

Abstract
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder characterized by accumulation of amyloid β-peptide (Aβ) plaques in the brain and decreased cognitive function leading to dementia. We tested if hydroxyurea (HU), a ribonucleotide reductase inhibitor known to activate adaptive cellular stress responses and ameliorate abnormalities associated with several genetic disorders, could protect rat hippocampal neurons against oxidative-, excitatory-, mitochondrial-, and Aβ-induced stress and if HU treatment could improve learning and memory in the APP/PS1 mouse model of AD. HU treatment attenuated the loss of cell viability induced by treatment of hippocampal neurons with hydrogen peroxide, glutamate, rotenone, and Aβ1–42. HU treatment attenuated reductions of mitochondrial reserve capacity, maximal respiration, and cellular ATP content induced by hydrogen peroxide treatment. In vivo, treatment of APP/PS1 mice with HU (45 mg/kg/day) improved spatial memory performance in the hippocampusdependent Morris water maze task without reducing Aβ levels. HU provides neuroprotection against toxic insults including Aβ, improves mitochondrial bioenergetics, and improves spatial memory in an AD mouse model. HU may offer a new therapeutic approach to delay cognitive decline in AD.
Keywords: hydroxyurea, amyloid beta, Alzheimer’s disease, APP/PS1, neurodegeneration, mitochondria
1. Introduction
Alzheimer’s disease (AD) is a debilitating neurodegenerative disorder that affects more than 5 million Americans age 65 or older and is expected to affect nearly 14 million Americans by 2050 (Hebert et al., 2013). Individuals with AD experience episodic memory loss and dementia (Association, 2016). AD histology is characterized by the accumulation of amyloid β-peptide (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau protein (Polanco et al., 2018). Brains from individuals with AD also exhibit an increase in inflammation characterized by activated astrocytes and microglial cells (Van Eldik et al., 2016). At the cellular level, neurons affected in AD exhibit mitochondrial dysfunction, impaired autophagy, increased oxidative stress, and an impaired ability to respond to and repair oxidative damage (Stranahan and Mattson, 2012).
Treatments are not available to prevent or slow the progression of AD. Currently, individuals with moderate to severe AD may be prescribed drugs such as memantine, an NMDA receptor antagonist, or acetylcholinesterase inhibitors, that have small effects on clinical symptoms (Aisen et al., 2012). However, there is currently no treatment available that substantially delays the onset or progression of AD. Due to a lack of prevention and an increase in prevalence, the current estimated cost of AD and other dementias in the United States alone is over 250 billion dollars annually. It is anticipated to rise to $1.1 trillion dollars by 2050 if an effective intervention(s) is not discovered (Association, 2016).
Previous studies in AD animal models have provided evidence that physiological and pharmacological interventions that activate adaptive cellular response signaling pathways, such as intermittent fasting and caloric restriction, can protect neurons against dysfunction and degeneration. For example, daily caloric restriction reduces Aβ pathology in amyloid precursor protein (APP) mutant mice and in APPswe695/PS1delE9 (APP/PS1) double mutant transgenic mice (Patel et al., 2005). Intermittent fasting (alternate day fasting) and 30% daily caloric restriction ameliorate cognitive decline during aging in 3xTg AD mice (Halagappa et al., 2007). Intermittent fasting induces components of the adaptive cellular stress response and leads to increased neuronal resistance to stress as well as improved synaptic plasticity (Mattson et al., 2018). Treatment of APP mutant mice with the mTOR inhibitor rapamycin stimulated neuronal autophagy, reduced Aβ pathology and lessened cognitive deficits (Spilman et al., 2010). Treatment of 3xTg AD mice with 2-deoxyglucose, which induces mild metabolic stress, bolstered mitochondrial function and reduced Aβ neuropathology (Yao et al., 2011). We previously found that hydroxyurea (HU), a ribonucleotide reductase inhibitor, improves cellular metabolic defects in fibroblasts from several genetic disorders and activates adaptive cellular stress responses in cultured human fibroblasts (Brose et al., 2012). These include up-regulation of antioxidant defenses, protein chaperones and autophagy, and stimulation of mitochondrial biogenesis. Here we show that HU treatment protects cultured hippocampal neurons against oxidative, metabolic and excitotoxic stress, and improves memory in the APP/PS1 mouse model of AD.
2. Methods
2.1. Rat hippocampal neuronal cultures
Primary hippocampal neuron cultures were prepared from embryonic day 18 Sprague-Dawley rat brains as described previously (Kaech and Banker, 2006; Mattson et al., 1989). Dissociated neurons were plated at a density of 40,000 neurons per well (96-well plate) or 1 × 106 cells per well (6-well plate) on polyethyleneimine-coated plastic dishes in MEM medium supplemented with 10% fetal bovine serum. Once the neurons attached to the plates, the cell plating medium was replaced with Neurobasal (NB) medium with 5% B27 supplement, 1% glutamax, and 1% antibiotic-antimycotic (Invitrogen, Waltham, MA) for 6–7 days. Prior to the addition of HU (5 μM unless otherwise specified; H8627, Sigma-Aldrich, St. Louis, MO), the neurons were washed with unsupplemented NB medium to remove traces of antioxidants in the B27 supplement, and the medium was replaced with NB medium supplemented with 5% B27 minus antioxidants (NB + B27 – AOX; Invitrogen), 1% glutamax, and 1% antibiotic-antimycotic. Experiments were performed between days in vitro (DIV) 6 and 9.
2.2. Neurotoxicity assays
On DIV 6 or 7, neurons were treated with HU (5 μM; 2.5 μM for Aβ1–42 experiment). The concentration of HU was titrated to allow 100% cell viability. After 24 hours, H2O2 (15 μM), glutamate (50 μM; Sigma-Aldrich), rotenone (75 nM; Sigma-Aldrich), or oligomerized Aβ1–42 peptide (5–10 μM; Bachem, Torrance, CA) was added and the neurons cultured for another 24 hours. For the Aβ1–42 peptide experiments, the neurons were incubated with HU (2.5 μM) in NB + B27 - AOX for the first 24 hours. The medium was then changed to NB medium without any B27 supplement plus HU (2.5 μM); and, Aβ1–42 peptide added for the last 24 hours of incubation. Without B27 supplements the neurons are more sensitive to perturbations. Therefore, the HU dosage was titrated to 2.5 μM to allow 100% cell viability. Cell viability was assessed using an MTS cell viability assay per manufacturer’s instructions (CellTiter Aqueous One Solution Cell Proliferation Assay; Promega, Madison, WI). Briefly, 20 μl of CellTiter Aqueous One Solution was added to cells in 100 μl of NB medium + B27 - AOX. Cells were incubated at 37o C in 5% CO2, and the A490 measured with a Synergy H1 Microplate Reader (Biotek, Winooski, VT). Since metabolically active cells reduce the MTS tetrazolium compound to a colored formazan product, the amount of formazan measured at A490 was proportional to the number of metabolically active cells. Cell viability was normalized to the untreated control cells (100% viability).
2.3. Mitochondrial activity measurements
Mitochondrial activity was assessed using a Seahorse XF-96 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA) according to the manufacturer’s instructions and as described previously (Divakaruni et al., 2014; Yao et al., 2017). Briefly, neurons were grown in Seahorse 96-well plates (40,000 cells/well). On DIV 6, the media was changed to NB + B27 minus AOX. Neurons were treated with HU (5 μM) on DIV 7. After 24 hours, the neurons were treated with H2O2 (35 μM) for one hour. The medium was changed to unbuffered XF assay medium (Seahorse Biosciences) supplemented with 5 mM glucose, 1 mM pyruvate, and 2 mM glutamax (Invitrogen). The neurons were incubated in a non-CO2 incubator at 37oC for one hour followed by measurement of basal oxygen consumption rate (OCR) and OCR after the sequential addition of 2 μM oligomycin, 1 μM FCCP and 5 μM rotenone/antimycin A. Oligomycin inhibits mitochondrial ATP synthase activity and facilitated the measurement of ATP-linked respiration. FCCP uncouples the mitochondrial proton gradient and disrupts the mitochondrial membrane potential allowing measurement of maximal respiration. Rotenone inhibits mitochondrial complex I and antimycin inhibits mitochondrial complex III. The use of rotenone and antimycin together shuts down mitochondrial respiration and allows measurement of non-mitochondrial respiration.
2.4. ATP content
Cellular adenosine triphosphate (ATP) content was determined using the ATP Determination Kit (#A22066; Molecular Probes, Eugene, OR) following the manufacturer’s instructions. Briefly, on DIV 7, hippocampal neurons were treated with HU (5 μM). Approximately 24 hours later, H2O2 peroxide was added (15 μM or 35 μM) for one hour. All medium was removed, 11 μl boiling water was added to each well, and 110 μl of ATP reaction mixture was added and mixed with the lysed cells. ATP content was determined by measuring luciferase activity compared to a standard curve. The concentration of ATP per treatment condition was normalized relative to the untreated control neurons. N ≥ 3 independent experiments.
2.5. RNA isolation and microarray analyses
RNA was isolated from rat hippocampal neurons treated with HU (5 μM) for 2, 6, 12, or 24 hours (n = 3 per time point) and from frozen mouse hippocampi isolated from 19 to 24-week-old nontransgenic control littermates of APP/PS1 mice treated with HU (45 mg/kg/day; n = 4) or vehicle control (water; n = 4) for 2.5 months using Trizol and following the manufacturer’s instructions (Thermo Fisher Scientific). Animal housing conditions are listed below in section 2.8. Total RNA was quality-controlled on the Agilent Bioanalyzer RNA 6000 Chip (Agilent, Santa Clara, CA), and 200 ng labeled using the Agilent Low-Input QuickAmp Labeling Kit, and purified and quantified according to the manufacturer’s instructions. A total of 600 ng Cy3labeled cRNA was hybridized for 17 hours to Agilent SurePrint G3 Rat and Mouse GE 8×60K microarrays. Following post-hybridization rinses, arrays were scanned using an Agilent SureScan microarray Scanner, and hybridization intensity data extracted from the scanned images using Agilent’s Feature Extraction Software. Raw data were analyzed by Z-normalization as described previously (Cheadle et al., 2003). Principal component analysis (PCA) was performed on the normalized Z-scores of all detectable probes using DIANE 6.0 software (http://www.grc.nia.nih.gov/branches/rrb/dna/diane_software.pdf). For the hippocampal samples, PCA analysis identified the data of one untreated wild type hippocampus and one HUtreated wild type hippocampus as outliers. Significance of gene expression differences between control and HU-treated neurons or hippocampi were based on a Z-test < 0.05, false discovery rate < 0.30, and a Z-ratio > 1.5 in either direction, and ANOVA p value < 0.05. The raw and normalized data sets are deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE111943 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111943), with the individual data sets available as GSE111941 (rat neurons) and GSE111940 (mouse hippocampi).
2.6. Western blot analyses
Neuronal cell lysates were collected in RIPA buffer [50 mM Tris-HCl, pH 7.4, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA. 50 mM NaF, 0.2 mM sodium orthovanadate, 1 mM PMSF, plus 1 x protease inhibitors (#11 836 153 001, Roche, Indianapolis, IN)] and stored at −80oC. Denatured LDS-PAGE and immunoblot analyses were performed using the following antibodies: total OXPHOS rodent WB antibody cocktail (ab110413, Abcam, Cambridge, MA), LC3 (NB100–2220, Novus Biologicals, Littleton, CO), β-actin (A5441, Sigma-Aldrich, St. Louis, MO), and SIRT3 (#5490, Cell Signaling, Danvers, MA). Blot imaging and protein quantification were performed using an Odyssey Imager (Li-Cor Biosciences, Lincoln, NE). β -actin levels were used for normalization.
2.7. Proteasome activity assay
20S and 26S proteasome activity levels were measured using a Proteasome Activity Assay Kit (#APT280; Millipore, Burlington, MA) following the manufacturer’s directions. The following modifications were made to measure 20S and 26S proteasome activity. Briefly, HU-treated neurons (6 × 106) were harvested on DIV 8 in 50 μl RIPA and stored at −80oC. The lysate was suspended in assay buffer supplemented with 10 mM MgCl2 and divided into two samples. Sample one contained 15 mM 2-deoxyglucose and hexokinase (0.1 mg/μl) to deplete ATP activity and measure 20S proteasome activity. Sample two was supplemented with 2 mM ATP to measure 26S proteasome activity. Proteasome activity was measured by the detection of free fluorophore 7-amino-4-methylcoumarin (AMC; fluorescence 380/460 nm) after cleavage from the proteasome substrate LLVY-AMC.
2.8. Animal behavior testing study design
All studies were approved by the National Institute of Aging Institute of Animal Care and Use Committee and all procedures followed the NIH Guide for the Care and Use of Laboratory Animals. All mice were female APPswe695/PSEN1dE9 mice [B6.Cg-Tg (APPswe,PSEN1dE9) 85Dbo/J; APP/PS1] or their nontransgenic (NTG) littermate controls. Original breeding pairs were purchased from the Jackson Laboratory, and were maintained on a C57BL/6J background in the National Institute of Aging vivarium under a 12-hour light/12-hour dark cycle. There were two to four mice per cage. HU-treated mice were given 45 mg/kg/day of HU in their drinking water. The dosage was based on the dosage used in sickle cell disease mouse models (50mg/kg/day; (Lebensburger et al., 2012). The dosage in mice is higher than in humans. Individuals with sickle cell disease, psoriasis, or HIV are administered 15 mg/kg/day and not to exceed 35 mg/kg/day (Medscape, 2018). Two cohorts were studied; cohort 2 was set-up after the completion of studies with cohort 1. Cohort 1 consisted of 11 untreated NTG control littermates (29.31g +/− 0.91 SEM), seven untreated APP/PS1 mice (27.44g +/− 0.72 SEM), and eight HUtreated APP/PS1 mice (26.66g +/− 1.16 SEM). One HU-treated APP/PS1 mouse was blind and excluded from behavioral testing. HU treatment began at two to four months of age. Behavioral testing started at six to eight months of age. The total treatment length was five months. Cohort 2 consisted of nine untreated NTG control mice (28.70 +/− 1.31 SEM), eight HU-treated NTG control mice (29.48 +/− 1.23 SEM), 13 untreated APP/PS1 mice (28.89g +/− 0.48 SEM), and 13 HU-treated APP/PS1 mice (30.36 +/− 0.74 SEM). HU treatment started at two and half to five months of age and behavioral testing began at 10.5 to 13 months of age. The total treatment time was nine months. Prior to each behavioral test, animals were habituated to the testing room for 12 hours, two days in a row. One week after behavioral testing, all animals were euthanized and brain tissues were rapidly frozen using dry ice. Within each cohort, the average weight per genotype and treatment group was similar (One-way ANOVA pcohort 1 = 0.137, pcohort 2 = 0.511).
2.9. Open field test
Mice were acclimated to the room for 30 minutes prior to the first test. The mice were placed in a clear acrylic arena (40.6 cm x 40.6 cm) housed in a sound-attenuating cubicle (Med Associates, St. Albans, VT) for 30 minutes to assess their locomotor and exploratory behavior. Data were collected using Activity Monitor software (Version 4.0, Med Associates). The total distance, total ambulatory time, distance in the center zone, and time in the center zone were measured.
2.10. Elevated plus maze
The elevated plus maze was used to assess anxiety-related behavior. The acrylic apparatus consisted of two open arms (30 cm x 5 cm) and two closed arms (30 cm x 5 cm x 16 cm) raised 60 cm above the floor. Each animal was placed in the center of the apparatus and the animal movement tracked for five minutes with ANY-maze software (Stoelting Co., Wood Dale, IL). The time spent in the open arms and the time spent in the closed arms plus the middle zone were measured.
2.11. Morris water maze
To assess spatial learning and memory, a circular pool (160 cm) was filled with water (22–25oC) and the water rendered opaque by adding enough non-toxic white tempura paint to hide a rectangular platform (14 cm x 14 cm) in the center of the target quadrant two centimeters below the water surface. Distal spatial cues were placed on the walls of the room. During the training phase, each mouse performed four consecutive 60-second swim trials for the specified number of days. For each trial, the mouse was placed facing the wall in a randomly predetermined quadrant such that the mouse started from all four quadrants throughout the four daily trials. If the mouse found the platform, the trial was stopped and the mouse given 15 seconds to sit on the platform before being removed from the water. If the mouse did not find the platform within the allotted 60 seconds, the mouse was guided visually by placing a finger on top of the platform or by manually guiding the mouse to the platform and allowed to sit on the platform for 15 seconds before being removed from the water. Probe trials were performed 24, 48, and 72 hours following the last training trial. For each probe trial, the platform was absent and each mouse was given one 60 second trial to swim in the pool. Following the 72-hour probe trial, a visual platform test was performed to identify any visual discrepancies in the mice. The test was performed in a separate room with a similar pool except paint was not added to the water, there were no distal cues on the walls, and the platform was identified with two flags. Each mouse was given four non-consecutive 60 second trials to locate the platform. Each mouse was tested with the platform in one quadrant before moving the platform to a different quadrant and performing the next trial round. All trials were video recorded and analyzed using ANY-maze software (Stoelting Co., Wood Dale, IL).
2.12. Aβ42 and Aβ40 Immunoassay
Aβ42 and Aβ40 peptide levels were measured by a V-PLEX immunoassay (4G8; Meso Scale Discovery, Rockville, MD) using 1 μg of mouse cortical homogenates and 2.5 μg of mouse hippocampal homogenates.
2.13. Statistical analyses
All data were analyzed using GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA). Data are represented as the mean ± standard error of the mean (SEM). Neuronal experiments were performed independently three or more times. Student t-tests were used to compare before and after treatments. One-way ANOVAs followed by post-hoc Bonferroni or post-hoc Dunnett’s multiple comparison tests were used to analyze data with three or more groups. MWM training parameters were analyzed using a two way repeated measures ANOVA (RM-ANOVA) followed by post-hoc Bonferroni analyses.
3. Results
3.1. Hydroxyurea treatment protects hippocampal neurons against neurotoxic stress
We hypothesized that HU treatment can upregulate one or more adaptive cellular stress response pathways and thereby protect hippocampal neurons against insults relevant to the pathogenesis of various neurodegenerative disorders. To test this, we investigated the ability of HU to protect wild type rat hippocampal neurons against four neurotoxic agents. The neurotoxic agents investigated included hydrogen peroxide to mimic increased cellular oxidative stress, glutamate to induce excitotoxicity, rotenone to impair mitochondrial function by inhibiting complex I of the mitochondrial electron transport chain, and Aβ1–42 to mimic the Aβ aggregation and accumulation that occurs in the brains of AD patients (Camandola and Mattson, 2017; Dumont and Beal, 2011). We examined these effects in wild type rat hippocampal neurons rather than mouse APP/PS1 hippocampal neurons to allow applicability of these results to various neurodegenerative disease models.
Rat hippocampal neurons were pretreated with HU for 24 hours, then co-treated with HU and a neurotoxin for an additional 24 hours. Each neurotoxin significantly reduced cell viability compared to untreated neurons (p < 0.05; Table 1): 39.6% reduction with 15 μM hydrogen peroxide, 22.7% reduction with 50 μM glutamate treatment, 17.6% reduction with 75 nM rotenone, and a 25.7%, 38.1%, and 49% reduction with 5, 7.5, and 10 μM Aβ1–42, respectively (Fig. 1). HU treatment significantly attenuated the reduction in cell viability by 20.5%, 17.5%, 13.5%, 9.8% and 11.2% for neurons treated with hydrogen peroxide, glutamate, rotenone, and 5 and 7.5 μM Aβ1–42, respectively (Table 1). These data suggest HU treatment protects hippocampal neurons, to some extent, against increased oxidative stress, increased excitotoxicity, decreased mitochondrial function, and insults caused by Aβ aggregation and accumulation.
Table 1.
Summary of MTS viability assay data.
| Toxin | Sample size (n) | RM-ANOVA p-valuetoxin |
Post-hoc Bonferroni p-valueHU+toxin vs toxin alone |
|---|---|---|---|
| 15 μM H2O2 | 6 | p = 0.0001 | p < 0.05 |
| 50 μM glutamate | 9 | p = 0.002 | p < 0.05 |
| 75 μM rotenone | 3 | p = 0.011 | p < 0.01 |
| 5 μM Aβ1–42 | 5 | p = 0.0001 | p < 0.05 |
| 7.5 μM Aβ1–42 | 5 | p = 0.0001 | p < 0.05 |
Figure 1. MTS viability assays.
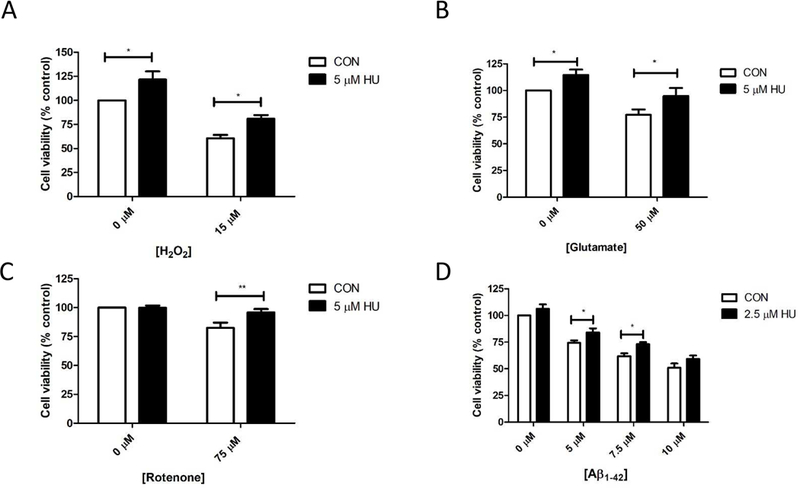
HU treatment of rat hippocampal neurons significantly attenuates the significant reduction in cell viability induced by: A) hydrogen peroxide (RM-ANOVA pH2O2 = 0.0001, pHU = 0.0007, post-hoc Bonferroni tests CON vs HU and HU + 15 μM H2O2 vs 15 μM H2O2 p < 0.05; n= 6); B) glutamate (RM-ANOVA pGLUT = 0.002, pHU = 0.004, post-hoc Bonferroni tests CON vs HU and HU + 50 μM glutamate vs 15 μM glutamate p < 0.05; n = 9); C) rotenone (RM-ANOVA pROT = 0.05, pHU = 0.011, post-hoc Bonferroni tests HU + 75 μM rotenone vs 75 μM rotenone p < 0.01; n = 3); and D) Aβ1–42 peptide (RM-ANOVA pAβ = 0.0001, pHU = 0.0001, post-hoc Bonferroni tests HU + 5 μM Aβ1–42 and HU + 7.5 μM Aβ1–42 vs 5 μM and 7.5 μM Aβ1–42 p < 0.05; n = 5). *p < 0.05, **p < 0.01. CON = untreated control neurons. Errors bars = SEM.
3.2. HU treatment attenuates H2O2-induced reductions in respiration measures
We chose to perform subsequent experiments with only one neurotoxic agent hydrogen peroxide. Hydrogen peroxide increases cellular oxidative stress, a component of multiple neurodegenerative diseases; and HU treatment attenuated the effects of hydrogen peroxide on cell viability to the greatest extent. Rotenone was specifically not chosen because it directly inhibits mitochondrial complex I function.
To determine the effects of HU treatment on neuronal energy metabolism, we measured cellular oxygen consumption rates (OCR) using the Seahorse XF-96 extracellular flux analyzer. HU treatment of hippocampal neurons did not significantly alter basal measurements of energy metabolism (Fig. 2A). To test whether HU treatment could instead precondition neurons against a toxic insult, we treated neurons with H2O2 (35 μM). H2O2 treatment significantly decreased basal respiration, ATP-linked respiration, maximal respiration, mitochondrial reserve capacity, and non-mitochondrial respiration (Fig. 2A; One-way ANOVAs p =0.014; post-hoc Bonferroni tests p < 0.05, p < 0.05, p < 0.001, p < 0.01, p < 0.01, respectively). HU treatment significantly attenuated the H2O2-induced reductions in maximal respiration, mitochondrial reserve capacity, and non-mitochondrial linked respiration (post-hoc Bonferroni tests p < 0.05). The mitochondrial reserve capacity is the difference between the ATP produced by oxidative phosphorylation at basal levels and that produced at maximal activity (Desler et al., 2012). The reserve is needed if a stressor, such as a neurotoxin, or an increased workload requires a sudden burst of energy. Notably, HU treatment did not attenuate H2O2-induced reductions in respiratory measures if basal levels of respiration in H2O2-treated neurons dropped below 50% of untreated control basal respiration levels. Together, these data suggest HU treatment improves theoxidative phosphorylation capacity in neurons under conditions of oxidative stress.
Figure 2. Mitochondrial measures.
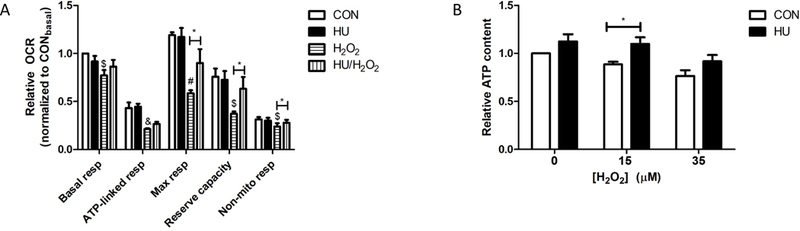
A) Relative oxygen consumption rates (OCR). HU treatment significantly attenuated the significant reduction in maximal respiration, energetic reserve capacity, and non-mitochondrial respiration caused by H2O2 treatment (35 μM; One-way ANOVAs within measurements p = 0.014, post-hoc Bonferroni tests CON vs H2O2 &p < 0.05, $p < 0.01, #p < 0.001, HU vs HU/H2O2 *p < 0.05). B) Relative ATP content. The significant reduction in ATP content by hydrogen peroxide treatment of hippocampal neurons was significantly attenuated by HU treatment (2-way ANOVA pH2O2= 0.002, pHU = 0.005, post-hoc Bonferroni tests 15 μM H2O2 vs 15 μM H2O2 + HU *p < 0.05). CON = untreated control neurons. Errors bars = SEM, n ≥ 3 independent experiments.
3.3. HU increases neuronal ATP content
HU treatment sustained maximal respiration, an ATP-dependent process, in neurons subjected to oxidative stress. Therefore, we measured the effect of HU on neuronal ATP content. HU treatment significantly increased neuronal ATP content compared to untreated control neurons and neurons treated with 15 μM H2O2 for one hour (Fig. 2B; 2-way ANOVA pH2O2= 0.002, pHU = 0.005, post-hoc Bonferroni tests 15 μM H2O2 vs 15 μM H2O2 + HU p < 0.05).
3.4. HU treatment increases expression of mitochondrial and autophagy related proteins
We performed microarray gene expression analyses on cultured rat hippocampal neurons treated with HU for 2, 6, 12, and 24 hours to gain further insight into the neuroprotective mechanisms of HU. Gene ontology and pathway enrichment analyses comparing neurons treated with HU for 2 hours or 24 hours demonstrated an increase in mitochondrial- and proteasomal-related genes compared to untreated control neurons (Supplementary Table S1).
Microarray pathway analyses suggested components of adaptive cellular stress response pathways were upregulated in HU-treated neurons. We used western blotting to examine the expression of various subunits of the mitochondrial electron transport chain complexes, the autophagy related protein LC3, and the mitochondrial deacetylase SIRT3 in hippocampal neurons treated with HU for 24 hours. HU treatment significantly increased expression of MTCO1, a cytochrome c oxidase subunit of mitochondrial electron transport complex IV (p < 0.05), and significantly increased total expression of the autophagy marker protein LC3 without affecting the ratio of LC3II/LC3I (Fig. 3A, B; p < 0.05). Although SIRT3 has been shown to mediate adaptive responses to oxidative and excitatory stress, HU treatment did not consistently increase SIRT3 protein expression (Fig. 3C). Since only expression of a subunit of mitochondrial complex IV, and not the markers of complexes I (NDUF8), II (SDHB), III (UQCRC2), and V (ATP5A), increased, HU treatment may increase oxidative phosphorylation capacity without increasing overall mitochondrial biogenesis.
Figure 3. Western blot analyses of mitochondrial and autophagy related protein expression.
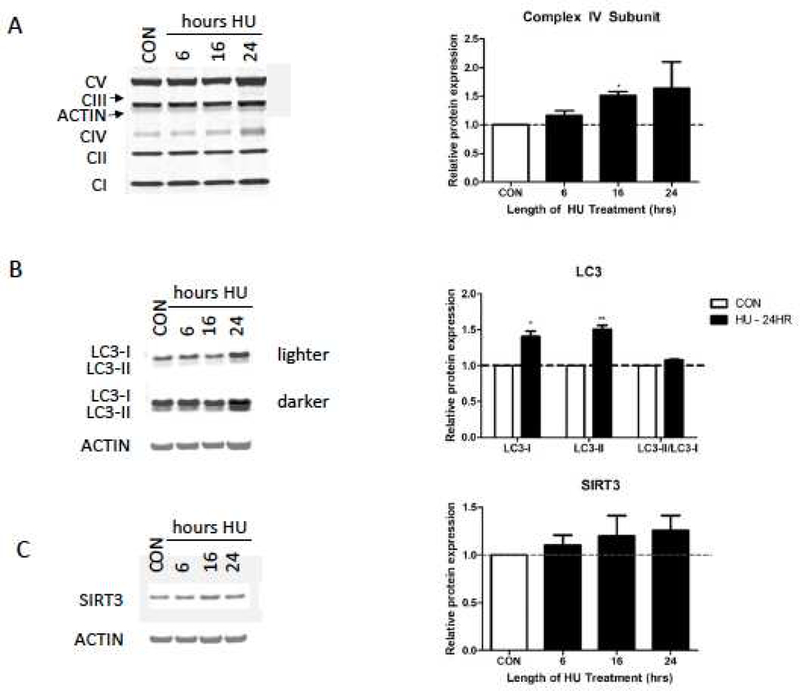
A) Western blot analyses of mitochondrial electron transport chain subunits (Complex I-V, CI-CV). Complex IV subunit MTCO1 significantly increased after 16 hours of HU treatment. B) Expression of LC3-I and LC3-II significantly increased after 24 hours of HU treatment. C) SIRT3 expression did not significantly increase after HU treatment. Relative protein expression normalized to untreated control neuron (CON) expression. Errors bars = SEM, n = 3 independent experiments. * p < 0.05, **p < 0.01.
Next, we assessed proteasome activity after HU treatment of wild type hippocampal neurons. In vitro measurements of 20S and 26S proteasome activities using cell lysates from neurons treated with HU for 2 or 24 hours indicated that HU treatment did not alter 20S or 26S proteasome activity (Fig. 4; One-way ANOVA p20S = 0.148, p26S = 0.544).
Figure 4. Proteasome activity.
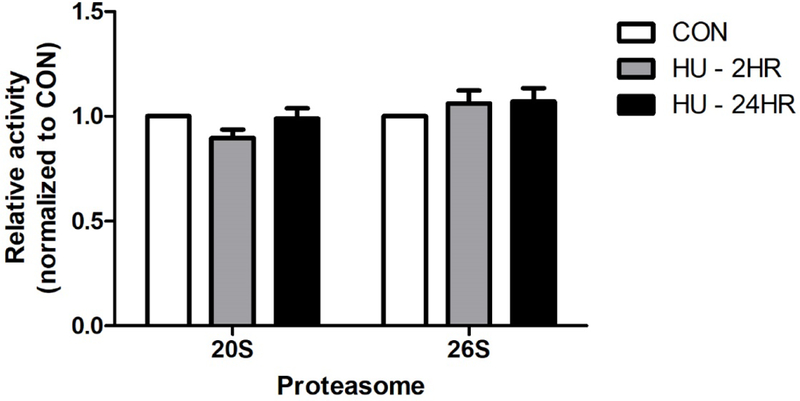
HU treatment did not significantly alter the activity of the 20S or 26S proteasome in rat hippocampal neurons (One-way ANOVA p20S = 0.148, p26S = 0.544, n ≥ 4). CON = untreated control neurons. Errors bars = SEM.
3.5. Impact of HU on behavior and Aβ pathology in APP/PS1 AD mice
Neuronal damage and memory loss are prominent features of AD (Association, 2016). Based on the ability of HU to attenuate the effects of hydrogen peroxide, glutamate, rotenone, and Aß1–42 peptide in wild type rat hippocampal neurons, we tested the possible neuroprotective effects of HU in an animal model of AD. Two cohorts of APP/PS1 mice and age-matched non-transgenic (NTG) control mice were treated with HU starting between 2–5 months of age. Cohort 1 was treated for a total of five months and cohort 2 for nine months. We first performed behavioral experiments on the younger cohort, cohort 1. Based on these results, we aged the APP/PS1 mice longer before performing behavioral testing in cohort 2. We reasoned that the cognitive deficits in the aged APP/PS1 mice would be greater than in the younger mice; and therefore, the effects of HU treatment on cognitive measures may be more readily discernable. Behavioral testing began between 6–8 months of age for cohort 1 and between 10.5–13 months of age for cohort 2.
3.6. APP/PS1 mice did not exhibit anxiety-related behaviors
The open field test was used to assess locomotion and exploratory behavior and the elevated plus maze was used to assess anxiety-related behavior in the mice (Bailey and Crawley, 2009). Mice that spend more time in the center zone of the open field or more time in the open arms of the elevated plus maze are considered to exhibit less anxiety than mice who spend less time in the open areas. The 6 to 8-month-old APP/PS1 mice and HU-treated APP/PS1 mice traveled a significantly greater distance and spent significantly more time moving than the NTG control mice in the open field test (Fig. 5A-B; One-way ANOVA pdistance = 0.006, post-hoc Bonferroni tests p < 0.05; One-way ANOVA ptime= 0.002, post-hoc Bonferroni tests p < 0.01). However, there was no significant difference between groups of mice in the distance traveled or the time spent in the center zone. There were no significant differences in any of the open field test measurements between the 10 to 13-month-old NTG mice, APP/PS1 mice, or either HU-treated group of mice (Fig. 5C-D). Similarly, there were no significant differences in either cohort between NTG, APP/PS1, or HU-treated mice in the time spent in the open arms or the time spent in the closed arms and middle zone of the elevated plus maze (Fig. 6). Neither age group of APP/PS1 mice exhibited anxiety-related behavior significantly different from NTG control mice in either behavioral test; and, HU-treatment did not significantly alter the behavior of mice in either test.
Figure 5. Open Field Test.
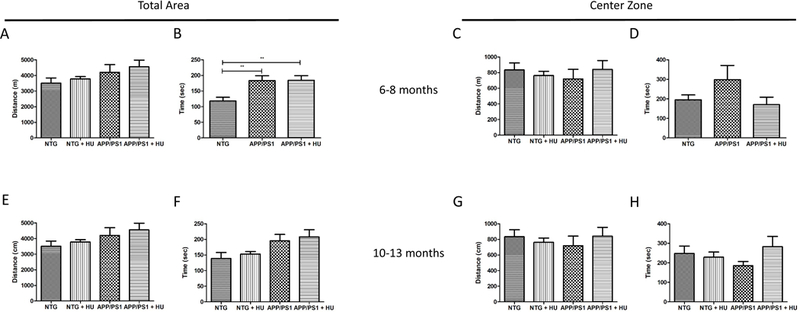
Six to eight-month-old APP/PS1 mice exhibit more hyperactivity in the open field test than non-transgenic (NTG) control mice. 6 to 8-month-old mice (A-D). NTG n =11, APP/PS1 and APP/PS1 + HU n = 7. A) Total distance traveled (One-way ANOVA p = 0.006, post-hoc Bonferroni tests * p < 0.05). B) Total ambulatory time (One-way ANOVA p = 0.002, post-hoc Bonferroni tests ** p < 0.01). C) Total distance traveled in the center zone (p = 0.140). D) Total time spent in the center zone (p = 0.151). 10 to 13-month-old mice (E-H). NTG n = 5, NTG + HU n = 8, APP/PS1 n = 13, APP/PS1 + HU n = 12. E) Total distance traveled (p = 0.468). F) Total ambulatory time (p = 0.119). C) Total distance traveled in the center zone (p = 0.836). D) Total time spent in the center zone (p = 0.267). Errors bars = SEM.
Figure 6. Elevated plus maze.
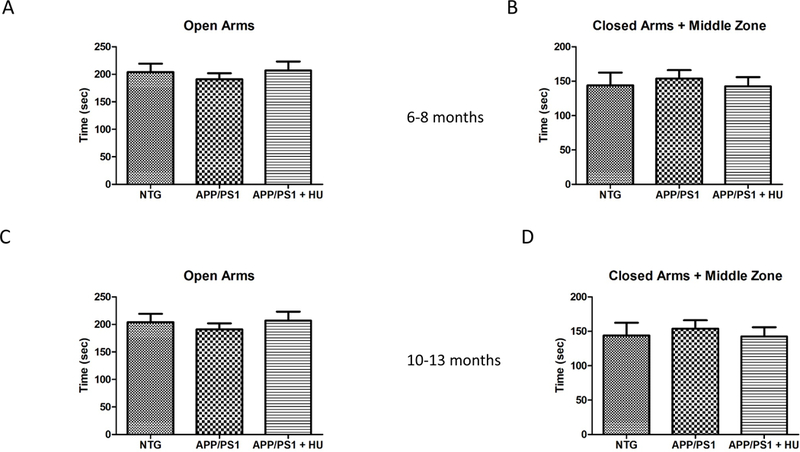
HU treatment did not affect performance in the elevated plus maze. 6 to 8-month-old mice (A-B). NTG n =11, APP/PS1 n = 6, and APP/PS1 + HU n = 7. A) Time in the open arms (p = 0.901). B) Time in the closed arms and middle zone (p = 0.782). 10 to 13-month-old mice (C-D). NTG n = 9, NTG + HU n = 8, APP/PS1 n = 13, APP/PS1 + HU n = 13. C) Time in the open arms (p = 0.782). D) Time in the closed arms and middle zone (p = 0.147). Errors bars = SEM.
3.7. Hydroxyurea treatment improved spatial reference memory in APP/PS1 mice
The MWM was used to assess the effects of HU-treatment on spatial learning and memory in APP/PS1 mice, a hippocampus-dependent task (Vorhees and Williams, 2006). During the training trials, there was a significant difference in the escape latency in the younger cohort (cohort 1), but not in the older cohort (cohort 2). The younger NTG control mice found the platform in significantly less time than the younger APP/PS1 mice on the seventh day of training (Fig. 7A; RM-ANOVA p = 0.032, post-hoc Bonferroni NTG vs APP/PS1 day 7 p < 0.05). HU treatment reduced the time the younger APP/PS1 mice took to find the platform on the seventh day of training in cohort 1, but had no effect on the ability of the NTG or APP/PS1 mice to learn the location of the hidden platform in cohort 2 (Fig. 8A). Additionally, there were no significant differences in the distance swam to locate the platform or the average swim speed between any of the groups in cohorts 1 or 2 (Fig. 7B-C and 8B-C).
Figure 7. Morris water maze cohort 1.
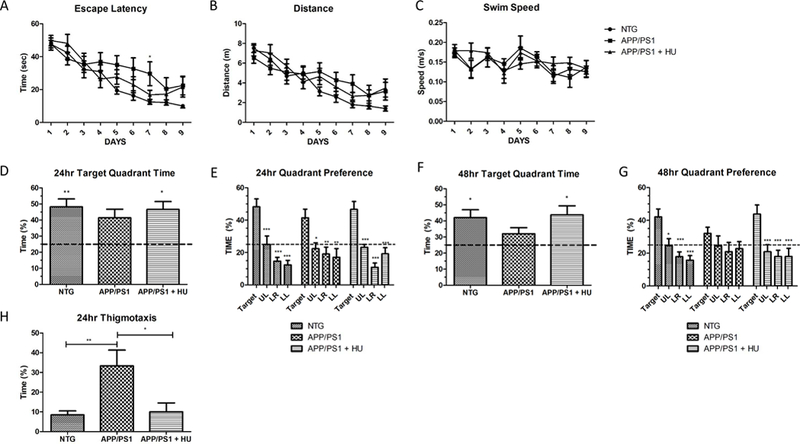
HU treatment improved spatial reference memory in 6 to 8-month-old APP/PS1 mice. A) Latency to reach the hidden platform across training days. There was a significant difference among groups in their latency to find the hidden platform. The NTG control mice found it in significantly less time than APP/PS1 mice on Day 7 (2-way RM-ANOVA p = 0.032, post-hoc Bonferroni analyses NTG vs APP/PS1 Day 7 *p < 0.05). B) Distance to reach the hidden platform across training days. C) Average swim speed across training days. There was no significant difference between groups in the distance traveled to reach the hidden platform or the group swim speeds. D) 24-hour probe trial time in target quadrant. NTG and HU-treated APP/PS1 mice spent significantly more time in the target quadrant compared to the 25% chance level (One-way ANOVA p = 0.007, post-hoc Bonferroni analyses pNTG < 0.01 and pAPP/PS1 + HU < 0.05). There was no statistical difference between groups (One-way ANOVA p = 0.634). E) 24-hour probe trial quadrant preference. NTG control, APP/PS1, and HU-treated APP/PS1 mice all spent significantly more time in the upper right target quadrant than in the non-target quadrants (UL = upper left, LR = lower right, LL = lower left; one way ANOVAs within groups, pNTG = 0.0001, pAPP/PS1 = 0.005, pAPP/PS1 + HU = 0.001, post –hoc Dunnett’s multiple comparison tests * p < 0.05, ** p < 0.01, *** p < 0.001). H). F) 48-hour probe trial time in target quadrant. NTG and HU-treated APP/PS1 mice spent significantly more time in the target quadrant compared to the 25% chance level (One-way ANOVA p = 0.023, post-hoc Bonferroni analyses p < 0.05). There was no statistical difference between groups (Oneway ANOVA p = 0.257). G) 48-hour probe trial quadrant preference. NTG and HU-treated APP/PS1 mice spent significantly more time in the target quadrant than in the non-target quadrants (One way ANOVA, pNTG = 0.001, pAPP/PS1 = 0.443, pAPP/PS1 + HU = 0.002, post –hoc Dunnett’s multiple comparison tests * p < 0.05, ** p < 0.01, *** p < 0.001). H) Thigmotaxis. HU-treatment significantly reduced thigmotaxis in APP/PS1 mice during the 24-hr probe trial (One way ANOVA p = 0.002, post-hoc Bonferroni analyses pWT vs APP/PS1 < 0.01, pAPP/PS1 vs APP/PS1 + HU < 0.05). NTG n = 11, APP/PS1 n = 7, APP/PS1 + HU n = 7. Errors bars = SEM.
Figure 8. Morris water maze cohort 2.
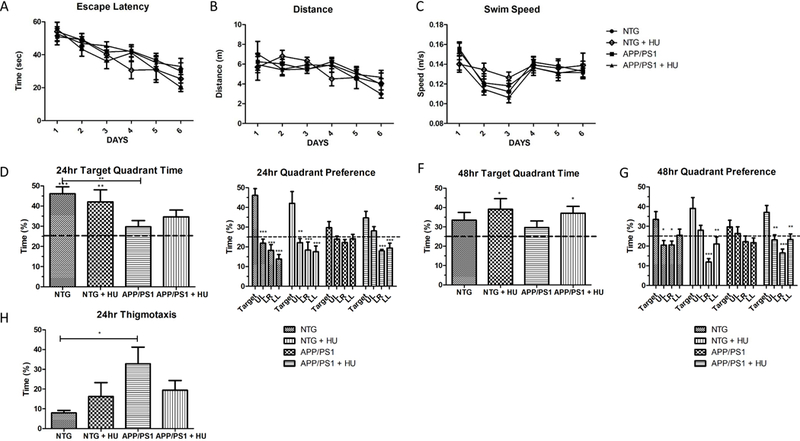
HU-treatment improved spatial reference memory in 10 to 13-month-old APP/PS1 mice. A) Latency to reach the hidden platform. B) Distance to reach the hidden platform. C) Average swim speed. There was no significant difference in escape latency, distance to find the platform, or swim speed between the four groups of mice during the six-day training period (2-way RM-ANOVA p = 0.533, p = 0.973, and p = 0.601, respectively). D) 24-hour probe trial time in target quadrant. NTG and HU-treated NTG mice spent significantly more time in the target quadrant compared to the 25% chance level (One-way ANOVA p = 0.0003, post-hoc Bonferroni analyses pNTG < 0.001 and pNTG + HU < 0.01). NTG mice spent significantly more time than APP/PS1 mice in the target quadrant (One-way ANOVA p = 0.021, post-hoc Bonferroni analyses NTG vs APP/PS1 **p < 0.01). E) 24-hour probe trial quadrant preference. NTG control, HU-treated NTG control, and HU-treated APP/PS1 mice spent significantly more time in the upper right (UR) target quadrant than in non-target quadrants (UL = upper left, LR = lower right, LL = lower left; one-way ANOVAs within groups NTG, NTG + HU, and APP/PS1 + HU p = 0.0001, pAPP/PS1 = 0.089, post –hoc Dunnett’s multiple comparison tests *p < 0.05, **p < 0.01, ***p < 0.001). F) 48-hour probe trial time in target quadrant. HU-treated NTG mice and HU-treated APP/PS1 mice spent significantly more time in the target quadrant compared to the 25% chance level (One-way ANOVA p = 0.023, post-hoc Bonferroni analyses p < 0.05). G) 48-hour probe trial quadrant preference. NTG control, HUtreated NTG control, and HU-treated APP/PS1 mice spent significantly more time in the upper right target quadrant than in the non-target quadrants (One-way ANOVA within group pNTG = 0.012, pNTG + HU = 0.001, pAPP/PS1 = 0.221, pAPP/PS1 + HU = 0.001, post –hoc Dunnett’s multiple comparison tests *p < 0.05, **p < 0.01, ***p < 0.001). H) 24-hr probe thigmotaxis. APP/PS1 mice exhibited significantly more thigmotaxis than NTG mice (One way ANOVA p = 0.064, post-hoc Bonferroni analyses NTG vs APP/PS1 p < 0.05). NTG n = 9, NTG + HU n = 8, APP/PS1 n = 13, APP/PS1 + HU n = 13. Bars = SEM.
Twenty-four and 48 hours after the last training trials the hidden platform was removed to test the spatial reference memory of the mice. Spatial reference memory was analyzed by 1) examining the percentage of time swam in the quadrant where the platform was located during training, the target quadrant, compared to the chance level of 25%, 2) comparing the percentage of time swam in the target quadrant between groups, and 3) comparing the percentage of time swam in the target quadrant versus the non-target quadrants within groups. These data are summarized in Table 2. For cohort 1, NTG control and HU-treated APP/PS1 mice spent significantly more time in the target quadrant compared to the 25% chance level during the 24hour and 48-hour probe trials (Fig. 7D and F). There were no statistical differences in the time spent in the target quadrant between groups during either probe trial. However, all three groups of younger mice, NTG control, APP/PS1, and HU-treated APP/PS1, spent significantly more time in the target quadrant compared to the non-target quadrants during the 24-hour probe trial (Fig. 7E). However, only the NTG control mice and HU-treated APP/PS1 mice spent significantly more time in the target quadrant compared to the time spent in each non-target quadrant during the 48-probe trial (Fig. 7G). This suggests HU treatment improved the spatial reference memory of the younger APP/PS1 mice.
Table 2. Summary of statistical analyses for Morris water maze probe trials.
Cohort 1: NTG n = 11, APP/PS1 n = 7, APP/PS1 + HU n = 7. Cohort 2: NTG n = 9, NTG + HU n = 8, APP/PS1 n = 13, APP/PS1 + HU n = 13.
| Measurement | Cohort | Probe trial | One-way ANOVA |
post-hoc p-value |
|---|---|---|---|---|
| Percent time in target quadrant compared to chance level (25%) |
1 | 24-hr | p = 0.007 | pNTG < 0.01 pAPP/PS1+HU< 0.05 |
| 1 | 48-hr | p = 0.023 | pNTG < 0.05 pAPP/PS1 + HU < 0.05 |
|
| 2 | 24-hr | p = 0.0003 | pNTG < 0.001 pNTG + HU < 0.01 |
|
| 2 | 48-hr | p = 0.034 | pNTG + HU < 0.05 pAPP/PS1 +HU < 0.05 |
|
| Percent time in target quadrant compared between groups |
1 | 24-hr | p = 0.634 | |
| 1 | 48-hr | p = 0.257 | ||
| 2 | 24-hr | p = 0.021 | pNTG vs APP/PS1 < 0.01 | |
| 2 | 48-hr | p = 0.353 | ||
| Percent time spent in target quadrant vs non-target quadrants |
1 | 24-hr | pNTG = 0.001 pAPP/PS1= 0.005 pAPP/PS1+ HU= 0.001 |
NTG: pUR vs UL pLR, LL < 0.001 APP/PS1:pUR vs LR, LL < 0.01, p UR vs UL < 0.05 APP/PS1+HU:pUR vs UL, LR, LL < 0.001 |
| 1 | 48-hr | pNTG = 0.001 pAPP/PS1+ HU= 0.001 |
NTG: pUR vs UL < 0.05, pLR, LL < 0.001 APP/PS1+HU:pUR vs UL, LR, LL < 0.001 |
|
| 2 | 24-hr | pNTG = 0.0001 pNTG + HU = 0.001 pAPP/PS1+ HU= 0.001 |
NTG: pUR vs UL pLR, LL < 0.001 NTG + HU: pUR vs LR, LL <0.001, pUR vs UL < 0.01 APP/PS1 + HU: pUR vs LR, LL < 0.001 |
Since the APP/PS1 mice spent significantly more time in the target quadrant than the non-target quadrants during the 24-hour probe trial, this suggested they remembered the location of the platform. Therefore, we decided to age the cohort 2 mice longer before behavioral testing to possibly increase their learning and memory deficits; and, we shorten the training period from nine days to six days for cohort 2. In the older cohort of mice (cohort 2), the NTG control mice and HU-treated NTG control mice spent significantly more time in the target quadrant compared to chance during the 24-hour probe trial (Fig. 8D); and the HU-treated NTG control mice and HU-treated APP/PS1 mice spent significantly more time in the target quadrant compared to chance during the 48-hour probe trial (Fig. 8F). During the 24-hour probe trials the NTG control mice spent significantly more time in the target quadrant compared to APP/PS1 mice (Fig. 8E), but not during the 48-hour probe trials (Fig. 8F). During the 24-hour and 48-hour probe trials, the older untreated APP/PS1 mice did not spent significantly more time in the target quadrant compared to the time spent in the three other non-target quadrants (Fig. 8E and8G). However, the older HU-treated APP/PS1 mice spent significantly more time in the target quadrant than in the lower right or lower left quadrants during the 24-hour probe trials (Fig. 8E) and spent significantly more time in the target quadrant compared to all three non-target quadrants during the 48-hour probe trial (Fig. 8G). Taken together, these data suggest that HU-treatment significantly improved the spatial reference memory of APP/PS1 mice in both the younger and older mice.
Analyses of thigmotaxic behavior, the tendency of some mice to swim around the perimeter of the pool, indicated that the younger APP/PS1 mice swam a significant amount of time in the perimeter of the pool during the 24-hour probe trial and that HU-treatment significantly reduced thigmotaxic behavior in the younger APP/PS1 mice (Fig. 7H; One-way ANOVA p = 0.002, posthoc Bonferroni analyses pWT vs APP/PS1 < 0.01, pAPP/PS1 vs APP/PS1 + HU < 0.05). The older APP/PS1 mice exhibited significant thigmotaxic behavior compared to the NTG control mice (Fig. 8H; One-way ANOVA p = 0.064, post-hoc Bonferroni’s pNTG vs. APP/PS1 < 0.05). However, in cohort 2 thigmotaxic behavior in the HU-treated APP/PS1 mice did not significantly differ from the APP/PS1 mice or NTG control mice. As a cued control, the task was performed with a visible platform; and, there were no significant differences in the latency to find a visible platform between any of the groups (pcohort 1 = 0.589, pcohort 2 = 0.101). In summary, HU-treatment decreased thigmotaxic behavior in the younger APP/PS1 mice.
3.8. Hydroxyurea treatment did not alter soluble Aβ levels
We measured soluble Aβ42 and Aβ40 levels in cortical and hippocampal samples from the younger (cohort 1) APP/PS1 and HU-treated APP/PS1 mice (Fig. 9). HU treatment did not significantly alter soluble Aβ42 and Aβ40 levels or the ratio of Aβ42/Aβ40 peptide levels in either brain tissue. However, there was a trend toward higher soluble levels of Aβ42 and Aβ40 in HUtreated APP/PS1 hippocampal samples (p = 0.063 and p = 0.066, respectively).
Figure 9. Aβ levels in cohort 1 mouse brains.
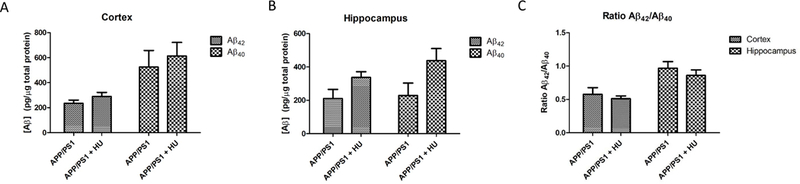
Aβ42 and Aβ40 peptide levels were measured in brain homogenates from the A) cortex and B) hippocampus of APP/PS1 (n = 7) and APP/PS1 + HU (n = 8) mice in cohort 1. There were no significant differences between Aβ42 and Aβ40 levels in the cortical (p ≥ 0.224) or hippocampal (p ≥ 0.063) samples or between the ratio of Aβ42 /Aβ40 (C; p ≥ 0.415) in the cortical or hippocampal samples.
3.9. Microarray analyses of gene expression in mouse hippocampi
To gain insight into the molecular mechanisms that underlie improved spatial reference memory in APP/PS1 mice following HU treatment, we performed microarray analyses on hippocampi isolated from NTG control mice and NTG control mice treated with HU for two and a half months. NTG control mice were treated so that the findings could potentially be applicable to other neurodegenerative diseases. Gene ontology (GO) and pathway enrichment analyses identified upregulation of multiple GO groups related to synaptic function and adaptive cellular stress responses including mitochondrial electron transport function, the lysosome, the heat shock response and unfolded protein response (GO0051082), and the antioxidant response (Table 3 and Supplementary Table S2). Further work will be necessary to confirm these data. This includes examining gene expression in hippocampi from HU-treated APP/PS1 mice.
Table 3. Gene ontology analysis of genes altered at the mRNA level by HU treatment of wild type mice.
Microarray analyses were performed on wild type mice treated with HU (45 mg/kg/day) for 2.5 months. GO term analysis identified the pathways above as upregulated by HU treatment. N=3 untreated wild type mice, n = 3 HU-treated wild type mice.
| Gene Ontology Term | Z_Score |
|---|---|
| GO0007608 SENSORY PERCEPTION OF SMELL | 10.72 |
| GO0004984 OLFACTORY RECEPTOR ACTIVITY | 10.71 |
| GO0004930 G PROTEIN COUPLED RECEPTOR ACTIVITY | 9.34 |
| GO0007186 G PROTEIN COUPLED RECEPTOR PROTEIN SIGNALING PATHWAY | 9.24 |
| GO0004871 SIGNAL TRANSDUCER ACTIVITY | 8.51 |
| GO0007165 SIGNAL TRANSDUCTION | 7.00 |
| GO0004872 RECEPTOR ACTIVITY | 6.42 |
| GO0031424 KERATINIZATION | 6.09 |
| GO0001533 CORNIFIED ENVELOPE | 5.03 |
| GO0003735 STRUCTURAL CONSTITUENT OF RIBOSOME | 4.67 |
| GO0005840 RIBOSOME | 4.63 |
| GO0006412 TRANSLATION | 4.61 |
| GO0016020 MEMBRANE | 4.45 |
| GO0030529 RIBONUCLEOPROTEIN COMPLEX | 4.40 |
| GO0016021 INTEGRAL TO MEMBRANE | 4.08 |
| GO0005179 HORMONE ACTIVITY | 3.70 |
| GO0005230 EXTRACELLULAR LIGAND GATED ION CHANNEL ACTIVITY | 3.33 |
| GO0005843 CYTOSOLIC SMALL RIBOSOMAL SUBUNIT | 3.23 |
| GO0005739 MITOCHONDRION | 3.13 |
| GO0050896 RESPONSE TO STIMULUS | 3.08 |
| GO0016469 PROTON TRANSPORTING TWO SECTOR ATPASE CO | 3.07 |
| GO0008137 NADH DEHYDROGENASE (UBIQUINONE) ACTIVITY | 2.98 |
| GO0046933 HYDROGEN ION TRANSPORTING ATP SYNTHASE ACTIVITY | 2.93 |
| GO0046961 HYDROGEN ION TRANSPORTING ATPASE ACTIVITY | 2.93 |
| GO0042254 RIBOSOME BIOGENESIS AND ASSEMBLY | 2.91 |
| GO0006754 ATP BIOSYNTHETIC PROCESS | 2.91 |
| GO0005830 CYTOSOLIC RIBOSOME (SENSU EUKARYOTA) | 2.89 |
| GO0045202 SYNAPSE | 2.85 |
| GO0015986 ATP SYNTHESIS COUPLED PROTON TRANSPORT | 2.74 |
| GO0015078 HYDROGEN ION TRANSMEMBRANE TRANSPORTER ACTIVITY | 2.73 |
| GO0060113 INNER EAR RECEPTOR CELL DIFFERENTIATION | 2.60 |
| GO0009887 ORGAN MORPHOGENESIS | 2.58 |
| GO0005743 MITOCHONDRIAL INNER MEMBRANE | 2.44 |
| GO0003924 GTPASE ACTIVITY | 2.36 |
| GO0008021 SYNAPTIC VESICLE | 2.31 |
| GO0001824 BLASTOCYST DEVELOPMENT | 2.24 |
| GO0045211 POSTSYNAPTIC MEMBRANE | 2.19 |
| GO0005842 CYTOSOLIC LARGE RIBOSOMAL SUBUNIT | 2.16 |
| GO0005764 LYSOSOME | 2.12 |
| GO0003954 NADH DEHYDROGENASE ACTIVITY | 2.10 |
| GO0007269 NEUROTRANSMITTER SECRETION | 2.04 |
| GO0004129 CYTOCHROME C OXIDASE ACTIVITY | 2.01 |
| GO0043234 PROTEIN COMPLEX | 1.98 |
| GO0030672 SYNAPTIC VESICLE MEMBRANE | 1.97 |
| GO0008180 SIGNALOSOME | 1.95 |
| GO0015935 SMALL RIBOSOMAL SUBUNIT | 1.86 |
| GO0016616 OXIDOREDUCTASE ACTIVITY ACTING ON THE CH-OH GROUP OF DONORS | 1.84 |
| GO0005765 LYSOSOMAL MEMBRANE | 1.80 |
| GO0051082 UNFOLDED PROTEIN BINDING | 1.80 |
| GO0005905 COATED PIT | 1.79 |
| GO0045261 PROTON TRANSPORTING ATP SYNTHASE COMPLEX | 1.79 |
| GO0045104 INTERMEDIATE FILAMENT CYTOSKELETON ORGAN | 1.79 |
| GO0017157 REGULATION OF EXOCYTOSIS | 1.77 |
| GO0008199 FERRIC IRON BINDING | 1.76 |
| GO0003723 RNA BINDING | 1.74 |
| GO0006120 MITOCHONDRIAL ELECTRON TRANSPORT NADH TO UBIQUINONE | 1.70 |
| GO0042734 PRESYNAPTIC MEMBRANE | 1.66 |
| GO0019717 SYNAPTOSOME | 1.65 |
| GO0019843 RRNA BINDING | 1.64 |
| GO0045263 PROTON TRANSPORTING ATP SYNTHASE COMPLEX | 1.59 |
| GO0043005 NEURON PROJECTION | 1.58 |
| GO0006099 TRICARBOXYLIC ACID CYCLE | 1.57 |
| GO0015031 PROTEIN TRANSPORT | 1.51 |
4. Discussion
In this study, we found that HU provides neuroprotection in vitro against neurotoxins that increase oxidative stress, increase excitotoxicity, reduce mitochondrial function, and mimic aspects of AD (exposure to aggregating Aβ). In vivo we demonstrated that HU treatment improves spatial reference memory in APP/PS1 mice independent from a reduction in soluble Aβ levels. Although we have not fully established the mechanism(s) by which HU provides neuroprotection, our primary neuronal data suggests HU increases oxidative phosphorylation capacity as evidenced by an increase in maximal respiration, mitochondrial reserve capacity, and ATP content in neurons treated with hydrogen peroxide. We further found that HU treatment results in increased levels of mitochondrial electron transport chain complex proteins and increases levels of LC3, a protein involved in autophagy. Microarray gene expression analyses of HU-treated wild type rat hippocampal neurons and hippocampal tissue from HU-treated NTG control mice further supported these findings and suggest that HU increases mitochondrial gene expression.
Studies of AD patients, and cell culture and animal models of AD, have provided evidence for the involvement of mitochondrial dysfunction (Mattson et al., 2008), oxidative stress (Texel and Mattson, 2011), excitotoxicity (Camandola and Mattson, 2011) and impaired lysosome function and autophagy (Kerr et al., 2017; Nixon, 2013), in the neurodegenerative process. Aggregating Aβ can trigger neuronal degeneration by inducing membrane lipid peroxidation which results in the impairment of membrane ion-motive ATPases, and glutamate and glucose transporters, thereby rendering neurons vulnerable to excitotoxicity (Keller et al., 1997; Mark et al., 1995; Mark et al., 1997; Mattson et al., 1992). We found that HU pretreatment protects primary hippocampal neurons against multiple insults involved in Aβ toxicity, results consistent with a hormesis-based mechanism that up-regulates multiple adaptive cellular stress response pathways (Calabrese and Mattson, 2017). HU is best known as a ribonucleotide reductase inhibitor that suppresses DNA synthesis and, by that mechanism, can prevent aberrant cell proliferation in patients with chronic myeloproliferative disorders and some types of cancer (Spivak and Hasselbalch, 2011). However, neurons are postmitotic, do not express ribonucleotide reductase (Zhu et al., 2003), and do not replicate their nuclear DNA. Therefore, the neuroprotective mechanism of action of HU is likely not the result of induction of replicative stress.
A major finding from the behavioral tests of the present study was that the deficit in memory retention in the water maze probe trials was ameliorated in HU-treated APP/PS1 mice compared to control APP/PS1 mice. This beneficial effect of HU on cognition was apparently not the result of an effect on the accumulation of Aβ in the brain because the concentrations of soluble Aβ42 and Aβ40 were unaffected by HU treatment. Previous studies have provided evidence that Aβ impairs synaptic function by mechanisms involving oxidative stress (Hensley et al., 1996; Texel and Mattson, 2011), mitochondrial dysfunction (Keller et al., 1997; Ma et al., 2011; Reddy and Beal, 2008), and impaired autophagy (Kerr et al., 2017; Nixon, 2007). The ability of HU to protect cultured hippocampal neurons against Aβ toxicity, and oxidative, metabolic and excitotoxic stress, suggests that HU may protect synapses and ameliorate learning and memory deficits by suppressing oxidative, metabolic and excitotoxic stress.
It is known that HU crosses the blood-brain barrier (Dogruel et al., 2003; Syvanen et al., 2007). A potential mechanism by which HU increases neuronal stress resistance in the brain is suggested by evidence that HU can induce a moderate elevation of intracellular Ca2+ levels and thereby increase the production of nitric oxide, which then activates soluble guanylate cyclase to generate cyclic GMP (Almeida et al., 2012; Cokic et al., 2006; Raththagala et al., 2010). Previous studies have shown that cyclic GMP can protect neurons against excitotoxicity, oxidative stress and Aβ toxicity (Barger et al., 1995; Keller et al., 1998; Orio et al., 2007). Nitric oxide and cyclic GMP also play important roles in synaptic plasticity and learning and memory (Hawkins et al., 1998; Prieto et al., 2017; Wincott et al., 2014). Future studies will be required to establish whether calcium- and NO-mediated signaling mediate the neuroprotective and cognition-preserving effects of HU documented in the primary neuronal culture and APP/PS1 experiments of the present study.
Supplementary Material
Gene ontology results for the microarray analyses of hippocampal neurons treated with HU for 2, 6, 12, and 24 hours (n = 3 independent samples).
Gene ontology results for the microarray analyses of mouse hippocampi isolated from untreated and HU-treated wild type mice (n = 3).
Highlights.
• Hydroxyurea treatment protects hippocampal neurons against neurotoxic stress
• Hydroxyurea attenuates reductions in mitochondrial respiration induced by H2O2
• Hydroxyurea increases neuronal ATP content
• HU treatment increases expression of mitochondrial and autophagy related proteins
• Hydroxyurea improves reference memory in the APP/PS1 Alzheimer’s mouse model
Acknowledgements:
We thank Sophia Rafesky and Simonetta Camandola for their assistance. This research was supported by the Intramural Research Program of the National Institute on Aging and by PHS award R01HD038384 (RHR).
Footnotes
Declarations of interest: The authors have no conflicts of interest to declare.
Declaration of interests: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aisen PS, Cummings J, Schneider LS, 2012. Symptomatic and nonamyloid/tau based pharmacologic treatment for Alzheimer disease. Cold Spring Harb Perspect Med 2(3), a006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CB, Scheiermann C, Jang JE, Prophete C, Costa FF, Conran N, Frenette PS, 2012. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vasoocclusive events in sickle cell disease mice. Blood 120(14), 2879–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association, A.s., 2016. 2016 Alzheimer’s Disease Facts and Figures. https://www.alz.org/documents_custom/2016-facts-and-figures.pdf. [DOI] [PubMed]
- Bailey KR, Crawley JN, 2009. Anxiety-Related Behaviors in Mice, in: nd, Buccafusco JJ (Eds.), Methods of Behavior Analysis in Neuroscience; Boca Raton (FL). [Google Scholar]
- Barger SW, Fiscus RR, Ruth P, Hofmann F, Mattson MP, 1995. Role of cyclic GMP in the regulation of neuronal calcium and survival by secreted forms of beta-amyloid precursor. J Neurochem 64(5), 2087–2096. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Mattson MP, 2017. How does hormesis impact biology, toxicology, and medicine? NPJ Aging Mech Dis 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camandola S, Mattson MP, 2011. Aberrant subcellular neuronal calcium regulation in aging and Alzheimer’s disease. Biochim Biophys Acta 1813(5), 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camandola S, Mattson MP, 2017. Brain metabolism in health, aging, and neurodegeneration. EMBO J 36(11), 1474–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle C, Vawter MP, Freed WJ, Becker KG, 2003. Analysis of microarray data using Z score transformation. J Mol Diagn 5(2), 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokic VP, Beleslin-Cokic BB, Tomic M, Stojilkovic SS, Noguchi CT, Schechter AN, 2006. Hydroxyurea induces the eNOS-cGMP pathway in endothelial cells. Blood 108(1), 184191. [DOI] [PubMed] [Google Scholar]
- Desler C, Hansen TL, Frederiksen JB, Marcker ML, Singh KK, Juel Rasmussen L, 2012. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J Aging Res 2012, 192503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Rogers GW, Murphy AN, 2014. Measuring Mitochondrial Function in Permeabilized Cells Using the Seahorse XF Analyzer or a Clark-Type Oxygen Electrode. Curr Protoc Toxicol 60, 25 22 21–16. [DOI] [PubMed] [Google Scholar]
- Dogruel M, Gibbs JE, Thomas SA, 2003. Hydroxyurea transport across the blood-brain and blood-cerebrospinal fluid barriers of the guinea-pig. J Neurochem 87(1), 76–84. [DOI] [PubMed] [Google Scholar]
- Dumont M, Beal MF, 2011. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med 51(5), 1014–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP, 2007. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 26(1), 212–220. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Son H, Arancio O, 1998. Nitric oxide as a retrograde messenger during longterm potentiation in hippocampus. Prog Brain Res 118, 155–172. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA, Evans DA, 2013. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80(19), 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, Butterfield DA, Hall N, Cole P, Subramaniam R, Mark R, Mattson MP, Markesbery WR, Harris ME, Aksenov M, et al. , 1996. Reactive oxygen species as causal agents in the neurotoxicity of the Alzheimer’s disease-associated amyloid beta peptide. Ann N Y Acad Sci 786, 120–134. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G, 2006. Culturing hippocampal neurons. Nat Protoc 1(5), 2406–2415. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Mattson MP, Markesbery WR, 1998. Cyclic nucleotides attenuate lipid peroxidation-mediated neuron toxicity. Neuroreport 9(16), 3731–3734. [DOI] [PubMed] [Google Scholar]
- Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP, 1997. 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience 80(3), 685696. [DOI] [PubMed] [Google Scholar]
- Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF, 2017. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci 40(3), 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensburger JD, Howard T, Hu Y, Pestina TI, Gao G, Johnson M, Zakharenko SS, Ware RE, Tuomanen EI, Persons DA, Rosch JW, 2012. Hydroxyurea therapy of a murine model of sickle cell anemia inhibits the progression of pneumococcal disease by down-modulating E-selectin. Blood 119(8), 1915–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Hoeffer CA, Wong H, Massaad CA, Zhou P, Iadecola C, Murphy MP, Pautler RG, Klann E, 2011. Amyloid beta-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci 31(15), 5589–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Hensley K, Butterfield DA, Mattson MP, 1995. Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci 15(9), 6239–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP, 1997. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci 17(3), 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE, 1992. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12(2), 376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A, 2008. Mitochondria in neuroplasticity and neurological disorders. Neuron 60(5), 748–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Guthrie PB, Kater SB, 1989. Intrinsic factors in the selective vulnerability of hippocampal pyramidal neurons. Prog Clin Biol Res 317, 333–351. [PubMed] [Google Scholar]
- Mattson MP, Moehl K, Ghena N, Schmaedick M, Cheng A, 2018. Intermittent metabolic switching, neuroplasticity and brain health. Nat Rev Neurosci 19(2), 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medscape, 2018.
- Nixon RA, 2007. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120(Pt 23), 4081–4091. [DOI] [PubMed] [Google Scholar]
- Nixon RA, 2013. The role of autophagy in neurodegenerative disease. Nat Med 19(8), 983997. [DOI] [PubMed] [Google Scholar]
- Orio M, Kunz A, Kawano T, Anrather J, Zhou P, Iadecola C, 2007. Lipopolysaccharide induces early tolerance to excitotoxicity via nitric oxide and cGMP. Stroke 38(10), 28122817. [DOI] [PubMed] [Google Scholar]
- Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J, Morgan DG, Morgan TE, Finch CE, 2005. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol Aging 26(7), 995–1000. [DOI] [PubMed] [Google Scholar]
- Polanco JC, Li C, Bodea LG, Martinez-Marmol R, Meunier FA, Gotz J, 2018. Amyloid-beta and tau complexity - towards improved biomarkers and targeted therapies. Nat Rev Neurol 14(1), 22–39. [DOI] [PubMed] [Google Scholar]
- Prieto GA, Trieu BH, Dang CT, Bilousova T, Gylys KH, Berchtold NC, Lynch G, Cotman CW, 2017. Pharmacological Rescue of Long-Term Potentiation in Alzheimer Diseased Synapses. J Neurosci 37(5), 1197–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raththagala M, Karunarathne W, Kryziniak M, McCracken J, Spence DM, 2010. Hydroxyurea stimulates the release of ATP from rabbit erythrocytes through an increase in calcium and nitric oxide production. Eur J Pharmacol 645(1–3), 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Beal MF, 2008. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med 14(2), 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V, 2010. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 5(4), e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak JL, Hasselbalch H, 2011. Hydroxycarbamide: a user’s guide for chronic myeloproliferative disorders. Expert Rev Anticancer Ther 11(3), 403–414. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP, 2012. Recruiting adaptive cellular stress responses for successful brain ageing. Nat Rev Neurosci 13(3), 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen S, Barletta J, Blomquist G, Langstrom B, Bergstrom M, 2007. PET-evaluated transport of [11C]hydroxyurea across the rat blood-brain barrier--lack of influence of cyclosporin and probenecid. Drug Metab Lett 1(3), 189–194. [DOI] [PubMed] [Google Scholar]
- Texel SJ, Mattson MP, 2011. Impaired adaptive cellular responses to oxidative stress and the pathogenesis of Alzheimer’s disease. Antioxid Redox Signal 14(8), 1519–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eldik LJ, Carrillo MC, Cole PE, Feuerbach D, Greenberg BD, Hendrix JA, Kennedy M, Kozauer N, Margolin RA, Molinuevo JL, Mueller R, Ransohoff RM, Wilcock DM, Bain L, Bales K, 2016. The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimers Dement (N Y) 2(2), 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT, 2006. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1(2), 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincott CM, Abera S, Vunck SA, Tirko N, Choi Y, Titcombe RF, Antoine SO, Tukey DS, DeVito LM, Hofmann F, Hoeffer CA, Ziff EB, 2014. cGMP-dependent protein kinase type II knockout mice exhibit working memory impairments, decreased repetitive behavior, and increased anxiety-like traits. Neurobiol Learn Mem 114, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Chen S, Mao Z, Cadenas E, Brinton RD, 2011. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS One 6(7), e21788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao PJ, Manor U, Petralia RS, Brose RD, Wu RT, Ott C, Wang YX, Charnoff A, Lippincott-Schwartz J, Mattson MP, 2017. Sonic hedgehog pathway activation increases mitochondrial abundance and activity in hippocampal neurons. Mol Biol Cell 28(3), 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wang ZY, Hansson HA, 2003. Visualization of proliferating cells in the adult mammalian brain with the aid of ribonucleotide reductase. Brain Res 977(2), 180–189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene ontology results for the microarray analyses of hippocampal neurons treated with HU for 2, 6, 12, and 24 hours (n = 3 independent samples).
Gene ontology results for the microarray analyses of mouse hippocampi isolated from untreated and HU-treated wild type mice (n = 3).