

Abstract
Diabetic polyneuropathy (DPN) continues to be generally considered as a “microvascular” complication of diabetes mellitus alongside nephropathy and retinopathy. The microvascular hypothesis, however, might be tempered by the concept that diabetes directly targets dorsal root ganglion sensory neurons. This neuron‐specific concept, supported by accumulating evidence, might account for important features of DPN, such as its early sensory neuron degeneration. Diabetic sensory neurons develop neuronal atrophy alongside a series of messenger ribonucleic acid (RNA) changes related to declines in structural proteins, increases in heat shock protein, increases in the receptor for advanced glycation end‐products, declines in growth factor signaling and other changes. Insulin is recognized as a potent neurotrophic factor, and insulin ligation enhances neurite outgrowth through activation of the phosphoinositide 3‐kinase–protein kinase B pathway within sensory neurons and attenuates phenotypic features of experimental DPN. Several interventions, including glucagon‐like peptide‐1 agonism, and phosphatase and tensin homolog inhibition to activate growth signals in sensory neurons, or heat shock protein overexpression, prevent or reverse neuropathic abnormalities in experimental DPN. Diabetic sensory neurons show a unique pattern of microRNA alterations, a key element of messenger RNA silencing. For example, let‐7i is widely expressed in sensory neurons, supports their growth and is depleted in experimental DPN; its replenishment improves features of DPN models. Finally, impairment of pre‐messenger RNA splicing in diabetic sensory neurons including abnormal nuclear RNA metabolism and structure with loss of survival motor neuron protein, a neuron survival molecule, and overexpression of CWC22, a splicing factor, offer further novel insights. The present review addresses these new aspects of DPN sensory neurodegeneration.
Keywords: Diabetic polyneuropathy, Neurodegeneration, Sensory ganglia
Introduction
Diabetes mellitus is a serious chronic disease characterized by hyperglycemia that results from insulin deficiency as a result of autoimmune‐mediated destruction of β‐cells of the pancreas, type 1 diabetes mellitus, or resistance to the actions of insulin, type 2 diabetes mellitus1, 2. The World Health Organization estimated that 422 million people worldwide were living with diabetes in 20143. The complications of diabetes mellitus include retinopathy, nephropathy, atherosclerosis and neuropathy. Diabetic neuropathies are amongst the most common chronic complications, targeting approximately 50% of persons with diabetes4. Diabetic neuropathies develop diverse clinical manifestations, such as sensory loss and pain, and put patients at high risk for foot ulcers, and amputation, an irreversible complication5, 6, 7. One‐third of patients with neuropathy experience positive symptoms, including spontaneous pain, paresthesia and allodynia, and this is often called painful diabetic neuropathy (PDN)8. Despite the global prevalence and severe complications of diabetes mellitus, the pathophysiological mechanisms of diabetic neuropathies have not been elucidated. Beyond strict control of glucose levels, therapy that can unequivocally arrest or reverse progressive neuropathy is still not available, although there are important symptomatic options in pain management (see other reviews of PDN9, 10, 11).
Diabetic neuropathies manifest in several different forms, including sensory, motor, focal/multifocal and autonomic neuropathies1, 5, 7, 12. The most common type is diabetic distal symmetric polyneuropathy (DPN), accounting for approximately 75% of diabetic neuropathies6, 13, 14, 15, 16. Patients with DPN develop gradual and insidious damage to the distal terminals of sensory neurons first, with symptoms of tingling, pain or loss of sensation in their toes. If their diabetes is poorly controlled, DPN advances with sensory loss involving more proximal extremities and even the central chest, the terminal portions of the intercostal nerves. There is generally involvement of motor nerves later17, 18. Epidermal biopsies of diabetes patients have confirmed that loss of sensory axon terminals in the skin of the distal extremity is greater than that observed at more proximal sites19. This pattern of disease onset might suggest that sensory neuronal cell bodies, or perikarya, in dorsal root ganglia (DRG) are targeted early by several forms of diabetic impairment and then undergo a “dying back” process of neurodegeneration (Figure 1). Investigations into DRG pathology in human diabetes are lacking, because human DRG biopsies are not ethical to carry out and because of their rapid degradation during the post‐mortem interval at autopsy20. Nevertheless, several animal models show neuronal atrophy, not necessarily associated with neuron dropout despite the loss of foot pad epidermal sensory axons21, 22, 23. Atrophic neurons in DRG also develop a series of gene expression changes related to structure, neuronal stress and protection24, 25. Taken together, we have emphasized the hypothesis that direct targeting of DRG by diabetes can account for the prominent and often early sensory neuron degeneration that patients with DPN develop6, 13, 18, 28, 29, 30, 31, 32.
Figure 1.
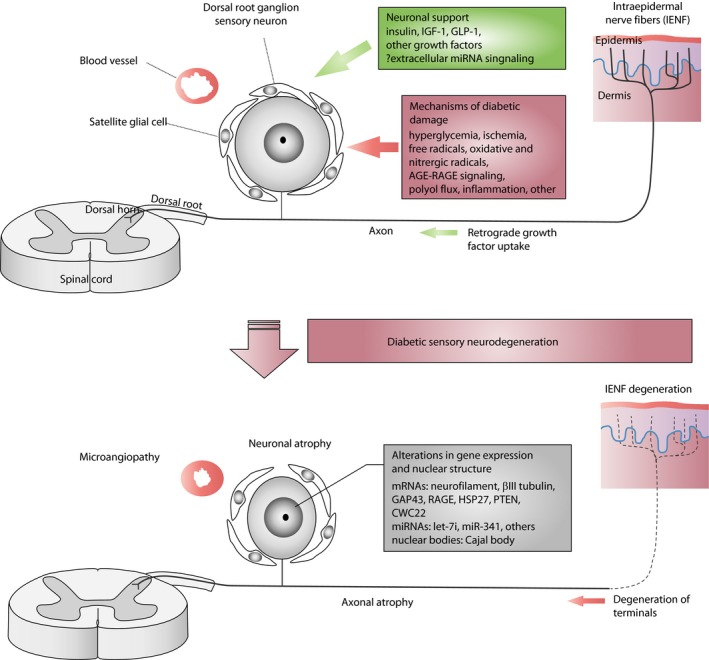
Simplified schematic drawing of sensory neurodegeneration during diabetes. AGE, advanced glycation end‐product; GAP43, growth‐associated protein 43; GLP‐1, glucagon‐like peptide‐1; HSP27, heat shock protein 27; IGF‐1, insulin‐like growth factor‐1; mRNA, messenger ribonucleic acid; miRNA, micro ribonucleic acid; PTEN, phosphatase and tensin homolog; RAGE, receptor for advanced glycation end‐product.
Diverse pathogenic etiologies, including microvascular‐induced ischemia, the formation of extracellular advanced glycation end‐products (AGEs), inflammatory cytokines, increased aldose reductase activity and oxidative stress, are considered in the development of DPN. These might target neuronal perikarya, axons, Schwann cells and nerve or ganglia microvessels. The present review begins with a discussion of the microvascular hypothesis, then addresses selective aspects of sensory neurodegeneration associated with the intracellular insulin signaling pathway. In addition, we provide new evidence that sensory neuron degeneration is linked not only to aberrant intracellular signaling with messenger ribonucleic acid (mRNA) expression changes, but also with dysregulated mRNA processing mediated by microRNA (miRNA) post‐transcriptional alterations. Understanding sensory neuron degeneration in the context of aberrant RNA processing might give rise to new therapeutic strategies.
Microvascular Hypothesis
DPN has long been described to be a microvascular complication of diabetes alongside nephropathy and retinopathy12, 28, 29, 30, 31, 32. Aberrant changes in endoneurial capillary morphology and vascular reactivity under diabetic conditions might contribute to the development of diabetic neuropathy through endoneurial ischemia. Pathological investigations of sural nerve biopsies from diabetes patients showed capillary basement membrane thickening, capillary pericyte degeneration and endothelial hyperplasia in endoneurial microvessels31, 33.The presence of endoneurial microangiopathy appeared to precede the development of peripheral neuropathy34. In addition, imaging of exposed sural nerves in patients with DPN has suggested the presence of microvascular abnormalities in the epineurial vessels35. A number of experimental DPN models have reported that both nerve blood flow (NBF) and endoneurial oxygen tension are reduced in the sciatic nerve, and their conduction velocities are reduced in proportion to the changes of NBF30, 36. Endothelial cell dysfunction is commonly considered to be a mainstay in the pathogenesis of diabetic microvascular diseases37, 38. For example, endothelium‐dependent vasodilation is impaired in the vasculature of experimental diabetic animals and humans with diabetes38, 39. Diabetes‐induced endothelial dysfunction is attributed to oxidative stress, impaired metabolic signal transduction pathways, impaired release of vasoactive molecules and decreased smooth muscle sensitivity32. The molecular mechanisms of microvascular damage in DPN might be mediated by intracellular signal transduction pathways in endothelial cells involving the polyol pathway40, 41, 42, 43, 44, 45, protein kinase C46, 47, AGEs48, 49, 50, 51, 52, 53, 54, 55, angiotensin II56, 57, 58, 59 and abnormal mitochondrial activity60, 61, 62, 63, 64, 65. Therapeutic approaches to improve vascular dysfunction by targeting these molecules have identified recovery of measures of DPN in diabetic animal models.
However, the selective involvement of sensory axons, or even nerve trunks more generally, is difficult to attribute to this hypothesis. Nerve trunks have an overlapping blood supply from end arteries that form multiple connections, or anastomoses, and only relatively severe ischemia provokes axonal degeneration. This is dramatically different from ischemia‐prone tissue, such as the brain and spinal cord66, 67. Furthermore, DPN can develop in children at early ages (e.g. 3 months) after the development of insulin‐dependent diabetes mellitus without the vascular complications of long‐term diabetes68. The Zochodne laboratory has also failed to identify convincing evidence for an initial microvascular trigger for polyneuropathy using a variety of diabetic experimental models in the hands of differing investigators. Morphological studies of the vasa nervorum in DPN models have not identified loss of vessels or decreased vessel calibers, but instead increased luminal caliber or angiogenesis69. Although some laboratories have identified reductions in NBF as mentioned, reductions in NBF are not observed in all models of diabetes mellitus, and some long‐term models in rats show normal NBF70 (several technical factors could contribute to these discrepant findings, summarized previously71). Sural nerve serial blood flow measures in patients with mild diabetic polyneuropathy, studied by laser Doppler flowmetry, did not decline over a 1‐year time‐period despite a mild ongoing reduction in nerve fiber density, whereas patients with more severe loss of nerve fibers tended to have higher rates of blood flow72. To resolve these contradictions, some investigators recently proposed an idea that disturbances in capillary flow patterns associated with microvascular changes, instead of a global reduction of blood flow in the whole nerve, can reduce the amount of oxygen and glucose to be extracted by the nerve31. In this hypothesis, there are consequent abnormalities of nerve function and perhaps frank axon damage. Individual capillary blood flows in the tissue might be variable, and capillary transit times across a vascular bed have a certain distribution with the standard deviation referred to as capillary transit time heterogeneity (CTH). Given this concept, the development of microangiopathy in DPN has been suggested to correspond to increases in CTH. Mild increases in CTH, which represents endothelial damage without loss of function, lead to the reduction of oxygen extraction accompanied by a compensatory increase of tissue blood flow. If CTH further increases with progression of microangiopathy, the compensatory flow response might be lost because of more advanced endothelial dysfunction, and the low tissue oxygen tension could contribute to nerve dysfunction or damage. Although there remain difficulties in linking microvascular changes with an initial trigger for DPN, it is likely that microangiopathy develops in parallel with early functional and structural changes of the nerve, and both are prominent later in the development of DPN. Direct targeting of DRG by diabetes could account for the apparent selective sensory abnormalities that patients with early DPN develop, discussed next.
Direct Neuronal Involvement of Diabetes: Insulin, Glucagon‐Like Peptide‐1, Receptor for Age and Heat Shock Protein 27
DRG contain the cell bodies of primary sensory neurons responsible for conveying sensory information from the periphery to the spinal cord (Figure 1). Sensory neurons in the DRG have an attenuated protective neurovascular barrier compared with the blood–brain or blood–nerve barrier, making them vulnerable to toxic circulating agents, unlike peripheral nerve trunks, the brain and spinal cord73, 74, 75. Blood capillaries are abundant within the DRG, and are more permeable to low and high molecular weight molecules than the brain or endoneurium74, 76, 77. In addition, DRGs have higher blood flow with features of partial autoregulation, unlike the endoneurium, as well as lower oxygen tensions78. These physiological features suggest that DRGs might be susceptible to microangiopathy in diabetes, leading to sensory neuron damage. In our laboratory, we identified that DRG blood flow had selective reductions in a rat model of diabetes mellitus, whereas endoneurial blood flow in the nerve trunk was preserved79. Despite these physiological differences, whether primary reductions in DRG blood flow render ischemic neuronal damage or are simply secondary reductions is unclear.
Diabetic sensory neurons have functional and structural alterations both at the level of perikarya in DRGs and of axons in the distal terminals of the epidermis (Figure 1). Neuronal and distal axon atrophy accompany functional deficits, such as conduction slowing and loss of sensation in long‐term experimental diabetes21, 23, 26. However, neuronal atrophy does not always evolve into overt neuron loss22, but is associated with a series of mRNA changes related to declines in structural proteins, such as neurofilament and βIII tubulin, increases in heat shock protein 27 (HSP27) and the receptor for AGE (RAGE), and declines in growth proteins, such as growth‐associated protein 4324, 26, 80, 81. Some of these changes depend on the DPN model and species. However, given these findings, we have examined the alteration of gene expression related to cell survival and growth in DRG sensory neurons, focusing on long‐term animal models of experimental DPN that might inform us about chronic human disease.
Insulin receptors (IRs), which display a critical role of glucose homeostasis, are expressed in sensory neurons82, 83, and for several decades insulin itself has been recognized as a potent growth or trophic factor for neurons (Figure 2)84. Most DRG sensory neurons appear to express IRs24. IR signaling, in turn, utilizes well established growth‐related downstream transduction cascades, such as the phosphoinositide 3‐kinase (PI3K)–protein kinase B (Akt) signaling pathway, including a gain in plasma membrane levels of glucose uptake transporters85. The PI3K–Akt pathway is a central pathway involved in cell survival, growth and proliferation, and its activation leads to increased axon growth86, 87. In summary, IRs undergo autophosphorylation by ligand binding, after which they develop tyrosine kinase activity, and finally activate through insulin receptor substrate proteins (IRS‐1 and IRS‐2). IRS proteins activate the PI3K–Akt pathway by recruiting and activating PI3K, leading to the generation of second messenger phosphatidylinositol (3,4,5)‐trisphosphate (PIP3). Membrane‐bound PIP3 recruits and activates 3‐phosphoinositide‐dependent protein kinase‐1, which phosphorylates and activates Akt. However, the conversion of PIP3 to PIP2 by phosphatase and tensin homolog (PTEN) reverses this growth pathway and thus antagonizes Akt signaling88. Activated Akt phosphorylates a large number of downstream targets related to cell proliferation, growth and survival, potentially including the mammalian target of rapamycin signal pathway, the CDK inhibitors p21 and p27, glycogen synthase kinase 3, transcription factors Forkhead box O3, and proapoptic Bcl‐2 family proteins BCL2‐associated X protein and Bcl‐2‐associated death promoter85, 89. Dissociated adult rat sensory neurons treated with insulin in vitro have enhanced dose‐dependent neurite outgrowth90. In the peripheral nervous system in vivo, nerve crush injury induces the upregulated expression of IRs in regenerating axons and cell bodies of DRG, and systemic or intrathecal insulin administration accelerates maturation of regenerating axons distal to a nerve crush injury91, 92.
Figure 2.
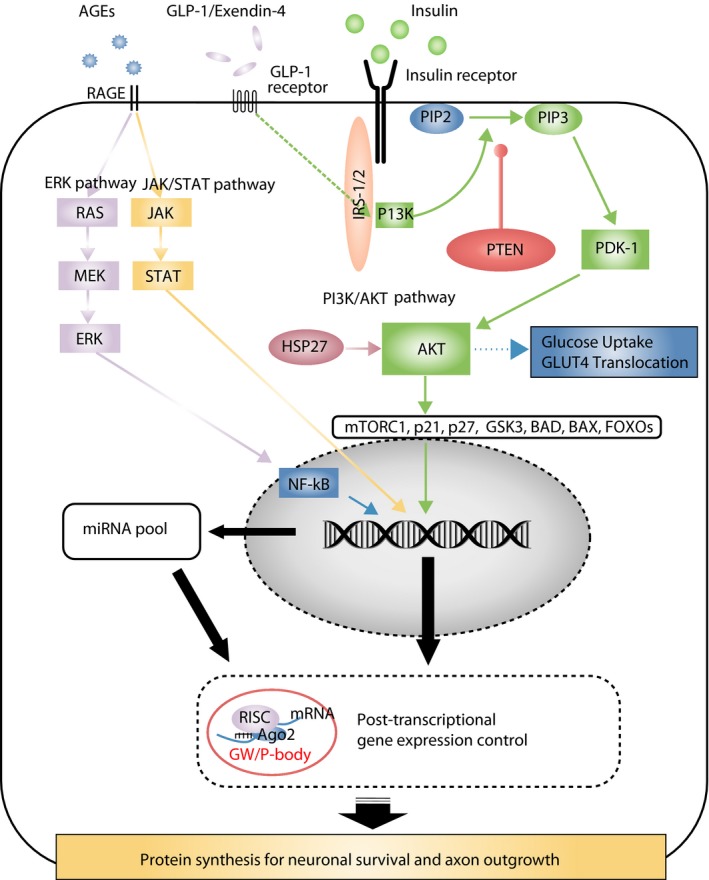
Signal transductions and epigenetics in diabetic sensory neurons; Insulin, glucagon‐like peptide‐1 (GLP‐1), receptor for advanced glycation end‐product (RAGE) and GW/P bodies. AGE, advanced glycation end‐product; AKT, protein kinase B; BAD, Bcl‐2‐associated death promoter; BAX, Bcl‐2‐associated X protein; ERK, extracellular‐signal‐regulated kinase; FOXO, Forkhead box O3; GLUT4, glucose transporter type 4; GSK3, glycogen synthase kinase 3; HSP27, heat shock protein 27; IGF‐1, insulin‐like growth factor‐1; IRS, insulin receptor substrate protein; JAK, c‐Jun N‐terminal kinase; MEK, mitogen‐activated protein kinase/extracellular‐signal‐regulated kinase kinase; mRNA, messenger ribonucleic acid; miRNA, micro ribonucleic acid; mTORC, mammalian target of rapamycin complex; PDK‐1, phosphoinositide‐dependent kinase 1; PI3K, phosphoinositide 3‐kinase; PIP3, phosphatidylinositol (3,4,5)‐trisphosphate; PTEN, phosphatase and tensin homolog; NF‐κB, nuclear factor kappa B; RAS, reactive oxygen species; RISC, ribonucleic acid‐induced silencing complex; STAT, signal transducers and activators of transcription.
Given these growth supportive roles of insulin in peripheral neurons, we have hypothesized that deficiency of insulin–PI3K–Akt trophic support during insulinopenic diabetes mellitus influences the development of DPN. For example, direct neuronal or axonal insulin administration, even if insufficient to alter blood glucose levels, reverses diabetic neuropathic changes in type 1 diabetes mellitus models that are characterized by the absence of the insulin ligand82, 93, 94, 95.
However, in type 2 diabetes mellitus models, even high‐dose insulin might fail to prevent or reverse DPN. For example, type 2 diabetes mellitus might be characterized by normal or elevated levels of circulating insulin associated with “insulin resistance” involving muscle, liver or adipose tissue. Several laboratories, including our own, have advanced the concept that neurons might also be susceptible to “insulin resistance” at the level of neurotrophic support96, 97, 98, 99, 100. Grote et al.98 showed that the DRG and sciatic nerve of ob/ob mice with type 2 diabetes mellitus had blunted Akt activation with insulin and insulin‐like growth factor‐1, including decreased DRG insulin receptor expression and upregulation of c‐Jun N‐terminal kinase activity, a mediator of insulin resistance in other tissues. Additional work has noted that insulin resistance in neurons might be linked to IRS‐2 serine phosphorylation99. Our laboratory showed that high‐dose insulin or repeated chronic low‐dose insulin blunted subsequent challenges of insulin to support growth. Blunted signaling in sensory neurons involved downregulation of the insulin receptor β‐subunit, upregulated glycogen synthase kinase 3β and downregulated phosphorylated Akt97. Thus, mechanisms of neuronal insulin resistance in type 2 diabetes mellitus include declines in IR expression, changes in IRS phosphorylation status and increases in glycogen synthase kinase 3β mRNA levels, all associated with impaired PI3K–phosphorylated Akt activation. Thus, taken together, impaired neurotrophic support might indeed contribute to the development of DPN.
The direct neurotrophic action of glucagon‐like peptide‐1 (GLP‐1) could offer further options for DPN treatment (Figure 2). GLP‐1 is an incretin peptide, secreted by the intestine in response to meal ingestion101. The GLP‐1 receptors are highly expressed on islet β‐cells, and their actions include enhancing insulin secretion. The GLP‐1 receptors are also widely expressed in non‐islet cells including those of the nervous system102. A GLP‐1 agonist, exendin‐4, like insulin, enhanced neurite outgrowth of sensory neurons and attenuated features of experimental DPN models of both type 1 and type 2 diabetes mellitus103, 104, 105.
Diabetes mellitus is associated with the production of AGEs resulting from non‐enzymatic glycation and oxidation of proteins and lipids. AGEs permanently accumulate in a variety of tissues and bind to specific receptors including RAGE. RAGE ligation in turn has been linked to the development of diabetic complications106, 107. AGEs and other ligands, including S100/calgranulin family of pro‐inflammatory molecules and high‐mobility group box 1 protein, trigger several signal transduction pathways (Figure 2). For example, binding of these ligands to RAGE results in the persistent activation of the transcription factor nuclear factor kappa B (NF‐κB)108. In sural nerve biopsies from patients with DPN, activated NF‐κB was colocalized with interkeukin‐6 and RAGE within the vasa nervorum109. Diabetes‐induced activation of NF‐κB was blunted in sciatic nerves of RAGE‐null mice, and loss of pain perception in DPN was prevented in RAGE‐null mice109. In addition, diabetic RAGE‐null mice had improved peripheral nerve regeneration, linked to altered macrophage responses110. Macrophages play an essential clearance role in the facilitation of regeneration in nerve.
RAGE ligation might generate diabetic complications through its impact on microvessels, whereas RAGE is also expressed by sensory neurons. AGE‐RAGE appears important for the support and growth of neurons. For example, its activation enhances the outgrowth of adult sensory neurons in vitro through the NF‐κB, c‐Jun N‐terminal kinase–signal transducer and activator of transcription–extracellular signal‐regulated kinase pathways111. Similarly, blockade of the ligand–RAGE axis suppressed nerve regeneration after crush injury in mice112. In diabetes mellitus, however, its overactivation could contribute to the DPN phenotype. In our laboratory, RAGE null mice showed protection from motor and sensory nerve conduction slowing at 8 weeks after diabetes induction, but the protection was less significant by 16 weeks113. Given these complexities, further investigation is required to clarify at what stage activation of the AGE–RAGE axis is protective or harmful to the peripheral nervous system during diabetes mellitus.
HSPs are molecular chaperones that mediate the repair or degradation of denatured proteins after stress114. Expression of one member of this extensive family, HSP27, is elevated in sensory neurons of experimental DPN models26. HSP27 knockdown or overexpression are respectively associated with attenuated or improved regenerative properties after nerve injury in mice115, 116. Overexpression of a human transgene of HSP27 in type 1 diabetes mellitus mice prevented loss of thermal sensation, mechanical allodynia, epidermal axon loss and sensory conduction slowing. RAGE, NF‐κB and activated caspase‐3 were attenuated by the transgene117. Another finding was that the protective impact by the HSP27 transgene was greater in female mice than in male mice. While we speculated on a possible role of estrogen related to HSP27 in that work117, in more recent work we have identified significant differences in electrophysiological features of DPN between male and female mice after diabetes induction113. Therefore, sex differences might be informative in sorting mechanisms in the pathogenesis of DPN.
Regeneration Strategies: Sensory Neurons and Tumor Suppressors
To reverse the neuropathic deficits of DPN, an important strategy might involve activation of intrinsic neurotrophic pathways including PI3K–Akt signaling. The pathogenesis of DPN involves degeneration, but also a deficit in regenerative capacity. The mechanisms of regenerative failure in diabetes might include an unsupportive microenvironment around axons or growth cones resulting from ischemia and microangiopathy of the local injury milieu, impaired macrophage clearance, altered basement membrane regenerative cues, Schwann cell dysfunction and lack of growth factors118. Removal of inhibitory extracellular matrix molecules and the addition of growth factors are potentially important strategies to accomplish regeneration outcomes. However, most growth factors offer selective support for only the neuron subclasses that express relevant receptors, such as TrkA, TrkB, TrkC, gp120, Ret and others. In diabetes, there is also evidence that specific growth factor receptors are downregulated in sensory neurons26. Manipulation of downstream growth signals, therefore, could be essential to enhance axonal plasticity and regeneration in the setting of diabetic abnormalities. From this point of view, our laboratory has focused on manipulating intrinsic “brake” molecules to regulate growth pathways. Such “brakes” include those within the class of “tumor suppressors” that help to inhibit oncogenic growth119, 120. PTEN is the first example of this type of therapeutic target studied in diabetes mellitus, a molecule that inhibits the PI3K–Akt signaling pathway (Figure 2). PTEN is mutated in a variety of human tumors121, 122, 123. The role of PTEN in the nervous system at both the central and peripheral levels has been recently elucidated, and its deletion has been suggested as a key regenerative strategy124, 125, 126, 127, 128, 129. PTEN is expressed in sensory neurons, prominently in IB4 non‐peptidergic sensory neurons that show restrained growth properties130. In sensory neurons in vitro, PTEN inhibition enhances neurite outgrowth, and, after nerve transection in rats in vivo, PTEN inhibition also accelerates the regrowth of axons from the proximal stump125. Furthermore, in mice with DPN, PTEN mRNA and protein expression are upregulated in sensory neurons, a surprising finding that identifies a new mechanism of regenerative failure in diabetes mellitus. Local DRG PTEN knockdown after focal nerve injuries in diabetic mice using non‐viral small interfering RNA delivery improved the recovery of motor compound action potential amplitudes, conduction velocities of motor and sensory axons, numbers and calibers of regenerating myelinated axons, and epidermial axon reinnervation131. These findings indicated that diabetes upregulates a regenerative “brake” and its release improves axon growth failure in DPN.
Sensory Neurodegeneration: New Epigenetic Therapeutic Targets
The term “neurodegeneration” describes a form of gradual neural deterioration in part characterized by progressive neuroaxonal atrophy and dysfunction. Neurodegeneration categorizes disorders of the nervous system including Alzheimer's disease, Parkinson's disease, Huntington's disease and amyotrophic lateral sclerosis. The molecular pathways of neurodegenerative diseases have been intensively studied, suggesting common mechanisms including oxidative stress, excitotoxicity, mitochondrial dysfunction, protein misfolding and aggregation, ubiquitin‐proteasome system dysfunction, and inflammation132, 133. Sensory neurodegeneration in diabetes might also share common molecular mechanisms with these diseases. In previous chapters, we described sensory neuronal atrophy, loss of terminal innervation and neuronal dysfunction, linked to aberrant intracellular growth through signaling pathways, such as insulin–PI3K–Akt. These intracellular alterations might involve not only a shift in gene expression at the transcriptional level, but also altered epigenetic mRNA processing at the post‐transcriptional level81. New evidence, recently explored in our laboratory, suggests that diabetes might promote sensory neuron dysfunction that involves aberrant mRNA splicing and that resembles some forms of motor neuron disease23.
To explore the role of epigenetic changes in diabetic sensory neurons, we first analyzed mRNA and miRNA profiles of DRGs in mice with type 1 diabetes mellitus compared with control mice by microarray81. The microarray examined 28,869 mRNAs, and identified 261 mRNAs that included 91 upregulated and 170 downregulated for a difference of at least 1.5‐fold change in diabetes mellitus samples. Of these, 24 (5 downregulated and 19 upregulated) achieved a statistically significant difference of P < 0.05 (Figure 3a). Most of these mRNAs were coded for proteins of unknown function in sensory neurons or diabetes. However, one prominently upregulated molecule, CWC22, was classified as a pre‐mRNA splicing factor.
Figure 3.
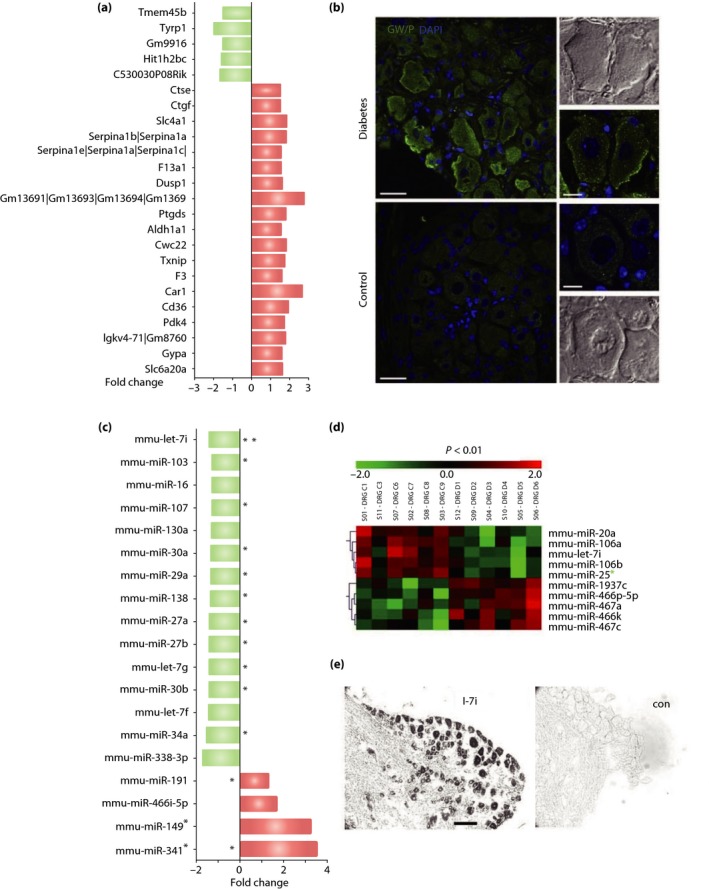
Evidence for gene expression changes and micro ribonucleic acid (miRNA) regulation in diabetic sensory neurons. (a) mRNA microarry analysis identifies 24 that are significantly different between diabetic and non‐diabetic mice at the P < 0.05 level with at least a 1.5‐fold change. (b) Diabetic dorsal root ganglion (DRG) sensory neurons had upregulation of GW/P bodies. Scale bar: 30 μm in large panels, 10 μm in insets. (c) Selected higher abundance miRNAs are shown indicating fold change and significance level at P < 0.10. (d) Heat map results in diabetic mice DRG miRNA microarray compared with non‐diabetic mice at P < 0.01. (e) In situ hybridization of mouse DRG identifies let‐7i expression in DRG sensory neurons. Reproduced from Cheng et al.81 with permission.
MicroRNAs (miRNAs) are small non‐coding RNAs of 18–23 nucleotides that bind to target sequences in mRNAs, resulting in suppressed gene expression, a key element of post‐transcriptional RNA silencing. Precursor miRNAs are exported from the nucleus and processed to form single‐stranded small RNA. Components of miRNA machinery, such as RNA‐induced silencing complex, a protein complex that cleaves mRNA, localizes in cytoplasmic structures called GW/P bodies, which function as sites of both mRNA degradation and storage of translationally repressed mRNAs134, 135, 136, 137. Diabetic sensory neurons had upregulated populations of GW/P bodies (Figure 3b)81, suggesting overt structural evidence of altered miRNA‐mediated mRNA processing by diabetes‐induced stress. As in the case of mRNAs, miRNA microarray analysis of chronic experimental diabetic DRGs identified 19 differentially expressed miRNAs (12 downregulated and 7 upregulated) of high‐abundance and 123 of low‐abundance (56 downregulated and 67 upregulated; Figure 3c,d)81. We focused on miRNAs with most prominent changes in the high‐abundance group, which included a 39% downregulation of mmu‐let‐7i and a 255% upregulation of mmu‐miR‐341. Let‐7i is an interesting miRNA that is predicted to target >900 conserved sequences in Ingenuity Pathway Analysis and TargetScan analysis, including 46 apoptotic cell death pathway mRNAs, 42 cardiovascular and diabetes‐related mRNAs, 84 growth pathway mRNAs, 80 inflammation‐related pathway mRNAs, 21 metabolism and diabetes pathway mRNAs, and 59 neurotransmitter and nervous system mRNAs. Using in situ hybridization, we noted that let‐7i was preferentially expressed in sensory neurons, rather than DRG satellite cells or vessels (Figure 3e). Administration of exogenous mmu‐let‐7i mimic enhanced neurite growth and branching in sensory neurons in vitro, and improved electrophysiological, structural and behavioral abnormalities in diabetic mice. In contrast to downregulation of let‐7i, a prominently upregulated miRNA was miR‐341. miR‐341 is also reported to be significantly upregulated in the injured DRG of rats with chronic constriction injury138. Although miR‐341 is only expressed in rodents, it was also expressed in sensory neurons, and the knock down of miR‐341 improved sensory nerve conduction slowing and thermal hyposensitivity of DPN mice. Taken together, it might be that as a single miRNA potentially regulates many target genes, targeting or replenishing a single miRNA might be a more interesting and potentially efficient strategy for gene therapy than targeting a single mRNA.
Among the differentially expressed mRNAs in diabetic neurons of uncertain significance, CWC22 was chosen as a starting molecule23. CWC22 protein was expressed in the nucleus of sensory neurons particularly, in nuclear speckles, a nuclear organelle of sensory neurons, and reverse transcription polymerase chain reaction also confirmed its mRNA upregulation of 2.5‐fold (Figure 4a). CWC22 is known to be required for pre‐mRNA spicing139, 140. A spliceosome is a large and complex molecular machine required to catalyze pre‐mRNA splicing in nuclear speckles141. It consists of small nuclear ribonucleoproteins (snRNPs) that contain RNA components (snRNAs: U1, U2, U4, U5 and U6) and additional splicing factors. Splicing factors bind to the pre‐mRNA in a sequential manner to thereby form the spliceosome, which catalyzes two consecutive steps of transesterification to excise the intron142, 143. We found new structural evidence of splicing abnormalities in diabetic sensory neurons. For example, snRNPs formed aggregated multiple nuclear foci (Figure 4b) and their associated snRNA expression was reduced. CWC22 is required for exon junction complex assembly, upstream of the exon–exon junction during pre‐mRNA splicing to regulate post‐transcriptional mRNA fate139, 140. Global defects of pre‐mRNA splicing and global downregulation of diverse gene expressions have been identified in CWC22 depleted cells144. We showed that CWC22 knockdown in sensory neurons in vitro enhanced neurite outgrowth, and CWC22 knockdown in vivo improved features of DPN in diabetes mellitus mice. These findings indicate that aberrant splicing associated with upregulated CWC22 might be included as a mediator of sensory neuron dysfunction. It is plausible that CWC22 upregulation reflects heightened forms of inappropriate spicing that ensue from diabetes, although this has not been established. For example, injured and regenerating non‐diabetic neurons require altered gene expression to support their growth. In diabetes, CWC22 overexpression appears to be harmful to spliceosome formation; its inhibition might reverse aberrant splicing, potentially normalizing gene expression critical for axon outgrowth.
Figure 4.
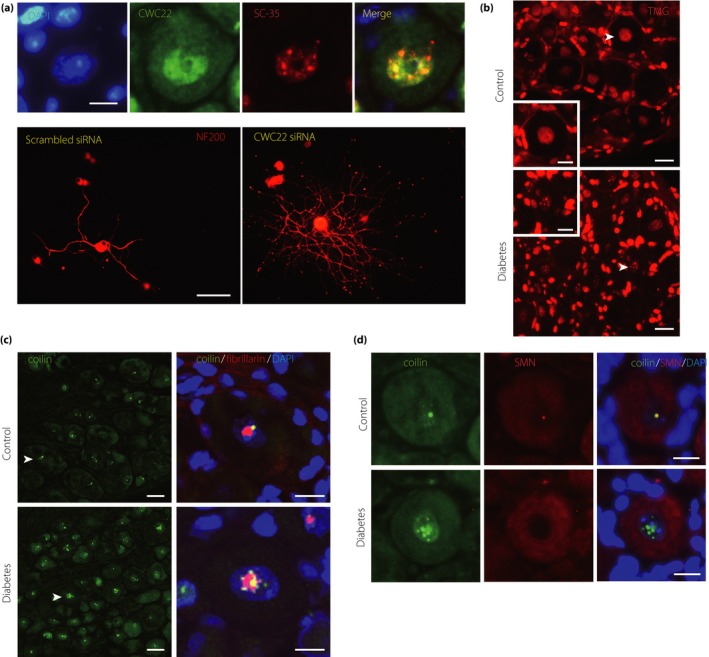
Abnormal messenger ribonucleic acid splicing in diabetic sensory neurodegeneration. (a) CWC22 is expressed in the nuclei of DRG sensory neurons, colocalized with a marker protein SC35 of nuclear speckles. Scale bar: 10 μm. CWC22 knockdown enhances DRG neurite outgrowth. Scale bar: 100 μm. (b) Small nuclear ribonucleoproteins form abnormally aggregated multiple foci in the nuclei of DRG sensory neurons. Scale bar: 20 μm, 10 μm in insets. (c) Cajal bodies number is increased in diabetic sensory neurons. Scale bar: 20 μm, 10 μm in magnified panels. (d) Cajal bodies lose their colocalization with survival motor neuron nuclear foci in diabetic sensory neurons. Scale bar: 10 μm. Reproduced from Kobayashi et al.23 with permission.
Furthermore, we identified additional unique alterations in nuclear structure that accompanied the aberrant splicing in diabetic sensory neurons. The key events of mRNA processing, including splicing, occur within the nucleus. In the nucleus, interchromatin structures, such as nucleoli, Cajal bodies (CBs), and nuclear speckles, could offer a cellular microenvironment that facilitates more efficient changes of gene expression145. CBs control transcriptional activity in cross‐talk with nucleoli on cellular stress, and emerge in proliferative and metabolically active cells, such as cancer cells or neurons146, 147, 148, 149. CBs concentrate snRNPs and increase the efficiency of gene expression through its sophisticated supply of snRNPs for the spliceosome150, 151. Nuclear speckles, colocalized with CWC22, also accumulate snRNPs and other non‐snRNP protein splicing factors, and provide a place to execute splicing141. However, the overall role of these nuclear bodies in diabetic sensory neurodegeneration has been otherwise unexplored152. In diabetic sensory neurons, we found that CBs were increased in number, but nucleoli and nuclear speckles were not structurally altered (Figure 4c). Another key molecule related to splicing, survival motor neuron (SMN) protein, localized in nuclear foci, functions in the assembly of snRNPs in collaboration with CBs142, 153, 154, 155. SMN mutations underlie spinal muscular atrophy (SMA), through defects in CB formation and the assembly of snRNPs in motor neurons156, 157, 158. In addition, SMN‐deficient sensory neurons in vitro are also abnormal with shorter neurites and small growth cones159. We identified that CBs lost their colocalization with SMN and abnormally aggregated snRNPs in DRG sensory neurons in diabetes mellitus mice, suggesting loss of recruitment of SMN proteins to CBs, similar to a key finding in the motor neuron degeneration of SMA (Figure 4d)160.
Taken together, our findings provide evidence that spliceosome dysregulation might be a key neurodegenerative mechanism of the development of DPN in type 1 diabetes mellitus patients, as summarized in Figure 5. It still remains unclear whether overexpressed CWC22 proteins in diabetic sensory neurons are blocking factors for spliceosome formation including SMN or snRNP recruitment, or independently inhibit the other growth signal pathways as “brakes.” In addition, further investigation is required to determine whether splicing abnormalities are also identifiable in type 2 diabetes mellitus, perhaps associated with “insulin resistance.”
Figure 5.
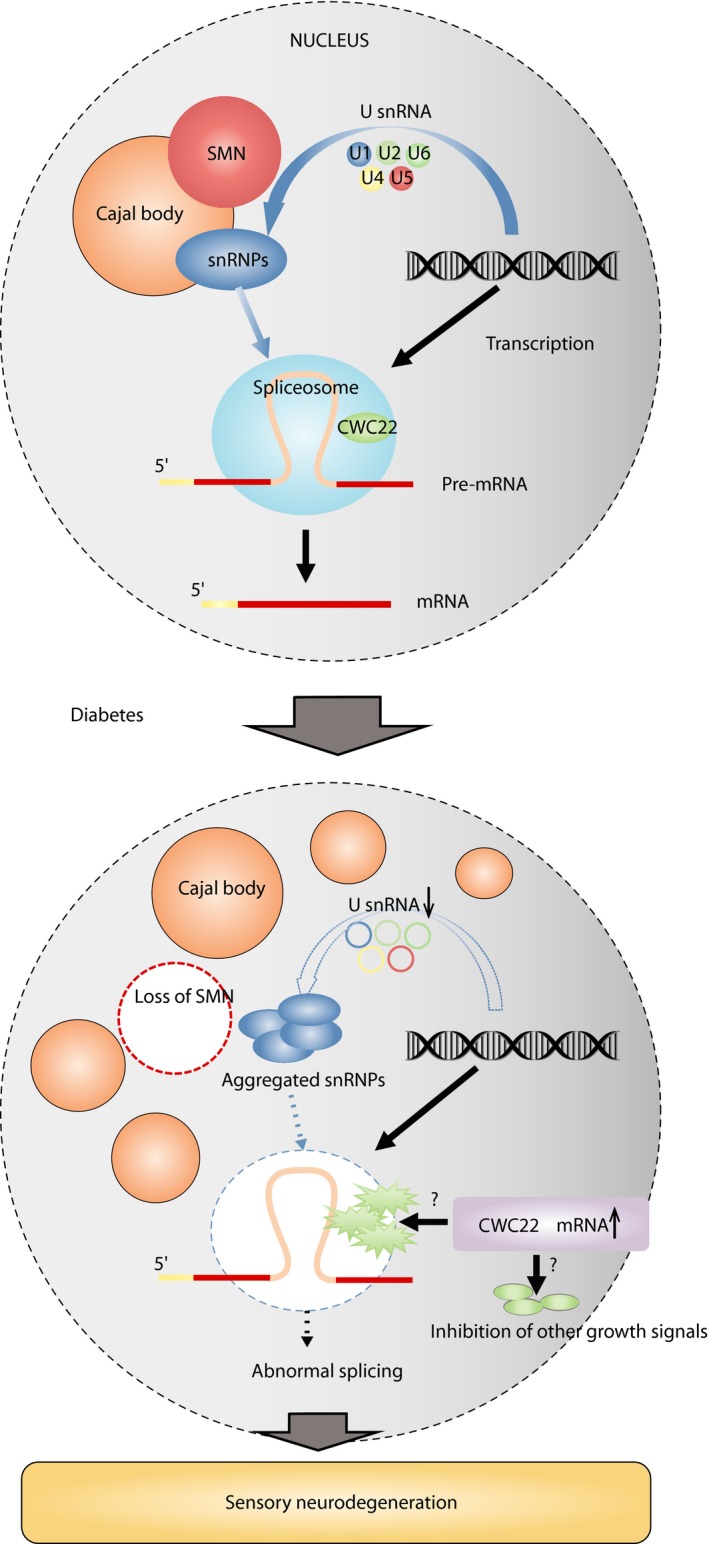
Schematic drawing of diabetic sensory neurodegeneration associated with splicing abnormalities. mRNA, messenger ribonucleic acid; SMN, survival motor neuron; snRNA, small nuclear ribonucleoproteins that contain ribonucleic acid components; snRNP, small nuclear ribonucleoproteins.
Conclusions
A microvascular hypothesis for the early development of DPN can be challenged, and molecular approaches to sensory neurodegeneration directly targeted by diabetes might offer a series of new therapeutic opportunities. Interventions to activate intrinsic neurotrophic pathways using approaches such as insulin, inactivation of growth suppressing tumor suppressors, GLP‐1 agonism and HSP overexpression might be new strategies to prevent or reverse neuropathic damage in DPN. Unfortunately DPN is currently an irreversible complication of diabetes mellitus. The pathogenesis of DPN might involve epigenetic changes mediated by miRNA that regulate gene expression in many biological processes including cell survival and growth. Sensory neurodegeneration in DPN could share common mechanisms with other neurological disorders, such as spliceosomal abnormalities, CB dysregulation and loss of SMN proteins.
Disclosure
The authors declares no conflict of interest.
Acknowledgments
The authors acknowledge the dedicated work of members of the Zochodne laboratory in generating experimental data described in this review including Dr Ambika Chandrasekhar, Dr Bhagat Singh, Dr Kim Christie, Dr Anand Krishnan, Dr Lawrence Korngut, Dr Chu Cheng and Dr Michelle Kan. MK was supported as the Denyse Lajoie Lake Fellow of the Hotchkiss Brain Institute, Faculty of Medicine, University of Calgary, 2013–2014. The Zochodne laboratory has been supported for this work by the Canadian Institutes of Health Research (184584), Canadian Diabetes Association (OG‐3‐12‐3669), Juvenile Diabetes Foundation, Alberta Heritage Foundation for Medical Research, Alberta Innovates‐Health Solutions and NIH (Diabetes Complications Consortium 12GHSU172).
J Diabetes Investig. 2018
References
- 1. Pop‐Busui R, Boulton AJM, Feldman EL, et al Diabetic neuropathy: a position statement by the American diabetes association. Diabetes Care 2017; 40: 136–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization . Definition, Diagnosis and Classification of Diabetes Mellitus and its Complications. Report of a WHO Consultation. Part1: diagnosis and classification of diabetes mellitus. Geneva: WHO, 1999. [Google Scholar]
- 3. World Health Organization . Global Report on Diabetes. Geneva: WHO, 2016. [Google Scholar]
- 4. Dyck PJ, Kratz KM, Karnes JL, et al The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population‐based cohort: the Rochester Diabetic Neuropathy Study. Neurology 1993; 43: 817. [DOI] [PubMed] [Google Scholar]
- 5. Brown MJ, Asbury AK. Diabetic neuropathy. Ann Neurol 1984; 15: 2–12. [DOI] [PubMed] [Google Scholar]
- 6. Zochodne DW. Diabetic neuropathies: features and mechanisms. Brain Pathol 1999; 9: 369–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boulton AJM, Vinik AI, Arezzo JC, et al Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 2005; 28: 956–962. [DOI] [PubMed] [Google Scholar]
- 8. Abbott CA, Malik RA, Van Ross ERE, et al Prevalence and characteristics of painful diabetic neuropathy in a large community‐based diabetic population in the U.K. Diabetes Care 2011; 34: 2220–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bril V, England JD, Franklin GM, et al Evidence‐based guideline: treatment of painful diabetic neuropathy‐report of the American Association of Neuromuscular and Electrodiagnostic Medicine, the American Academy of Neurology, and the American Academy of Physical Medicine & Rehabilitation. Muscle Nerve 2011; 43: 910–917. [DOI] [PubMed] [Google Scholar]
- 10. Javed S, Petropoulos IN, Alam U, et al Treatment of painful diabetic neuropathy. Ther Adv Chronic Dis 2015; 6: 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Todorovic SM. Is diabetic nerve pain caused by dysregulated ion channels in sensory neurons? Diabetes 2015; 64: 3987–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes 2011; 29: 116–122. [Google Scholar]
- 13. Zochodne DW. Diabetes mellitus and the peripheral nervous system: manifestations and mechanisms. Muscle Nerve 2007; 36: 144–166. [DOI] [PubMed] [Google Scholar]
- 14. Zochodne DW. Diabetic polyneuropathy: an update. Curr Opin Neurol 2008; 21: 527–533. [DOI] [PubMed] [Google Scholar]
- 15. Bansal V. Diabetic neuropathy. Postgrad Med J 2006; 82: 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dyck PJ, Albers JW, Andersen H, et al Diabetic polyneuropathies: update on research definition, diagnostic criteria and estimation of severity. Diabetes Metab Res Rev 2011; 27: 620–628. [DOI] [PubMed] [Google Scholar]
- 17. Ramji N, Toth C, Kennedy J, et al Does diabetes mellitus target motor neurons? Neurobiol Dis 2007; 26: 301–311. [DOI] [PubMed] [Google Scholar]
- 18. Zochodne DW, Ramji N, Toth C. Neuronal targeting in diabetes mellitus: a story of sensory neurons and motor neurons. Neuroscientist 2008; 14: 311–318. [DOI] [PubMed] [Google Scholar]
- 19. Polydefkis M, Hauer P, Griffin JW, et al Skin biopsy as a tool to assess distal small fiber innervation in diabetic neuropathy. Diabetes Technol Ther 2001; 3: 23–28. [DOI] [PubMed] [Google Scholar]
- 20. Li XG, Zochodne DW. Microvacuolar neuronopathy: a postmortem artifact of sensory neurons. J Neurocytol 2003; 32: 393–398. [DOI] [PubMed] [Google Scholar]
- 21. Kishi M, Tanabe J, Schmelzer JD, et al Morphometry of dorsal root ganglion in chronic experimental diabetic neuropathy. Diabetes 2002; 51: 819–824. [DOI] [PubMed] [Google Scholar]
- 22. Cheng C, Zochodne DW. Sensory neurons with activated caspase‐3 survive long‐term experimental diabetes. Diabetes 2003; 52: 2363–2371. [DOI] [PubMed] [Google Scholar]
- 23. Kobayashi M, Chandrasekhar A, Cheng C, et al Diabetic polyneuropathy, sensory neurons, nuclear structure and spliceosome alterations: a role for CWC22. Dis Model Mech 2017; 10: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zochodne DW. Diabetes and the plasticity of sensory neurons. Neurosci Lett 2015; 596: 60–65. [DOI] [PubMed] [Google Scholar]
- 25. Zochodne DW. Sensory neurodegeneration in diabetes: beyond glucotoxicity. Int Rev Neurobiol 2016; 127: 151–180. [DOI] [PubMed] [Google Scholar]
- 26. Zochodne DW, Verge VM, Cheng C, et al Does diabetes target ganglion neurones? Progressive sensory neurone involvement in long‐term experimental diabetes. Brain 2001; 124: 2319–2334. [DOI] [PubMed] [Google Scholar]
- 27. Toth C, Brussee V, Cheng C, et al Diabetes mellitus and the sensory neuron. J Neuropathol Exp Neurol 2004; 63: 561–573. [DOI] [PubMed] [Google Scholar]
- 28. Feener EP, King GL. Vascular dysfunction in diabetes mellitus. Lancet 1997; 350: S9–S13. [DOI] [PubMed] [Google Scholar]
- 29. Tesfaye S, Malik R, Ward JD. Vascular factors in diabetic neuropathy. Acta Med Scand 1994; 37: 847–854. [DOI] [PubMed] [Google Scholar]
- 30. Cameron NE, Eaton SEM, Cotter MA, et al Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia 2001; 44: 1973–1988. [DOI] [PubMed] [Google Scholar]
- 31. Østergaard L, Finnerup NB, Terkelsen AJ, et al The effects of capillary dysfunction on oxygen and glucose extraction in diabetic neuropathy. Diabetologia 2015; 58: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yorek MA. Vascular impairment of epineurial arterioles of the sciatic nerve: implications for diabetic peripheral neuropathy. Rev Diabet Stud 2015; 12: 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giannini C, Dyck PJ. Basement membrane reduplication and pericyte degeneration precede development of diabetic polyneuropathy and are associated with its severity. Ann Neurol 1995; 37: 498–504. [DOI] [PubMed] [Google Scholar]
- 34. Thrainsdottir S, Malik RA, Dahlin LB, et al Endoneurial capillary abnormalities presage deterioration of glucose tolerance and accompany peripheral neuropathy in man. Diabetes 2003; 52: 2615–2622. [DOI] [PubMed] [Google Scholar]
- 35. Tesfaye S, Selvarajah D. Advances in the epidemiology, pathogenesis and management of diabetic peripheral neuropathy. Diabetes Metab Res Rev 2012; 28(Suppl 1): 8–14. [DOI] [PubMed] [Google Scholar]
- 36. Tuck RR, Schmelzer JD, Low PA. Endoneurial blood flow and oxygen tension in the sciatic nerves of rats with experimental diabetic neuropathy. Brain 1984; 107: 935–950. [DOI] [PubMed] [Google Scholar]
- 37. Sena CM, Pereira AM, Seiça R. Endothelial dysfunction – a major mediator of diabetic vascular disease. Biochim Biophys Acta 2013; 1832: 2216–2231. [DOI] [PubMed] [Google Scholar]
- 38. De Vriese AS, Verbeuren TJ, Van De Voorde J, et al Endothelial dysfunction in diabetes. Br J Pharmacol 2000; 130: 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pieper GM, Siebeneich W. Diabetes‐induced endothelial dysfunction is prevented by long‐term treatment with the modified iron chelator, hydroxyethyl starch conjugated‐deferoxamine. J Cardiovasc Pharmacol 1997; 30: 734–738. [DOI] [PubMed] [Google Scholar]
- 40. Gabbay KH. Hyperglycemia, polyol metabolism, and complications of diabetes mellitus. Annu Rev Med 1975; 26: 521–536. [DOI] [PubMed] [Google Scholar]
- 41. Kinoshita JH. A thirty year journey in the polyol pathway. Exp Eye Res 1990; 50: 567–573. [DOI] [PubMed] [Google Scholar]
- 42. Goto Y, Hotta N, Shigeta Y, et al Effects of an aldose reductase inhibitor, epalrestat, on diabetic neuropathy. Clinical benefit and indication for the drug assessed from the results of a placebo‐controlled double‐blind study. Biomed Pharmacother 1995; 49: 269–277. [DOI] [PubMed] [Google Scholar]
- 43. Pfeifer MA, Schumer MP, Gelber DA. Aldose reductase inhibitors: the end of an era or the need for different trial designs? Diabetes 1997; 46: 11–12. [DOI] [PubMed] [Google Scholar]
- 44. Hotta N, Akanuma Y, Kawamori R, et al Long‐term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3‐year, multicenter, comparative aldose reductase inhibitor‐diabetes complications trial. Diabetes Care 2006; 29: 1538–1544. [DOI] [PubMed] [Google Scholar]
- 45. Hotta N, Kawamori R, Atsumi Y, et al Stratified analyses for selecting appropriate target patients with diabetic peripheral neuropathy for long‐term treatment with an aldose reductase inhibitor, epalrestat. Diabet Med 2008; 25: 818–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coppey LJ, Gellett JS, Davidson EP, et al Preventing superoxide formation in epineurial arterioles of the sciatic nerve from diabetic rats restores endothelium‐dependent vasedilation. Free Radic Res 2003; 37: 33–40. [DOI] [PubMed] [Google Scholar]
- 47. Ryan GJ. New pharmacologic approaches to treating diabetic retinopathy. Am J Health Syst Pharm 2007; 64: 28–29. [DOI] [PubMed] [Google Scholar]
- 48. Cameron NE, Gibson TM, Nangle MR, et al Inhibitors of advanced glycation end product formation and neurovascular dysfunction in experimental diabetes. Ann N Y Acad Sci 2005; 1043: 784–792. [DOI] [PubMed] [Google Scholar]
- 49. Yamagishi S, Nakamura K, Imaizumi T. Advanced glycation end products (AGEs) and diabetic vascular complications. Curr Diabetes Rev 2005; 1: 93–106. [DOI] [PubMed] [Google Scholar]
- 50. Wada R, Yagihashi S. Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Ann N Y Acad Sci 2005; 1043: 598–604. [DOI] [PubMed] [Google Scholar]
- 51. Yamagishi S, Nakamura K, Matsui T, et al Agents that block advanced glycation end product (AGE)‐RAGE (receptor for AGEs)‐oxidative stress system: a novel therapeutic strategy for diabetic vascular complications. Expert Opin Investig Drugs 2008; 17: 983–996. [DOI] [PubMed] [Google Scholar]
- 52. Yamagishi S, Nakamura K, Matsui T, et al Receptor for advanced glycation end products (RAGE): a novel therapeutic target for diabetic vascular complication. Curr Pharm Des 2008; 14: 487–495. [DOI] [PubMed] [Google Scholar]
- 53. Sugimoto K, Yasujima M, Yagihashi S. Role of advanced glycation end products in diabetic neuropathy. Curr Pharm Des 2008; 14: 953–961. [DOI] [PubMed] [Google Scholar]
- 54. Takeuchi M, Takino J, Yamagishi S. Involvement of the toxic AGEs (TAGE)‐RAGE system in the pathogenesis of diabetic vascular complications: a novel therapeutic strategy. Curr Drug Targets 2010; 11: 1468–1482. [DOI] [PubMed] [Google Scholar]
- 55. Chilelli NC, Burlina S, Lapolla A. AGEs, rather than hyperglycemia, are responsible formicrovascular complications in diabetes: a “glycoxidation‐centric” point of view. Nutr Metab Cardiovasc Dis 2013; 23: 913–919. [DOI] [PubMed] [Google Scholar]
- 56. Cameron NE, Cotter MA, Robertson S. Angiotensin converting enzyme inhibition prevents development of muscle and nerve dysfunction and stimulates angiogenesis in streptozotocin‐diabetic rats. Diabetologia 1992; 35: 12–18. [DOI] [PubMed] [Google Scholar]
- 57. Maxfield EK, Cameron NE, Cotter MA, et al Angiotensin II receptor blockade improves nerve function, modulates nerve blood flow and stimulates endoneurial angiogenesis in streptozotocin‐diabetic rats. Diabetologia 1993; 36: 1230–1237. [DOI] [PubMed] [Google Scholar]
- 58. Yorek MA. The potential role of angiotensin converting enzyme and vasopeptidase inhibitors in the treatment of diabetic neuropathy. Curr Drug Targets 2008; 9: 77–84. [DOI] [PubMed] [Google Scholar]
- 59. Coppey LJ, Davidson EP, Rinehart TW, et al Diabetic neuropathy in streptozotocin‐induced diabetic rats. Diabetes 2006; 55: 341–348. [DOI] [PubMed] [Google Scholar]
- 60. Nishikawa T, Edelstein D, Du XL, et al Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787–790. [DOI] [PubMed] [Google Scholar]
- 61. Cameron NE, Tuck Z, McCabe L, et al Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia 2001; 44: 1161–1169. [DOI] [PubMed] [Google Scholar]
- 62. Coppey LJ, Gellett JS, Davidson EP, et al Effect of antioxidant treatment of streptozotocin‐induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes 2001; 50: 1927–1937. [DOI] [PubMed] [Google Scholar]
- 63. Coppey LJ, Gellett JS, Davidson EP, et al Effect of treating streptozotocin‐induced diabetic rats with sorbinil, myo‐inositol or aminoguanidine on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Int J Exp Diabetes Res 2002; 3: 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Obrosova IG, Drel VR, Oltman CL, et al Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin‐diabetic rats. Am J Physiol Endocrinol Metab 2007; 293: E1645–E1655. [DOI] [PubMed] [Google Scholar]
- 65. Stavniichuk R, Shevalye H, Lupachyk S, et al Peroxynitrite and protein nitration in the pathogenesis of diabetic peripheral neuropathy. Diabetes Metab Res Rev 2014; 30: 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schmelzer JD, Zochodne DW, Low PA. Ischemic and reperfusion injury of rat peripheral nerve. Proc Natl Acad Sci USA 1989; 86: 1639–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zochodne DW. Neurobiology of Peripheral Nerve Regeneration. New York, NY:Cambridge University Press, 2008. [Google Scholar]
- 68. Rundles RW. Diabetic neuropathy‐ general review with report of 125 cases. Medicine 1945; 24: 111–160. [Google Scholar]
- 69. Zochodne DW, Nguyen C. Increased peripheral nerve microvessels in early experimental diabetic neuropathy: quantitative studies of nerve and dorsal root ganglia. J Neurol Sci 1999; 166: 40–46. [DOI] [PubMed] [Google Scholar]
- 70. Zochodne DW, Ho LT. Normal blood flow but lower oxygen tension in diabetes of young rats: microenvironment and the influence of sympathectomy. Can J Physiol Pharmacol 1992; 70: 651–659. [DOI] [PubMed] [Google Scholar]
- 71. Zochodne DW. Nerve and ganglion blood flow in diabetes: an appraisal. Int Rev Neurobiol 2002; 50: 161–202. [DOI] [PubMed] [Google Scholar]
- 72. Theriault M, Dort J, Sutherland G, et al Local human sural nerve blood flow in diabetic and other polyneuropathies. Brain 1997; 120: 1131–1138. [DOI] [PubMed] [Google Scholar]
- 73. Abram SE, Yi J, Fuchs A, et al Permeability of injured and intact peripheral nerves and dorsal root ganglia. Anesthesiology 2006; 105: 146–153. [DOI] [PubMed] [Google Scholar]
- 74. Jimenez‐Andrade JM, Herrera MB, Ghilardi JR, et al Vascularization of the dorsal root ganglia and peripheral nerve of the mouse: implications for chemical‐induced peripheral sensory neuropathies. Mol Pain 2008; 4: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sapunar D, Kostic S, Banozic A, et al Dorsal root ganglion – a potential new therapeutic target for neuropathic pain. J Pain Res 2012; 5: 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jacobs JM. Vascular permeability and neurotoxicity. Environ Health Perspect 1978; 26: 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Arvidson B, Tjälve H. Distribution of 109Cd in the nervous system of rats after intravenous injection. Acta Neuropathol 1986; 69: 111–116. [DOI] [PubMed] [Google Scholar]
- 78. Zochodne DW, Ho LT. Unique microvascular characteristics of the dorsal root ganglion in the rat. Brain Res 1991; 559: 89–93. [DOI] [PubMed] [Google Scholar]
- 79. Zochodne DW, Ho LT, Allison JA. Dorsal root ganglia microenvironment of female BB Wistar diabetic rats with mild neuropathy. J Neurol Sci 1994; 127: 36–42. [DOI] [PubMed] [Google Scholar]
- 80. Scott JN, Clark AW, Zochodne DW. Neurofilament and tubulin gene expression in progressive experimental diabetes. Failure of synthesis and export by sensory neurons. Brain 1999; 122: 2109–2118. [DOI] [PubMed] [Google Scholar]
- 81. Cheng C, Kobayashi M, Martinez JA, et al Evidence for epigenetic regulation of gene expression and function in chronic experimental diabetic neuropathy. J Neuropathol Exp Neurol 2015; 74: 804–817. [DOI] [PubMed] [Google Scholar]
- 82. Brussee V, Cunningham FA, Zochodne DW. Direct insulin signalling of neurons reverses diabetic neuropathy. Diabetes 2004; 53: 1824–1830. [DOI] [PubMed] [Google Scholar]
- 83. Sugimoto K, Murakawa Y, Sima AA. Expression and localization of insulin receptor in rat dorsal root ganglion and spinal cord. J Peripher Nerv Syst 2002; 7: 44–53. [DOI] [PubMed] [Google Scholar]
- 84. Frazier WA, Angeletti RH, Bradshaw RA. Nerve growth factor and insulin. Science 1972; 176: 482–488. [DOI] [PubMed] [Google Scholar]
- 85. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin‐resistant states. Cold Spring Harb Perspect Biol 2014; 6: a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Korhonen JM, Saïd FA, Wong AJ, et al Gab1 mediates neurite outgrowth, DNA synthesis, and survival in PC12 cells. J Biol Chem 1999; 274: 37307–37314. [DOI] [PubMed] [Google Scholar]
- 87. Soltoff SP, Rabin SL, Cantley LC, et al Nerve growth factor promotes the activation of phosphatidylinositol 3‐kinase and its association with the trk tyrosine kinase. J Biol Chem 1992; 267: 17472–17477. [PubMed] [Google Scholar]
- 88. Stambolic V, Suzuki A, de la Pompa JL, et al Negative regulation of PKB/Akt‐dependent cell survival by the tumor suppressor PTEN. Cell 1998; 95: 29–39. [DOI] [PubMed] [Google Scholar]
- 89. Hemmings BA, Restuccia DF. PI3K‐PKB/Akt pathway. Cold Spring Harb Perspect Biol 2012; 4: a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fernyhough P, Willars GB, Lindsay RM, et al Insulin and insulin‐like growth factor I enhance regeneration in cultured adult rat sensory neurones. Brain Res 1993; 607: 117–124. [DOI] [PubMed] [Google Scholar]
- 91. Xu QG, Li XQ, Kotecha SA, et al Insulin as an in vivo growth factor. Exp Neurol 2004; 188: 43–51. [DOI] [PubMed] [Google Scholar]
- 92. Toth C, Brussee V, Martinez JA, et al Rescue and regeneration of injured peripheral nerve axons by intrathecal insulin. Neuroscience 2006; 139: 429–449. [DOI] [PubMed] [Google Scholar]
- 93. Singhal A, Cheng C, Sun H, et al Near nerve local insulin prevents conduction slowing in experimental diabetes. Brain Res 1997; 763: 209–214. [DOI] [PubMed] [Google Scholar]
- 94. Aghanoori MR, Smith DR, Roy Chowdhury S, et al Insulin prevents aberrant mitochondrial phenotype in sensory neurons of type 1 diabetic rats. Exp Neurol 2017; 297: 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Guo G, Kan M, Martinez JA, et al Local insulin and the rapid regrowth of diabetic epidermal axons. Neurobiol Dis 2011; 43: 414–421. [DOI] [PubMed] [Google Scholar]
- 96. Singh B, Xu Y, Guo G, et al Insulin signaling and insulin resistance in sensory neurons. J Peripher Nerv Syst 2009; 14: 254. [Google Scholar]
- 97. Singh B, Xu Y, McLaughlin T, et al Resistance to trophic neurite outgrowth of sensory neurons exposed to insulin. J Neurochem 2012; 121: 263–276. [DOI] [PubMed] [Google Scholar]
- 98. Grote CW, Groover AL, Ryals JM, et al Peripheral nervous system insulin resistance in ob/ob mice. Acta Neuropathol Commun 2013; 1: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Grote CW, Morris JK, Ryals JM, et al Insulin receptor substrate 2 expression and involvement in neuronal insulin resistance in diabetic neuropathy. Exp Diabetes Res 2011; 2011: 212571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kim B, McLean LL, Philip SS, et al Hyperinsulinemia induces insulin resistance in dorsal root ganglion neurons. Endocrinology 2011; 152: 3638–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Baggio LL, Drucker DJ. Biology of incretins: GLP‐1 and GIP. Gastroenterology 2007; 132: 2131–2157. [DOI] [PubMed] [Google Scholar]
- 102. Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 2013; 17: 819–837. [DOI] [PubMed] [Google Scholar]
- 103. Kan M, Guo G, Singh B, et al Glucagon‐like peptide 1, insulin, sensory neurons, and diabetic neuropathy. J Neuropathol Exp Neurol 2012; 71: 494–510. [DOI] [PubMed] [Google Scholar]
- 104. Himeno T, Kamiya H, Naruse K, et al Beneficial effects of exendin‐4 on experimental polyneuropathy in diabetic mice. Diabetes 2011; 60: 2397–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Jolivalt CG, Fineman M, Deacon CF, et al GLP‐1 signals via ERK in peripheral nerve and prevents nerve dysfunction in diabetic mice. Diabetes Obes Metab 2011; 13: 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci 2011; 1243: 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ramasamy R, Vannucci SJ, Yan SS, et al Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005; 15: 16R–28R. [DOI] [PubMed] [Google Scholar]
- 108. Bierhaus A, Schiekofer S, Schwaninger M, et al Diabetes‐associated sustained activation of the transcription factor nuclear factor‐kappaB. Diabetes 2001; 50: 2792–2808. [DOI] [PubMed] [Google Scholar]
- 109. Bierhaus A, Haslbeck KM, Humpert PM, et al Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J Clin Invest 2004; 114: 1741–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Juranek JK, Geddis MS, Song F, et al RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes 2013; 62: 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Saleh A, Smith DR, Tessler L, et al Receptor for advanced glycation end‐products (RAGE) activates divergent signaling pathways to augment neurite outgrowth of adult sensory neurons. Exp Neurol 2013; 249: 149–159. [DOI] [PubMed] [Google Scholar]
- 112. Rong LL, Trojaborg W, Qu W, et al Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J 2004; 18: 1812–1817. [DOI] [PubMed] [Google Scholar]
- 113. de la Hoz CL, Cheng C, Fernyhough P, et al A model of chronic diabetic polyneuropathy: benefits from intranasal insulin are modified by sex and RAGE deletion. Am J Physiol Endocrinol Metab 2017; 312: E407–E419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Whitley D, Goldberg SP, Jordan WD. Heat shock proteins: a review of the molecular chaperones. J Vasc Surg 1999; 29: 748–751. [DOI] [PubMed] [Google Scholar]
- 115. Lewis SE, Mannion RJ, White FA, et al A role for HSP27 in sensory neuron survival. J Neurosci 1999; 19: 8945–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ma CH, Omura T, Cobos EJ, et al Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Invest 2011; 121: 4332–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Korngut L, Ma CH, Martinez JA, et al Overexpression of human HSP27 protects sensory neurons from diabetes. Neurobiol Dis 2012; 47: 436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kennedy JM, Zochodne DW. Impaired peripheral nerve regeneration in diabetes mellitus. J Peripher Nerv Syst 2005; 10: 144–157. [DOI] [PubMed] [Google Scholar]
- 119. Krishnan A, Duraikannu A, Zochodne DW. Releasing “brakes” to nerve regeneration: intrinsic molecular targets. Eur J Neurosci 2016; 43: 297–308. [DOI] [PubMed] [Google Scholar]
- 120. Zochodne DW. Neurons and tumor suppressors. ACS Chem Neurosci 2014; 5: 618–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hopkins BD, Hodakoski C, Barrows D, et al PTEN function: the long and the short of it. Trends Biochem Sci 2014; 39: 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chalhoub N, Baker SJ. PTEN and the PI3‐kinase pathway in cancer. Annu Rev Pathol 2009; 4: 127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cully M, You H, Levine AJ, et al Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 2006; 6: 184–192. [DOI] [PubMed] [Google Scholar]
- 124. Luikart BW, Schnell E, Washburn EK, et al Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci 2011; 31: 4345–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Christie KJ, Webber CA, Martinez JA, et al PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J Neurosci 2010; 30: 9306–9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Liu K, Lu Y, Lee JK, et al PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 2010; 13: 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Park KK, Liu K, Hu Y, et al Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008; 322: 963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sun F, Park KK, Belin S, et al Sustained axon regeneration induced by co‐deletion of PTEN and SOCS3. Nature 2011; 480: 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Little D, Valori CF, Mutsaers CA, et al PTEN depletion decreases disease severity and modestly prolongs survival in a mouse model of spinal muscular atrophy. Mol Ther 2015; 23: 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Guo G, Singh V, Zochodne DW. Growth and turning properties of adult glial cell‐derived neurotrophic factor coreceptor α1 nonpeptidergic sensory neurons. J Neuropathol Exp Neurol 2014; 73: 820–836. [DOI] [PubMed] [Google Scholar]
- 131. Singh B, Singh V, Krishnan A, et al Regeneration of diabetic axons is enhanced by selective knockdown of the PTEN gene. Brain 2014; 137: 1051–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Bossy‐Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med 2004; 10: S2–S9. [DOI] [PubMed] [Google Scholar]
- 133. Ramanan VK, Saykin AJ. Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer's disease, Parkinson's disease, and related disorders. Am J Neurodegener Dis 2013; 2: 145–175. [PMC free article] [PubMed] [Google Scholar]
- 134. Liu J, Valencia‐Sanchez MA, Hannon FJ, et al MicroRNA‐dependent localization of targeted mRNAs to mammalian P‐bodies. Nat Cell Biol 2005; 7: 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Krützfeldt J, Poy MN, Stoffel M. Strategies to determine the biological function of microRNAs. Nat Genet 2006; 38: S14–S19. [DOI] [PubMed] [Google Scholar]
- 136. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 2012; 13: 271–282. [DOI] [PubMed] [Google Scholar]
- 137. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15: 509–524. [DOI] [PubMed] [Google Scholar]
- 138. Li H, Shen L, Ma C, et al Differential expression of miRNAs in the nervous system of a rat model of bilateral sciatic nerve chronic constriction injury. Int J Mol Med 2013; 32: 219–226. [DOI] [PubMed] [Google Scholar]
- 139. Barbosa I, Haque N, Fiorini F, et al Human CWC22 escorts the helicase eIF4AIII to spliceosomes and promotes exon junction complex assembly. Nat Struct Mol Biol 2012; 19: 983–990. [DOI] [PubMed] [Google Scholar]
- 140. Steckelberg AL, Boehm V, Gromadzka AM, et al CWC22 connects pre‐mRNA splicing and exon junction complex assembly. Cell Rep 2012; 2: 454–461. [DOI] [PubMed] [Google Scholar]
- 141. Girard C, Will CL, Peng J, et al Post‐transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat Commun 2012; 3: 994. [DOI] [PubMed] [Google Scholar]
- 142. Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 2014; 15: 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Han J, Xiong J, Wang D, et al Pre‐mRNA splicing: where and when in the nucleus. Trends Cell Biol 2011; 21: 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Steckelberg AL, Altmueller J, Dieterich C, et al CWC22‐dependent pre‐mRNA splicing and eIF4A3 binding enables global deposition of exon junction complexes. Nucleic Acids Res 2015; 43: 4687–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Mao YS, Zhang B, Spector DL. Biogenesis and function of nuclear bodies. Trends Genet 2011; 27: 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Boulon S, Westman BJ, Hutten S, et al The nucleolus under stress. Mol Cell 2010; 40: 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Machyna M, Heyn P, Neugebauer KM. Cajal bodies: where form meets function. Wiley Interdiscip Rev RNA 2013; 4: 17–34. [DOI] [PubMed] [Google Scholar]
- 148. Strzelecka M, Oates AC, Neugebauer KM. Dynamic control of Cajal body number during zebrafish embryogenesis. Nucleus 2010; 1: 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Palanca A, Casafont I, Berciano MT, et al Reactive nucleolar and Cajal body responses to proteasome inhibition in sensory ganglion neurons. Biochim Biophys Acta 2014; 1842: 848–859. [DOI] [PubMed] [Google Scholar]
- 150. Klingauf M, Stanek D, Neugebauer KM. Enhancement of U4/U6 small nuclear ribonucleoprotein particle association in Cajal bodies predicted by mathematical modeling. Mol Biol Cell 2006; 17: 4972–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Strzelecka M, Trowitzsch S, Weber G, et al Coilin‐dependent snRNP assembly is essential for zebrafish embryogenesis. Nat Struct Mol Biol 2010; 17: 403–409. [DOI] [PubMed] [Google Scholar]
- 152. Morimoto M, Boerkoel CF. The role of nuclear bodies in gene expression and disease. Biology (Basel) 2013; 2: 976–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J 1996; 15: 3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 154. Hebert MD, Szymczyk PW, Shpargel KB, et al Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev 2001; 15: 2720–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Narayanan U, Achsel T, Lührmann R, et al Coupled in vitro import of U snRNPs and SMN, the spinal muscular atrophy protein. Mol Cell 2004; 16: 223–234. [DOI] [PubMed] [Google Scholar]
- 156. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008; 371: 2120–2133. [DOI] [PubMed] [Google Scholar]
- 157. Girard C, Neel H, Bertrand E, et al Depletion of SMN by RNA interference in HeLa cells induces defects in Cajal body formation. Nucleic Acids Res 2006; 34: 2925–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Zhang Z, Lotti F, Dittmar K, et al SMN deficiency causes tissue‐specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008; 133: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Jablonka S, Karle K, Sandner B, et al Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy. Hum Mol Genet 2006; 15: 511–518. [DOI] [PubMed] [Google Scholar]
- 160. Lefebvre S, Burlet P, Liu Q, et al Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997; 16: 265–269. [DOI] [PubMed] [Google Scholar]