Google Scholar]
- 48.Andrews LD, Zalatan JG & Herschlag D Probing the origins of catalytic discrimination between phosphate and sulfate monoester hydrolysis: comparative analysis of alkaline phosphatase and protein tyrosine phosphatases. Biochemistry 53, 6811–6819 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brune M, Hunter JL, Corrie JET & Webb MR Direct, Real-Time Measurement of Rapid Inorganic Phosphate Release Using a Novel Fluorescent Probe and Its Application to Actomyosin Subfragment 1 ATPase. Biochemistry 33, 8262–8271 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Zalatan JG, Fenn TD & Herschlag D Comparative enzymology in the alkaline phosphatase superfamily to determine the catalytic role of an active-site metal ion. J. Mol. Biol. 384, 1174–1189 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.