

Abstract
Pathological observations show that cancer cells frequently invade the surrounding stroma in collective groups rather than through single cell migration. Here, we studied the role of the actin‐binding protein Girdin, a specific regulator of collective migration of neuroblasts in the brain, in collective cancer cell migration. We found that Girdin was essential for the collective migration of the skin cancer cell line A431 on collagen gels as well as their fibroblast‐led collective invasion in an organotypic culture model. We provide evidence that Girdin binds to β‐catenin that plays important roles in the Wnt signaling pathway and in E‐cadherin‐mediated cell‐cell adhesion. Girdin‐depleted cells displayed scattering and impaired E‐cadherin‐specific cell‐cell adhesion. Importantly, Girdin depletion led to impaired cytoskeletal association of the β‐catenin complex, which was accompanied by changes in the supracellular actin cytoskeletal organization of cancer cell cohorts on collagen gels. Although the underlying mechanism is unclear, this observation is consistent with the established role of the actin cytoskeletal system and cell‐cell adhesion in the collective behavior of cells. Finally, we showed the correlation of the expression of Girdin with that of the components of the E‐cadherin complex and the differentiation of human skin cancer. Collectively, our results suggest that Girdin is an important modulator of the collective behavior of cancer cells.
Keywords: actin cytoskeleton, cell adhesion, collective invasion, collective migration, Girdin
1. INTRODUCTION
Years of study have identified the general mechanisms of cancer invasion and metastasis. These processes are mostly mediated by genomic and epigenetic alterations and dysregulated cell signaling that activates cytoskeletal organization.1 A classical view of cancer invasion is that cancer cells undergo a phenotypic change called the epithelial‐mesenchymal transition (EMT) to downregulate cell‐cell adhesion molecules such as E‐cadherin and gain mesenchymal morphology and motility.2, 3, 4 Indeed, many studies that used single cells in culture have extended our knowledge of the mechanisms of cancer invasion, including those that regulate the EMT and changes in cytoskeletal reorganization.5
In contrast, pathological observations of tissue sections from cancer patients have long suggested that most of the cancer cells from malignant epithelial tumors form variable sized groups that collectively invade surrounding tissues.6, 7, 8 This is most evident in squamous cell carcinomas but holds as well in adenocarcinomas and non‐epithelial malignancies, such as melanoma.7 Pancreatic ductal adenocarcinoma, which is one of the most devastating invasive cancers, also keeps a moderately differentiated morphology to form glands and groups of cells that invade the stroma.9 Notably, cancer cells that invade lymphatic vessels also form groups.8 These observations question whether the EMT program is always required for the intravasation of cancer cells into those vessels and subsequent metastasis to distant sites.7, 8
Recent studies have begun to reveal the mechanisms of collective invasion of cancer by using in vitro culture models as well as studies of collective migration of cells during embryonic development.6, 7, 10, 11 These include the contact inhibition of locomotion (CIL) that keeps the integrity of cell‐cell contact and spatially regulated trafficking of cell adhesion proteins such as N‐cadherin.12, 13, 14, 15, 16, 17 We previously reported that the collective movement of cancer cell groups requires the expression of integrin β1 by the leading cells but not the following cells.18 Despite this progress, our knowledge of the mechanisms of collective invasion of cancer is far from complete. The primary problems are due to the difficulty of understanding the regulators of “supracellular” cytoskeletal organization in cell groups, the complex interactions between cancer cells and the stroma and the lack of identification of proteins that are specifically involved in the collective behavior of cells.6, 19
Since the identification of the actin‐binding hub protein Girdin (also known as Gα‐interacting vesicle‐associated protein; GIV), we have been particularly interested in its function in neural development, angiogenesis, and cancer progression.20, 21, 22, 23, 24, 25, 26 We reported that conventional Girdin knockout mice showed severe deficiency in the collective movement (termed “chain migration”) of neuroblasts born in the subventricular zone of the lateral ventricle of postnatal and adult brains.8, 27, 28 The data indicated specific involvement of Girdin in the collective behavior of cells, which is partly explained by the regulation of actin remodeling and cell polarity by Girdin.20, 29, 30 However, the precise manner in which Girdin fine‐tunes and maintains the integrity of cell‐cell adhesion to allow for cell rearrangements that drive collective cell migration is unclear. A recent report on Drosophila embryogenesis showed that a fly orthologue of Girdin coordinates collective epithelial migration by promoting the anchorage of the cadherin‐catenin complex to the cytoskeleton, suggesting a conserved role of Girdin between species.31
We and others have previously shown that Girdin is expressed by multiple types of cancers, where its expression correlated with cancer progression.22, 32, 33, 34, 35 In this study, we showed a role of Girdin in the collective invasion of skin cancer cells, where it interacts with β‐catenin, a component of the E‐cadherin complex.36 Our data showed that Girdin is indispensable for stable cell‐cell interaction, supracellular cytoskeletal organization, and the collective migration of cancer. Finally, we also examined the clinical relevance of Girdin expression in the progression of human skin cancer.
2. MATERIALS AND METHODS
2.1. Cell culture and time‐lapse imaging
The human cancer cell lines A431 and HeLa, the Madin‐Darby canine kidney epithelial cell line MDCK, and the human embryonic kidney epithelial cell line 293FT were cultured in DMEM supplemented with 10% FBS (Thermo Fisher Scientific, San Jose, CA, USA). The human colorectal cancer cell line DLD1 was cultured in RPMI‐1640 (Thermo Fisher Scientific) supplemented with 10% FBS. Cell line authentication was assessed using a short tandem repeat (STR) DNA profiling method (BEX Co., Ltd, Tokyo, Japan), and Mycoplasma contamination was tested by staining with DAPI every 3 months. Immortalized cancer‐associated fibroblasts (CAFs) that were established from primary CAFs of human vulvar cancer and A431 cells stably expressing E‐cadherin‐Ruby were provided by Takuya Kato (The Francis Crick Institute, London, UK) and maintained in DMEM supplemented with 10% FBS. For time‐lapse imaging of cells, we prepared a 1.6 mg/mL collagen type I gel with Cellmatrix type I‐P (Nitta Gelatin, Osaka, Japan) in a 6‐well plate as described previously,37, 38 followed by seeding A431 cells and time‐lapse imaging with an IncuCyte Zoom microscope (×20 objective lens; Essen Bioscience, Ann Arbor, MI, USA).
For the analysis of the trajectories of cells, single solitary cells (Figure 1A) or single leading cells of the clusters (≥5 cells) of control and Girdin‐depleted cells (Figure 1C‐G) were randomly chosen by an investigator blinded to the groups and manually tracked using Manual Tracking plugin for ImageJ 1.52a software (US NIH, Bethesda, MD, USA). The data were exported to a Microsoft Excel (Microsoft, Redmond, WA, USA) spreadsheet for analysis. Cell clusters that became <5 cells by cell detachment during the observational period were excluded from the analysis.
Figure 1.

Girdin regulates the collective migration of A431 cells on collagen gels. A, The behavior of single cells on collagen gels is different from that on conventional plastic dishes. The directionality of migrating single cells was calculated as the ratio (d/D), that is, the distance between the starting and ending points (d) divided by the actual trajectory (D). Ten and five dishes were evaluated for the collagen gel group and plastic dish group, respectively, and 25 cells in each dish were manually tracked, followed by quantification. B, Representative images of A431 cell groups cultured on plastic dishes (upper panel) and a collagen gel (lower panel). Note that the cell groups (dotted circles) undergo proliferation without movement on plastic dishes, whereas those seeded on the collagen gel collectively migrate with directionality. Arrows indicate the direction of the movement of the cell group. See also Movies S1 and S2. C, Schematic illustration showing the measurement of collective migration of A431 cells cultured on a collagen gel. We focused on one single cell on the edge of migrating cell groups and tracked its trajectory by tracing the nuclear centroid. D, shRNA‐mediated depletion of Girdin in A431 cells. MW, molecular weight. E‐G, Representative images of control and Girdin‐depleted A431 cell groups cultured on a collagen gel. Time interval between each panel is 2 hours. Note that the control cell group underwent collective directional migration, whereas the Girdin‐depleted cells tended to remain in place. Shown in (F) are the representative paths of the migration of control shRNA‐ (red) and Girdin shRNA (1; green)‐transduced cell groups, as determined by tracing the nuclear centroid over a period of 5 hours (n = 25 for each group). The directionality of migrating cell groups was quantified and shown in G (n = 50 for each group)
2.2. Plasmids, antibodies, and western blot analysis
The cDNAs for human β‐catenin and α‐catenin were generously provided by Frank Costantini (Columbia University, New York, NY, USA) and Takashi Watanabe (Nagoya University, Nagoya, Japan), respectively. The isolation of human Girdin as well as the subcloning of Girdin domains was described previously.24 The construction of plasmids encoding GFP‐Girdin fragments has been described.24 cDNA fragments encoding the fragments of β‐catenin were inserted into the pEF‐GST‐BOS vector. For the production and purification of recombinant proteins in the Escherichia coli expression system, α‐ and β‐catenin cDNAs were inserted into the pGEX‐4T‐2 vector.
The following antibodies were used for western blotting and immunofluorescent studies: GFP (598; MBL, Nagoya, Japan); GST (sc‐459; Santa Cruz Biotechnology, Santa Cruz, CA, USA); β‐actin (A5316; Cell Signaling Technology, Beverly, MA, USA); Girdin (AF5345; R&D Systems, Minneapolis, MN, USA); β‐catenin (610153) and E‐cadherin (610181; BD Biosciences, San Jose, CA, USA); α‐catenin (ALX‐804‐101‐C100; Enzo Life Sciences, Farmingdale, NY, USA); epidermal growth factor receptor (Ab‐5, clone H11; Thermo Fisher Scientific); c‐jun (9156; Cell Signaling Technology); vimentin (M0725, clone V9; Dako, Glostrup, Denmark); and heat shock protein 70 (sc‐24; Santa Cruz Biotechnology).
For western blot analysis, cells were treated with lysis buffer containing 50 mmol/L Tris‐HCl (pH 7.4), 150 mmol/L NaCl, and 0.2% NP40 supplemented with Complete Protease Inhibitor and PhosSTOP Phosphatase Inhibitor cocktails (Roche, Mannheim, Germany). Lysates were clarified by centrifugation at 12 000 g for 10 minutes at 4°C, followed by the addition of SDS sample buffer (10 mmol/L Tris‐HCl, 2% SDS, 2 mmol/L EDTA, 0.02% bromophenol blue, and 6% glycerol; pH 6.8) and separation by SDS‐PAGE. Proteins were transferred to nitrocellulose membranes, blocked in 5% milk in PBS containing 0.05% Tween‐20, incubated with primary antibodies, and detected by HRP‐conjugated secondary antibodies (Dako, Denmark). All of the experiments were replicated at least twice, including preliminary experiments. We showed representative data from the repeated experiments.
2.3. RNA interference
Target sequences for shRNA‐mediated depletion of Girdin were described.24 A set of single‐stranded oligonucleotides encoding the Girdin target sequences and their complements were synthesized as follows (only the sense sequence is shown): human Girdin shRNA (1), 5′‐GGAACAAACAAGATTAGAA‐3′ (nucleotides 3837‐3855); human Girdin shRNA (7), 5′‐GAAGGAGAGGCAACTGGAT‐3′ (nucleotides 4166‐4184). The oligonucleotide pair was annealed and inserted into the pSIREN‐RetroQ retroviral shRNA expression vector (Clontech, Palo Alto, CA, USA). To produce retroviral supernatants, GP2‐293 packaging cells were transfected with the pVSV‐G (vesicular stomatitis virus G protein) vector and either control or Girdin shRNA‐containing pSIREN‐RetroQ vector using Lipofectamine 2000 reagent (Thermo Fisher Scientific). The medium was replaced 24 hours later, and virus‐containing supernatants were harvested 48 hours post‐transfection and used for infection of HeLa and A431 cells.
The siRNA‐mediated depletion of Girdin was carried out as previously described.20 The siRNAs for E‐cadherin and α‐ and β‐catenins were purchased from Qiagen (Hilden, Germany). The targeted sequences that effectively mediated the silencing of the expressions of the indicated genes are as follows (only sense sequences are shown): Girdin‐1, 5′‐AAGAAGGCTTAGGCAGGCAGGAATT‐3′; Girdin‐2, 5′‐AACCAGGTCATGCTCCAAATT‐3′; E‐cadherin, 5′‐GAATCTATCATTTTGAAGCCA‐3′; α‐catenin, 5′‐AAGTGGATAAGCTGAACATTA‐3′; β‐catenin, 5′‐CTCGGGATGTTCACAACCGAA‐3′. Negative control siRNA (AllStars Negative Control siRNA) was purchased from Qiagen (Hilden, Germany).
2.4. Cell dissociation assay
We followed a standard protocol previously described that measures the strength of cell‐cell adhesion.39, 40 Confluent cultured HeLa or MDCK cells (4 × 106 cells per 6‐cm dish) were treated with 0.01% trypsin in HCMF (10 mmol/L HEPES, 140 mmol/L NaCl, 5 mmol/L NaOH, 5 mmol/L KCl, 3.5 mmol/L Na2HPO4·7H2O, and 5.55 mmol/L glucose; pH 7.4) supplemented with either 0.1 mmol/L CaCl2 (TC treatment) or HCMF supplemented with 1 mmol/L EDTA (pH 7.5; TE treatment) for 30 minutes at 37°C, followed by dissociation by pipetting 10 times. The numbers of cell particles that included cell clusters and single cells were counted after dissociating A431 and HeLa cells in the TC and TE treatment conditions. The extent of cell dissociation was represented by the index TC/TE, where TC and TE are the total particle numbers after the TC and TE treatment, respectively.
2.5. Cell fractionation
The isolation of nuclear, membrane, and cytoskeletal fractions of A431 cells was done using an S‐PEK cell fractionation kit (539790; Merck, Kenilworth, NJ, USA), which is based on the different solubility of subcellular compartments in proprietary detergents, following the manufacturer's instructions.
2.6. Organotypic culture model
The organotypic cultivation of A431 and CAF was carried out as described elsewhere.41 Immortalized CAFs derived from a human vulvar cancer (5 × 105) were embedded in a mixture of type I collagen and Matrigel (BD Biosciences), yielding a final collagen concentration of 4 mg/mL and a final Matrigel concentration of 2 mg/mL. The gel was incubated at 37°C for 30 minutes in 24‐well plates, on top of which A431 cells (5 × 105) were plated in a serum‐free medium for 24 hours. Gels were then mounted on 6‐well chambers and fed from underneath with a complete medium. After 10 days, the cultures were fixed with 4% paraformaldehyde plus 0.25% glutaraldehyde in PBS, followed by paraffin embedding, sectioning, and H&E staining or immunohistochemistry.
2.7. Immunohistochemical staining of mouse and human tissues
We followed a standard protocol for immunohistochemical staining of mouse brain and human skin cancer tissues. Antigen retrieval was carried out by microwave treatment in antigen retrieval buffer (pH 6 or pH 9; Dako) at 95°C for 10 minutes. Human skin cancer tissues were obtained with informed patient consent at the time of surgery in Nagoya University Hospital (Nagoya, Japan). We also used tissue arrays of human skin cancers and matched normal adjacent tissues that were purchased from US Biomax (SK802b; Rockville, MD, USA). To evaluate the expression levels of Girdin and the components of the E‐cadherin/catenin complex, cases with total scores (the sum of intensity and proportion scores) of more than 3 were considered positive. The study was carried out in accordance with the Helsinki Declaration for Human Research and approved by the Ethics Committee of Nagoya University Graduate School of Medicine (protocol number 2017‐0127).
2.8. Data analysis
All statistical analyses were undertaken using GraphPad Prism 6 software (GraphPad, San Diego, CA, USA). Data are presented as the means ± SD. Statistical significance was evaluated with Student's t test. The χ²‐test was used to analyze correlations between Girdin expression and clinicopathological parameters. P values <.05 were considered statistically significant.
3. RESULTS
3.1. Significance of Girdin in the collective migration of cancer cells on collagen gels
To address the involvement of Girdin in the collective migration of cancer cells, we first undertook experiments to visualize collective migration in vitro. Our previous studies reported that a highly invasive skin cancer cell line (A431) seeded on a collagen gel initiated collective migration that was different from that observed when the cell line was plated on plastic dishes.37, 38, 42 We reproducibly observed that the behavior of A431 single cells and cell groups on plastic dishes was significantly different from that on collagen gels (Figure 1A,B). We found that the distance between start and end points (d), but not the actual trajectory (D), of single A431 cells on collagen gels was statistically longer than those plated on plastic dishes. Furthermore, the directionality (persistency) of migrating single cells, which was calculated as the ratio of d to D (d/D), was much higher in cells on collagen gels than those on plastic dishes (Figure 1A). This was also the case when we observed the behavior of A431 cancer cell cohorts (Figure 1B, Movies S1, S2). In contrast to cell groups on plastic dishes that proliferated in place, cells on the collagen gel dynamically moved with directionality.
We next depleted Girdin with shRNA to determine the impact on the collective behavior of A431 cells seeded on collagen gels (Figure 1C‐G). We focused on leading cells located at the front of the collectively migrating cell groups (≥5 cells) and calculated directionality indices (Figure 1C). The results showed that, in contrast to control cell groups collectively migrating with high persistency, Girdin‐depleted cells were not migratory and tended to stay in place (Figure 1D‐G, Movies [Link], [Link], [Link]). We found that d and d/D, rather than D, were affected by Girdin depletion in the collective migration of A431 cell groups (Figure 1G). It was noted that the number of cell groups was affected by Girdin depletion over the early part of the observation period but later it did not depend on Girdin (Figure S1A,B). Girdin depletion also had a modest effect on cell proliferation of A431 cells (Figure S1C). Taken together, although not conclusive, these data suggested that Girdin plays an essential role in collective cancer cell migration.
3.2. Identification of β‐catenin as a Girdin‐interacting protein
To examine the mechanism by which Girdin mediated collective cell migration, we exploited a mass spectrometric shotgun approach to identify Girdin immunocomplexes isolated by tandem affinity purification from the lysate of HeLa cervical cancer cells (Figure S2A). Among the identified proteins, we focused on β‐catenin, a critical regulator of the Wnt signaling pathway and cell‐cell adhesion that was reportedly involved in the function of Girdin in Drosophila.31, 36
Immunoprecipitation (IP) experiments showed that β‐catenin was co‐immunoprecipitated by Girdin antibody in several types of cancer cells or immortalized epithelial cells including A431, HeLa, DLD1 (colon cancer), and MDCK (renal epithelium) cells (Figures 2A, S2B‐D). α‐Catenin was also co‐immunoprecipitated with Girdin, leading to the speculation that Girdin, α‐catenin, β‐catenin, and E‐cadherin might form a complex in these cells. The interaction was also confirmed by a reciprocal IP test that showed that β‐catenin immunoprecipitates contained Girdin, α‐catenin, and E‐cadherin in A431 cells (Figure 2B).
Figure 2.
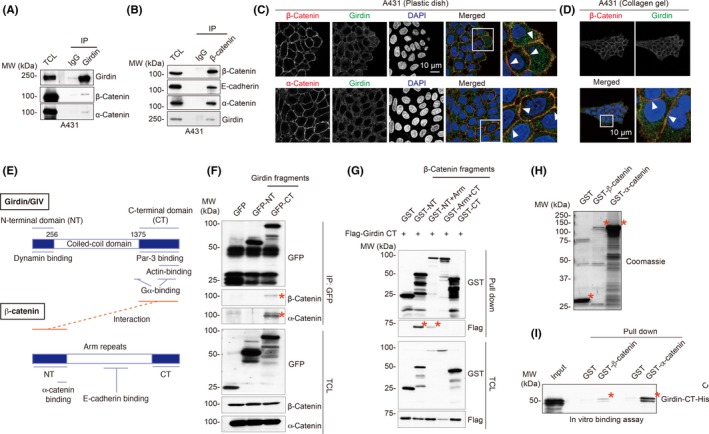
Interaction of Girdin with the β‐catenin complex. A, B, Immunoprecipitation (IP) with anti‐Girdin antibody showed that Girdin interacts with β‐ and α‐catenins in A431 cells (A). Interaction between Girdin and catenins was also shown by reciprocal IP with anti‐β‐catenin antibody (B). TCL, total cell lysates. C, D, Immunofluorescence staining showed the colocalization of Girdin (green) with α‐ and β‐catenins (red) in A431 cells seeded on plastic dishes (C) and collagen gels (D). E, Domain structures and interacting proteins of human Girdin and β‐catenin. Fragments and domains of Girdin and β‐catenin used in the study are shown. The domains responsible for the interaction are shown in red. F, Mapping of interacting domains of Girdin and β‐catenin. β‐Catenin‐binding site maps to the C‐terminal (CT) domain of Girdin. Lysates from 293FT cells transfected with the Girdin fragments fused with GFP were immunoprecipitated with anti‐GFP antibody, followed by western blot analyses using β‐ and α‐catenin antibodies. Bound catenins are indicated by asterisks. G, Girdin‐binding site maps to the N‐terminal (NT) domain of β‐catenin. Lysates from 293FT cells transfected with the Flag tag‐fused Girdin CT domain and the β‐catenin fragments fused with GST were precipitated with glutathione‐Sepharose beads, followed by western blot analyses using anti‐Flag antibody. Bound Flag‐Girdin CT is indicated by asterisks. H, I, Direct interaction of Girdin and β‐catenin. Coomassie brilliant blue staining showing recombinant GST, GST‐fused β‐ and α‐catenins that were expressed and purified from the Escherichia coli expression system (H). Purified recombinant Girdin CT domain was incubated with the recombinant catenins fused with GST (60‐90 pmol) for 1 hour at 4°C, followed by precipitation with glutathione‐Sepharose beads and western blot analysis (I). Girdin CT domain that bound to GST‐catenins is indicated by asterisks. MW, molecular weight
Immunofluorescent staining on A431 and MDCK cells clearly showed the colocalization of Girdin and β‐ and α‐catenins at cell‐cell adhesion sites on both plastic dishes or collagen gels, suggesting that Girdin plays a role in the regulation of intercellular adhesion (Figures 2C,D and S2E). Of note, Girdin/β‐catenin interaction was not obvious in 293FT cells by IP tests or by immunofluorescent staining (Figure S2F,G). Given that 293FT cells are neither migratory nor invasive by nature, these data suggested that Girdin/β‐catenin interaction could be vital for cell motility.
3.3. Girdin carboxyl‐terminal domain interacts with β‐catenin
We and others have shown that Girdin's amino‐ (NT) and carboxyl‐terminal (CT) domains flank the central coiled‐coil domain and interact with multiple proteins including actin filaments, the subunits of the tripartite G proteins, the cell polarity regulator Par‐3, and Dynamin guanosine triphosphatase (GTPase; Figure 2E).20, 25, 29, 43, 44 To address which of Girdin's domains is responsible for β‐catenin interaction, we expressed the domains of Girdin and β‐catenin in 293FT cells, and mapped interacting domains by IP tests (Figure 2F,G). The data showed that Girdin's CT domain interacts with the N‐terminal domain of β‐catenin. An in vitro binding assay using purified recombinant GST‐fused β‐catenin and the His‐fused Girdin CT domain showed their direct interaction (Figure 2H,I). Interestingly, the Girdin CT domain also bound to recombinant α‐catenin, suggesting the possibility that the Girdin CT domain possessed multiple interfaces to bind to multiple proteins.
3.4. Involvement of Girdin in the strength of cell‐cell adhesion
It was important to determine how Girdin was involved in E‐cadherin‐mediated adhesion between cells. We found that Girdin localization at cell‐cell contact sites was more prominent in high‐density cultured A431 cells that formed mature cell‐cell contacts than in low‐density cultured cells with immature contacts (Figure 3A). We used siRNA to deplete the endogenous levels of Girdin in HeLa cells. This treatment induced HeLa cells to scatter such that the cells could not adhere to each other tightly or regularly (Figure 3B,C). This observation was consistent with our previous finding that the Girdin‐depleted SH‐SY5Y neuroblastoma cell line showed a similar response.28 Together, these data indicated that Girdin played an essential role in cell‐cell adhesion.
Figure 3.
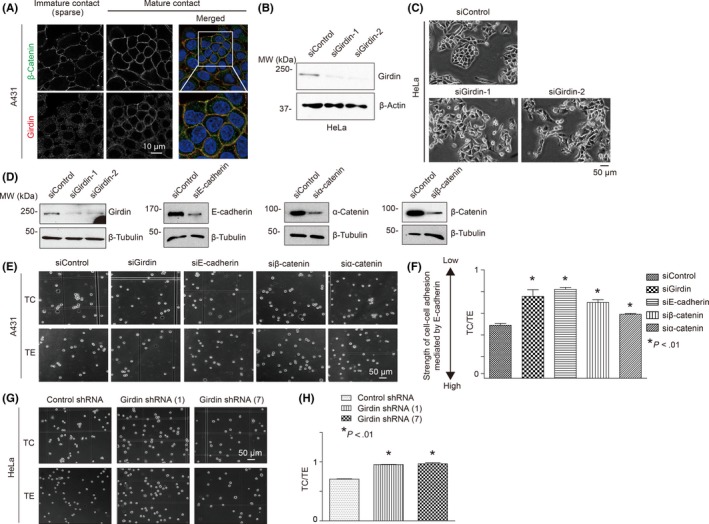
Girdin controls the strength of cell‐cell adhesion. A, Girdin localized at cell‐cell contacts. Its colocalization with β‐catenin was not evident in immature cell‐cell adhesion of A431 cells sparsely plated on dishes (left), whereas it was clearly observed in confluent cells with mature cell‐cell adhesion (right). B, C, HeLa cells depleted of Girdin showed morphology of scattered cells. D, Efficiency of siRNA‐mediated knockdown of Girdin, E‐cadherin, and α‐ and β‐catenins in A431 cells was shown by western blot analyses using the indicated antibodies. E, F, A431 cells transfected with the indicated siRNA were treated with trypsin for 30 minutes in the presence of either 0.1 mmol/L Ca2+ (TC) or 1 mmol/L EDTA (TE) at 37°C. The cells were dissociated by pipetting, and the number of particles was counted. Cells of each group were seeded in three 6‐cm dishes, followed by counting the numbers of all particles in the dishes and quantification. Representative images are shown in (E). The extent of cell dissociation was represented by the index TC/TE, where TC and TE are the total particle numbers after the TC and TE treatment, respectively (F). The TC/TE ratio represents the inverse strength of cadherin activity. G, H, shRNA‐mediated Girdin depletion decreased cadherin activity in HeLa cells, which is shown by high TC/TE values in the cell dissociation assay
Mammalian cells express many types of adhesion molecules and a number of cell surface proteins that mediate cell‐cell adhesion. Thus, we next examined the role of Girdin in calcium‐dependent, E‐cadherin‐mediated adhesion between cells as determined by a cell dissociation assay (Figure 3D‐H).39, 40 Thus, we assessed the numbers of cell particles that included cell clusters and single cells after dissociating A431 and HeLa cells in the presence of 0.1 mmol/L calcium (TC treatment) or 1 mmol/L EDTA (TE treatment; see Materials and Methods; Figure S3A). We found that the depletion of Girdin as well as E‐cadherin and β‐ and α‐catenins significantly increased the number of cell particles after the TC treatment, indicating that these molecules are involved in the strength of cell‐cell adhesion mediated by E‐cadherin (Figure 3D‐H). These results were reproduced when MDCK cells were stably depleted of Girdin and examined in the cell dissociation assay (Figure S3B,C). These data suggested the involvement of Girdin in E‐cadherin‐mediated cell‐cell adhesion.
3.5. Girdin regulates the association between the E‐cadherin complex and the cytoskeleton and supracellular cytoskeletal organization
Many studies have shown that E‐ and N‐cadherin and their complexes undergo remodeling during cell migration.15, 45, 46 Those observations are supported by our experiment in which E‐cadherin interaction with β‐catenin was attenuated by inducing collective cell migration by scratching a monolayer of confluent A431 cells (Figure S4A). We investigated the effect of Girdin depletion on the formation of the E‐cadherin/catenin complex (Figure S4B). The results showed that there was no obvious difference between control and Girdin‐depleted cells, suggesting that Girdin had limited, if any, impact on the assembly of this core adhesion complex. We also examined the localization of E‐cadherin by time‐lapse imaging in A431 cells stably expressing E‐cadherin‐Ruby on collagen gels (Figure S4C). Again, we did not find any obvious changes in E‐cadherin localization or its intracellular trafficking, even in control cells. All of these data showed that the mechanism of Girdin‐mediated collective migration cannot be explained by dysregulated formation or dynamics of the E‐cadherin/catenin complex, which might not be the same as those reported on other cell types and experimental systems.
Another possible mechanism supporting collective migration involves the dynamic regulation of supracellular cytoskeletal organization and its link to cell‐cell adhesion.6, 47 Interestingly, a fractionation experiment showed that the amounts of β‐ and α‐catenins and E‐cadherin that reside in the cytoskeletal fraction were decreased, whereas those in a membrane fraction were increased by Girdin depletion, suggesting a role of Girdin in cytoskeletal association of the catenin protein complex (Figure 4A,B). Another difference between control and Girdin‐depleted cells was revealed by the staining of the actin filaments by phalloidin (Figure 4C). Under these conditions, we found obvious differences in the rearrangement of the actin cytoskeleton during collective migration of A431 cells on collagen gels (Figure 4C). Specifically, filopodia formation at the edges and cell‐cell adhesion sites at the basal, but not apical, plane of the cell groups was significantly attenuated in Girdin‐depleted cells. Given the importance of supracellular cytoskeletal organization, we speculate that Girdin regulates collective migration by facilitating the cytoskeletal association of β‐catenin and related cytoskeletal reorganization (Figure 4D). Furthermore, the overexpression of the NT domain of β‐catenin, which is responsible for binding with Girdin, led to a competitive disruption of endogenous Girdin/β‐catenin interaction (Figure 5A) and directional migration of A431 cell groups on collagen gels (Figure 5B‐D, Movies S6, S7), supporting the physiological role of Girdin/β‐catenin interaction in collective cell migration.
Figure 4.
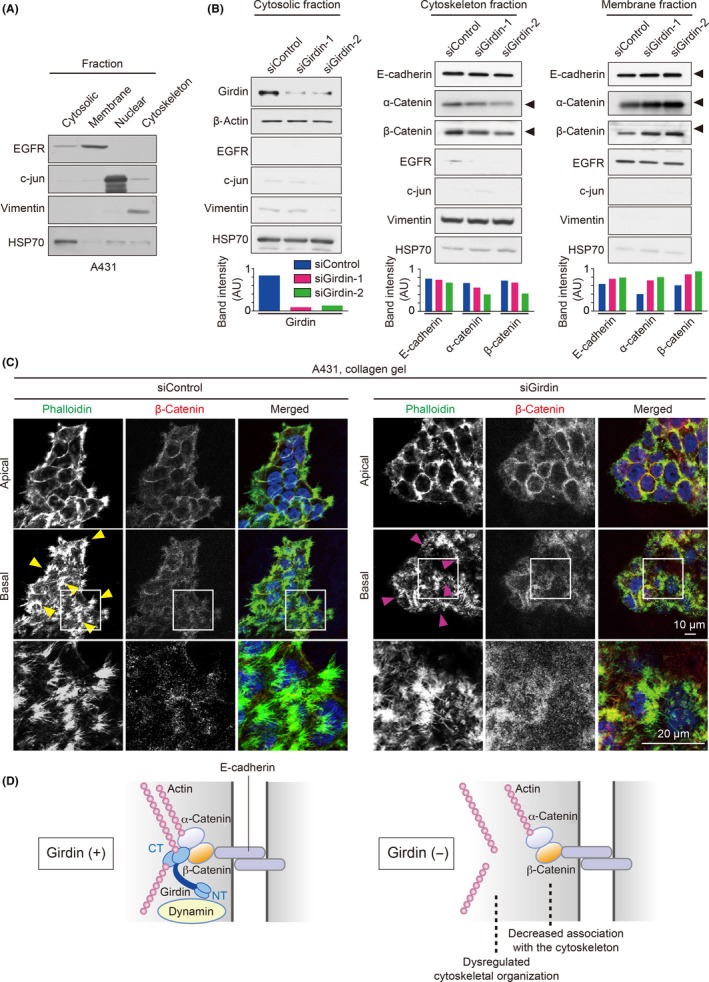
Girdin participates in the cytoskeletal association of the E‐cadherin/catenin complex and cytoskeletal organization. A, Representative data showing successful fractionation of various subcellular fractions of A431 cells. B, Cytosolic, cytoskeletal, and membrane fractions from A431 cells transfected with control or Girdin siRNA were examined by western blot analyses using the indicated antibodies. Lower panel, expression levels of the proteins in each fraction were quantified and presented as arbitrary unit (AU). EGFR, epidermal growth factor receptor; HSP70, heat shock protein 70. C, Control cells (left panel) and Girdin‐depleted cells (right panel) were stained by β‐catenin (red) and phalloidin to visualize actin filaments (green). Yellow arrowheads denote filopodia formation found in the edges and cell‐cell contact sites at the basal planes of control cell groups, whereas filopodia formation was weak or disrupted in Girdin‐depleted cells (magenta arrowheads). D, Schematic illustration of the speculated function of Girdin at cell‐cell adhesions. Girdin mediates the link of β‐catenin and the actin cytoskeleton (left panel), which is lost by the absence of Girdin accompanied by dysregulated cytoskeletal organization (right panel). A previous study showed that Girdin interaction with Dynamin is essential for E‐cadherin endocytosis, which might also be involved in collective cell migration. CT, C‐terminal domain; NT, N‐terminal domain
Figure 5.

Effect of overexpression of the β‐catenin N‐terminal (NT) domain on collective migration of A431 cells. A, Inhibition of endogenous Girdin/β‐catenin interaction by expression of the β‐catenin NT domain. Lysates from A431 cells transfected with the indicated combination of expression plasmids were immunoprecipitated (IP) with anti‐Girdin antibody. After washing, bound proteins were detected by western blot analyses using the indicated antibodies. β‐Catenin precipitated by Girdin antibody is shown by asterisk. B, A431 cells were transfected with GFP (as a fill) and either GST or GST‐β‐catenin NT at the ratio of 1:4, and were sorted for GFP‐positive cells. They were cultured on collagen gels and assessed by tracking of single cells on the edge of migrating cell groups (≥5 cells). C, Representative images of GST and GST‐β‐catenin NT‐transduced A431 cell groups cultured on collagen gels. Time interval between each panel is 2 hours. D, Quantification of the directionality of migrating cell groups (n = 18 for each group)
3.6. Role of Girdin in 3D collective invasion of cancer cells
Our data on A431 cells cultured on collagen gels suggested the importance of Girdin in their collective invasion. To support this hypothesis, we adopted an organotypic culture model in which we prepared Matrigel containing CAFs on which we seeded A431 cells, rendering them air‐exposed (Figure 6A).18, 41 In the model, CAFs remodel the ECM in Matrigel to make a path for the cancer cells, thus allowing us to observe their collective invasion. We found that the depletion of Girdin, as well as that of the other components of the E‐cadherin/catenin complex, attenuated the number of collective cell groups (clusters) invading into Matrigel, but not the cell number in each group, further confirming a role of Girdin in the collective invasion of cancer cells (Figure 6B,C). The depth of invasion was regulated by Girdin and E‐cadherin, but not β‐ and α‐catenins (Figure 6D), suggesting that Girdin and E‐cadherin promote collective invasion with high persistency, but not elucidating the role of the E‐cadherin protein complex.
Figure 6.

Girdin is essential for collective invasion of cancer cells. A, Illustration showing the experimental setup to recapitulate the collective invasion of cancer cells in vitro. CAF, cancer‐associated fibroblast. B, C, Depletion of Girdin and the components of the E‐cadherin complex impedes the collective invasion of A431 cells. Representative H&E‐stained images of sections through Matrigel invaded by A431 cells. Boxed regions are magnified in lower panels, where arrows denote the groups of cells collectively invading into Matrigel. In (C), the numbers of cell groups (clusters; ≥3 cells) invading into Matrigel and cell numbers in each cluster were counted and quantified (n = 3). D, Depth of invasion for each cluster found in each group was calculated and quantified (n = 3). E, Images of representative Girdin staining intensity for each intensity score (IS; 0‐3). Cases with total scores (TS), which represents the sum of IS and proportion scores (PS), of >3 were considered positive. F, Representative images of immunohistochemical staining for Girdin and the components of the E‐cadherin/catenin complex in cases A and B of invasive squamous cell carcinoma of the skin. Invasive lesions of the tumors (lower panels) and the adjacent normal skin (upper panels) are shown. Note that Girdin expression in the carcinomas is more evident than in normal skin
3.7. Clinical relevance of Girdin expression in the progression of skin cancer
Finally, we investigated the significance of Girdin expression in the progression of squamous cell carcinoma of the skin that is known to collectively invade the stroma. To do this, we developed a scoring system by which the expression levels were evaluated by both intensity scores and proportion scores (Figure 6E). Statistical analysis of 76 cases of skin cancer showed that the expression level of Girdin was upregulated in most cases of squamous cell carcinoma of the skin (Figure 6F, Table 1), suggesting a role of Girdin in cancer progression, as already shown for other types of cancer including breast, colon, and brain tumors.22, 33, 34, 35 We also found that Girdin expression level correlated well with the differentiation of the cancer (Table 1) and the expression of β‐ and α‐catenins as well as E‐cadherin (Table 2), suggesting that Girdin synergizes with its partner proteins in the progression of skin cancer. One unresolved issue, however, was that Girdin expression levels had a tendency to correlate with the depth of tumor infiltration, but not with statistically significant difference (P value, 0.1824; Table 1). We also did not find a correlation between Girdin expression and the presence of isolated cell clusters comprised of 1‐4 cancer cells (tumor budding) at the invasive front of tumors (Table S1). Thus, the present study did not show the significance of Girdin in the tumor budding of human skin cancer. This might partially be attributed to the involvement of various microenvironmental factors or synergistic effects of Girdin with other intrinsic factors that could influence the invasion of the cancer cells.
Table 1.
Girdin expression in human squamous cell carcinoma of the skin and its relation to clinical and pathological characteristics
| Total number, n | Girdin positive (TS ≥ 3), n (%) | P value | |
|---|---|---|---|
| Age, years | |||
| ≥60 | 34 | 21 (61.7) | .2929 |
| >60 | 42 | 22 (52.3) | |
| Sex | |||
| Male | 42 | 21 (50.0) | .1621 |
| Female | 34 | 22 (67.6) | |
| WHO classification | |||
| Well diff. | 49 | 37 (75.6) | <.0001 |
| Mod. diff. | 19 | 6 (31.5) | |
| Poor. diff. | 5 | 0 (0.0) | |
| Tumor invasion | |||
| T1/T2 | 64 | 36 (56.2) | .1824 |
| T3/T4 | 11 | 9 (81.8) | |
diff., differentiated; Mod., moderately; Poor., poorly.
Table 2.
Correlation of Girdin expression with the expression of the components of E‐cadherin/catenin complex in human squamous cell carcinoma of the skin
| Total number, n | Girdin positive (TS ≥ 3), n (%) | P value | |
|---|---|---|---|
| E‐cadherin | |||
| Positive (TS ≥ 3) | 49 | 40 (81.6) | <.0001 |
| Negative (TS < 2) | 27 | 3 (11.1) | |
| β‐Catenin | |||
| Positive (TS ≥ 3) | 44 | 35 (79.5) | <.0001 |
| Negative (TS < 2) | 32 | 8 (25) | |
| α‐Catenin | |||
| Positive (TS ≥ 3) | 63 | 43 (68.2) | <.0001 |
| Negative (TS < 2) | 13 | 0 (0.0) | |
TS, total score = sum of intensity score and proportion score.
4. DISCUSSION
In the present study, which was based on the defective collective migration of neuroblasts in mice that lacked the actin‐binding hub protein Girdin,27, 28 we dissected the role of Girdin in collective migration and the invasion of cancer cells. We showed that Girdin is involved in the function of the E‐cadherin/catenin complex, where it controls the stability of cell‐cell adhesion, but at the same time regulates supracellular cytoskeletal organization.
An intriguing finding from this study is that the role of Girdin might be shared by both developing neuroblasts and cancer cells. The role of Girdin in chain migration of subventricular zone neuroblasts has been shown in whole mice, but not in tissue‐ or cell‐specific conditional Girdin knockouts, indicating the specificity of Girdin's function in neuroblasts’ collective migration.8, 27 We and others reported the aberrant expression of Girdin in several types of human cancers, findings that suggest similar roles for Girdin in neuroblasts and cancer cells.22, 33, 34, 35 Given the identification of β‐catenin as a Girdin‐interacting protein in this study, we examined β‐catenin expression in migrating neuroblasts. Interestingly, β‐catenin and N‐cadherin (with which it interacts) were robustly expressed in the neuroblasts in control animals, whereas the expression of those proteins was apparently downregulated or dysregulated in Girdin knockout mice (Figure S5). These data suggested that Girdin might cooperate with β‐catenin in migratory processes of both neuroblasts and cancer cells.
One should consider a previous study that showed retrograde flow of N‐cadherin along the cell‐cell contact sites during collective migration of primary cultured astrocytes.15 Our observation using A431 cells expressing E‐cadherin‐Ruby, however, did not reveal any significant movement along the cell‐cell junctions or intracellular trafficking of E‐cadherin during collective migration (Figure S4C). Those results suggest that the mechanism underlying cadherin dynamics in cancer cells might be distinct from that in neural cells. Further studies are needed to clarify the cell‐ or tissue‐specific mechanisms for collective migration.
At present, the precise mechanism by which Girdin controls cell‐cell adhesion together with actin remodeling cannot be explained solely by Girdin/β‐catenin interaction. We speculate that other binding partner(s) of Girdin are also involved in this process. One of those may be the large GTPase Dynamin that we previously reported binds to the Girdin NT domain.44 The binding of Girdin to Dynamin activates its GTPase activity to pinch off clathrin‐coated vesicles and stimulate E‐cadherin‐specific endocytosis.44 It will be interesting if Girdin/Dynamin interaction and its regulation of E‐cadherin endocytosis are also involved in Girdin‐mediated collective migration, which will be a subject for future research (Figure 4D).
Another limitation of the study is that we could not clearly show whether Girdin forms a four‐protein complex containing α‐ and β‐catenins and E‐cadherin at cell‐cell adhesion sites. We showed their colocalization by immunofluorescent staining. However, those data did not necessarily show evidence of a four‐protein complex or its biological significance. Also, there are no Girdin mutants that do not specifically interact with β‐catenin, making it difficult to solve the specific importance of Girdin/β‐catenin interaction. A future challenge is to identify upstream regulators of Girdin/β‐catenin interaction, preferably kinases. However, as far as we tested, neither Akt, nor cyclin‐dependent kinase 5, nor Src, all of which are known to phosphorylate the Girdin CT domain,20, 48, 49 had any effects on Girdin/β‐catenin interaction. Further investigations of the mechanisms of Girdin/β‐catenin interaction and its coupling to Girdin‐mediated Dynamin GTPase activity will clarify the mechanisms of collective cell migration.
Collective cell migration has been studied for years and several mechanisms have been proposed. However, it remains unclear how proteins involved in collective migration differ from those involved in single cell migration. One attractive model for collective migration argued the importance of supracellular cytoskeletal organization, where the dynamics of the cytoskeleton is shared between multiple cells to jointly generate force for migration and polarization.6, 11 Our finding that Girdin is involved in actin remodeling at the cell‐cell interface in A431 cancer cell groups (Figure 4C) supports the model, but it is unknown at present whether it was a direct or indirect effect of Girdin depletion. Another intriguing mechanism for collective migration is that cells maintain CIL to avoid interfering with their neighbors.14, 16, 17 We previously showed that the Girdin CT domain also binds the cell polarity regulator partitioning‐defective gene 3, which is known to be critical for collective migration and CIL by downregulating actomyosin contractility at cell‐cell contact (Figure 2E).29, 50 Further sophisticated experimental approaches and imaging methods could reveal the involvement of Girdin in CIL in the future.
CONFLICT OF INTEREST
No potential conflict of interests were disclosed.
Supporting information
ACKNOWLEDGMENTS
We gratefully thank Takashi Watanabe for providing the α‐catenin constructs, Kozo Uchiyama for help in the organotypic culture experiments, Kaori Ushida for support in immunostaining, and Eri Yorifuji for support in time‐lapse imaging. We also thank Takuya Kato for providing CAF and A431‐E‐cadherin‐Ruby and support on the organotypic culture model experiment. This work was supported by a Grant‐in‐Aid for Scientific Research (S; 26221304 to M.T.) and Grant‐in‐Aid for Scientific Research (B; 15H04719 to A.E.) commissioned by the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Kobayashi Foundation for Cancer Research (to A.E.), and AMED‐CREST (Japan Agency for Medical Research and Development, Core Research for Evolutional Science and Technology; to A.E. and H.H.).
Wang X, Enomoto A, Weng L, et al. Girdin/GIV regulates collective cancer cell migration by controlling cell adhesion and cytoskeletal organization. Cancer Sci. 2018;109:3643–3656. 10.1111/cas.13795
Contributor Information
Atsushi Enomoto, Email: enomoto@iar.nagoya-u.ac.jp.
Masahide Takahashi, Email: mtakaha@med.nagoya-u.ac.jp.
REFERENCES
- 1. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559‐1564. [DOI] [PubMed] [Google Scholar]
- 2. Yang J, Weinberg RA. Epithelial‐mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818‐829. [DOI] [PubMed] [Google Scholar]
- 3. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15‐33. [DOI] [PubMed] [Google Scholar]
- 4. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:1781‐1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445‐457. [DOI] [PubMed] [Google Scholar]
- 7. Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14:777‐783. [DOI] [PubMed] [Google Scholar]
- 8. Wang X, Enomoto A, Asai N, Kato T, Takahashi M. Collective invasion of cancer: perspectives from pathology and development. Pathol Int. 2016;66:183‐192. [DOI] [PubMed] [Google Scholar]
- 9. McDonald OG, Maitra A, Hruban RH. Human correlates of provocative questions in pancreatic pathology. Adv Anat Pathol. 2012;19:351‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell. 2008;14:570‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedl P, Mayor R. Tuning collective cell migration by cell‐cell junction regulation. Cold Spring Harb Perspect Biol. 2017;9:a029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cui Y, Yamada S. N‐cadherin dependent collective cell invasion of prostate cancer cells is regulated by the N‐terminus of α‐catenin. PLoS ONE. 2013;8:e55069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li L, Hartley R, Reiss B, et al. E‐cadherin plays an essential role in collective directional migration of large epithelial sheets. Cell Mol Life Sci. 2012;69:2779‐2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carmona‐Fontaine C, Matthews HK, Kuriyama S, et al. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 2008;456:957‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peglion F, Llense F, Etienne‐Manneville S. Adherens junction treadmilling during collective migration. Nat Cell Biol. 2014;16:639‐651. [DOI] [PubMed] [Google Scholar]
- 16. Coburn L, Lopez H, Caldwell BJ, et al. Contact inhibition of locomotion and mechanical cross‐talk between cell‐cell and cell‐substrate adhesion determine the pattern of junctional tension in epithelial cell aggregates. Mol Biol Cell. 2016;27:3436‐3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bahm I, Barriga EH, Frolov A, Theveneau E, Frankel P, Mayor R. PDGF controls contact inhibition of locomotion by regulating N‐cadherin during neural crest migration. Development. 2017;144:2456‐2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kato T, Enomoto A, Watanabe T, et al. TRIM27/MRTF‐B‐dependent integrin β1 expression defines leading cells in cancer cell collectives. Cell Rep. 2014;7:1156‐1167. [DOI] [PubMed] [Google Scholar]
- 19. Zegers MM, Friedl P. Rho GTPases in collective cell migration. Small GTPases. 2014;5:e28997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Enomoto A, Murakami H, Asai N, et al. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9:389‐402. [DOI] [PubMed] [Google Scholar]
- 21. Enomoto A, Ping J, Takahashi M. Girdin, a novel actin‐binding protein, and its family of proteins possess versatile functions in the Akt and Wnt signaling pathways. Ann N Y Acad Sci. 2006;1086:169‐184. [DOI] [PubMed] [Google Scholar]
- 22. Jiang P, Enomoto A, Jijiwa M, et al. An actin‐binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 2008;68:1310‐1318. [DOI] [PubMed] [Google Scholar]
- 23. Kitamura T, Asai N, Enomoto A, et al. Regulation of VEGF‐mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008;10:329‐337. [DOI] [PubMed] [Google Scholar]
- 24. Enomoto A, Asai N, Namba T, et al. Roles of disrupted‐in‐schizophrenia 1‐interacting protein girdin in postnatal development of the dentate gyrus. Neuron. 2009;63:774‐787. [DOI] [PubMed] [Google Scholar]
- 25. Weng L, Enomoto A, Ishida‐Takagishi M, Asai N, Takahashi M. Girding for migratory cues: roles of the Akt substrate Girdin in cancer progression and angiogenesis. Cancer Sci. 2010;101:836‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakai T, Nagai T, Tanaka M, et al. Girdin phosphorylation is crucial for synaptic plasticity and memory: a potential role in the interaction of BDNF/TrkB/Akt signaling with NMDA receptor. J Neurosci. 2014;34:14995‐15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Y, Kaneko N, Asai N, et al. Girdin is an intrinsic regulator of neuroblast chain migration in the rostral migratory stream of the postnatal brain. J Neurosci. 2011;31:8109‐8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muramatsu A, Enomoto A, Kato T, et al. Potential involvement of kinesin‐1 in the regulation of subcellular localization of Girdin. Biochem Biophysic Res Commun. 2015;463:999‐1005. [DOI] [PubMed] [Google Scholar]
- 29. Ohara K, Enomoto A, Kato T, et al. Involvement of Girdin in the determination of cell polarity during cell migration. PLoS ONE. 2012;7:e36681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sasaki K, Kakuwa T, Akimoto K, Koga H, Ohno S. Regulation of epithelial cell polarity by PAR‐3 depends on Girdin transcription and Girdin‐Gαi3 signaling. J Cell Sci. 2015;128:2244‐2258. [DOI] [PubMed] [Google Scholar]
- 31. Houssin E, Tepass U, Laprise P. Girdin‐mediated interactions between cadherin and the actin cytoskeleton are required for epithelial morphogenesis in Drosophila. Development. 2015;142:1777‐1784. [DOI] [PubMed] [Google Scholar]
- 32. Jiang P, Enomoto A, Takahashi M. Cell biology of the movement of breast cancer cells: intracellular signalling and the actin cytoskeleton. Cancer Lett. 2009;284:122‐130. [DOI] [PubMed] [Google Scholar]
- 33. Garcia‐Marcos M, Jung BH, Ear J, Cabrera B, Carethers JM, Ghosh P. Expression of GIV/Girdin, a metastasis‐related protein, predicts patient survival in colon cancer. FASEB J. 2011;25:590‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Natsume A, Kato T, Kinjo S, et al. Girdin maintains the stemness of glioblastoma stem cells. Oncogene. 2012;31:2715‐2724. [DOI] [PubMed] [Google Scholar]
- 35. Ghosh P, Tie J, Muranyi A, et al. Girdin (GIV) Expression as a prognostic marker of recurrence in mismatch repair‐proficient stage II colon cancer. Clin Cancer Res. 2016;22:3488‐3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nelson WJ, Nusse R. Convergence of Wnt, ß‐catenin, and cadherin pathways. Science. 2004;303:1483‐1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haga H, Irahara C, Kobayashi R, Nakagaki T, Kawabata K. Collective movement of epithelial cells on a collagen gel substrate. Biophys J . 2005;88:2250‐2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamaguchi N, Mizutani T, Kawabata K, Haga H. Leader cells regulate collective cell migration via Rac activation in the downstream signaling of integrin β1 and PI3K. Sci Rep. 2015;5:7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagafuchi A, Ishihara S, Tsukita S. The roles of catenins in the cadherin‐mediated cell adhesion: functional analysis of E‐cadherin‐alpha catenin fusion molecules. J Cell Biol. 1994;127:235‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuroda S, Fukata M, Nakagawa M, et al. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E‐cadherin‐mediated cell‐cell adhesion. Science. 1998;281:832‐835. [DOI] [PubMed] [Google Scholar]
- 41. Gaggioli C, Hooper S, Hidalgo‐Carcedo C, et al. Fibroblast‐led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392‐1400. [DOI] [PubMed] [Google Scholar]
- 42. Ishida S, Tanaka R, Yamaguchi N, et al. Epithelial sheet folding induces lumen formation by Madin‐Darby canine kidney cells in a collagen gel. PLoS ONE. 2014;9:e99655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Le‐Niculescu H, Niesman I, Fischer T, DeVries L, Farquhar MG. Identification and characterization of GIV, a novel Galpha i/s‐interacting protein found on COPI, endoplasmic reticulum‐Golgi transport vesicles. J Biol Chem. 2005;280:22012‐22020. [DOI] [PubMed] [Google Scholar]
- 44. Weng L, Enomoto A, Miyoshi H, et al. Regulation of cargo‐selective endocytosis by dynamin 2 GTPase‐activating protein girdin. EMBO J. 2014;33:2098‐2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Theveneau E, Mayor R. Cadherins in collective cell migration of mesenchymal cells. Curr Opin Cell Biol. 2012;24:677‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mayor R, Etienne‐Manneville S. The front and rear of collective cell migration. Nat Rev Mol Cell Biol. 2016;17:97‐109. [DOI] [PubMed] [Google Scholar]
- 47. Haeger A, Wolf K, Zegers MM, Friedl P. Collective cell migration: guidance principles and hierarchies. Trends Cell Biol. 2015;25:556‐566. [DOI] [PubMed] [Google Scholar]
- 48. Bhandari D, Lopez‐Sanchez I, To A, et al. Cyclin‐dependent kinase 5 activates guanine nucleotide exchange factor GIV/Girdin to orchestrate migration‐proliferation dichotomy. Proc Natl Acad Sci USA. 2015;112:E4874‐E4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin C, Ear J, Pavlova Y, et al. Tyrosine phosphorylation of the Galpha‐interacting protein GIV promotes activation of phosphoinositide 3‐kinase during cell migration. Sci Signal. 2011;4:ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hidalgo‐Carcedo C, Hooper S, Chaudhry SI, et al. Collective cell migration requires suppression of actomyosin at cell‐cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol. 2011;13:49‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials