

Abstract
Introduction
We evaluated the efficacy and safety of DS‐8500a as add‐on therapy to sitagliptin in Japanese type 2 diabetes mellitus patients.
Materials and Methods
This multicenter, randomized, double‐blind, placebo‐controlled, phase 2 trial randomized patients aged ≥20 years with hemoglobin A1c ≥7.0% and <9.0%, and inadequate glycemic control with sitagliptin 50‐mg monotherapy to receive 25 or 75 mg DS‐8500a, or a placebo, orally. The primary end‐point was change from baseline to day 28 in 24‐h weighted mean glucose. Secondary end‐points included change from baseline in fasting plasma glucose, 2‐h postprandial plasma glucose and lipid profiles.
Results
Overall, 29, 28 and 27 patients in the placebo, 25‐ and 75‐mg groups, respectively, were analyzed. A significant dose‐dependent reduction was observed in 24‐h weighted mean glucose (linear: P = 0.0006, saturated at 25 mg: P = 0.0003, responded from 75 mg: P = 0.0176) when compared with the placebo (25 mg: −13.19 mg/dL [−0.73 mmol/L], P = 0.0044 vs placebo and 75 mg: −16.12 mg/dL [−0.89 mmol/L], P = 0.0006 vs placebo). A significant reduction in fasting plasma glucose at 75 mg vs placebo was observed (P < 0.001). At 25 and 75 mg, significant reductions of 2‐h postprandial plasma glucose (after breakfast), total cholesterol, low‐cholesterol and triglycerides were observed (all P < 0.05), with a (non‐significant) trend towards increased high‐density lipoprotein cholesterol. Both doses of DS‐8500a were well tolerated. There were no significant treatment‐emergent adverse events leading to discontinuation during the study.
Conclusions
DS‐8500a was well tolerated, and showed significant glycemic benefits and favorable changes in lipid profile in Japanese type 2 diabetes mellitus patients with inadequate glycemic control with sitagliptin therapy.
Keywords: Japan, Randomized controlled trial, Type 2 diabetes mellitus
Introduction
Type 2 diabetes mellitus is associated with various risks, including microvascular and macrovascular complications1. The prevalence of type 2 diabetes mellitus is increasing worldwide, particularly in Asian countries, possibly as a result of an increase in the aging population and a Westernized lifestyle2. In 2015, the estimated number of people with diabetes (including both type 1 diabetes mellitus and type 2 diabetes mellitus) was 7.2 million, placing Japan among the top 10 countries with the highest number of people with diabetes1.
Current treatment options seek to control glycemia and include a wide range of classes of oral antidiabetic drugs, such as sulfonylureas, short‐acting insulin secretagogues, dipeptidyl peptidase‐4 (DPP‐4) inhibitors, thiazolidines, biguanides, sodium‐glucose transporter 2 inhibitors and α‐glucosidase inhibitors. These drugs exert their effects through a variety of mechanisms, including compensation for impaired insulin secretion, reversal of insulin resistance, suppression of gluconeogenesis in the liver, enhancement of urinary glucose excretion and suppression or delay of glucose absorption.
Patients with poor glycemic control can be treated with combination therapy targeting these different pathways. However, as pancreatic β‐cell dysfunction is a major contributor to type 2 diabetes mellitus progression, new drugs that improve insulin secretion in pancreatic β‐cells might become an important addition to available treatment approaches3.
G protein‐coupled receptor 119 (GPR119) is expressed in small intestinal L cells in the human gastrointestinal tract and pancreatic β‐cells4, 5. Studies in mice have shown that it promotes glucagon‐like peptide‐1 (GLP‐1) secretion and glucose‐dependent insulinotropic effects, and improves glucose tolerance5, 6. Furthermore, GPR119 agonists were reported to increase the concentration of cyclic adenosine monophosphate in cells expressing human GPR119 in a concentration‐dependent manner6. Another study reported that GPR119 agonists upregulated insulin and genes essential for controlling pancreatic β‐cells in a mouse type 2 diabetes mellitus model7. Based on these physiological roles, GPR119 has been considered a novel treatment target for type 2 diabetes mellitus, prompting the development of GPR119 agonists. Unfortunately, the development of several GPR119 agonists was terminated after the initial clinical studies because of weak glucose‐lowering effects or lack of efficacy after repeated dosing of up to 14 days8, 9. GPR119 agonists are expected to improve pancreatic β‐cell function and reduce blood glucose levels by promoting insulin secretion.
DS‐8500a is a novel selective GPR119 agonist discovered by Daiichi Sankyo Co., Ltd., and it is being developed as a treatment for type 2 diabetes mellitus. Our preliminary study showed that DS‐8500a had agonistic activity in Chinese hamster ovary‐K1 cells expressing human GPR119 by promoting GLP‐1 secretion and enhancing glucose‐stimulated insulin secretion (Matsumoto K, ADA 2016: 1124‐P, unpublished data). We also found that it reversed glucose tolerance in Zucker fatty rats, an animal model of glucose intolerance. Furthermore, DS‐8500a had a significant glucose‐lowering effect when co‐administered with the DPP‐4 inhibitor, sitagliptin, in Zucker fatty rats.
Our recent randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter phase 2 study (J201 study, JapicCTI‐142597, NCT02222350) investigated the use of DS‐8500a administered orally at 10 or 75 mg for 4 weeks in type 2 diabetes mellitus patients10. We found that the difference in the change from baseline in 24‐h weighted mean glucose (WMG) at the completion of the study was −13.3 mg/dL (−0.74 mmol/L) in the DS‐8500a 10‐mg group and −18.9 mg/dL (−1.05 mmol/L) in the 75‐mg group vs placebo.
The DPP‐4 inhibitor, sitagliptin, prolongs the action of GLP‐1 and glucose‐dependent insulinotropic polypeptide, thus enhancing the effect of incretins involved in glucose homeostasis11. We hypothesized that DS‐8500a would show synergistic activity with sitagliptin. We therefore investigated the efficacy of DS‐8500a at 25 and 75 mg after 28‐day multiple oral administration in type 2 diabetes mellitus patients receiving sitagliptin. Safety and pharmacodynamics were also assessed.
Methods
Trial design
The present multicenter, randomized, placebo‐controlled, double‐blind, parallel‐group study was carried out between 1 January 2016 and 30 September 2016 at six institutions in Japan (Appendix S1). Patients were enrolled on day −28, observed for 14 days (observation period 1) and then underwent a 14‐day run‐in period during which all participants received a placebo. Next, the patients were randomized to one of three groups (DS‐8500a 25 mg, DS‐8500a 75 mg or placebo once daily), received treatment for 28 days and were followed up for 14 days (Figure S1).
The present study was carried out in compliance with the relevant standards of the Pharmaceutical Affairs Law, and by the Ordinance Regarding Good Clinical Practice and the ethical standards of the Declaration of Helsinki. The study received institutional review board approval and all participants provided written informed consent. This trial was registered at ClinicalTrials.gov (NCT02685345) and JapicCTI (JapicCTI‐163136).
Participants
The main inclusion criteria were Japanese type 2 diabetes mellitus patients aged ≥20 years at the time of informed consent; having received monotherapy with sitagliptin 50 mg for ≥12 weeks before the observation period; and having hemoglobin A1c (HbA1c) ≥7.0% and <9.0%. The main exclusion criteria were patients with type 1 diabetes mellitus or with a history of diabetic coma, precoma or ketoacidosis; clinically significant diabetic retinopathy (e.g., preproliferative retinopathy), diabetic nephropathy (overt nephropathy at stage 3 or worse) or diabetic neuropathy; receiving insulin; poorly controlled blood pressure; body mass index <18.5 kg/m2 or ≥35.0 kg/m2; clinically evident liver damage or disease; clinically evident renal impairment or disease (estimated glomerular filtration rate <45 mL/min per 1.73 m2); anemia; fasting plasma glucose of ≥240 mg/dL (13.33 mmol/L); acute coronary syndrome, stroke or transient ischemic attack within 6 months before informed consent; heart failure; malignant tumor; history of severe drug allergy; serious disease (e.g., central nervous, cardiovascular and respiratory diseases); those who underwent sampling of whole blood; infectious disease; pregnant or breast‐feeding; those who previously participated in another clinical study and received a study drug (DS‐8500a or other); and those judged ineligible for participation in the study by the investigator.
Interventions
Tablets were administered orally to each group as follows: one DS‐8500a 25‐mg tablet plus two placebo tablets in the 25‐mg group; three DS‐8500a 25‐mg tablets in the 75‐mg group; and three placebo tablets in the placebo group. Doses were administered once daily after breakfast.
After obtaining informed consent, diet and exercise therapy remained unchanged throughout the study period. Each participant's diet was verified during the study at each visit and documented in the medical records.
All patients were hospitalized from days −2 to 1 and from days 27 to 29, so that food intake could be controlled and the 24‐h WMG determined. During the two in‐hospitalization periods, patients received standardized meals that provided 1,600 kcal per day in three meals (500 kcal for breakfast and lunch, and 600 kcal for dinner). Each meal comprised 60% carbohydrates, 25% fat and 15% protein.
Pharmacodynamic assessments were carried out on day 28 using the total GLP‐1 assay kit (ToK150JVC; Meso Scale Discovery, Rockville, MD, USA), active GLP‐1 enzyme‐linked immunosorbent assay kit (EGLP‐35K; Merck, Kenilworth, NJ, USA), total human peptide YY (PYY) RIA kit (EMD Millipore, Billerica, MA, USA) and total human glucose‐dependent insulinotropic peptide (GIP) enzyme‐linked immunosorbent assay kit (EMD Millipore) according to manufacturers’ instructions.
The doses used were selected based on our phase 2 study in type 2 diabetes mellitus patients, where DS‐8500a was administered at 10 or 75 mg for 4 weeks10. The results of the previous study showed a significant decrease in 24‐h WMG in both DS‐8500a 10‐ and 75‐mg groups vs placebo; furthermore, both doses of DS‐8500a were well tolerated without significant treatment‐related adverse events, hypoglycemia or discontinuations due to adverse events (AEs)10.
End‐points
The primary end‐point was change in 24‐h WMG from baseline (day −1) to day 28. This measure was reported to be strongly associated with HbA1c levels obtained after 3 months12, 13; thus, we consider this to be a valid marker of glucose control in the present study. The 24‐h WMG was calculated as the area under the plasma concentration–time curve (AUC)0–24 h of plasma glucose/24, in accordance with a previous study14 (Appendix S2).
The secondary end‐points were change in fasting plasma glucose (FPG) from baseline (day −1) to day 28; change in serum insulin and C‐peptide from baseline (day −1) to day 28; change in pharmacodynamic parameters, including plasma glucose, serum insulin, PYY, total GLP‐1, active GLP‐1, total GIP, glucagon, area under the concentration‐time curve from 0 to 4 h (AUC0–4 h) for glucose, insulin AUC0–4 h, C‐peptide AUC0–4 h, insulin AUC0–4 h/glucose AUC0–4 h and C‐peptide AUC0–4 h/glucose AUC0–4 h from baseline (day −1) to day 28; change in total cholesterol, high‐density lipoprotein cholesterol, low‐density lipoprotein (LDL) cholesterol and triglycerides from baseline (day −1) to day 28; change in 2‐h postprandial plasma glucose (PPG; after breakfast, after lunch, after dinner); and safety (AEs, laboratory values, bodyweight, vital signs [blood pressure and pulse rate] and 12‐lead electrocardiogram).
Sample size
The rationale for the target sample size (90 participants; 30 per group) was based on the expected change in 24‐h WMG from baseline to day 28, which was assumed to be 0 mg/dL, −15 mg/dL (0.83 mmol/L) and −15 mg/dL (0.83 mmol/L) in the placebo, 25‐ and 75‐mg groups, respectively, with a common standard deviation of 22 mg/dL. The power of test was expected to show a dose–response relationship, ‘saturated at 25 mg,’ of 82% in 28 participants per treatment group at a significance level of 2.5% (one‐sided). This power was determined to be sufficient, assuming the exclusion of two participants per group from the analysis.
Randomization and blinding
Randomization was carried out using an interactive web response system. Participants were randomized by applying the permuted block method with HbA1c (<8.0% or ≥8.0%) in observation period 1 as a stratification factor to the DS‐8500a 25‐mg, DS‐8500a 75‐mg or placebo groups at a ratio of 1:1:1 according to the schedule. The assignment of groups and treatments was blinded to all participants, investigators and the sponsor, excluding the independent statistician. Placebos were indistinguishable from the investigational drug in terms of appearance, packaging and other features.
Statistical analysis
Efficacy was analyzed in the full analysis set (FAS) and sensitivity analysis in the per‐protocol set. The FAS included randomized individuals who met the following criteria: (i) individuals who met the inclusion criteria; (ii) individuals treated with at least one dose of the study drug in the treatment period; and (iii) individuals from whom data on 24‐h WMG on day −1 or after administration were obtained. The per‐protocol set included individuals who were included in the FAS and who did not meet any of the following criteria: (i) individuals meeting any of the exclusion criteria; (ii) individuals with treatment compliance in the treatment period of <75%; and (iii) individuals with a serious protocol deviation. The safety analysis set included randomized individuals treated with at least one dose of the study drug in the treatment period.
The analyses for the primary end‐point were as follows: change in 24‐h WMG from baseline (day −1) to day 28 using an analysis of covariance (ancova) model defining ‘treatment group’ as the fixed effect and ‘24‐h WMG at day −1’ as the covariate. Sensitivity analyses were also carried out in the per‐protocol set. In the FAS, each DS‐8500a group was compared with the placebo group, and the difference in the least squares mean (each DS‐8500a group – the placebo group) and the 95% confidence interval (CI) were calculated and the corresponding P‐value obtained. The statistical methods for the secondary end‐point sensitivity analyses were the same as above, but applied in the FAS. The pre‐dose value of each end‐point was used as a covariate in an ancova model.
For safety analyses, AEs were tabulated using the Medical Dictionary for Regulatory Activities Version 19.0 by preferred terms. Treatment‐emergent AEs (TEAEs) were defined as AEs that occurred after the initiation of the treatment period. TEAE incidence was calculated per treatment group and tabulated by event and severity. Adverse drug reactions were also tabulated in a similar manner. For laboratory tests, bodyweight and vital signs, summary statistics were calculated per treatment group at each time‐point.
Missing values were not supplemented with an estimated or calculated value. A P‐value <0.05 was considered statistically significant. No multiplicity adjustments were carried out because this was an exploratory study. Statistical analyses were carried out using SAS system release 9.2 (SAS Inc., Cary, NC, USA).
Results
Figure S2 shows the participant disposition. Overall, 29, 28 and 27 participants in the placebo, 25‐ and 75‐mg groups were analyzed. Most participants were men in all treatment groups, and the overall mean age was 62.0 ± 9.4 years. Table 1 shows that patient baseline characteristics were comparable between the placebo, DS‐8500a 25‐mg and DS‐8500a 75‐mg groups: mean baseline HbA1c (7.51 ± 0.51%, 7.63 ± 0.51% and 7.61 ± 0.59%, respectively); FPG (156.7 ± 21.3 mg/dL [8.71 ± 1.18 mmol/L], 156.9 ± 26.4 mg/dL [8.72 ± 1.47 mmol/L] and 157.1 ± 31.6 mg/dL [8.73 ± 1.76 mmol/L], respectively); and 24‐h WMG (191.9 ± 31.5 mg/dL [10.66 ± 1.75 mmol/L], 195.0 ± 33.2 mg/dL [10.83 ± 1.84 mmol/L] and 193.2 ± 38.6 mg/dL [10.74 ± 2.14 mmol/L], respectively).
Table 1.
Participant baseline demographic characteristics
| Placebo (n = 29) | DS‐8500a 25 mg (n = 28) | DS‐8500a 75 mg (n = 28) | |
|---|---|---|---|
| Age (years) | 60.7 ± 10.3 | 62.4 ± 8.0 | 62.9 ± 10.0 |
| Sex | |||
| Male | 21 (72.4) | 21 (75.0) | 25 (89.3) |
| Female | 8 (27.6) | 7 (25.0) | 3 (10.7) |
| BMI (kg/m2) | 25.29 ± 4.22 | 23.55 ± 2.79 | 24.02 ± 2.82 |
| Duration of DM (years) | 9.87 ± 6.35 | 9.48 ± 7.49 | 8.79 ± 6.00 |
| HbA1c (%) | 7.51 ± 0.51 | 7.63 ± 0.51 | 7.61 ± 0.59 |
|
FPG (mg/dL) [mmol/L] |
156.7 ± 21.3 [8.71 ± 1.18] |
156.9 ± 26.4 [8.72 ± 1.47] |
157.1 ± 31.6 [8.73 ± 1.76] |
|
24‐h WMG (mg/dL) [mmol/L] |
191.9 ± 31.5 [10.65 ± 1.75] |
195.0 ± 33.2 [10.82 ± 1.84] |
193.23 ± 38.6 [10.72 ± 2.14] |
Data are presented as the mean ± standard deviation or n (%). BMI, body mass index; DM, diabetes mellitus; FPG, fasting plasma glucose; HbA1c, hemoglobin A1c; WMG, weighted mean glucose.
Regarding the study primary end‐point, there was a significant dose‐dependent reduction in 24‐h WMG from baseline to day 28 (linear: P = 0.0006, saturated at 25 mg: P = 0.0003, responded from 75 mg: P = 0.0176) when compared with the placebo (change from baseline to day 28: −0.41 mg/dL [−0.02 mmol/L], −13.19 mg/dL [−0.73 mmol/L] and −16.12 mg/dL [−0.89 mmol/L] in the placebo, 25‐mg and 75‐mg groups, respectively; DS‐8500a 25 mg vs placebo, P = 0.0044; DS‐8500a 75 mg vs placebo, P = 0.0006; Figure 1a).
Figure 1.
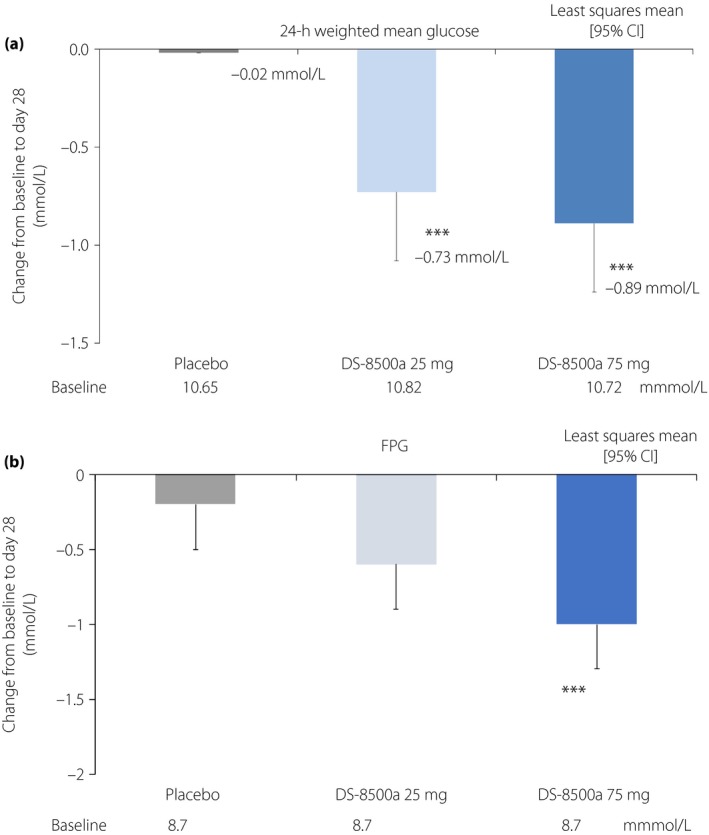
Change from baseline in (a) 24‐h weighted mean glucose and (b) fasting plasma glucose (FPG) at day 28 (full analysis set). ***P < 0.001 vs placebo, analysis of covariance (ancova). CI, confidence interval.
Regarding the secondary end‐points, FPG was significantly reduced from baseline to day 28 at the 75‐mg dose compared with the placebo (Figure 1b). The 2‐h PPG values after breakfast (Table 2) were significantly reduced from baseline to day 28 at both doses compared with the placebo. The decrease in 2‐h PPG after lunch was significantly greater only in the 25‐mg group vs placebo (Table 2), and no significant difference was observed between both the DS‐8500a and placebo groups for the 2‐h PPG values after dinner (Table 2). Several markers linked with β‐cell function and insulin secretion were measured, including the AUC0–4 h for glucose, C‐peptide and insulin, as well as the ratios of insulin AUC0–4 h/glucose AUC0–4 h and C‐peptide AUC0–4 h/glucose AUC0–4 h. Glucose AUC0–4 h was significantly reduced from baseline to day 28 compared with the placebo at both doses, and the largest difference was observed in the 75‐mg group. Consequently, C‐peptide AUC0–4 h/glucose AUC0–4 h was significantly increased in the DS‐8500a 75‐mg group compared with the placebo. However, no significant changes in insulin AUC0–4 h, C‐peptide AUC0–4 h or insulin AUC0–4 h/glucose AUC0–4 h were observed (Table 2). Total cholesterol, LDL cholesterol and triglycerides were significantly reduced from baseline compared with the placebo at both doses (Figure 2). High‐density lipoprotein cholesterol tended to be increased, although this was not statistically significant (Figure 2).
Table 2.
Pharmacodynamics
| Placebo (n = 29) | 25 mg DS‐8500a (n = 28) | 75 mg DS‐8500a (n = 27) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Day –1 | Day 28 | Change from day −1 to day 28 | Day −1 | Day 28 | Change from day −1 to day 28 | LSM difference vs placebo | Day −1 | Day 28 | Change from day −1 to day 28 | LSM difference vs placebo (full analysis set) | |
| 2 h‐PPG after breakfast (mg/dL) | 220.5 | 223.1 | 1.4 | 234.9 | 209.4 | −23.5*** | −24.8†† | 222.4 | 205.0 | −18.2** | −19.6† |
| 2 h‐PPG after lunch (mg/dL) | 216.1 | 213.4 | −5.3 | 223.8 | 197.2 | −26.2*** | −20.9† | 229.2 | 205.2 | −21.5*** | −16.2 |
| 2 h‐PPG after dinner (mg/dL) | 234.2 | 233.7 | −1.7 | 239.4 | 227.1 | −12.1* | −10.4 | 242.3 | 225.1 | −16.1** | −14.4 |
| Fasting insulin (μU/mL) | 7.91 | 7.59 | −0.27 | 6.53 | 6.30 | −0.26 | 0.01 | 7.19 | 6.63 | −0.56 | −0.29 |
| Fasting C‐peptide (ng/mL) | 1.44 | 1.43 | −0.01 | 1.30 | 1.29 | −0.02 | −0.01 | 1.47 | 1.44 | −0.03 | −0.02 |
| Glucose AUC0–4 h (mg/dL · h) | 827.6 | 829.2 | −1.39 | 862.57 | 783.72 | −74.16*** | −72.77†† | 832.86 | 756.34 | −78.28*** | −76.89††† |
| Insulin AUC0–4 h (μU/mL · h) | 139.5 | 134.0 | −4.3 | 118.1 | 114.4 | −5.3 | −1.0 | 133.8 | 131.8 | −1.5 | 2.8 |
| C‐peptide AUC0–4 h (ng/mL · h) | 14.97 | 14.6 | −0.36 | 13.98 | 13.86 | −0.16 | 0.21 | 15.26 | 15.56 | 0.32 | 0.69 |
| PYY AUC0–4 h (pg/mL · h) | 461.8 | 447.0 | −23.7 | 464.6 | 469.4 | −2.8 | 20.9 | 520.5 | 549.4 | 46.1* | 69.8† |
| Active GLP‐1 AUC0–4 h (pmol/L · h) | 41.9 | 43.4 | 0.5 | 49.6 | 54.2 | 5.0 | 4.4 | 51.0 | 53.5 | 3.1 | 2.5 |
| Total GLP‐1 AUC0–4 h (pmol/L · h) | 48.96 | 48.43 | −2.2 | 53.0 | 55.01 | 1.3 | 3.5 | 66.7 | 63.71 | −0.4 | 1.8 |
| Glucagon AUC0–4 h (pg/mL · h) | 522.8 | 521.7 | −1.0 | 506.5 | 512.9 | 2.8 | 3.8 | 528.5 | 528.0 | 1.9 | 2.8 |
| GIP AUC0–4 h (pg/mL · h) | 1047.4 | 978.7 | −74.4 | 996.8 | 1292.7 | 281.4*** | 355.8††† | 1193.2 | 1486.7 | 315.6*** | 390.0††† |
| Insulin AUC0–4 h/glucose AUC0–4 h | 21.45 | 20.36 | −1.09 | 17.93 | 19.07 | 1.14 | 1.69 | 21.29 | 22.52 | 1.23 | 2.30 |
| C‐peptide AUC0–4 h/glucose AUC0–4 h | 0.110 | 0.107 | −0.003 | 0.100 | 0.109 | 0.009* | 0.011 | 0.114 | 0.127 | 0.013** | 0.016†† |
Data are presented as the change from baseline to day 28, or least squares mean (LSM) difference vs placebo. *P < 0.05, **P < 0.01 and ***P < 0.001 vs baseline; † P < 0.05, †† P < 0.01 and ††† P < 0.001 vs placebo. AUC0–4 h, area under the concentration‐time curve from 0 to 4 h; GIP, glucose‐dependent insulinotropic peptide; GLP‐1, glucagon‐like peptide‐1; PYY, peptide YY.
Figure 2.
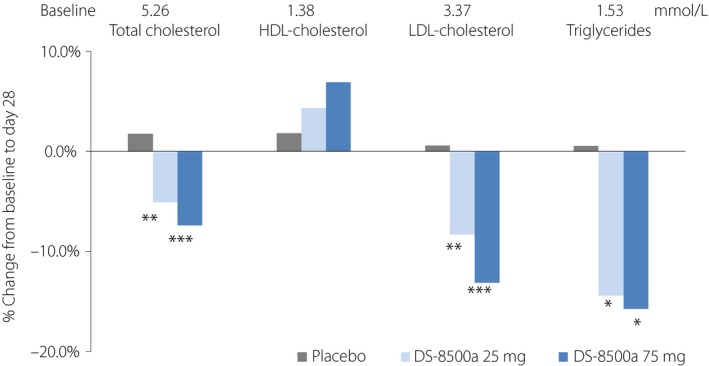
Change from baseline in total cholesterol, high‐density lipoprotein (HDL) cholesterol, low‐density lipoprotein (LDL) cholesterol and triglycerides at day 28 (full analysis set). *P < 0.05, **P < 0.01, ***P < 0.001 vs placebo, ancova.
Analysis of other secondary end‐points at day 28 showed that DS‐8500a at 25 and 75 mg significantly reduced glucose AUC0–24 h from baseline compared with the placebo (change from baseline to day 28: −9.9, −316.5 and −386.8 mg/dL · h in the placebo, 25‐ and 75‐mg groups, respectively; DS‐8500a 25 mg vs placebo P = 0.0044; DS‐8500a 75 mg vs placebo P = 0.0006). Furthermore, there were no significant changes in fasting insulin or fasting C‐peptide between both the DS‐8500a and placebo groups from baseline to day 28, or differences in insulin, active GLP‐1, total GLP‐1 or glucagon in response to meals between the DS‐8500a and placebo groups. However, DS‐8500a at 75 mg significantly increased GIP and PYY at day 28 compared with the placebo, whereas DS‐8500a at 25 mg significantly increased GIP, but not PYY at day 28 compared with the placebo (Table 2).
Safety analysis showed that TEAEs occurred in four patients in the placebo group, and in one and two patients in the 25‐ and 75‐mg DS‐8500a groups, respectively (Table 3). None were considered serious or severe, and none caused study discontinuation. All AEs were resolved. Furthermore, there were no apparent safety signals in terms of laboratory values, bodyweight (mean ± standard deviation change from baseline to day 28: placebo −0.11 ± 0.80 kg; 25‐mg group −0.04 ± 0.85 kg; 75‐mg group 0.05 ± 0.68 kg), vital signs (blood pressure and pulse rate) or 12‐lead electrocardiogram. One TEAE (headache) occurred in one patient in the DS‐8500a 25‐mg group; and one TEAE each of hypoglycemia and toothache occurred in the 75‐mg group. The hypoglycemia event in the 75‐mg group was related to the study drug and thus considered an adverse drug reaction. However, all TEAEs were resolved, none were considered serious or severe, and none of the patients discontinued because of a TEAE.
Table 3.
Treatment‐emergent adverse events during the study
| Placebo (n = 29) | 25 mg DS‐8500a (n = 28) | 75 mg DS‐8500a (n = 28) | |
|---|---|---|---|
| Patients with any TEAE | 4 (13.8) | 1 (3.6) | 2 (7.1) |
| Type of TEAE | |||
| Nasopharyngitis | 1 (3.4) | 0 (0.0) | 0 (0.0) |
| Tinea cruris | 1 (3.4) | 0 (0.0) | 0 (0.0) |
| Hypoglycemia | 0 (0.0) | 0 (0.0) | 1 (3.6) |
| Headache | 0 (0.0) | 1 (3.6) | 0 (0.0) |
| Abdominal pain | 1 (3.4) | 0 (0.0) | 0 (0.0) |
| Toothache | 0 (0.0) | 0 (0.0) | 1 (3.6) |
| Eczema | 1 (3.4) | 0 (0.0) | 0 (0.0) |
| Blood urine present | 1 (3.4) | 0 (0.0) | 0 (0.0) |
Data are presented as number of patients (%). Events were coded using Medical Dictionary for Regulatory Activities version 19.0. TEAE, treatment‐emergent adverse events.
Discussion
We evaluated the effect of DS‐8500a on blood glucose reduction and insulin secretion when administered as add‐on therapy to the DPP‐4 inhibitor, sitagliptin, in Japanese type 2 diabetes mellitus patients with inadequate glycemic control with sitagliptin. As DS‐8500a was co‐administered, synergistic or additive effects cannot be confirmed in the present study. We observed a significant dose‐dependent reduction in 24‐h WMG with both doses of DS‐8500a vs placebo. A significant reduction in FPG was also seen at 75 mg vs placebo, and significant reductions in 2‐h PPG, total cholesterol, LDL cholesterol and triglycerides at both 25‐ and 75‐mg doses, with a tendency toward changes in high‐density lipoprotein cholesterol levels. Importantly, both doses of DS‐8500a were well tolerated without significant TEAEs or discontinuations because of AEs. Additionally, neither dose of DS‐8500a was associated with significant changes in bodyweight, blood pressure or pulse.
Despite maintaining an adequate plasma exposure level to reduce 24‐h WMG, DS‐8500a administration did not significantly improve the 2‐h PPG after dinner compared with the placebo in the present study. This finding was not consistent with that of a previous study in which DS‐8500a was given as monotherapy10. The reason for this discrepancy might be related to differences in disease states or baseline HbA1c between the two studies.
Compared with sitagliptin monotherapy, we observed further reductions in blood glucose levels with the use of DS‐8500a as add‐on therapy. However, although DS‐8500a was expected to further enhance insulin secretion when co‐administered with a DPP‐4 inhibitor, this was not observed. Rather, the insulin secretion for glucose reduction relatively decreased because of the decrease in plasma glucose owing to DS‐8500a.
The data from a pre‐clinical study of DS‐8500a using Zucker fatty rats (Matsumoto K, Yoshitomi T, Takahashi K, Tanaka N, Namiki H, Ishizuka T, Chiba K, Kuroha M, and Shimada T, unpublished data, 2018) and other pre‐clinical studies of other GPR119 compounds5, 15, 16 showed an increase in active GLP‐1 levels. However, data from previous clinical studies of JNJ‐384310558 and GSK2639, and of other GPR119 compounds in type 2 diabetes mellitus patients and present data showed no significant increase in active GLP‐1 levels after breakfast. The difference in the GLP‐1 secretion profile between non‐clinical and clinical studies is considered attributable to species differences between humans and other animals rather than ethnic differences.
In an early phase 2 study (the J201 study, JapicCTI‐142597, NCT02222350), no assay was carried out for GIP, so no comparison can be made in terms of combined effects with sitagliptin10. However, data from the present study showed that GIP levels significantly increased in the DS‐8500a group vs the placebo group. Additionally, data from a non‐clinical study showed that DS‐8500a administered as monotherapy increased GIP levels (Matsumoto K, Yoshitomi T, Takahashi K, Tanaka N, Namiki H, Ishizuka T, Chiba K, Kuroha M, and Shimada T, unpublished data, 2018). GIP has been suggested to be involved in insulin secretion in humans17, 18. However, the involvement of GIP in insulin secretion was not confirmed in the J201 study or the present study. Regarding its involvement in adipocyte activity, GIP was suggested to contribute to blood glucose reduction by facilitating the cellular uptake of nutrients in peripheral tissues19. In transgenic mice using the GIP promoter, GIP secretion was restricted to K cells, and GPR119 messenger ribonucleic acid was expressed in K cells20. Therefore, we believe that in humans, DS‐8500a promotes GIP secretion through GPR119 expression in K cells.
Peptide YY was reported to be degraded by DPP‐421. In the J201 study, no difference in the PYY level was observed in the DS‐8500a group vs the placebo group. However, our present findings showed a significant increase in the PYY level in the 75‐mg group vs the placebo group. This increase was not considered clinically meaningful.
Even though PYY and GLP‐1 are coexpressed, co‐stored and released together from intestinal L cells, only PYY secretion was increased in the present study. A possible explanation is that not all GLP‐1 and PYY secretory granules are the same22. There might be PYY‐predominant or GLP‐1‐predominant L cells that respond differently to cell surface GPR activation23.
Regarding the effect of DS‐8500a on lipid profile, the present study showed that treatment with DS‐8500a significantly improved total cholesterol, LDL cholesterol and TG levels. The favorable changes in the lipid profile suggest that long‐term treatment with DS‐8500a might have a positive effect on cardiovascular events. In rats, GPR119 agonists slowed the systemic appearance of cholesterol after an oral cholesterol load, and they might promote the clearance of triglyceride‐rich lipoproteins from the circulation24. The mechanism of action of the above‐mentioned changes in lipids is unknown. However, we expect this to be clarified by comparative analyses of data on DS‐8500a monotherapy and combination therapy with DPP‐4 inhibitors in future non‐clinical studies.
Regarding safety, both doses of DS‐8500a were well tolerated in the present study, which had a relatively short duration. Of note, neither dose of DS‐8500a was associated with significant changes in blood pressure or pulse rate, or with serious or severe AEs. Furthermore, as DS‐8500a did not affect bodyweight in the present study, DS‐8500a might not provide a potential benefit in terms of bodyweight reduction. All AEs and TEAEs were resolved and none led to study discontinuation.
Some limitations of the present study warrant mention. Although the number of patients included was relatively small, the sample size was deemed to be sufficient to evaluate efficacy and safety based on the results of a preliminary study10. Second, the 28 days of treatment administration might not have been sufficient to show long‐term efficacy and safety of DS‐8500a, although the duration was selected based on safety demonstrated in an earlier study10. Third, the present results were obtained from a well‐specified and highly selected cohort, which included a higher proportion of men than women, so they should not be extrapolated to other populations. Given the benefits to glycemic and lipid profiles and the good tolerability, longer‐term studies are warranted.
In conclusion, in Japanese type 2 diabetes mellitus patients with inadequate glycemic control with sitagliptin therapy, additive blood glucose‐lowering effects were observed with combination therapy of DS‐8500a with sitagliptin. DS‐8500a was well tolerated, and showed favorable changes in lipid profile over the administration period.
Disclosure
Yasuo Terauchi received research support from Astellas Pharma Inc., AstraZeneca K.K., Bayer Yakuhin, Ltd., Daiichi Sankyo Co., Ltd., Dainippon Sumitomo Pharma Co., Ltd., Eli Lilly Japan K.K., Kowa Pharmaceutical Company Ltd., Merck Sharp & Dohme K.K., Mitsubishi Tanabe Pharma Corporation, Nippon Boehringer Ingelheim Co., Ltd., Novo Nordisk Pharma Ltd., Ono Pharmaceutical Co., Ltd., Pfizer Japan Inc., Sanwa Kagaku Kenkyusho Co., Ltd., Sanofi K.K., Shionogi & Co., Ltd., Taisho Toyama Pharmaceutical Co., Ltd. and Takeda Pharmaceutical Co., Ltd.; and speakers’ bureau fees from Astellas Pharma Inc., AstraZeneca K.K., Bayer Yakuhin, Ltd., Daiichi Sankyo Co., Ltd., Dainippon Sumitomo Pharma Co., Ltd., Eli Lilly Japan K.K., Merck Sharp & Dohme K.K., Mitsubishi Tanabe Pharma Corporation, Nippon Boehringer Ingelheim Co., Ltd., Novo Nordisk Pharma Ltd., Ono Pharmaceutical Co., Ltd., Sanwa Kagaku Kenkyusho Co., Ltd., Sanofi K.K., Shionogi & Co., Ltd., Taisho Toyama Pharmaceutical Co., Ltd. and Takeda Pharmaceutical Co., Ltd. Yuichiro Yamada received honoraria from Dainippon Sumitomo Pharma Co., Ltd., Kowa Pharmaceutical Company Ltd. and Sanofi K.K.; and other fees from Dainippon Sumitomo Pharma Co., Ltd., Kowa Pharmaceutical Company Ltd. Hirotaka Watada received research support from Novartis Pharma K.K., Eli Lilly Japan K.K., Taisho Toyama Pharmaceutical Co., Ltd., MSD K.K., Astellas Pharma Inc., AstraZeneca K.K., Ono Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Sanofi K.K., Daiichi Sankyo Co., Ltd., Dainippon Sumitomo Pharma Co., Ltd., Takeda Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Terumo Corporation, Nippon Boehringer Ingelheim Co., Ltd., Novo Nordisk Pharma Ltd., Pfizer Japan Inc., Benefit One Health Care Inc., Mochida Pharmaceutical Co., Ltd. and Nitto Boseki Co., Ltd.; and speakers’ bureau fees from Astellas Pharma Inc., AstraZeneca K.K., Kowa Pharmaceutical Co., Ltd., Sanofi K.K., Takeda Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Novartis Pharma K.K., Novo Nordisk Pharma Ltd., Nippon Boehringer Ingelheim Co., Ltd., Merck Sharp & Dohme K.K. and Dainippon Sumitomo Pharma Co., Ltd. Yasuhiko Nakatsuka, Kazuhito Shiosakai, Takuo Washio and Takashi Taguchi are employees of Daiichi Sankyo Co., Ltd.
Supporting information
Figure S1 ¦ Study design. HbA1c, hemoglobin A1c; R, randomization; T2DM, type 2 diabetes mellitus.
Figure S2 ¦ Participant disposition. †‘Other’ denotes that the condition of blood vessels was poor at day 28 and drawing blood was not possible.
Appendix S1 ¦ List of participating institutions.
Appendix S2 ¦ Details of the calculation of 24‐h weighted mean glucose.
Acknowledgments
This study was funded by Daiichi Sankyo Co., Ltd. The authors thank Helen Roberton and Ludovic Croxford, PhD, of Edanz Medical Writing for providing medical writing services.
J Diabetes Investig. 2018
Clinical Trial Registry
ClinicalTrials.gov and Japan Pharmaceutical Information Center
NCT02685345 and JapicCTI‐163136
References
- 1. International Diabetes Federation . IDF Diabetes Atlas, 7th edn Brussels, Belgium: International Diabetes Federation, 2015. http://www.diabetesatlas.org [Google Scholar]
- 2. Ramachandran A, Snehalatha C, Shetty AS, et al Trends in prevalence of diabetes in Asian countries. World J Diabetes 2012; 3: 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Standards of medical care in diabetes‐2016: summary of revisions. Diabetes Care 2016; 39(Suppl 1): S1–S106. [DOI] [PubMed] [Google Scholar]
- 4. Soga T, Ohishi T, Matsui T, et al Lysophosphatidylcholine enhances glucose‐dependent insulin secretion via an orphan G‐protein‐coupled receptor. Biochem Biophys Res Commun 2005; 326: 744–751. [DOI] [PubMed] [Google Scholar]
- 5. Chu ZL, Carroll C, Alfonso J, et al A role for intestinal endocrine cell‐expressed G protein‐coupled receptor 119 in glycemic control by enhancing glucagon‐like Peptide‐1 and glucose‐dependent insulinotropic Peptide release. Endocrinology 2008; 149: 2038–2047. [DOI] [PubMed] [Google Scholar]
- 6. Chu ZL, Jones RM, He H, et al A role for β‐cell‐expressed G protein‐coupled receptor 119 in glycemic control by enhancing glucose‐dependent insulin release. Endocrinology 2007; 148: 2601–2609. [DOI] [PubMed] [Google Scholar]
- 7. Yoshida S, Ohishi T, Matsui T, et al The role of small molecule GPR119 agonist, AS1535907, in glucose‐stimulated insulin secretion and pancreatic β‐cell function. Diabetes Obes Metab 2011; 13: 34–41. [DOI] [PubMed] [Google Scholar]
- 8. Katz LB, Gambale JJ, Rothenberg PL, et al Effects of JNJ‐38431055, a novel GPR119 receptor agonist, in randomized, double‐blind, placebo‐controlled studies in subjects with type 2 diabetes. Diabetes Obes Metab 2012; 14: 709–716. [DOI] [PubMed] [Google Scholar]
- 9. Nunez DJ, Bush MA, Collins DA, et al Gut hormone pharmacology of a novel GPR119 agonist (GSK1292263), metformin, and sitagliptin in type 2 diabetes mellitus: results from two randomized studies. PLoS ONE 2014; 9: e92494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Inagaki N, Chou HS, Tsukiyama S, et al Glucose‐lowering effects and safety of DS‐8500a, a G protein‐coupled receptor 119 agonist, in Japanese patients with type 2 diabetes: results of a randomized, double‐blind, placebo‐controlled, parallel‐group, multicenter, phase II study. BMJ Open Diabetes Res Care 2017; 5: e000424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goldstein BJ, Feinglos MN, Lunceford JK, et al Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase‐4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30: 1979–1987. [DOI] [PubMed] [Google Scholar]
- 12. Rohlfing CL, Wiedmeyer HM, Little RR, et al Defining the relationship between plasma glucose and HbA1c: analysis of glucose profiles and HbA1c in the diabetes control and complications trial. Diabetes Care 2002; 25: 275–278. [DOI] [PubMed] [Google Scholar]
- 13. Nathan DM, Kuenen J, Borg R, et al Translating the A1C assay into estimated average glucose values. Diabetes Care 2008; 31: 1473–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nonaka K, Tsubouchi H, Okuyama K, et al Effects of once‐daily sitagliptin on 24‐h glucose control following 4 weeks of treatment in Japanese patients with type 2 diabetes mellitus. Horm Metab Res 2009; 41: 232–237. [DOI] [PubMed] [Google Scholar]
- 15. Bahirat UA, Shenoy RR, Goel RN, et al APD668, a G protein‐coupled receptor 119 agonist improves fat tolerance and attenuates fatty liver in high‐trans fat diet induced steatohepatitis model in C57BL/6 mice. Eur J Pharmacol 2017; 801: 35–45. [DOI] [PubMed] [Google Scholar]
- 16. Park EY, Kim EH, Kim CY, et al Angelica dahurica extracts improve glucose tolerance through the activation of GPR119. PLoS ONE 2016; 11: e0158796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Preitner F, Ibberson M, Franklin I, et al Gluco‐incretins control insulin secretion at multiple levels as revealed in mice lacking GLP‐1 and GIP receptors. J Clin Invest 2004; 113: 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 2003; 26: 2929–2940. [DOI] [PubMed] [Google Scholar]
- 19. Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev 2008; 60: 470–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parker HE, Habib AM, Rogers GJ, et al Nutrient‐dependent secretion of glucose‐dependent insulinotropic polypeptide from primary murine K cells. Diabetologia 2009; 52: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aaboe K, Knop FK, Vilsbøll T, et al Twelve weeks treatment with the DPP‐4 inhibitor, sitagliptin, prevents degradation of peptide YY and improves glucose and non‐glucose induced insulin secretion in patients with type 2 diabetes mellitus. Diabetes Obes Metab 2010; 12: 323–333. [DOI] [PubMed] [Google Scholar]
- 22. Grunddal KV, Ratner CF, Svendsen B, et al Neurotensin is coexpressed, coreleased, and acts together with GLP‐1 and PYY in enteroendocrine control of metabolism. Endocrinology 2016; 157: 176–194. [DOI] [PubMed] [Google Scholar]
- 23. Engelstoft MS, Egerod KL, Holst B, et al A gut feeling for obesity: 7TM sensors on enteroendocrine cells. Cell Metab 2008; 8: 447–449. [DOI] [PubMed] [Google Scholar]
- 24. Brown KK, Shadoan MK, Croom DK, et al Activation of GPR119 reduces the appearance of labeled cholesterol in an oral fat tolerance test. Diabetes 2012; 61(Suppl 1): A160. (abstract 631‐P). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 ¦ Study design. HbA1c, hemoglobin A1c; R, randomization; T2DM, type 2 diabetes mellitus.
Figure S2 ¦ Participant disposition. †‘Other’ denotes that the condition of blood vessels was poor at day 28 and drawing blood was not possible.
Appendix S1 ¦ List of participating institutions.
Appendix S2 ¦ Details of the calculation of 24‐h weighted mean glucose.