

Abstract
Resibufogenin (RB), one of the major active compounds of the traditional Chinese medicine Chansu, has received considerable attention for its potency in cancer therapy. However, the anticancer effects and the underlying mechanisms of RB on pancreatic cancer remain elusive. Here, we found that RB inhibited the viability and induces caspase‐dependent apoptosis in human pancreatic cancer cells Panc‐1 and Aspc. Resibufogenin‐induced apoptosis was through inhibition of constitutive nuclear factor‐κB (NF‐κB) activity and its target genes’ expression, which was caused by downregulation of transforming growth factor‐β‐activated kinase 1 (TAK1) levels and suppression of IκB kinase activity in Panc‐1 and Aspc cells. This induction of TAK1‐mediated NF‐κB inactivation by RB was associated with increased glycogen synthase kinase‐3 (GSK‐3) phosphorylation and subsequent suppression of its activity. Moreover, RB‐induced GSK‐3 phosphorylation/inactivation acted through activation of protein kinase C but not Akt. Finally, RB suppressed human pancreatic tumor xenograft growth in athymic nude mice. Thus, our findings reveal a novel mechanism by which RB suppresses TAK1‐mediated NF‐κB activity through protein kinase C‐dependent inhibition of GSK‐3. Our findings provide a rationale for the potential application of RB in pancreatic cancer therapy.
Keywords: glycogen synthase kinase‐3, nuclear factor‐κB, protein kinase C, resibufogenin, transforming growth factor‐β‐activated kinase 1
Abbreviations
- GS
glycogen synthase
- GSK‐3
glycogen synthase kinase 3
- IB
immunoblotting
- IHC
immunohistochemistry
- IKK
IκB kinase
- NF‐κB
nuclear factor κB
- PARP
poly(ADP‐ribose) polymerase
- PKC
protein kinase C
- RB
Resibufogenin
- TAB 1/2
TAK‐1‐binding proteins1/2
- TAK1
transforming growth factor‐β‐activated kinase 1
1. INTRODUCTION
Pancreatic cancer is the fourth most common cause of cancer‐related deaths worldwide. The first year survival rate is as low as 20% and fifth year survival rate decreases to 6% due to the aggressiveness of the disease and the lack of effective therapies. Although identification of the most frequently mutated genes in pancreatic cancer (KRAS, SMAD4, P53, and P16), the poor prognosis has not improved over the past 40 years.1 Pancreatic cancer is still highly resistant to current chemotherapeutic agents.2, 3 Therefore, the identification of novel therapeutic strategies to improve the outcome in this deadly disease is urgently needed.4
Deregulated NF‐κB signaling has been linked to the pathogenesis of pancreatic cancer.5 Nuclear factor‐κB contains 5 evolutionarily conserved mammalian transcription factors: RelA (p65), c‐Rel, RelB, NF‐κB1 (p50/p105), and NF‐κB2 (p52/p100).6, 7 The activation of NF‐κB can be induced by canonical and noncanonical pathways. The canonical NF‐κB pathway is controlled by activation of the p65/p50 heterodimer,8 which is dependent on the activation of TAK1 and IKKβ.7 Following activation, TAK1 promotes the activity of IKKβ and subsequently phosphorylates the IκB proteins. The phosphorylation triggers rapid ubiquitination‐dependent degradation of IκB proteins, thus liberating NF‐κB for nuclear translocation.9 By contrast, the alternative noncanonical NF‐κB pathway is controlled by the activation of RelB/p52 dimer, which relies on the activation of NF‐κB‐inducing kinase and IKKα.10 Activated NF‐κB‐inducing kinase stimulates IKKα phosphorylation and induces proteolytic processing of p100, resulting in generation of the active p52 subunit. Therefore, RelB/p52 heterodimers translocate into the nucleus and activate a distinct set of genes.11
Glycogen synthase kinase‐3 is a ubiquitous serine/threonine kinase that exists as 2 similar isoforms (GSK‐3α and GSK‐3β).12 A recent study suggested that GSK‐3 plays a critical role in maintaining constitutive NF‐κB activity and cell survival in cancer cells.5 In addition, inactivation of GSK‐3 has been shown to inhibit the growth of various cancers, including pancreatic cancer.13, 14, 15 Glycogen synthase kinase‐3 regulates growth and survival in pancreatic cancer cells by stabilizing the TAK1‐TAB complex to promote IKK activity and subsequent NF‐κB DNA binding and activity.16, 17 Moreover, it is reported that GSK‐3 also plays an important role in promoting noncanonical NF‐κB signaling in pancreatic cancer cells.15 Therefore, GSK‐3 has been considered as a potential therapeutic target for pancreatic cancer.
Huachansu, the dried venom secreted from the skin of the giant toad (containing Bufo bufo gargarizans Cantor and Bufo melanostictus Schneider), has long been used in China and other Asian countries for cancer treatment, including pancreatic cancer.18, 19, 20, 21 Resibufogenin is one of the major active components in Huachansu, and it is regarded as a representative compound for the quality control of Huachansu.22 Resibufogenin shows strong cytotoxic activities against human cancer cells through induction of apoptosis or G1‐phase arrest.23, 24 Moreover, the growth inhibition effect of RB against human cancer cells was comparable to or stronger than paclitaxel.19 However, the effect and precise mechanism of RB on pancreatic cancer has not been elucidated. In this study, we investigated the anticancer effects and molecular mechanisms of RB on pancreatic cancer cells and nude mice bearing Aspc tumor xenografts. We found that RB induced PKC‐dependent inhibition of GSK‐3 activation and, subsequently, suppression of noncanonical NF‐κB activity and TAK1‐mediated canonical NF‐κB activity.
2. MATERIALS AND METHODS
2.1. Cell culture
Panc‐1 and Aspc cells were procured from ATCC (Manassas, VA, USA). The cells were cultured in DMEM and RPMI (HyClone, Logan, UT, USA) supplemented with 10% FBS (HyClone), 100 U/mL penicillin, and100 μg/mL streptomycin sulfate, and incubated at 37°C in a humidified atmosphere with 5% CO2. All cells used in this study were within 20 passages after receipt or resuscitation.
2.2. Cell transfection
Cells were transfected with Vigofect (Vigorous Biotechnology, Beijing, China) according to the manufacturer's protocols. For siRNA‐mediated silencing, cells were transfected with 100 nmol/L of siPKCα or siPKCβ (GenePharma, Shanghai, China) siRNAs and a control siRNA. Forty‐eight hours after transfection, the protein expression was analyzed by IB.
2.3. Reagents and antibodies
Resibufogenin was purchased from Shanghai Standard Technology (Shanghai, China) and dissolved in DMSO. Go6983, Go6976, Go6850, Ro31‐8220, and LY294002 were purchased from Selleck (Beijing, China). Rottlerin and Z‐VAD‐FMK were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The GAPDH antibody was obtained from Sigma‐Aldrich (St. Louis, MO, USA). Antibodies against p‐p65 (Ser536), p65, p‐GSK‐3α (Ser21), GSK‐3α, p‐GSK‐3β (Ser9), GSK‐3β, p‐IKKα/β (Ser176/180), IKKα, TAK1, TAB 1, TAB 2, cleaved PARP‐1, cleaved caspase9, cleaved caspase3, p‐Akt (Ser473), p‐PKCα/β (Thr638/641), PKCα, p100/p52, p‐GS (Ser641), GS, and Notch1 were purchased from Cell Signaling (Danvers, MA, USA). Anti‐c‐FLIP antibody was purchased from ENZO (Farmingdale, NY, USA).
2.4. Measurement of cell viability and apoptosis
The effect of RB on the cell viability of pancreatic cancer cells was determined by MTT assay as previously described.25, 26 Observation of the chromatin shrinking in pancreatic cancer cells induced by RB was carried out by Hoechst 33342 staining assay.27 Detection of apoptotic cell rate was measured using an annexin V‐FITC/propidium iodide apoptosis detection kit (KeyGen, Nanjing, China) as described previously.25, 27
2.5. Microarray analysis
Total RNA was isolated using miRNeasy Mini Kit (Qiagen, Valencia, CA, USA), reverse‐transcribed, labeled, and hybridized to an Agilent SurePrint G3 Human Gene Expression v3.0 microarray 8 × 60K (Agilent Technologies, Santa Clara, CA, USA). The microarray slides were scanned using an Agilent Microarray Scanner, and Feature Extraction software (version 10.7.1.1, Agilent Technologies) was used to analyze array images to obtain raw data. GeneSpring (version 13.1, Agilent Technologies) was employed to finish the basic analysis with the raw data. The probes that had at least 100% of the values in any one out of all conditions flagged as ‘detected’ were chosen for further data analysis. Differentially expressed genes were then identified through fold change. The threshold set for up‐ and downregulated genes was a fold change ≥2.0. Afterwards, Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analyses were applied to determine the roles of these differentially expressed mRNAs. Hierarchical clustering analyses were carried out using Python (version 3.6, https://www.python.org/).
2.6. Cell fractionation and IB analysis
Cell fraction, whole‐cell protein lysates were prepared and analyzed by IB as described previously.25, 27
2.7. Dual luciferase assays
Experiments were undertaken as described previously.26 Briefly, Panc‐1 or Aspc cells were cotransfected with p5× NF‐κB, pM50 TOPFlash reporter plasmid with pRL‐TK or pM50 FOPFlash plasmid (both kindly provided by Dr. Ortwin Naujok, Hannover Medical School, Hannover, Germany) using Vigofect transfection reagent. After 24 hours of transfection, cells were pretreated with RB for 10 hours. Following RB treatment, cells were lysed, and the luciferase activity was determined using a dual‐luciferase reporter assay system (Promega, Madison, WI, USA).
2.8. Real‐time PCR
Total RNAs were isolated by TRIzol reagent (Invitrogen, Carlsbad, CA, USA). First‐strand cDNA synthesis and SYBR green PCR reaction were carried out as described before.27 Total RNA was normalized in each reaction using GAPDH cDNA as an endogenous loading control. The primer sequences of target genes were as following: c‐FLIP L (sense 5′‐GCTCACCATCCCTGTACCTG‐3′, anti‐sense 5′‐CAGGAGTGGGCGTTTTCTT‐3′); Bcl‐2 (sense 5′‐GGCAGTGTGGTCTCC GAATGTC‐3′, anti‐sense 5′‐CCATTGCCTCTCCTCACGTTCC‐3′); and Gapdh (sense 5′‐TGCAC CACCAACTGCTTAGC‐3′, anti‐sense 5′‐GGCATGGACTGTGGTCATGAG‐3′).
2.9. Tumour xenografts in nude mice
The animal procedures were carried out with the approval of the Animal Ethics Committee of the Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences (Beijing, China). Two‐month‐old female athymic nude mice (BALB/c‐nu/nu mice) weighing 18‐22 grams were obtained from Vital River Laboratory Animal Technology (Beijing, China). The nude mice were inoculated s.c. with 5 × 106 Aspc cells. After 7 days, 98% of mice grew visible tumors. The mice were randomly divided into 3 groups: vehicle, RB (10 mg/kg), and RB (20 mg/kg). The drugs were given by intragastric injection every day. The vehicle group was treated with 0.5% sodium carboxymethyl cellulose. Body weight and tumor volume were measured every other day, and the tumor volumes were calculated using the following equation: tumor volume (mm3) = 1/2 × (tumor length) × (tumor width)2. After 20 days of treatment, tumors were excised and weighed.
2.10. Immunohistochemistry analysis
For IHC, tumor tissues were fixed in fresh 10% formaldehyde and cut to 4‐μm‐thick paraffin sections. After incubated with primary antibodies against p‐PKCα/β (Thr638/641), p‐GSK3β (Ser9), and p‐NF‐κB p65 (Ser536) (1:300) for the determination of relative protein expression, PBS was used to replace the primary antibody for the negative control. The slides were probed with an HRP‐labeled secondary antibody for 30 minutes. Subsequently, these slides were counterstained with DAB. Images were obtained using fluorescence microscopy (Axio Vert.A1; Zeiss, Oberkochen, Germany).
2.11. Statistical analysis
Results are presented as mean values ± SE of independent triplicate experiments. All statistical analyses were carried out by using Prism software (GraphPad Prism, La Jolla, CA, USA), and P values of less than .05 were considered statistically significant.
3. RESULTS
3.1. Resibufogenin inhibits the viability of human pancreatic cancer cells
To investigate the antitumor activity of RB (Figure 1A) on pancreatic cancer, we monitored the proliferation of two pancreatic cancer cell lines, Panc‐1 and Aspc, by MTT assays. Treatment with RB in Panc‐1 and Aspc cells significantly increased growth inhibition of cells in a concentration and time‐dependent manner (Figure 1B), The IC50 values for cell viability inhibition of RB were 2.88 μmol/L in Panc‐1 cells and 4.76 μmol/L in Aspc cells at 48 hours. However, the IC50 value for cell viability inhibition in nontransformed pancreatic epithelial HPDE cells was 58.12 μmol/L, approximately 10–20‐fold more than those in pancreatic cancer cells, which fully suggests that RB displayed selective cytotoxicity against tumor cells. In addition, RB could markedly reduce cell‐to‐cell contact and induce cell shrinkage compared with the HPDE cells (Figure 1C). Moreover, RB highly inhibited colony formation and resulted in a remarkable decrease at colony formation ratio in Panc‐1 and Aspc cells (Figure 1D). These results suggest that RB inhibits the viability of pancreatic cancer cells.
Figure 1.
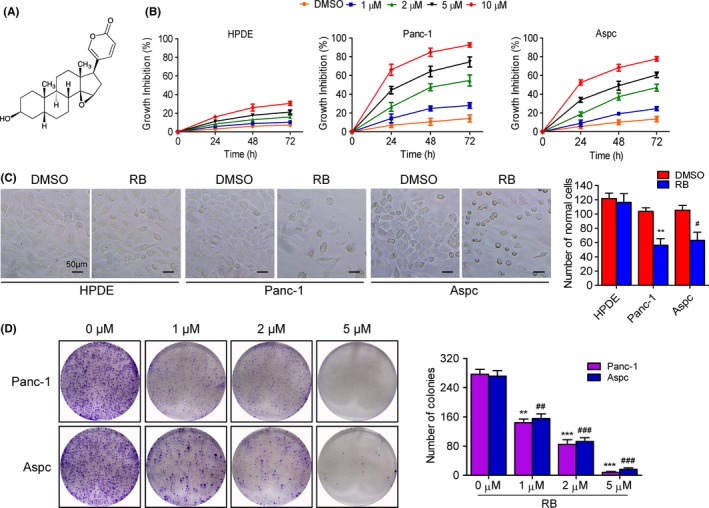
Resibufogenin (RB) inhibits cell viability and changes cell morphology. A, Chemical structure of RB. B, Cell viabilities in normal HPDE, Panc‐1, and Aspc cells treated with various concentrations of RB for the indicated times, determined by MTT. C, Cell morphology changes in HPDE, Panc‐1, and Aspc cells treatment with RB (5 μmol/L) for 48 hours were observed by microscope. Total numbers of live cells were counted and quantified as mean ± SD of three samples of each group. D, Colony formation showed the proliferation of Panc‐1 and Aspc cells treated with the indicated concentrations of RB. **P < .005, ***P < .0005; ## P < .005, ### P < .0005
3.2. Pancreatic cancer cells undergo caspase‐dependent apoptosis following RB treatment
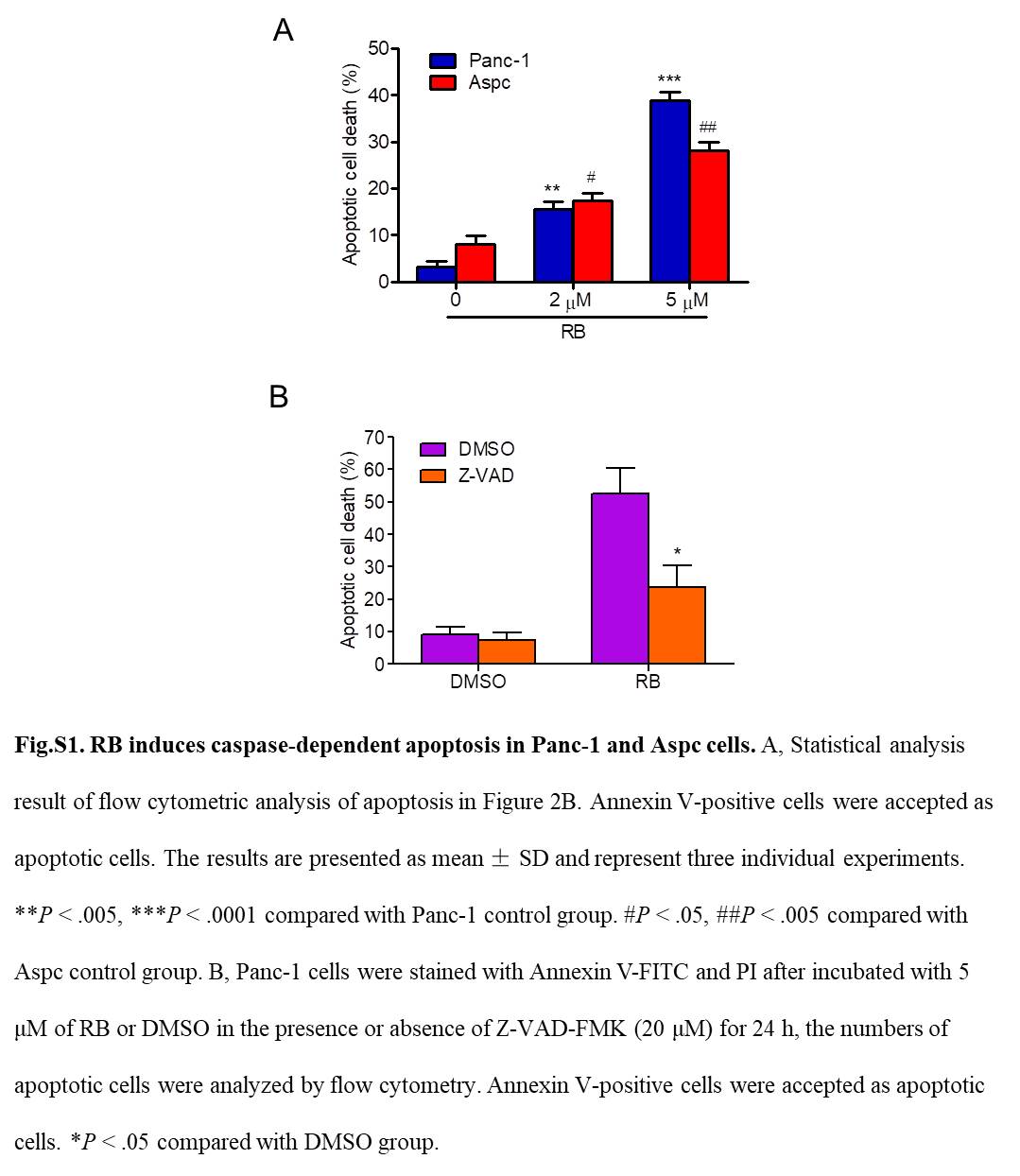
Next, we investigated whether cell apoptosis was involved in RB‐induced pancreatic cancer cell death. Hoechst 33342 staining assay revealed the apoptotic characteristics in cells treated with RB (Figure 2A, arrows). Annexin V/propidium iodide staining further confirmed that RB induced phosphatidylserine plasma membrane externalization in Panc‐1 and Aspc cells in a dose‐dependent manner (Figures 2B, S1A). In addition, RB‐induced apoptosis was inhibited by Z‐VAD‐FMK, a pancaspase inhibitor, suggesting that RB‐induced apoptotic cell death associated with caspase activation (Figure S1B). Furthermore, IB results showed that RB treatment resulted in obvious activation of cleaved PARP1 and cleaved caspase 9 and caspase 3 in a concentration‐dependent manner (Figure 2C). However, the addition of Z‐VAD‐FMK completely prevented the RB action (Figure 2D). Moreover, we use necrostatin‐1, a specific inhibitor of necroptosis, to clarify whether necroptosis is involved in RB‐induced cell death. As shown in Figure 2E, necrostatin‐1 had no effect on RB‐induced pancreatic cancer cell death. These results indicated that caspase‐mediated apoptosis is the major process involved in RB‐induced pancreatic cancer cell death.
Figure 2.
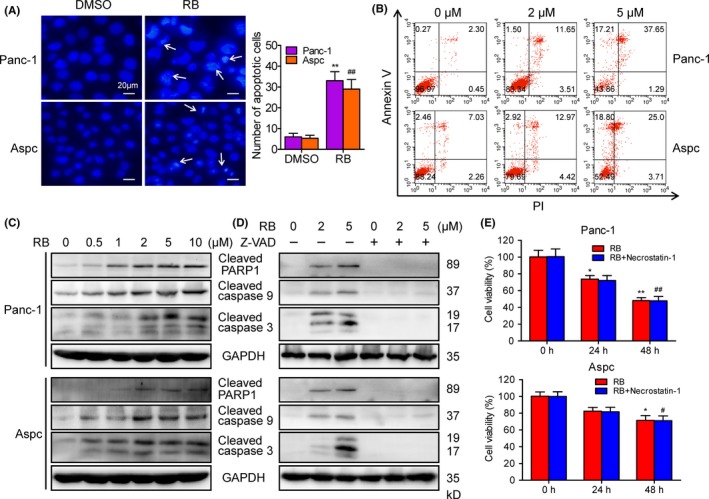
Resibufogenin (RB) induces caspase‐dependent apoptosis in pancreatic cancer cells. A, Hoechst 33342 staining was used to analyze apoptotic cells in Panc‐1 and Aspc cells treated with RB (5 μmol/L) for 24 hours. B, Apoptosis of Panc‐1 and Aspc cells treated with RB for 24 hours, measured by flow cytometry. C, D, Levels of cleaved poly(ADP‐ribose) polymerase (PARP)1, caspase 9, and caspase 3 in Panc‐1 and Aspc cells treated with RB for 24 hours in the presence or absence of Z‐VAD‐FMK (20 μmol/L) were detected by immunoblotting. E, MTT was used to determine the cell viability in Panc‐1 and Aspc cells treated with RB (5 μmol/L) for 24 or 48 hours in the presence or absence of necrostatin‐1 (50 μmol/L). *P < .05, **P < 0.005; # P < .05, ## P < .005
3.3. Resibufogenin induces apoptosis through inhibition of canonical and noncanonical NF‐κB activity in pancreatic cancer cells
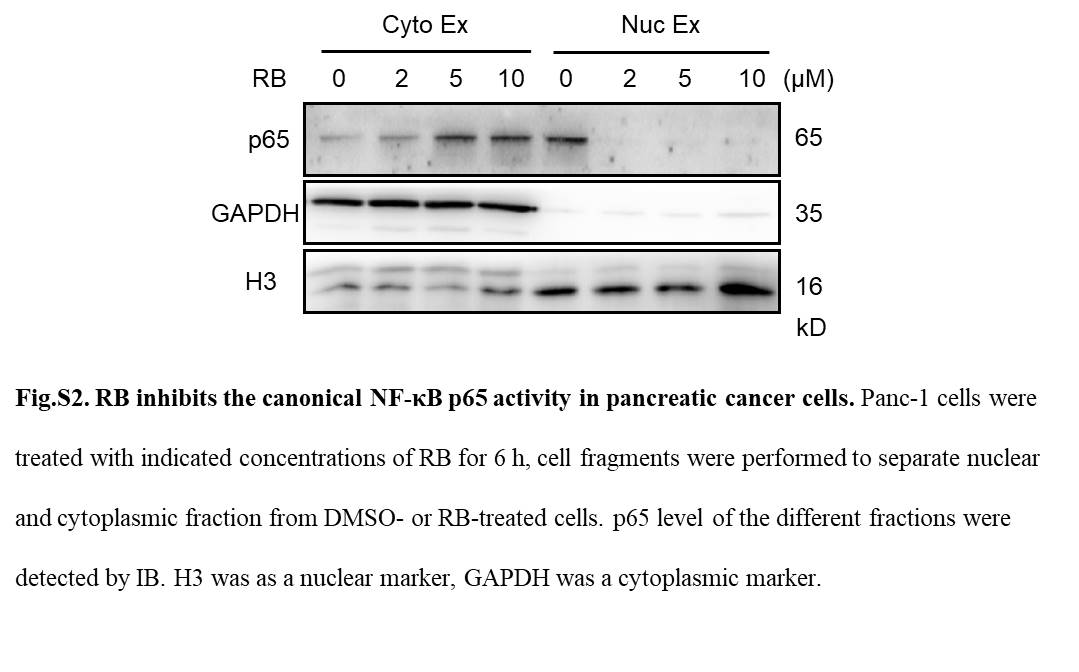
To investigate whether RB‐induced apoptosis in pancreatic cancer cells is through the NF‐κB pathway, we measured the effect of RB on basal NF‐κB‐luciferase reporter activity. Our data showed that treatment with RB led to a significant reduction of constitutive NF‐κB‐luciferase activity in Panc‐1 and Aspc cells (Figure 3A). Next, we explored whether RB‐dependent regulation of reporter activity correlated with the expression of 2 NF‐κB‐targeted antiapoptotic genes, c‐FLIP L and Bcl‐2. Treatment with RB markedly decreased mRNA expression of c‐FLIPL and Bcl‐2 in Panc‐1 cells (Figure 3B). The protein levels of c‐FLIPL and Bcl‐2 were also downregulated by RB in Panc‐1 and Aspc cells (Figure 3C). In addition, RB treatment induced a time‐dependent decrease in levels of NF‐κB p65 phosphorylation (Figure 3D). In accordance with the reduction of phosphorylated NF‐κB p65 activity in Panc‐1 and Aspc cells, we also observed a growth of p65 in the cytoplasm after treatment with RB for 6 hours (Figure S2). These data suggest that RB inhibits canonical NF‐κB transcriptional activity in pancreatic cancer cells.
Figure 3.
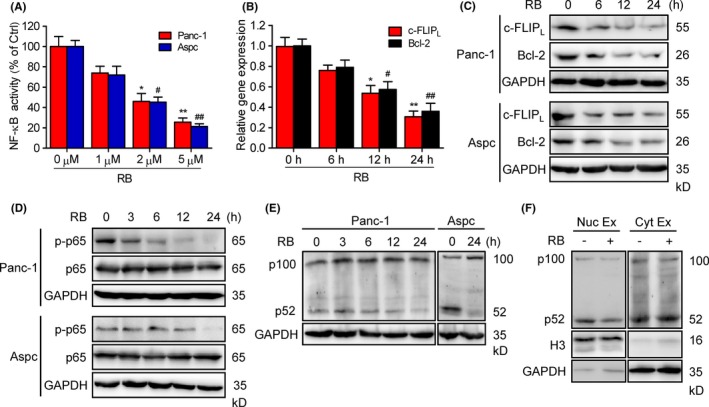
Resibufogenin (RB) inhibits canonical and noncanonical nuclear factor‐κB (NF‐κB) activity. A, Dual luciferase assay verified the NF‐κB activity in RB‐treated Panc‐1 and Aspc cells. B, C, Quantitative RT‐PCR and immunoblotting (IB) analysis of the mRNA and protein expression of c‐FLIPL and Bcl‐2 in Panc‐1 cells treated with 5 μmol/L RB for indicated the times. *P < .05, **P < .005; # P < .05, ## P < .005. D, E, Levels of phosphorylated (p‐)p65 and p65, p100, and p52 in Panc‐1 and Aspc cells treated with RB (5 μmol/L) for the indicated times, detected by IB. F, IB analysis of the nuclear (Nuc) and cytoplasmic (Cyt) fraction of p100 and p52 in Panc‐1 cells treated with 5 μmol/L RB for 24 hours
The noncanonical NF‐κB pathway has been shown to be constitutively active in pancreatic cancer cells, which relies on processing of NF‐κB2 precursor protein p100 to p52.28 Therefore, we determined whether RB also inhibits the noncanonical NF‐κB pathway in pancreatic cancer cells. Indeed, RB treatment suppressed the processing of p100 to p52 in Panc‐1 and Aspc cells (Figure 3E). Moreover, cell fraction assays indicated that RB decreased p52 levels only in the nuclear fraction but not in the cytoplasmic fraction (Figure 3F), suggesting that RB regulates the processing of p100 in the nuclear fraction. Taken together, the above data indicate that RB inhibits both canonical and noncanonical NF‐κB activity in pancreatic cancer cells.
3.4. Effect of RB on gene expression in human pancreatic cancer cells
To further demonstrate that RB modulates NF‐κB signaling, we next sought to determine the effect of RB on the change of gene expression in pancreatic cancer cells. Resibufogenin‐induced global changes in gene expression in Panc‐1 cells were examined by gene expression microarrays. Treatment with RB led to the differential expression (based on fold change analysis) of 14 508 genes that changed more than 2‐fold in Panc‐1 cells (Figure 4A). Given that the NF‐κB pathway is inhibited by RB, we next investigated whether known or suspected NF‐κB target genes were regulated by RB. Indeed, RB induced the downregulation of 49 genes in NF‐κB signaling after 24 hours of treatment (Figure 4B). Thirty‐one of these genes were downregulated more than 3.5‐fold. Moreover, the expression of prosurvival genes such as PARP1, c‐FLIP, and TRAF5 in pancreatic cancer were dramatically decreased with RB treatment (Figure 4C). These results further show that RB inhibits NF‐κB signaling in pancreatic cancer.
Figure 4.
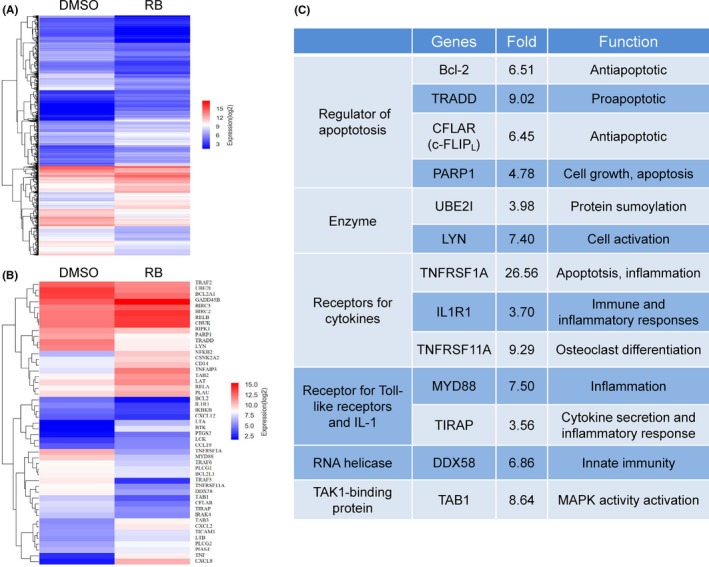
Resibufogenin (RB) treatment led to changes in nuclear factor‐κB (NF‐κB) target gene expression. A, Microarray analysis of genes differentially expressed in Panc‐1 cells treated with DMSO or RB (5 μmol/L). B, Differential changes in known NF‐κB target genes are shown in the heat map. C, NF‐κB target genes that were downregulated more than 3.5‐fold are listed with their known function in pancreatic cancer
3.5. Involvement of TAK1 in RB‐induced NF‐κB inactivation in pancreatic cancer cells
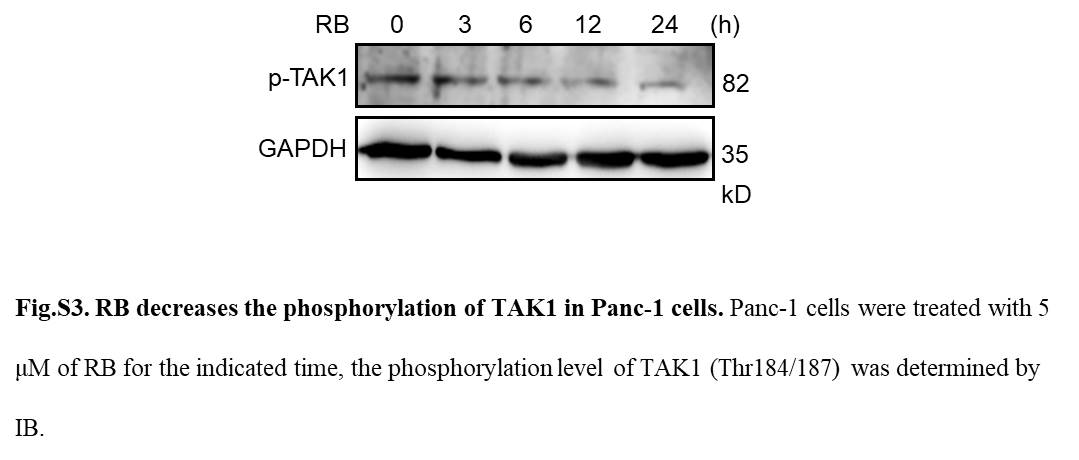
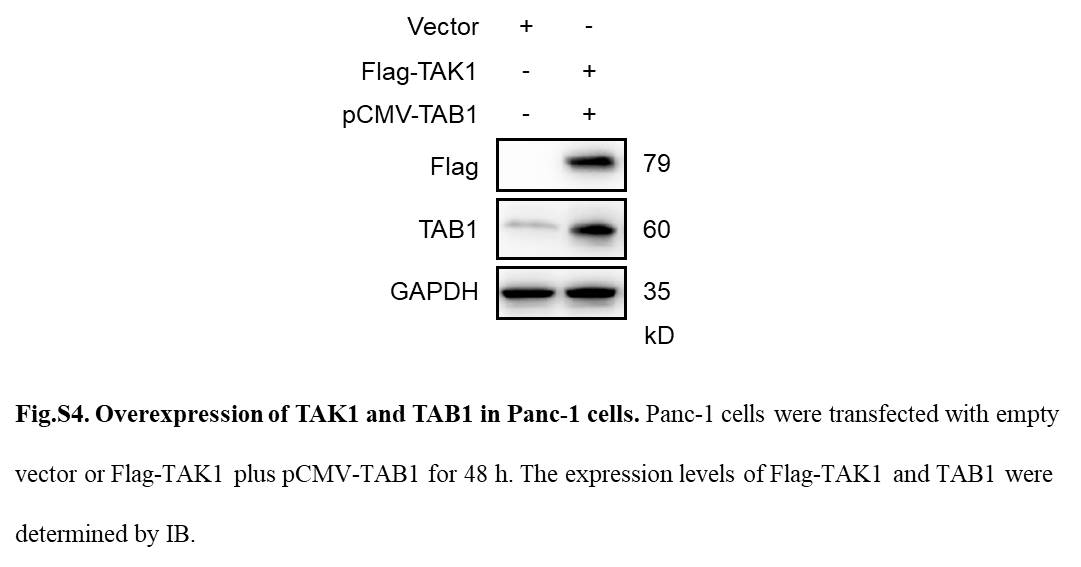
Transforming growth factor‐activated kinase 1 is crucial for survival of pancreatic cancer cells.29, 30 The activation of TAK1 is tightly regulated by TAK‐1‐binding proteins (TAB 1, TAB 2, and TAB 3).31 To examine whether TAK1 is involved in RB‐mediated NF‐κB inactivation, pancreatic cancer cells were treated with RB for a 24‐hour time course. Immunoblotting results revealed that RB induced a decrease in the phosphorylation of TAK1 at threonine 184/187 (Figure S3), and the expression levels of TAK1, TAB 1, and TAB 2 in a time‐dependent manner (Figure 5A). Moreover, the phosphorylation of TAK1 downstream proteins such as IKKα/β and IκBα, markers of canonical NF‐κB signaling, were also decreased after RB treatment (Figure 5B). To examine whether TAK1 directly mediates RB‐induced growth inhibition in pancreatic cancer cells, we enforced expression of TAK1 (Flag‐TAK1) and TAB 1 in Panc‐1 cells and then examined their impact on RB‐induced cell death. Immunoblotting analysis showed the presence of overexpressed TAK1 and TAB 1 proteins (Figure S4). The TAK1/TAB 1 overexpressed RB‐treated Panc‐1 cells showed less apoptotic cell death compared with cells transfected with the empty vector (Figure 5C). Altogether, these results suggest that TAK1 is involved in RB‐mediated NF‐κB inactivation in pancreatic cancer cells.
Figure 5.
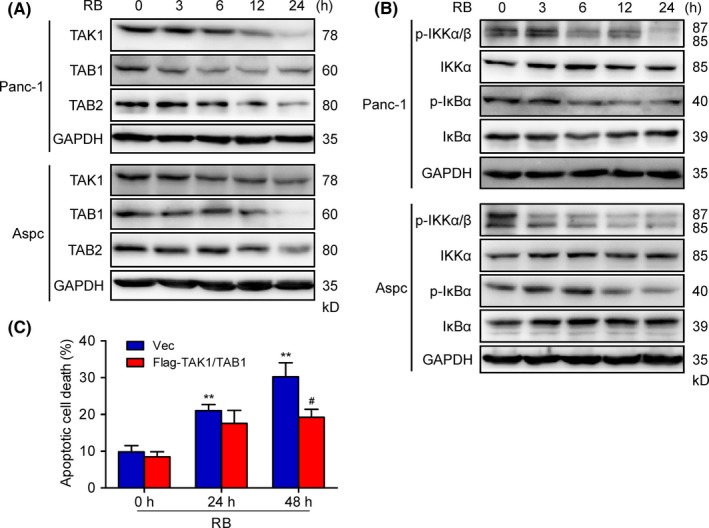
Resibufogenin (RB) suppresses transforming growth factor‐β‐activated kinase 1 (TAK1) and IκB kinase (IKK) activities in pancreatic cancer cells. A, B, Immunoblotting determined the levels of TAK1, TAB 1, and TAB 2 (A) and p‐IKKα/β, IKKα, p‐IκBα, and IκBα (B) in Panc‐1 and Aspc cells treated with 5 μmol/L RB for the indicated times. C, Flow cytometry analysis of apoptosis in Panc‐1 cells transfected with Flag‐TAK1 and TAB 1 following 5 μmol/L RB treatment for 24 hours. # P < .05, **P < .005
3.6. Resibufogenin inhibits GSK‐3 activity in pancreatic cancer cells
To investigate the mechanism by which RB induced inactivation of TAK1, we examined whether RB could regulate GSK‐3 activity in pancreatic cancer cells. Immunoblotting analysis confirmed that treatment with RB in Panc‐1 and Aspc cells resulted in a significant increase of phosphorylated GSK‐3α and GSK‐3β (Figure 6A). In addition, the phosphorylation of GS, a primary GSK‐3β substrate, was also decreased after RB treatment (Figure 6B). The level of active Notch1 (Notch1 intracellular domain), another substrate protein of GSK‐3,32, 33 also showed a significant reduction following RB treatment (Figure 6B). Moreover, treatment of Panc‐1 and Aspc cells with RB stimulated the expression of TCF reporter gene, M50‐TOPFlash (Figure 6C), which reflects GSK‐3 activity.34 To further investigate the effects of GSK‐3 on RB‐induced pancreatic cancer cell death, we enforced expression of WT, constitutively active (S9A), and kinase dead (K85A) GSK‐3β in Panc‐1 cells and then examined their impact on RB‐induced cell death. Transfection with GSK‐3β WT and S9A restored reduced cell viability by RB treatment compared with GSK‐3β K85A mutant (Figure 6D). Taken together, these results indicate that RB inhibits GSK‐3 activity in pancreatic cancer cells.
Figure 6.
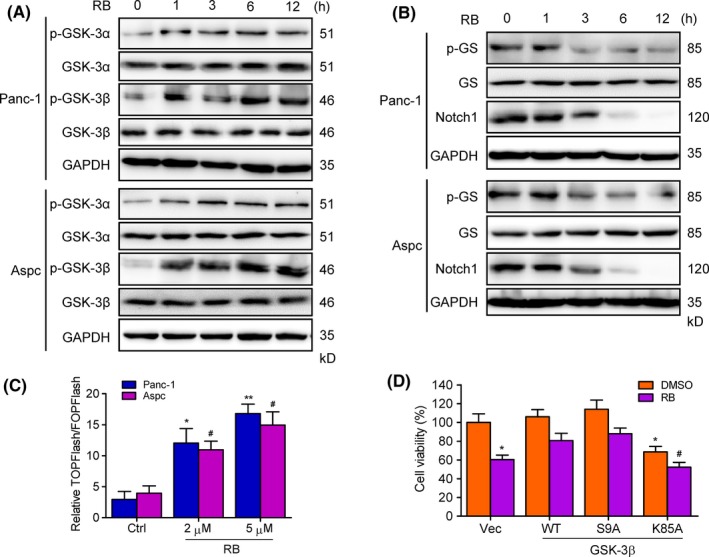
Resibufogenin (RB) inhibits glycogen synthase kinase‐3 (GSK‐3) activity in pancreatic cancer cells. A, B, Immunoblotting determined the levels of p‐GSK‐3α, GSK‐3α, p‐GSK‐3β, and GSK‐3β (A), glycogen synthase (GS), p‐GS, and Notch1 (B) in Panc‐1 and Aspc cells incubated with 5 μmol/L RB for the indicated times. C, Dual luciferase assay verified the TOPFlash and FOPFlash activity in RB‐treated Panc‐1 and Aspc cells. D, Viability in Panc‐1 cells transfected with GSK‐3β WT, S9A, and K85A following RB (5 μmol/L) treatment for 24 hours was measured by MTT. *P < .05, **P < .005; # P < .05
3.7. Resibufogenin induces PKC‐dependent GSK‐3 phosphorylation
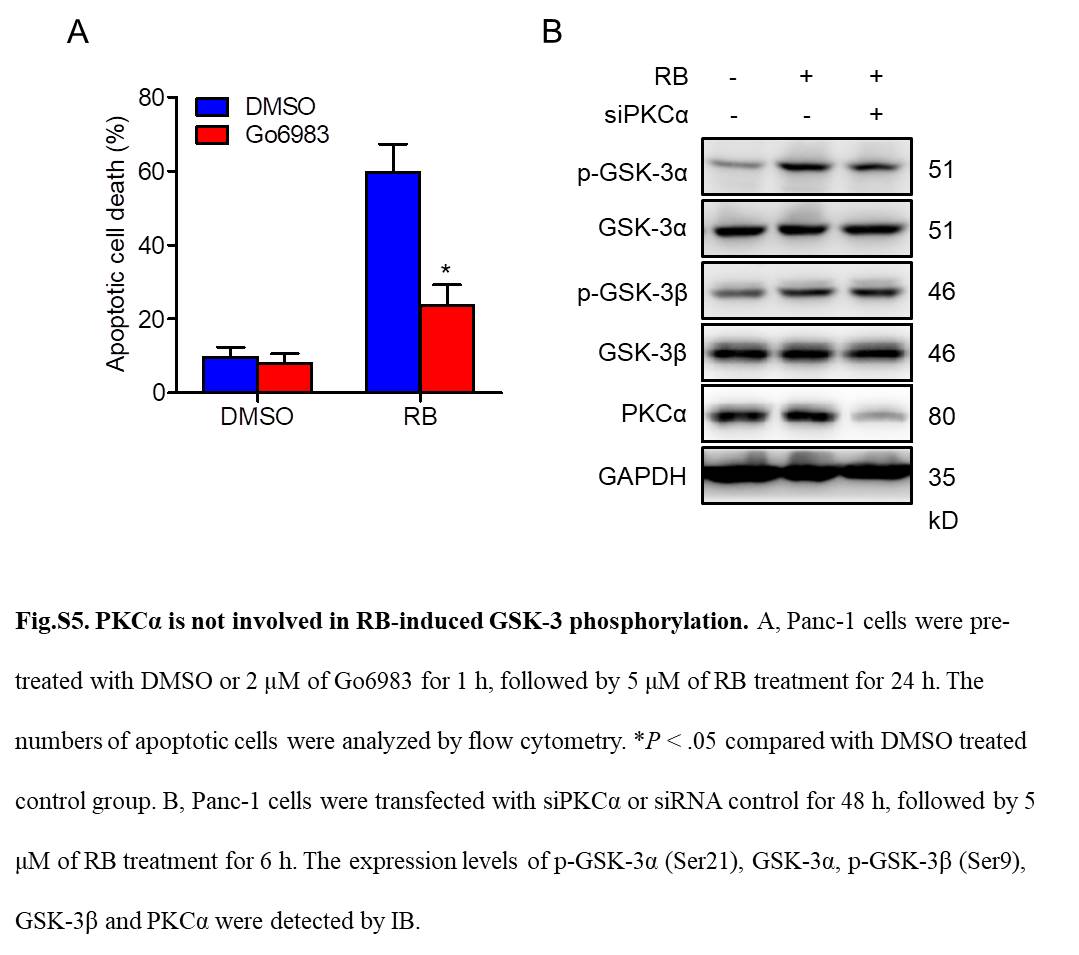
It has been suggested that PI3K/Akt phosphorylates GSK‐3, resulting in its inactivation.35, 36 To examine whether PI3K/Akt is involved in RB‐induced GSK‐3 phosphorylation, we test the effects of RB on GSK‐3 phosphorylation in the presence or absence of the PI3K/Akt‐specific inhibitor LY294002. As shown in Figure 7A, LY294002 abolished RB‐induced Akt phosphorylation but failed to suppress RB‐induced GSK‐3α and GSK‐3β phosphorylation, indicating that RB‐induced GSK‐3 phosphorylation is independent of Akt. Protein kinase C has been reported to phosphorylate GSK‐3.37, 38, 39 Therefore, we next examined whether PKC is involved in RB action. Interestingly, Go6983, a panPKC inhibitor, abolished RB‐induced GSK‐3α and GSK‐3β phosphorylation in Panc‐1 cells (Figure 7B). In addition, Go6983 also inhibited RB‐induced apoptotic cell death in Panc‐1 cells (Figure S5A). Moreover, other panPKC inhibitors Ro31‐8220, the PKCα‐γ inhibitor Go6850, and the PKCα/β inhibitor Go6976 could abolish RB‐induced GSK‐3α/β phosphorylation (Figure 7C). In contrast, the PKCδ inhibitor rottlerin did not inhibit RB‐induced GSK‐3 phosphorylation (Figure 7C), indicating that PKCα/β is involved in RB‐induced GSK‐3 inactivation. Indeed, RB treatment induced a time‐dependent PKCα/β phosphorylation in Panc‐1 cells (Figure 7D). Furthermore, silencing of PKCβ, but not PKCα, abolished RB‐induced GSK‐3 phosphorylation, and increased RB‐induced TAK1 expression and IKK phosphorylation (Figures 7E, S5B). Together, these results suggest that RB induces GSK‐3 phosphorylation through a PKC‐mediated mechanism, likely involving PKCβ.
Figure 7.
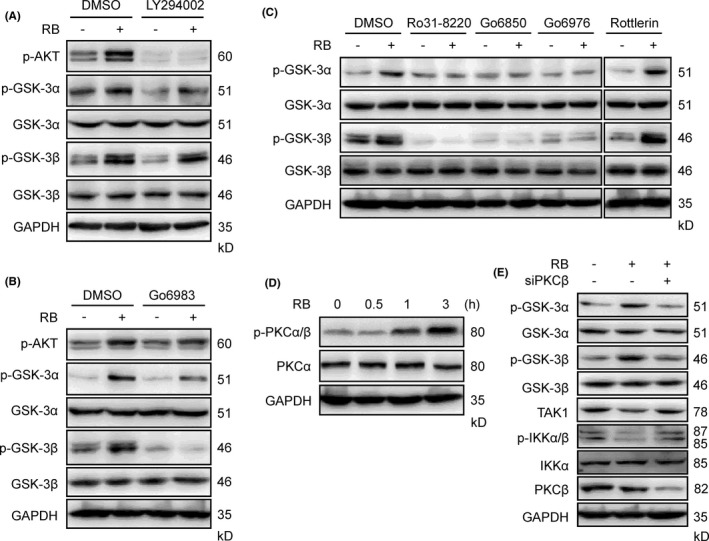
Resibufogenin (RB) induces protein kinase C (PKC)‐mediated glycogen synthase kinase‐3 (GSK‐3) phosphorylation. A, B, Immunoblotting (IB) analyzing the levels of p‐Akt, p‐GSK‐3α, and p‐GSK‐3β in Panc‐1 cells pretreated with LY294002 (A) or Go6983 (B) for 1 hour following RB (5 μmol/L) treatment for 6 hours. C, IB determined the levels of p‐GSK‐3α and p‐GSK‐3β in Panc‐1 cells pretreated with indicated PKC inhibitors for 1 hour following RB (5 μmol/L) treatment for 6 hours. D, p‐PKCα/β levels in Panc‐1 cells treated with 5 μmol/L RB for the indicated times were determined by IB. E, IB analysis of p‐GSK‐3α, p‐GSK‐3β, p‐IKKα/β, and transforming growth factor‐β‐activated kinase 1 (TAK1) expression post‐siRNA knockdown of PKCβ in Panc‐1 cells
3.8. Resibufogenin inhibits human pancreatic tumor xenograft growth in athymic nude mice
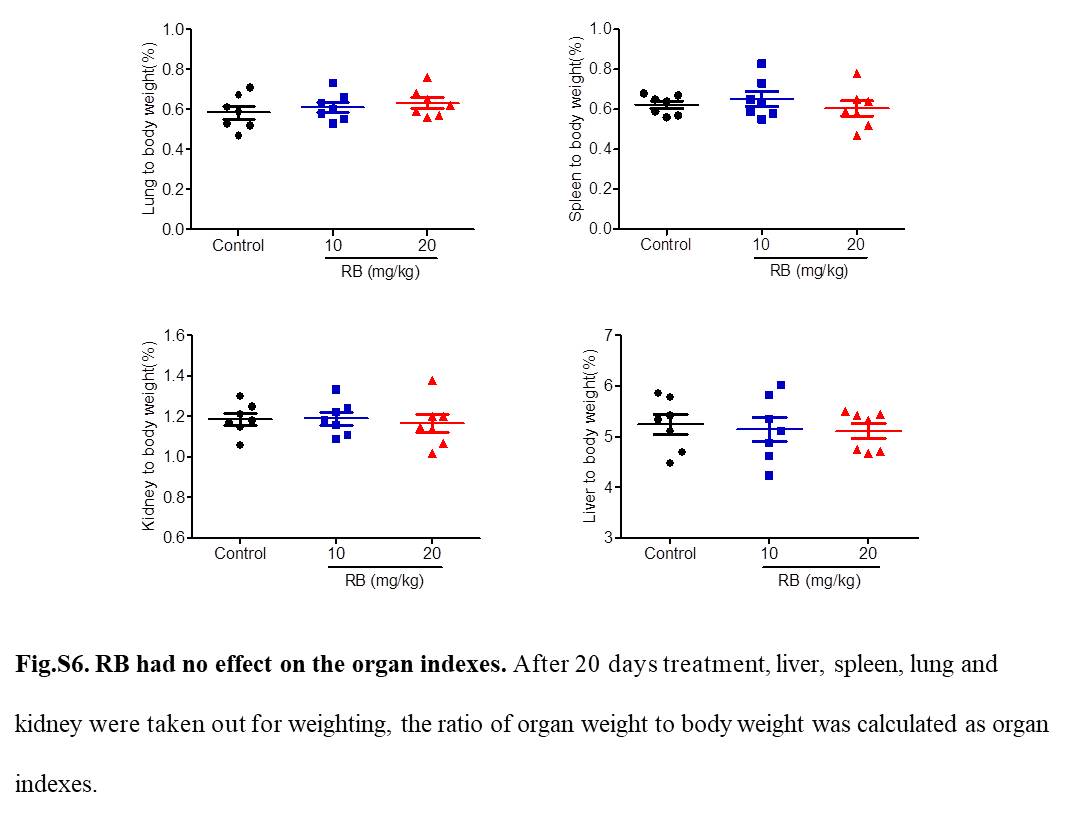
To determine the antipancreatic cancer activity of RB in vivo, an Aspc xenograft model was established. We treated nude mice transplanted with tumor growth produced by Aspc cells with vehicle, RB (10 mg/kg), or RB (20 mg/kg) by intragastric administration once every day for 20 days. The body weight of the mice between the control and RB‐treated groups had no significant different during the treatment period (Figure 8A). In addition, RB (10 mg/kg or 20 mg/kg) did not remarkably alter organ indexes, including liver, spleen, lung, and kidney (Figure S6), suggesting no systemic toxicity was observed after RB treatment.
Figure 8.
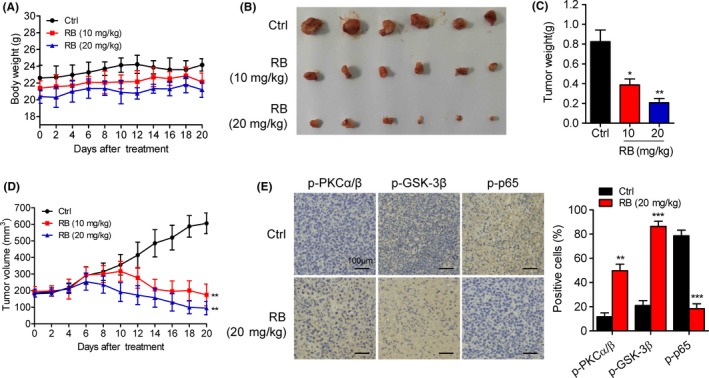
Resibufogenin (RB) inhibits Aspc xenograft tumor growth in nude mice as a single‐agent therapy. A, Mean body weight of RB‐treated mice measured at the indicated number of days. B‐D, Gross morphology of tumors, average xenograft tumor weight, and tumor volume were measured over 20 days (n = 6). E, Immunohistochemical staining was carried out for the determination of phosphorylated protein kinase C (p‐PKC)α/β, glycogen synthase kinase (p‐GSK)3β, and p‐p65 in mice tumor samples. Columns are expressed as mean ± SD of 6 samples in each group. *P < .05, **P < .005, ***P < .0005
Notably, the growth of Aspc tumor xenografts was dramatically inhibited following RB treatment (Figure 8B). The average tumor masses following 10 and 20 mg/kg RB treatment were 0.39 ± 0.11 g and 0.21 ± 0.07 g, respectively, which were dramatically lower than that of the control group (0.82 ± 0.21 g) (Figure 8C). The tumor volume was also inhibited significantly following the injection of RB at the dose levels 10 and 20 mg/kg (Figure 8D). Moreover, IHC assay indicated that the phosphorylation levels of PKCα/β and GSK‐3β were increased, whereas the phosphorylation level of p65 was decreased, in tumors of RB‐treated mice (Figure 8E). Therefore, RB suppresses human pancreatic tumor xenograft growth in vivo.
4. DISCUSSION
Recently, the search for natural products from traditional Chinese medicine has become a promising approach for novel drug development.40, 41 The present investigation revealed the potent antipancreatic cancer effects of RB both in vivo and in vitro. We provided evidence that RB possessed a significant inhibitory effect on the viability of pancreatic cancer cells in a dose‐ and time‐dependent manner; the inhibition was less remarkable in non‐transformed HPDE cells. These data suggest that RB displayed selective cytotoxicity against tumor cells. In addition, RB treatment significantly increased the apoptotic rates and resulted in obvious activation of cleaved PARP1, caspase 9, and caspase 3 in pancreatic cancer cells. These results clearly indicate that RB provoked caspase‐dependent apoptosis in pancreatic cancer cells.
Inhibition of NF‐κB activity has become a novel chemotherapeutic approach in pancreatic and other cancers.42 Our results showed that RB suppressed Panc‐1 and Aspc cell growth and induced apoptosis, at least partly, through downregulation of NF‐κB activity. In addition to inhibition of p65‐mediated canonical NF‐κB activity, RB also inhibited noncanonical NF‐κB activity in pancreatic cancer cells. This was revealed by significantly reduced p100 processing and nuclear accumulation of p52 following RB treatment. Our finding is consistent with a previous study that showed that the noncanonical NF‐κB pathway was constitutively active and contributed to survival in pancreatic cancer cells.28 Therefore, the inhibition of both canonical and noncanonical NF‐κB pathways might explain the anticancer effect of RB.
To identify the mechanisms involved in the inhibition of NF‐κB activation of RB, we tested the effect of RB on TAK1 activation signals.43 We found that RB induced a remarkable decrease in levels of TAK1 and its binding partners TAB 1 and TAB 2. Emerging reports imply that silencing the expression or inhibiting the activity of TAK1 dramatically suppresses NF‐κB activity, which leads to a proapoptotic phenotype in pancreatic cancer cells.29, 44 This is consistent with our result, as our findings indicate a reduction in the phosphorylation of TAK1 downstream protein IKKα/β following RB treatment. Furthermore, enforced expression of TAK1/TAB 1 also suppressed RB‐induced apoptosis in Panc‐1 cell. Thus, our data support a role for TAK1 downregulation in mediating RB‐induced apoptosis in pancreatic cancer cells.
Glycogen synthase kinase‐3 has been shown to regulate the stability of TAK1 and noncanonical NF‐κB signaling in pancreatic cancer cells.15 To examine the mechanism by which TAK1 and noncanonical NF‐κB activity is downregulated by RB, we measured the effect of RB on GSK‐3 activity. Interestingly, our results showed that the phosphorylation of GSK‐3, including both α and β isoforms, was increased by RB. These data indicate that RB actually inhibits GSK‐3 function.36, 45 Complementarily, enforced expression of GSK‐3 suppressed RB‐induced apoptosis in Panc‐1 cells. Thus, our findings clearly show that RB downregulates TAK1 levels through inhibition of GSK‐3 activity, supporting the concept that GSK‐3 acts as a tumor promoter by enhancing TAK1 expression in pancreatic cancer cells.
To clarify which upstream signal is involved in RB‐induced GSK‐3 phosphorylation (inactivation), Akt activity was inhibited by a specific inhibitor, as it is well known that Akt phosphorylates GSK‐3, resulting in its inactivation.35, 36 Although RB increased the phosphorylation of both Akt and GSK‐3, PI3K/AKT inhibitor LY294002 failed to abrogate RB‐induced GSK‐3α/β phosphorylation, indicating that RB induces Akt‐independent GSK‐3 inactivation. In addition to Akt, p70S6K and PKC can also phosphorylate GSK‐3.37, 38, 39, 46, 47 As the mTOR/p70S6K inhibitor rapamycin did not affect RB‐induced GSK‐3 phosphorylation (data not shown), we examined whether PKC is involved in RB‐mediated GSK‐3 phosphorylation. Our results revealed that RB‐induced GSK‐3 phosphorylation was dramatically abolished by two panPKC inhibitors, Go6983 and Ro31‐8220, suggesting that RB induces PKC‐dependent GSK‐3 phosphorylation/inactivation. Using specific PKC isoform inhibitors, we found that PKCα/β is involved in RB‐induced GSK‐3 phosphorylation. In addition, silencing of PKCβ, but not PKCα, abolished RB‐induced GSK‐3 phosphorylation. These results thus suggest that the PKCβ isoforms could be important for mediating RB‐induced GSK‐3 phosphorylation or inactivation. Our discovery regarding RB suppression of TAK1 and GSK‐3 activity in a PKC‐dependent manner is an important finding of this work. To the best of our knowledge, this is the first report of RB inducing PKC‐dependent GSK‐3 inactivation in pancreatic cancer cells.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
We thank Dr. Ortwin Naujok (Hannover Medical School, Hannover, Germany) for kindly supplying M50‐TOPFlash/FOPFlash plasmids, Dr. James Woodgett (University of Toronto, Toronto, Canada) for kindly providing GSK‐3β (WT, K85A, and S9A) plasmids, and Dr. Jun Ninomiya‐Tsuji (North Carolina State University, Raleigh, NC, USA) for kindly providing pCMV‐Flag‐TAK1 and pCMV‐TAB 1 plasmids. This work was supported by grants from the National Natural Science Foundation of China (81773782, 81473248, and 81771520), and CAMS Major Collaborative Innovation Project (2016‐12M‐1‐011).
Liu L, Liu Y, Liu X, et al. Resibufogenin suppresses transforming growth factor‐β‐activated kinase 1‐mediated nuclear factor‐κB activity through protein kinase C‐dependent inhibition of glycogen synthase kinase 3. Cancer Sci. 2018;109:3611–3622. 10.1111/cas.13788
Lu Liu and Yang Liu contributed equally to this work.
REFERENCES
- 1. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605‐1617. [DOI] [PubMed] [Google Scholar]
- 3. Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218‐1249. [DOI] [PubMed] [Google Scholar]
- 4. Neuzillet C, Tijeras‐Raballand A, Bourget P, et al. State of the art and future directions of pancreatic ductal adenocarcinoma therapy. Pharmacol Ther. 2015;155:80‐104. [DOI] [PubMed] [Google Scholar]
- 5. Wilson W 3rd, Baldwin AS. Maintenance of constitutive IkappaB kinase activity by glycogen synthase kinase‐3alpha/beta in pancreatic cancer. Cancer Res. 2008;68:8156‐8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature. 2006;441:431‐436. [DOI] [PubMed] [Google Scholar]
- 7. Hayden MS, Ghosh S. NF‐kappaB, the first quarter‐century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baud V, Karin M. Is NF‐kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baldwin AS. Regulation of cell death and autophagy by IKK and NF‐kappaB: critical mechanisms in immune function and cancer. Immunol Rev. 2012;246:327‐345. [DOI] [PubMed] [Google Scholar]
- 10. Sun SC. The noncanonical NF‐kappaB pathway. Immunol Rev. 2012;246:125‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kew RR, Penzo M, Habiel DM, Marcu KB. The IKKalpha‐dependent NF‐kappaB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J Immunol. 2012;188:2380‐2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doble BW, Woodgett JR. GSK‐3: tricks of the trade for a multi‐tasking kinase. J Cell Sci. 2003;116:1175‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Domoto T, Pyko IV, Furuta T, et al. Glycogen synthase kinase‐3beta is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016;107:1363‐1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang JS, Herreros‐Villanueva M, Koenig A, et al. Differential activity of GSK‐3 isoforms regulates NF‐kappaB and TRAIL‐ or TNFalpha induced apoptosis in pancreatic cancer cells. Cell Death Dis. 2014;5:e1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bang D, Wilson W, Ryan M, Yeh JJ, Baldwin AS. GSK‐3alpha promotes oncogenic KRAS function in pancreatic cancer via TAK1‐TAB stabilization and regulation of noncanonical NF‐kappaB. Cancer Discov. 2013;3:690‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ougolkov AV, Fernandez‐Zapico ME, Bilim VN, Smyrk TC, Chari ST, Billadeau DD. Aberrant nuclear accumulation of glycogen synthase kinase‐3beta in human pancreatic cancer: association with kinase activity and tumor dedifferentiation. Clin Cancer Res. 2006;12:5074‐5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ougolkov AV, Fernandez‐Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase‐3beta participates in nuclear factor kappaB‐mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65:2076‐2081. [DOI] [PubMed] [Google Scholar]
- 18. Yang T, Shi R, Chang L, et al. Huachansu suppresses human bladder cancer cell growth through the Fas/Fasl and TNF‐ alpha/TNFR1 pathway in vitro and in vivo. J Exp Clin Cancer Res. 2015;34:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qi F, Li A, Inagaki Y, et al. Antitumor activity of extracts and compounds from the skin of the toad Bufo bufo gargarizans Cantor. Int Immunopharmacol. 2011;11:342‐349. [DOI] [PubMed] [Google Scholar]
- 20. Yin JH, Zhu XY, Shi WD, Liu LM. Huachansu injection inhibits metastasis of pancreatic cancer in mice model of human tumor xenograft. BMC Complement Altern Med. 2014;14:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meng Z, Garrett CR, Shen Y, et al. Prospective randomised evaluation of traditional Chinese medicine combined with chemotherapy: a randomised phase II study of wild toad extract plus gemcitabine in patients with advanced pancreatic adenocarcinomas. Br J Cancer. 2012;107:411‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma XC, Zhang BJ, Xin XL, et al. Simultaneous quantification of seven major bufadienolides in three traditional Chinese medicinal preparations of chansu by HPLC‐DAD. Nat Prod Commun. 2009;4:179‐184. [PubMed] [Google Scholar]
- 23. Wang DL, Qi FH, Xu HL, et al. Apoptosis‐inducing activity of compounds screened and characterized from cinobufacini by bioassay‐guided isolation. Mol Med Rep. 2010;3:717‐722. [DOI] [PubMed] [Google Scholar]
- 24. Ichikawa M, Sowa Y, Iizumi Y, Aono Y, Sakai T. Resibufogenin Induces G1‐Phase Arrest through the Proteasomal Degradation of Cyclin D1 in Human Malignant Tumor Cells. PLoS ONE. 2015;10:e0129851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu L, Zhang N, Dou Y, et al. Lysosomal dysfunction and autophagy blockade contribute to IMB‐6G‐induced apoptosis in pancreatic cancer cells. Sci Rep. 2017;7:41862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deng H, Zhang N, Wang Y, et al. S632A3, a new glutarimide antibiotic, suppresses lipopolysaccharide‐induced pro‐inflammatory responses via inhibiting the activation of glycogen synthase kinase 3beta. Exp Cell Res. 2012;318:2592‐2603. [DOI] [PubMed] [Google Scholar]
- 27. Zhang N, Liu X, Liu L, et al. Glycogen synthase kinase‐3β inhibition promotes lysosome‐dependent degradation of c‐FLIPL in hepatocellular carcinoma. Cell Death Dis. 2018;9:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wharry CE, Haines KM, Carroll RG, May MJ. Constitutive non‐canonical NFkappaB signaling in pancreatic cancer cells. Cancer Biol Ther. 2009;8:1567‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Melisi D, Xia Q, Paradiso G, et al. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J Natl Cancer Inst. 2011;103:1190‐1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin‐dependent kinase of MKK and IKK. Nature. 2001;412:346‐351. [DOI] [PubMed] [Google Scholar]
- 31. Omori E, Inagaki M, Mishina Y, Matsumoto K, Ninomiya‐Tsuji J. Epithelial transforming growth factor beta‐activated kinase 1 (TAK1) is activated through two independent mechanisms and regulates reactive oxygen species. Proc Natl Acad Sci USA. 2012;109:3365‐3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han X, Ju JH, Shin I. Glycogen synthase kinase 3‐beta phosphorylates novel S/T‐P‐S/T domains in Notch1 intracellular domain and induces its nuclear localization. Biochem Biophys Res Commun. 2012;423:282‐288. [DOI] [PubMed] [Google Scholar]
- 33. Kunnimalaiyaan S, Gamblin TC, Kunnimalaiyaan M. Glycogen synthase kinase‐3 inhibitor AR‐A014418 suppresses pancreatic cancer cell growth via inhibition of GSK‐3‐mediated Notch1 expression. HPB (Oxford). 2015;17:770‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Naujok O, Lentes J, Diekmann U, Davenport C, Lenzen S. Cytotoxicity and activation of the Wnt/beta‐catenin pathway in mouse embryonic stem cells treated with four GSK3 inhibitors. BMC Res Notes. 2014;7:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vivanco I, Sawyers CL. The phosphatidylinositol 3‐Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489‐501. [DOI] [PubMed] [Google Scholar]
- 36. Hermida MA, Dinesh Kumar J, Leslie NR. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv Biol Regul. 2017;65:5‐15. [DOI] [PubMed] [Google Scholar]
- 37. Moore SF, van den Bosch MT, Hunter RW, Sakamoto K, Poole AW , Hers I. Dual regulation of glycogen synthase kinase 3 (GSK3)alpha/beta by protein kinase C (PKC)alpha and Akt promotes thrombin‐mediated integrin alphaIIbbeta3 activation and granule secretion in platelets. J Biol Chem. 2013;288:3918‐3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Q, Zhou Y, Evers BM. Neurotensin phosphorylates GSK‐3alpha/beta through the activation of PKC in human colon cancer cells. Neoplasia. 2006;8:781‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vilimek D, Duronio V. Cytokine‐stimulated phosphorylation of GSK‐3 is primarily dependent upon PKCs, not PKB. Biochem Cell Biol. 2006;84:20‐29. [DOI] [PubMed] [Google Scholar]
- 40. Kim DH, Suh J, Surh YJ, Na HK. Regulation of the tumor suppressor PTEN by natural anticancer compounds. Ann N Y Acad Sci. 2017;1401:136‐149. [DOI] [PubMed] [Google Scholar]
- 41. Hua F, Shang S, Hu ZW. Seeking new anti‐cancer agents from autophagy‐regulating natural products. J Asian Nat Prod Res. 2017;19:305‐313. [DOI] [PubMed] [Google Scholar]
- 42. Carbone C, Melisi D. NF‐kappaB as a target for pancreatic cancer therapy. Expert Opin Ther Targets. 2012;16(Suppl 2):S1‐S10. [DOI] [PubMed] [Google Scholar]
- 43. Liu Q, Busby JC, Molkentin JD. Interaction between TAK1‐TAB 1‐TAB 2 and RCAN1‐calcineurin defines a signalling nodal control point. Nat Cell Biol. 2009;11:154‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang FT, Peng JF, Cheng WJ, et al. MiR‐143 targeting TAK1 attenuates pancreatic ductal adenocarcinoma progression via MAPK and NF‐kappaB pathway in vitro. Dig Dis Sci. 2017;62:944‐957. [DOI] [PubMed] [Google Scholar]
- 45. McCubrey JA, Davis NM, Abrams SL, et al. Diverse roles of GSK‐3: tumor promoter‐tumor suppressor, target in cancer therapy. Adv Biol Regul. 2014;54:176‐196. [DOI] [PubMed] [Google Scholar]
- 46. Deng H, Hershenson MB, Lei J, Anyanwu AC, Pinsky DJ, Bentley JK. Pulmonary artery smooth muscle hypertrophy: roles of glycogen synthase kinase‐3beta and p70 ribosomal S6 kinase. Am J Physiol Lung Cell Mol Physiol. 2010;298:L793‐L803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang HH, Lipovsky AI, Dibble CC, Sahin M, Manning BD. S6K1 regulates GSK3 under conditions of mTOR‐dependent feedback inhibition of Akt. Mol Cell. 2006;24:185‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials