

Abstract
Kinesin family member 20B (KIF20B, also known as MPHOSPH1) is a kinesin protein that plays a critical role in cytokinesis. Previously, we and others have demonstrated the oncogenic role of KIF20B in several cancers; however, the exact mechanisms underlying its tumorigenic effects remain unclear. Herein, we showed overexpression of KIF20B in human hepatocellular carcinoma (HCC) and reported a negative correlation between KIF20B level and prognosis of patients. Mechanistically, reducing KIF20B blockades mitotic exit of HCC cells at telophase in a spindle assembly checkpoint independent way. Importantly, reducing KIF20B acts synergistically with three microtubule‐associated agents (MTA) to p53‐ or p14ARF‐dependently suppress p53‐wt or p53‐null HCC cells. In addition to taxol, reducing KIF20B also enhanced the toxicity of two chemotherapeutic drugs, hydroxycamptothecin and mitomycin C. In conclusion, we found a novel mechanism in that blocking cytokinesis by KIF20B inhibition increases the efficacy of MTA; our results thus suggested a dual‐mitotic suppression approach against HCC by combining MTA with KIF20B inhibition, which simultaneously blocks mitosis at both metaphase and telophase.
Keywords: cytokinesis defect, hepatocellular carcinoma, KIF20B, metaphase and telophase inhibition, microtubule‐targeting agent resistance
Abbreviations
- CDK
cyclin dependent kinase
- HCC
hepatocellular carcinoma
- IHC
immunohistochemistry
- KIF20B
kinesin family member 20B
- MPHOSPH1
M‐phase phosphoprotein 1
- MTA
microtubule‐targeting agent
- rec‐Ads
recombinant adenoviruses
- SAC
spindle assembly checkpoint
- shKIF20B
shRNAs targeting KIF20B
1. INTRODUCTION
Hepatocellular carcinoma causes 600 000 deaths annually.1 As a result of the lack of effective drug therapy, HCC patients usually have a poor prognosis. Most of the chemotherapeutic drugs, such as taxol, epirubicin, cisplatin, 5‐fluorouracil and etoposide, either individually or in combination, have relatively low efficacy in treating HCC.2 Great efforts have been made to unveil the mechanisms underlying the insensitivity of HCC cells to chemotherapies, such as MTA.3, 4 One main cause that confers MTA resistance to HCC cells is the mitotic checkpoint bypass.5 For example, the regulation of SAC, which controls cell cycle progression during mitosis and synchronizes mitosis by attaching chromosomes to spindle microtubules,6 is critical for taxol‐mediated cell death.7, 8 However, reduced or malfunctioned SAC is commonly seen in cancer cells,9 and such SAC defects usually correlate with MTA resistance.10 Therefore, new approaches targeting alternative checkpoints besides SAC would help treat MTA‐resistant HCC.
Kinesin family member 20B (KIF20B, also known as MPHOSPH1) belongs to the kinesin superfamily (KIF), which shares a highly conserved motor domain that enables its microtubule‐dependent plus‐end motion ability through its ATPase activity.11 KIF20B is a microtubule‐associated protein specifically phosphorylated at M‐phase and plays a critical role in cytokinesis.12 In addition to its physiological roles in mitosis, KIF20B has also been found to play roles in solid tumors such as breast and bladder cancers.13, 14 Recently, we also reported the oncogenic role of KIF20B in HCC.15 However, the mechanism by which reducing KIF20B suppresses HCC remains unresolved.
Herein, we showed that reducing KIF20B suppresses HCC proliferation through blocking cytokinesis in a SAC‐independent way. We designed and tested a dual‐mitotic suppression strategy against HCC by combining MTA with KIF20B blockade. Synergistic antitumor effects were achieved through KIF20B reduction combined with multiple MTA in cultured HCC cells, suggesting the potential of this dual‐mitotic suppression strategy against HCC, which simultaneously blockades metaphase and cytokinesis.
2. MATERIALS AND METHODS
2.1. Compounds
Microtubule‐targeting agents taxol, epothilone B and vincristine, Aurora inhibitor VX‐680, spindle assembly checkpoint inhibitor AZ3146, and cytokinesis inhibitor blebbistatin were from Sigma‐Aldrich (St Louis, MO, USA). Hydroxycamptothecin and mitomycin C were from Selleck Chemicals (Houston, TX, USA).
2.2. Cell lines and tissue specimens
Human HCC cell lines Hep3B, HepG2, and HuH‐7 were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (CBTCC; Shanghai, China) or China Center for Type Culture Collection (CCTCC; Wuhan, China). HEK293 was obtained from Microbix Biosystems (Toronto, Ontario, Canada). Cell lines were grown in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% FBS as previously reported.16, 17 Human samples, including 14 from non‐malignant liver diseases (cirrhosis, choledocholithiasis, hepatapostema, cholangitis and cavernous hemangioma), as well as 50 from HCC and 36 adjacent tissues were obtained from the Union Hospital, Tongji Medical College of Huazhong University of Science and Technology (Wuhan, China). The protocol for acquisition of tissue specimens was approved by the Institutional Review Board of Union Hospital; human sample collection procedures were in accordance with the established guidelines.
2.3. Recombinant adenoviral vectors
Recombinant adenoviruses were generated by respective homologous recombination between the shuttle plasmids and the packaging plasmid pBHGE3 in HEK293 cells. Generation, identification, production, purification, and titration of the recombinant adenovirus were carried out as previously described.18, 19
2.4. Lentiviral vectors
Stably expressing shKIF20B cell lines using the lentiviral system were constructed as previously reported.16
2.5. Immunohistochemical staining
Paraffin‐embedded tissue samples from patients were cut into 5‐μm sections and placed on poly‐lysine‐coated slides. Samples were deparaffinized in xylene and rehydrated using a series of graded alcohol. Antigen retrieval was carried out by heat mediation in citrate buffer. Samples were blocked with 10% goat serum before incubation with primary antibody. Samples were incubated overnight using primary antibodies a humidified container at 4°C. Immunohistochemical staining was carried out with a DAB Horseradish Peroxidase Color Development Kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer's instructions. Intensity of staining was carried out and analyzed using Histo‐score by two individuals through a double‐blind procedure as reported.20
2.6. Cell viability assays
MTT assays were carried out using standard procedures as previously reported.21 For colony formation assays, cells were counted and seeded in six‐well plates in growth medium containing 0.7% agar (2 mL/well) on top of a layer of growth medium containing 1.4% agar (1 mL/well). Growth medium (1 mL) with 10% FBS was added on top of the agar. The cell suspension was plated and cultured in a 37°C incubator for 20 days, then the colonies were fixed with 4% paraformaldehyde, stained with 0.005% (w/v) crystal violet in 25% (v/v) methanol, and the colonies were then pictured and counted.
2.7. Cell synchronization
Cells were treated with 150 nmol/L VX‐680 or 2 nmol/L nocodazole for 16 hours (−16 hours); the adenoviruses were added to the media 8 hours before the completion of synchronization (−8 hours); 8 hours later, fresh VX‐680/nocodazole‐free media with or without taxol/epothilone B/vincristine replaced the media to release the cells from synchronization (0 hours). To reinduce mitotic exit, 2 or 3 hours after release from synchronization (2 hours/3 hours), the cells were retreated with VX‐680.22
2.8. Evaluation for synergistic combination
Cells were seeded into 96‐well plates, and different concentrations of MTA (taxol, epothilone B or vincristine) or Ad‐shKIF20B (adenoviral vector expressing shRNAs targeting KIF20B) in complete medium were added into each well for individual drug concentration with 100 μL of the final volume. At the end of the experiment, cell viability was measured, and the number of cells in each drug treatment condition was compared to the untreated negative control. Half maximal inhibitory concentration (LC50) of taxol and Ad‐shKIF20B were analyzed for synergistic effect by plotting an isobologram. The assay was carried out in triplicate.
2.9. Western blot
Western blots were carried out with standard procedures as previously reported.23 The following primary antibodies were used: rabbit anti‐KIF20B (ab122165; Abcam, Cambridge, UK), mouse anti‐β‐actin (A5316; Sigma‐Aldrich), mouse anti‐p53 (sc‐263; Santa Cruz Biotechnology, Dallas, TX, USA), mouse anti‐p21 (#2946; Cell Signaling Technology, Danvers, MA, USA), mouse anti‐p14ARF (#2407; Cell Signaling Technology) and rabbit anti‐p‐H3 (#53348; Cell Signaling Technology). Secondary antibodies m‐IgGκ BP‐HRP and mouse antirabbit IgG‐HRP were from Santa Cruz Biotechnology.
2.10. In vitro nuclei and microtubule staining
Nuclei and microtubule staining were carried out as previously reported.24 Cells were seeded on glass coverslips, and 24 hours later they were infected with adenovirus at MOI of 1. Cells were then fixed with 4% paraformaldehyde, and costained with Phalloidin Alexa Fluor 555 (Molecular Probes, Eugene, OR, USA) and DAPI (Sigma‐Aldrich). Cells were observed and pictured using a Leica TCS SP2 confocal microscope (Leica Microsystems, Wetzlar, Germany).
2.11. Statistics
All data are expressed as the mean ± SD and were analyzed using independent sample t tests and one‐way analyses of variance (ANOVA) using SPSS Base 10.0. Results were considered statistically significant when P < .05.
3. RESULTS
3.1. Kinesin family member 20B overexpression is correlated with poor prognosis in HCC
Upregulated KIF20B has been reported in some solid tumors, such as breast and bladder cancers.13 Here, we observed a significant upregulation of KIF20B in HCC tissues (n = 50) compared with adjacent (n = 36) or non‐tumor (n = 14) tissues (Figure 1A,B). We further analyzed the data of 336 HCC and 42 non‐tumor patients available in The Cancer Genome Atlas (TCGA) database to investigate whether KIF20B expression correlates with HCC prognosis. Consistent with the IHC results, the mRNA levels of KIF20B were much higher in HCC tissues compared with normal tissues (Figure 1C). Importantly, HCC patients with higher than median KIF20B expression showed significantly shorter overall (P = .036, left panel, Figure 1D) and disease‐free survival duration (P = .022, right panel, Figure 1D), especially in the early period, which has a higher confidence level compared to the late stage. Together, these results suggest upregulated KIF20B in HCC tissues, and its expression level is negatively correlated with the prognosis of patients.
Figure 1.

Overexpression of kinesin family member 20B (KIF20B) in hepatocellular carcinoma (HCC) samples. A, Representative pictures of KIF20B immunohistochemical staining on clinical samples. B, H‐score of KIF20B for different groups; presented as mean + SD. C, mRNA levels of KIF20B in HCC and para‐HCC tissues (data from The Cancer Genome Atlas [TCGA]). D, Overall (left panel) and disease‐free (right panel) survival rates of HCC patients with high KIF20B expression levels (red) and low levels (blue) (the cut‐off for determining high vs low levels of KIF20B is the midpoint, data from TCGA). (*P < .05, ***P < .001)
3.2. Reducing KIF20B sensitizes HCC cells to taxol
Evidence has suggested that some KIF proteins are correlated with taxol resistance of cancer cells.25 To fully address whether reducing KIF20B increases the taxol sensitivity of HCC cells, we applied Ad‐shKIF20B, a recombinant adenoviral vector expressing shRNAs against KIF20B,15 to HCC cell lines. As shown in Figure 2A, Ad‐shKIF20B significantly reduced mRNA levels of KIF20B in HepG2, Hep3B and HuH‐7 cell lines. Significantly enhanced taxol cytotoxicity was observed in all three cell lines receiving Ad‐shKIF20B (Figure 2B,C). Furthermore, soft agar colony formation assay indicated that HCC cells receiving Ad‐shKIF20B/taxol combined treatment showed markedly reduced colony numbers compared with the respective shKIF20B or taxol mono‐treated cells (Figure 2D). Moreover, isobologram analysis suggested that the shKIF20B/taxol combination brings synergistic effects on suppressing the viability of these cell lines (Figure 2E).
Figure 2.

Adenoviral vector expressing small hairpin RNAs targeting kinesin family member 20B (Ad‐shKIF20B) enhances taxol toxicity to hepatocellular carcinoma cells. A, Quantification of KIF20B mRNA levels in HepG2, Hep3B and HuH‐7 cells 48 h after infection. MOI = 1. B, Relative cell viability of HepG2, Hep3B and HuH‐7 cells by MTT assays 72 h after indicated treatments. C, Relative cell viability of HepG2, Hep3B and HuH‐7 cells with indicated treatments by MTT assays. MOI = 1, taxol concentration = 1 μmol/L. B,C, Value of control group was arbitrarily set at 1. Three independent experiments were carried out. D, Colony formation assays with indicated treatments. MOI = 1, taxol concentration = 1 μmol/L. E, MTT assays were carried out after cells received adenoviral vector expressing shRNAs targeting KIF20B (Ad‐shKIF20B) and taxol for 72 h. Standard isobolograms are shown. IC 50 values for each drug are plotted on the axes; the solid line represents the additive effect, whereas the points represent the concentrations of each drug resulting in 50% inhibition of proliferation. Points falling below the line indicate synergism, whereas those above the line indicate antagonism
3.3. Reducing KIF20B suppresses mitosis of HCC cells at telophase
At metaphase/anaphase transition, SAC activation is essential for the efficacy of taxol;7 however, some cancer cells can bypass its surveillance to escape from taxol inhibition.9 To address whether shKIF20B suppresses HCC cells in a SAC‐dependent way, a SAC inhibitor AZ3146 was applied.26 As expected, AZ3146 significantly attenuated the cytotoxicity of taxol to HepG2 cells (Figure 3A). In contrast, the viability of cells receiving Ad‐shKIF20B or Ad‐shKIF20B/taxol combination was unaffected by AZ3146 treatment (Figure 3A), indicating that the suppressive function of shKIF20B is independent of SAC activation.
Figure 3.

Adenoviral vector expressing shRNAs targeting kinesin family member 20B (Ad‐shKIF20B) blocks mitotic exit of HepG2 cells at telophase. A, MTT results 72 h after indicated treatments. MOI = 1, AZ3146 concentration = 1 μmol/L, taxol concentration = 1 μmol/L. Three independent experiments were carried out. B,C, Temporal variation in phosphorylated H3 after synchronization by VX‐680 measured using western blots. D, Representative confocal microscopy images of cells at different phases of mitosis following different treatments. Typical cell morphologies at different mitotic phases are presented in the top row. Blue, DAPI stained nuclei; green, phalloidin stained microfilaments; yellow arrows, cells at cytokinesis; white arrows, cells at pro‐metaphase. Magnification, ×40. E, Percentage of cells arrested at telophase, pro‐metaphase or M‐phase under different treatments (epo B, epothilone B). Results refer to three slides of each group. (*P < .05, **P < .01); ns, not significant
As SAC does not affect shKIF20B‐induced mitotic and proliferation inhibition, and KIF20B plays a critical role in cytokinesis,12 consequently, we investigated at which mitotic phase shKIF20B suppressed HCC cells. First, we synchronized HepG2 cells at M‐phase by VX‐680 and measured the length of mitosis by checking the phosphorylation level of histone H3 (p‐H3), an M‐phase indicator.27 Significantly delayed mitotic exit of cells receiving shKIF20B was observed as shown by decreased p‐H3 levels (12 vs 5 hours, Figure 3B), suggesting a prolonged mitotic arrest induced by KIF20B knockdown. In addition, consistent with a previous report,22 VX‐680 re‐administration at 3 hours released cells from mitotic arrest (shown by decreased p‐H3 levels, Figure 3C). Next, confocal microscopy was applied to examine the cell phase. At 4 hours after release from synchronization, shKIF20B induced a significant increase in the number of cells arrested at telophase (shown as cytokinesis, yellow arrows, Figure 3D,E); this result was similar to the cells receiving blebbistatin, an agent that induces mitotic defects of cytokinesis (Figure 3D,E).28 In contrast, taxol and epothilone B (epo B), the latter is an MTA in clinical trials for breast and ovarian cancers,29 mainly caused arrest at pro‐metaphase/metaphase (shown as pro‐metaphase, white arrows, Figure 3D,E). Combined treatment of Ad‐shKIF20B with either taxol or epo B significantly increased the number of cells with defects in cytokinesis, compared with cells treated with taxol or epo B alone (P < .05, Figure 3D,E). Moreover, consistent with the results shown in Figure 3C, VX‐680 retreatment released mitotic arrest, indicated by the reduced number of cells at M‐phase of groups receiving taxol, epo B, Ad‐shKIF20B, or their combinations (right panel, Figure 3E). Together, these results indicated that reducing KIF20B blockaded mitotic exit at telophase, whereas taxol/epo B mainly suppressed mitosis at metaphase.
3.4. Reduction of KIF20B sensitizes HCC cells to taxol through mitotic arrest and p53/p14ARF
Tumor suppressor p53 plays a critical role in the development of drug resistance in cancer cells, and its accumulation during mitotic arrest triggers apoptosis following mitotic slippage.30 To explore whether the reduced cell viability by shKIF20B/taxol depends on the p53 pathway in p53‐wt cells, a p53‐specific inhibitor pifithrin‐α was used.31 Levels of p53 and its downstream cell cycle inhibitor p21 were upregulated by shKIF20B, whereas pifithrin‐α reversed these upregulations (Figure 4A). MTT assays indicated that pifithrin‐α significantly attenuated the toxicity of shKIF20B/taxol combined treatment on HepG2 cells (Figure 4B). Moreover, VX‐680 treatment, which releases cells from mitotic arrest (Figure 3C,E), also significantly attenuated the inhibitory effects of shKIF20B/taxol on cell viability (Figure 4B). These results suggested that combined shKIF20B/taxol treatment suppressed cell growth in p53‐wt HCC cells through a p53‐ and mitotic arrest‐dependent method.
Figure 4.

Reducing Adenoviral vector expressing small hairpin RNAs targeting kinesin family member 20B (Ad‐shKIF20B) attenuates the proliferation of hepatocellular carcinoma cells in a p53/p14 dependent method. A, Western blots for indicated proteins of HepG2 cells 24 h after receiving adenoviral vector expressing shRNAs targeting KIF20B (Ad‐shKIF20B) or pifithrin‐α. MOI = 1, pifithrin‐α concentration = 400 nmol/L. B, MTT results of HepG2 cells 72 h after indicated treatments. MOI = 1, pifithrin‐α concentration = 400 nmol/L, VX‐680 concentration = 150 nmol/L for 2 h to induce mitotic slippage. Three independent experiments were carried out. C, Western blots for indicated proteins of Hep3B cells 24 h after receiving Ad‐shKIF20B or shp14 transfection. MOI = 1. D, MTT results of Hep3B 72 h after indicated treatments. MOI = 1, VX‐680 concentration = 150 nmol/L for 2 h to induce mitotic slippage. Three independent experiments were carried out. (***P < .001)
In p53‐null and p53‐mutated cancer cells, p14ARF may partially substitute for p53 in inhibiting the proliferation;32 we thus hypothesized that reduction of cell viability by shKIF20B in p53‐null cells may depend on p14ARF. To test this possibility, we knocked down p14ARF in Hep3B cells using a shp14 plasmid (Figure 4C). As expected, the levels of p14ARF and p21 were increased by Ad‐shKIF20B infection and decreased after knockdown of p14ARF by shp14 (Figure 4C). MTT assays showed that p14ARF knockdown attenuated the Ad‐shKIF20B/taxol combination‐induced viability suppression (Figure 4D), suggesting that shKIF20B‐enhanced taxol efficacy may depend on p14ARF. Similarly, in Hep3B cells, release from mitotic arrest also attenuated the combined shKIF20B/taxol treatment‐induced proliferation inhibition, showing a mitotic arrest‐dependent mechanism.
3.5. Microtubule‐targeting agent and KIF20B reduction show synergistic suppressive effects on HCC cells
Taxol and epo B suppress mitosis at metaphase (Figure 3D,E), whereas shKIF20B induces cytokinesis inhibition at telophase. We therefore hypothesized that a strategy simultaneously blocking metaphase and telophase of mitosis may achieve better outcomes. Besides taxol, we studied two additional common MTA (epo B and vincristine) on HCC cell lines. Cell viability assays showed that shKIF20B increased the toxicity of epo B to all three cell lines (Figure 5A), and Ad‐shKIF20B acted synergistically with epo B to suppress proliferation (Figure 5B). Similarly, shKIF20B significantly enhanced the efficacy of vincristine in synergy with all HCC cells (Figure 5C,D). Together, these results indicated that shKIF20B and MTA synergistically inhibit proliferation of HCC cells.
Figure 5.
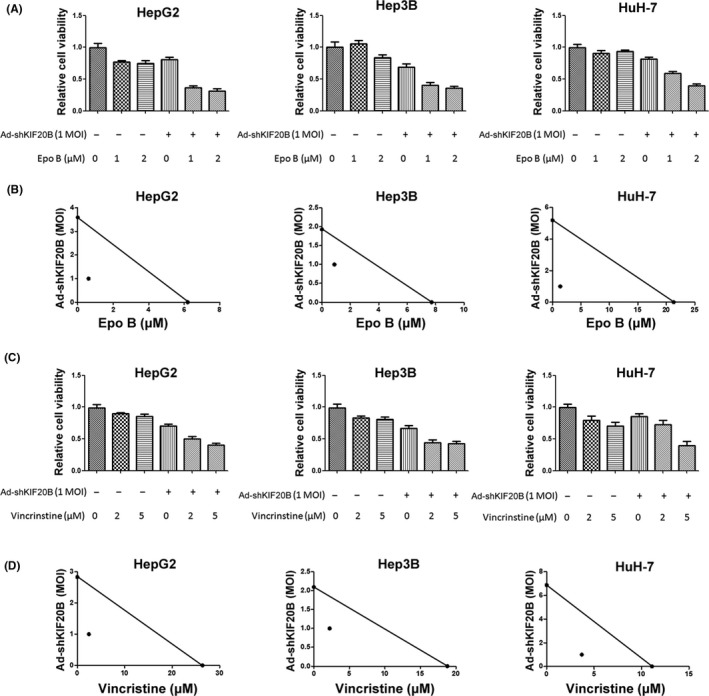
Adenoviral vector expressing shRNAs targeting kinesin family member 20B (Ad‐shKIF20B) enhances taxol toxicity to hepatocellular carcinoma (HCC) cells with a synergistic effect. A, MTT results of three HCC cell lines 72 h after indicated treatments. Three independent experiments were carried out. B, MTT assays were done after cells received Ad‐shKIF20B and epothilone B (epo B) for 72 h. Standard isobolograms are shown. C, MTT results of three HCC cell lines 72 h after indicated treatments. Three independent experiments were carried out. D MTT assays were done after cells receiving Ad‐shKIF20B and vincristine for 72 h. Standard isobolograms are shown
3.6. Reducing KIF20B sensitizes HCC cells to two chemotherapeutic agents
In the clinic, transarterial chemoembolization (TACE) is a first‐line therapeutic option for HCC treatment, and multiple chemotherapeutic drugs have been used in TACE, such as mitomycin and camptothecin.33 To determine whether reducing KIF20B sensitizes HCC cells to these chemotherapeutic drugs, we detected the viability of HepG2, Hep3B and HuH‐7 cells treated with Ad‐shKIF20B and hydroxycamptothecin or mitomycin C. Results suggested that shKIF20B increased the toxicity of hydroxycamptothecin or mitomycin C (Figure 6A). For example, at 10 μM concentration of mitomycin, the inhibitory ratio is 23.3% vs 52.8% in HepG2 (mitomycin mono‐treatment vs mitomycin/shKIF20B combined treatment, similarly hereinafter), 41.4% vs 58.2% in Hep3B, and 44.7% vs 61.9% in HuH‐7 cells (Figure 6A). Furthermore, by using a lentiviral vector system that we previously used,16 we constructed two HCC cell lines that stably expressed shKIF20B (HepG2‐KIF20BKD and Hep3B‐KIF20BKD, Figure 6B). Both KIF20B knockdown cell lines were much more sensitive to hydroxycamptothecin or mitomycin C than the control cells (Figure 6C), suggesting that reducing KIF20B also elevates the cytotoxicity of these TACE chemotherapeutic agents.
Figure 6.

Reducing Adenoviral vector expressing small hairpin RNAs targeting kinesin family member 20B (Ad‐shKIF20B) enhanced the toxicity of hydroxycamptothecin (HCPT) or mitomycin C (Mito) to hepatocellular carcinoma (HCC) cells. A, MTT results of three HCC cell lines at 72 h after indicated treatments. Three independent experiments were carried out. B, Western blots for indicated proteins in stable KIF20B knockdown of HepG2 and Hep3B cells. C, MTT results of three HCC cell lines at 72 h after indicated treatments. Three independent experiments were carried out. KD, KIF20B stably knocked down
4. DISCUSSION
Many studies have indicated SAC as a critical checkpoint that strictly surveils the process of mitosis and controls the metaphase/anaphase transition.34, 35 Including taxol, many MTAs cause cell death via activating SAC, which induces mitotic arrest and subsequent apoptosis (Figure 7).36 However, some cancer cells can bypass its surveillance by reducing or inactivating SAC and therefore escape from mitotic arrest to become insensitive to SAC‐activating MTA.37 To effectively blockade mitosis of MTA‐resistant cancer cells, targeting additional checkpoints besides SAC may achieve beneficial outcomes. Here, we simultaneously blockaded metaphase and telophase of HCC cells by MTA/shKIF20B dual treatment, and the results indicated potent and synergistic antitumor effects. We also showed a p53/p14ARF dependent method through which KIF20B knockdown induces mitotic arrest in HCC cells; further test with additional cell lines will strengthen this conclusion. Moreover, we also showed that transiently or stably reducing KIF20B (using adenoviral or lentiviral vectors, respectively) significantly increased the efficacy of hydroxycamptothecin and mitomycin C, two antimitotic drugs commonly used in TACE against HCC, thus suggesting a plausible choice for HCC therapy.
Figure 7.

Potential mechanism for dual‐mitotic suppression strategy for treating hepatocellular carcinoma (HCC). Reduction of kinesin family member 20B (KIF20B) induces cytokinesis defects, which triggers mitotic arrest and leads to growth arrest of HCC cells. Additionally, taxol and other microtubule‐associated agents (MTA) induce spindle assembly checkpoint (SAC) to limit cancer cells from entering anaphase. However, some cancer cells bypass SAC surveillance to become MTA resistant. Thus, reducing KIF20B, which blocks cytokinesis, together with MTA can act synergistically by suppressing mitosis at both telophase and metaphase. Epo B, epothilone B
Previous studies reported that some overexpressed KIF proteins represented increased MTA resistance of cancer cells. For example, upregulated microtubule‐binding motor proteins KIFC3 and MCAK (KIF2C) are correlated with MTA resistance in breast cancer.25, 38, 39 In addition to KIF, some agents with cytokinesis inhibition ability, such as mulberry water extract, have been shown to increase the efficacy of taxol to bladder cancer with a synergistic effect.40 However, it is not clear whether a cytokinesis defect is responsible for the synergistic antitumor effect. Herein, we demonstrate that KIF20B reduction increases HCC sensitivity to multiple MTA by blocking cytokinesis. Moreover, our work also raises the possibility that besides KIF20B, additional cytokinesis‐associated proteins may also serve as potential therapeutic targets. Future studies on the roles and molecular mechanisms of these molecules in MTA‐resistant cancers will be interesting.
Cell cycle is strictly and precisely regulated to assure that all events are finely orchestrated throughout the cycle progression. A number of regulators that function at different cell cycle events, in addition to mitotic checkpoints such as SAC and cytokinesis, have been indicated as potential targets for cancer therapy. For instance, therapies targeting CDK1/2, which abrogates mitotic entry and blockades G2 phase, have entered clinical trials for acute leukemia and colon cancer treatments.41, 42 As such regulators are activated in a broad range of cancers and control different stages of cell cycle, we speculate that triggering different checkpoints, which simultaneously blockades multiple cell cycle events, may achieve potent efficacy against MTA‐resistant cancers. In the present study, we tested the combinatorial HCC therapy as proof of concept, and it will be interesting to apply this dual suppressive strategy to other MTA‐resistant cancers, such as colon and breast cancers.11, 41, 43
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
This work was supported by the Natural Science Foundation of China (81573461, 31500941, 31471208, 31671195, 31500706 and 81703552), the Natural Science Foundation of Hubei Province (2014CFA021 and 2017CFB651), the Academic Frontier Youth Team Project of HUST, Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College (HUST) and the Natural Science Foundation of the Self‐dependent Innovation of HUST (2016YXMS144). The authors thank the Analytical and Testing Center of HUST, and the Analytical & Testing Facility of College of Life Sciences, Wuhan University, for technical assistance.
Liu X, Li Y, Zhang X, et al. Inhibition of kinesin family member 20B sensitizes hepatocellular carcinoma cell to microtubule‐targeting agents by blocking cytokinesis. Cancer Sci. 2018;109:3450–3460. 10.1111/cas.13794
Xinran Liu and Yangkai Li contributed equally to this work.
REFERENCES
- 1. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245‐1255. [DOI] [PubMed] [Google Scholar]
- 2. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63:844‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuo TC, Lu HP, Chao CC. The tyrosine kinase inhibitor sorafenib sensitizes hepatocellular carcinoma cells to taxol by suppressing the HURP protein. Biochem Pharmacol. 2011;82:184‐194. [DOI] [PubMed] [Google Scholar]
- 4. Minero VG, De Stefanis D, Costelli P, Baccino FM, Bonelli G. In vitro and in vivo conditional sensitization of hepatocellular carcinoma cells to TNF‐induced apoptosis by taxol. Cell Cycle. 2015;14:1090‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi M, Min YH, Pyo J, Lee CW, Jang CY, Kim JE. TC Mps1 12, a novel Mps1 inhibitor, suppresses the growth of hepatocellular carcinoma cells via the accumulation of chromosomal instability. Br J Pharmacol. 2017;174:1810‐1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shah JV, Cleveland DW. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell. 2000;103:997‐1000. [DOI] [PubMed] [Google Scholar]
- 7. Giovinazzi S, Bellapu D, Morozov VM, Ishov AM. Targeting mitotic exit with hyperthermia or APC/C inhibition to increase paclitaxel efficacy. Cell Cycle. 2013;12:2598‐2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gascoigne KE, Taylor SS. Cancer cells display profound intra‐ and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111‐122. [DOI] [PubMed] [Google Scholar]
- 9. McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, microtubules and chemoresistant breast cancer. Biochem Biophys Acta. 2008;1785:96‐132. [DOI] [PubMed] [Google Scholar]
- 10. Orth JD, Tang Y, Shi J, et al. Quantitative live imaging of cancer and normal cells treated with Kinesin‐5 inhibitors indicates significant differences in phenotypic responses and cell fate. Mol Cancer Ther. 2008;7:3480‐3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X, Gong H, Huang K. Oncogenic role of kinesin proteins and targeting kinesin therapy. Cancer Sci. 2013;104:651‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Janisch KM, McNeely KC, Dardick JM, Lim SH, Dwyer ND. Kinesin‐6 KIF20B is required for efficient cytokinetic furrowing and timely abscission in human cells. Mol Biol Cell. 2018;29:166‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanehira M, Katagiri T, Shimo A, et al. Oncogenic role of MPHOSPH1, a cancer‐testis antigen specific to human bladder cancer. Can Res. 2007;67:3276‐3285. [DOI] [PubMed] [Google Scholar]
- 14. Liu XR, Cai Y, Cao X, et al. A new oncolytic adenoviral vector carrying dual tumour suppressor genes shows potent anti‐tumour effect. J Cell Mol Med. 2012;16:1298‐1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu X, Zhou Y, Liu X, et al. MPHOSPH1: a potential therapeutic target for hepatocellular carcinoma. Can Res. 2014;74:6623‐6634. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y, Guo X, Yan W, et al. ANGPTL8 negatively regulates NF‐kappaB activation by facilitating selective autophagic degradation of IKKgamma. Nat Commun. 2017;8:2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen H, Wang L, Wang W, et al. ELABELA and an ELABELA Fragment Protect against AKI. J Am Soc Nephrol. 2017;28:2694‐2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cai Y, Liu X, Huang W, Zhang K, Liu XY. Synergistic antitumor effect of TRAIL and IL‐24 with complete eradication of hepatoma in the CTGVT‐DG strategy. Acta Biochim Biophys Sin. 2012;44:535‐543. [DOI] [PubMed] [Google Scholar]
- 19. Liu X, Cao X, Wei R, et al. Gene‐viro‐therapy targeting liver cancer by a dual‐regulated oncolytic adenoviral vector harboring IL‐24 and TRAIL. Cancer Gene Ther. 2012;19:49‐57. [DOI] [PubMed] [Google Scholar]
- 20. Budwit‐Novotny DA, McCarty KS, Cox EB, et al. Immunohistochemical analyses of estrogen receptor in endometrial adenocarcinoma using a monoclonal antibody. Can Res. 1986;46:5419‐5425. [PubMed] [Google Scholar]
- 21. Liu S, Sun Y, Jiang M, et al. Glyceraldehyde‐3‐phosphate dehydrogenase promotes liver tumorigenesis by modulating phosphoglycerate dehydrogenase. Hepatology. 2017;66:631‐645. [DOI] [PubMed] [Google Scholar]
- 22. Zhu Y, Zhou Y, Shi J. Post‐slippage multinucleation renders cytotoxic variation in anti‐mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle. 2014;13:1756‐1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol. 2012;56:900‐907. [DOI] [PubMed] [Google Scholar]
- 24. Liu X, Li Y, Meng L, et al. Reducing protein regulator of cytokinesis 1 as a prospective therapy for hepatocellular carcinoma. Cell Death Dis. 2018;9:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan MH, De S, Bebek G, et al. Specific kinesin expression profiles associated with taxane resistance in basal‐like breast cancer. Breast Cancer Res Treat. 2012;131:849‐858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hewitt L, Tighe A, Santaguida S, et al. Sustained Mps1 activity is required in mitosis to recruit O‐Mad2 to the Mad1‐C‐Mad2 core complex. J Cell Biol. 2010;190:25‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell. 2012;23:567‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Straight AF, Cheung A, Limouze J, et al. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003;299:1743‐1747. [DOI] [PubMed] [Google Scholar]
- 29. Perez EA, Patel T, Moreno‐Aspitia A. Efficacy of ixabepilone in ER/PR/HER2‐negative (triple‐negative) breast cancer. Breast Cancer Res Treat. 2010;121:261‐271. [DOI] [PubMed] [Google Scholar]
- 30. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701‐713. [DOI] [PubMed] [Google Scholar]
- 31. Derdak Z, Villegas KA, Harb R, Wu AM, Sousa A, Wands JR. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non‐alcoholic fatty liver disease. J Hepatol. 2013;58:785‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Forys JT, Kuzmicki CE, Saporita AJ, Winkeler CL, Maggi LB Jr, Weber JD. ARF and p53 coordinate tumor suppression of an oncogenic IFN‐beta‐STAT1‐ISG15 signaling axis. Cell Rep. 2014;7:514‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vogl TJ, Mohamed SA, Albrecht MH, et al. Transarterial chemoembolization in pancreatic adenocarcinoma with liver metastases: MR‐based tumor response evaluation, apparent diffusion coefficient (ADC) patterns, and survival rates. Pancreatology. 2018;18:94‐99. [DOI] [PubMed] [Google Scholar]
- 34. Courtheoux T, Diallo A, Damodaran AP, Reboutier D, Watrin E, Prigent C. Aurora A kinase activity is required to maintain the spindle assembly checkpoint active during pro‐metaphase. J Cell Sci. 2018. 10.1242/jcs.191353. [DOI] [PubMed] [Google Scholar]
- 35. Yu L, Shang ZF, Abdisalaam S, et al. Tumor suppressor protein DAB2IP participates in chromosomal stability maintenance through activating spindle assembly checkpoint and stabilizing kinetochore‐microtubule attachments. Nucleic Acids Res. 2016;44:8842‐8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Silva PM, Ribeiro N, Lima RT, et al. Suppression of spindly delays mitotic exit and exacerbates cell death response of cancer cells treated with low doses of paclitaxel. Cancer Lett. 2017;394:33‐42. [DOI] [PubMed] [Google Scholar]
- 37. Bargiela‐Iparraguirre J, Prado‐Marchal L, Pajuelo‐Lozano N, Jimenez B, Perona R, Sanchez‐Perez I. Mad2 and BubR1 modulates tumourigenesis and paclitaxel response in MKN45 gastric cancer cells. Cell Cycle. 2014;13:3590‐3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De S, Cipriano R, Jackson MW, Stark GR. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Can Res. 2009;69:8035‐8042. [DOI] [PubMed] [Google Scholar]
- 39. Ganguly A, Yang H, Cabral F. Overexpression of mitotic centromere‐associated Kinesin stimulates microtubule detachment and confers resistance to paclitaxel. Mol Cancer Ther. 2011;10:929‐937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen NC, Chyau CC, Lee YJ, Tseng HC, Chou FP. Promotion of mitotic catastrophe via activation of PTEN by paclitaxel with supplement of mulberry water extract in bladder cancer cells. Sci Rep. 2016;6:20417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dudgeon C, Wang P, Sun X, et al. PUMA induction by FoxO3a mediates the anticancer activities of the broad‐range kinase inhibitor UCN‐01. Mol Cancer Ther. 2010;9:2893‐2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karp JE, Smith BD, Resar LS, et al. Phase 1 and pharmacokinetic study of bolus‐infusion flavopiridol followed by cytosine arabinoside and mitoxantrone for acute leukemias. Blood. 2011;117:3302‐3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rath O, Kozielski F. Kinesins and cancer. Nat Rev Cancer. 2012;12:527‐539. [DOI] [PubMed] [Google Scholar]