

SUMMARY
Until recently, high-throughput gene expression technology, such as RNA-Sequencing (RNA-seq) required hundreds of thousands of cells to produce reliable measurements. Recent technical advances permit genome-wide gene expression measurement at the single-cell level. Single-cell RNA-Seq (scRNA-seq) is the most widely used and numerous publications are based on data produced with this technology. However, RNA-seq and scRNA-seq data are markedly different. In particular, unlike RNA-seq, the majority of reported expression levels in scRNA-seq are zeros, which could be either biologically-driven, genes not expressing RNA at the time of measurement, or technically-driven, genes expressing RNA, but not at a sufficient level to be detected by sequencing technology. Another difference is that the proportion of genes reporting the expression level to be zero varies substantially across single cells compared to RNA-seq samples. However, it remains unclear to what extent this cell-to-cell variation is being driven by technical rather than biological variation. Furthermore, while systematic errors, including batch effects, have been widely reported as a major challenge in high-throughput technologies, these issues have received minimal attention in published studies based on scRNA-seq technology. Here, we use an assessment experiment to examine data from published studies and demonstrate that systematic errors can explain a substantial percentage of observed cell-to-cell expression variability. Specifically, we present evidence that some of these reported zeros are driven by technical variation by demonstrating that scRNA-seq produces more zeros than expected and that this bias is greater for lower expressed genes. In addition, this missing data problem is exacerbated by the fact that this technical variation varies cell-to-cell. Then, we show how this technical cell-to-cell variability can be confused with novel biological results. Finally, we demonstrate and discuss how batch-effects and confounded experiments can intensify the problem.
Keywords: Sparsity, Censoring, Missing not at random (MNAR), Genomics, Single-cell RNA-Sequencing, Confounding
1. Introduction
Single-cell RNA-Sequencing (scRNA-seq) has become the primary tool for profiling the transcriptomes of hundreds or even thousands of individual cells in parallel. In contrast to the standard RNA-seq approach, which is applied to samples containing hundreds of thousands of cells and therefore measures average gene expression level across cells, scRNA-seq measures gene expression in a single cell. To distinguish these two technologies we refer to the latter as bulk RNA-seq. Today scRNA-seq is increasingly being used across a diverse set of biomedical applications such as profiling the transcriptomes of differentiated cell types (Macosko and others, 2015; Treutlein and others, 2014; Zeisel and others, 2015; Wilson and others, 2015), profiling the changes in cell states (Trapnell and others, 2014; Shalek and others, 2014), identifying allele-specific expression (Borel and others, 2015; Deng and others, 2014), spatial reconstruction (Satija and others, 2015; Achim and others, 2015), and the classification of subtypes (Patel and others, 2014; Jaitin and others, 2014; Usoskin and others, 2015).
While scRNA-seq data provides a new level of data resolution, it also results in a larger number of genes reporting the expression level to be zero, or practically zero, as compared to using bulk RNA-seq (Bacher and Kendziorski, 2016). A gene reporting the expression level to be zero can arise in two ways: (i) the gene was not expressing any RNA at the time the cell was experimentally isolated and processed prior sequencing (referred to as structural zeros Zhu and others, 2016) or (ii) the gene was expressing RNA in the cell at the time of isolation, but due to limitations of current experimental protocols to detect low amounts of RNA in a cell, the gene was not detected (referred to as dropoutsKharchenko and others, 2014; Zhu and others, 2016). While the former is a type of biological event, the latter is purely technical. Possible technical reasons for a dropout to occur in the experimental procedure to capture and process the RNA prior to sequencing includes mRNA degradation after cell lysis, capture efficiency to convert mRNA to cDNA, variability in amplification efficiency, and dilution of cell libraries or sequencing depth (Lun and others, 2016; Vallejos and others, 2017).
Batch effects are commonly found in high-throughput data (Leek and others, 2010) and given the way that scRNA-seq experiments are conducted, there is much room for concern regarding confounding (Stegle and others, 2015). Specifically, batch effects in scRNA-seq experiments occur when cells from one biological group or condition are cultured, captured, and sequenced separate from cells in a second condition (Figure S1 of supplementary material available at Biostatistics online). However, due to the nature of certain experimental scRNA-seq protocols, which restrict the way cells are captured and sequenced separately, sometimes standard balanced experimental designs are not possible (Bacher and Kendziorski, 2016; Stegle and others, 2015; Grün and van Oudenaarden, 2015; Saliba and others, 2014). This reality makes it particularly important to be cautious about the potential for correlated variability induced by technical factors.
The unwanted variability introduced by batch effects can be particularly troublesome in scRNA-seq data because one of the most common applications has been data exploration after applying unsupervised learning methods (Pearson, 1901; Tipping and Bishop, 1999; Torgerson, 1952; Lafon and Lee, 2006; Nadler and others, 2006; van der Maaten, 2008) such as dimensionality reduction or clustering to identify novel or rare subpopulations of cells (Patel and others, 2014; Jaitin and others, 2014; Usoskin and others, 2015). Although a diverse set of techniques are used in these papers, both linear dimensionality reduction techniques, such as principal component analysis (PCA) (Pearson, 1901), and non-linear ones, such as t-Stochastic Neighbor Embedding (t-SNE) (van der Maaten, 2008), rely on computing distances between the cell expression profiles. Given that the majority of genes in a cell report the expression level to be zero and that the proportion of zeros varies greatly from cell to cell, it is not surprising that the distance estimates between cells are greatly influenced by the proportion of zeros (Finak and others, 2014; Pierson and Yau, 2015). However, it remains unclear to what extent this cell-to-cell variation is being driven by technical rather than biological variation.
We begin this article by describing the publicly available scRNA-seq data sets we used, which includes studies with only scRNA-seq data and studies with scRNA-seq and a matched bulk RNA-seq sample measured on the same population of cells. In the next section, we survey a large number of published scRNA-seq studies and illustrate the wide range of variation in the proportion of genes reporting the expression level to be zero across cells and studies (Section 3.1). Then, we present evidence that some of these reported zeros are driven by technical variation by demonstrating that scRNA-seq produces more zeros than expected and that this bias is greater for lower expressed genes (Section 3.2). In addition, we show that the consequences of this missing data problem are exacerbated by the fact that the technical variation of the probability of a gene being detected varies from cell to cell. Then, we illustrate that the proportion of genes reporting the expression level to be zero is a major source of cell-to-cell variation and this variability is partly driven by a mathematical artifact related to the transforming data in the original scale, but computing distances in the log scale (Section 3.3). Finally, we consider several case studies showing how differences in the cell-specific detection rates can be driven by batch effects, which in turn can result in the false discovery of new groups (Section 3.4).
2. Data Description
A scRNA-seq experiment typically involves randomly sampling and capturing single cells from a population of cells, isolating the mRNA from the individual cells, reverse transcribing the RNA into cDNA, and sequencing the cDNA using massively parallel sequencing technologies (Grün and van Oudenaarden, 2015; Kolodziejczyk and others, 2015; Shapiro and others, 2013). Strengths and weaknesses of different scRNA-seq experimental protocols vary (Combs and Eisen, 2015; Svensson and others, 2017; Ziegenhain and others, 2017) in the cost per cell, the sensitivity to capture and convert RNA to cDNA, and the accuracy to quantify the concentration of RNA, leading to differences in the number of cells sequenced per study and the number of features detected per cell. This experimental process is particularly challenging, and laboratory protocols are still under intense development.
2.1. scRNA-seq data sets
We examine 15 publicly available scRNA-seq data sets that included at least 200 samples with preprocessed and normalized expression data available on Gene Expression Omnibus (GEO) (Edgar and others, 2002) (Table 1). Different gene annotations were used including RefSeq (O’Leary and others, 2016), UCSC (Tyner and others, 2017), and Gencode (Harrow and others, 2006), and different gene-level and cell-level filtering criteria were used in each study leading to different numbers of genes included in each study. These data sets were created using six different scRNA-seq protocols for sequencing (Macosko and others, 2015; Jaitin and others, 2014; Tang and others, 2009; Islam and others, 2011; Ramsköld and others, 2012; Picelli and others, 2013; Hashimshony and others, 2012; Zheng and others, 2017) and five studies include the use of unique molecular identifiers (Kivioja and others, 2011) (UMIs) for counting specific mRNA molecules. For the 10 studies not using UMIs, we examine the data as submitted to GEO, with one exception (Patel and others, 2014). These 10 studies reported measurements in either Transcripts per Million (TPM) (Li and Dewey, 2011), Reads Per Kilobase of transcript per Million mapped reads (RPKM) (Mortazavi and others, 2008), or Fragments Per Kilobase of transcript per Million mapped reads (FPKM) (Trapnell and others, 2010), so each sample was corrected for gene length and library size. The one exception (Patel and others, 2014) uploaded data that was de-trended so that measurements for each gene averaged to zero across cells. For this particular study, we downloaded the raw sequencing files data from the Sequence Read Archive (SRA) (Leinonen and others, 2011) and computed expression in TPM units using Kallisto (Bray and others, 2016). In the studies that used UMIs for molecule counting (Macosko and others, 2015; Zeisel and others, 2015; Satija and others, 2015; Jaitin and others, 2014; Zheng and others, 2017), the data uploaded to GEO was not normalized for library size, so to assure that these data were in similar units to the rest of our studies, we followed a published procedure (Macosko and others, 2015) that normalizes each gene or transcript count by dividing by the total number of UMIs per cell and multiplies by a scaling factor (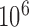 ). We refer to this unit as Counts Per Million (CPM).
). We refer to this unit as Counts Per Million (CPM).
Table 1.
Column 1 shows the reference. Column 2 shows the organism. Column 3 shows the single-cell protocol used. Column 4 shows the number of cells (samples) included in the study. Column 5 shows the number of genes included in the data uploaded to the public repository with varying gene annotations used. Column 6 indicates the units in which the values were reported. Column 7 shows the level of confounding between biological condition and batch effect quantified using the standardized Pearson contingency coefficient as a measure of association. The percentage ranges from 0
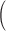 no confounding
no confounding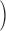 to 100
to 100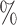
 completely confounded
completely confounded
| Study | Organism | scRNA-seq protocol | Number of cells | Number of genes | Processed data available | Confounding ( ) ) |
|---|---|---|---|---|---|---|
| Deng and others (2014) | Mouse | SMART-Seq | 286 | 22 958 | RPKM | 96.6‡ |
| Guo and others (2015) | Human | Tang and others (2009) | 154 | 23 394 | FPKM | 82.1 |
| Kowalczyk and others (2015) | Mouse | SMART-Seq | 533 | 8422 | TPM | 84.8 |
| Kumar and others (2014) | Mouse | SMART-Seq | 361 | 22 443 | TPM | 97.1 |
| Patel and others (2014) | Human | SMART-Seq | 430 | 5948 | TPM | 98.9 |
| Treutlein and others (2014) | Mouse | SMART-Seq | 198 | 23 745 | FPKM | 92.8 |
| Shalek and others (2014) | Mouse | SMART-Seq | 383 | 27 723 | TPM | 100 |
| Trapnell and others (2014) | Human | SMART-Seq | 306 | 47 192 | FPKM | 100 |
| Burns and others (2015) | Mouse | SMART-Seq | 249 | 26 585 | TPM | NA |
| Leng and others (2015) | Human | SMART-Seq | 458 | 19 804 | TPM | NA |
| Jaitin and others (2014) | Mouse | MARS-Seq | 4466 | 20 190 | UMI | NA |
| Macosko and others (2015) | Mouse | Drop-Seq | 49 300 | 16 961 | UMI | NA |
| Satija and others (2015) | Zebrafish | SMART-Seq | 1152 | 13 902 | UMI | NA |
| Zeisel and others (2015) | Mouse | STRT-Seq | 3004 | 19 972 | UMI | NA |
| Zheng and others (2017) | Human | Chromium Single Cell | 20 000 | 27 998 | UMI | NA |
| (10X Genomics) |
The main purpose of this study was to investigate monoallelic gene expression in mouse embryos, but here we consider the different developmental stages (oocyte to blastocyst) as the biological condition as an example
Although details of the experimental protocols, which can help define groupings that may lead to technical batch effects, are not always included in the annotations that are publicly available, one can extract informative variables from the raw sequencing (FASTQ) files (Gilad and Mizrahi-Man, 2015). Namely, the sequencing instrument used, the run number from the instrument and the flow cell lane. Although the sequencing is unlikely to be a major source of unwanted variability, it serves as a surrogate for other experimental procedures that very likely do have an effect, such as the starting amount of RNA in a cell, capture efficiency, PCR amplification reagents/conditions, and cell cycle stage of the cells (Shalek and others, 2014; Brennecke and others, 2013; Buettner and others, 2015; Tung and others, 2017). Here, we will refer to the resulting differences induced by different groupings of these sources of variability as batches.
2.2. scRNA-seq data sets with matched bulk RNA-seq data
To help determine if the increased proportion of zeros in scRNA-seq is explained by biology or technical biases, we examined three publicly available scRNA-seq data sets (Trapnell and others, 2014; Shalek and others, 2013; Wu and others, 2014) that included a matched bulk RNA-seq sample measured on the same population of cells with preprocessed and normalized expression data available on GEO. One of these studies (Trapnell and others, 2014) is one of the 15 studies described in the previous subsection. The other two studies (Shalek and others, 2013; Wu and others, 2014) sequenced only 18 and 96 cells, respectively, thus were not included in the 15 large studies.
3. Results
3.1. The proportion of reported zeros varies from cell to cell and from study to study
We define the cell-specific detection rate as the proportion of genes in a cell reporting the expression levels greater than a predetermined threshold 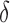 . The unit of expression for each cell in each study is either TPM, RPKM, FPKM, or CPM using UMIs (Table 1). In this article, we used
. The unit of expression for each cell in each study is either TPM, RPKM, FPKM, or CPM using UMIs (Table 1). In this article, we used  based on exploratory data analysis as this revealed two clear modes in the gene expression distribution (Figure S2 of supplementary material available at Biostatistics online), which we interpreted to be associated with background noise and signal respectively, with the lower mode defined as values below or equal to a TPM, FPKM, RPKM, or CPM threshold of
based on exploratory data analysis as this revealed two clear modes in the gene expression distribution (Figure S2 of supplementary material available at Biostatistics online), which we interpreted to be associated with background noise and signal respectively, with the lower mode defined as values below or equal to a TPM, FPKM, RPKM, or CPM threshold of  . This threshold has been previously used by Shalek and others (2014) and accommodates the bimodality (Bacher and Kendziorski, 2016; Kharchenko and others, 2014; Shalek and others, 2013; Finak and others, 2015; Korthauer and others, 2016) of scRNA-seq data that is not found in bulk RNA-Seq. We found wide variation in the detection rate across cells in all studies: from
. This threshold has been previously used by Shalek and others (2014) and accommodates the bimodality (Bacher and Kendziorski, 2016; Kharchenko and others, 2014; Shalek and others, 2013; Finak and others, 2015; Korthauer and others, 2016) of scRNA-seq data that is not found in bulk RNA-Seq. We found wide variation in the detection rate across cells in all studies: from  1% detected to 65
1% detected to 65 (Figure 1). Similar results were obtained if we set
(Figure 1). Similar results were obtained if we set 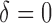 (Figure S3 of supplementary material available at Biostatistics online). For studies including groups known to have different gene expression profiles, we stratified by biological group to minimize the possibility of a biological explanation and also found wide variation (Figures S4–S9 of supplementary material available at Biostatistics online). In addition, we compared the relationship between the detection rate and sequencing depth using data from two studies and found the correlation is 0.096 and 0.139 for the Patel and others (2014) and Trapnell and others (2014) data (Figure S10 of supplementary material available at Biostatistics online), respectively, implying that over 95
(Figure S3 of supplementary material available at Biostatistics online). For studies including groups known to have different gene expression profiles, we stratified by biological group to minimize the possibility of a biological explanation and also found wide variation (Figures S4–S9 of supplementary material available at Biostatistics online). In addition, we compared the relationship between the detection rate and sequencing depth using data from two studies and found the correlation is 0.096 and 0.139 for the Patel and others (2014) and Trapnell and others (2014) data (Figure S10 of supplementary material available at Biostatistics online), respectively, implying that over 95 of the variability in the detection rates is not explained by sequencing depth.
of the variability in the detection rates is not explained by sequencing depth.
Fig. 1.

Boxplots of the detection rate, or the proportion of genes in a cell reporting expression values greater 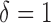 calculated for each cell across 15 publicly available scRNA-seq studies (the number of genes (G) included in each study). Boxplots are ordered by median detection rate across cells within a study. The detection rate across cells and studies ranges from less than 1
calculated for each cell across 15 publicly available scRNA-seq studies (the number of genes (G) included in each study). Boxplots are ordered by median detection rate across cells within a study. The detection rate across cells and studies ranges from less than 1 to 65
to 65 . For Patel and others (2014)
. For Patel and others (2014) , the data submitted to GEO were de-trended so that measurements for each gene averaged to zero across cells and authors applied heavy gene filtering resulting in only
, the data submitted to GEO were de-trended so that measurements for each gene averaged to zero across cells and authors applied heavy gene filtering resulting in only 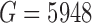 genes. Therefore, the detection rate for this study was calculated by downloading raw sequencing files and quantifying gene-level expression for
genes. Therefore, the detection rate for this study was calculated by downloading raw sequencing files and quantifying gene-level expression for 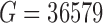 genes. (see Section 2.1 for complete details).
genes. (see Section 2.1 for complete details).
3.2. scRNA-seq data contains more zeros than expected from biological variation
To demonstrate that there are more zeros in scRNA-seq data than expected from biological variation, we examined the gene expression of cells measured both on scRNA-seq and bulk RNA-seq. We found a bias consistent with a technical explanation. The details follow.
Denote the expression level for the 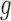 th gene and
th gene and 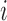 th cell as
th cell as  where
where 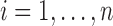 . The expression for the
. The expression for the 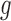 th gene in bulk tissue composed of these cells will then be:
th gene in bulk tissue composed of these cells will then be:
 |
Measurement technology, such as bulk RNA-seq, gives us an estimate proportional to  . Here, we denote this random variable with
. Here, we denote this random variable with 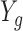 and model is using:
and model is using:
 |
where  represents measurement error. Here,
represents measurement error. Here,  is a normalizing constant needed to account for the fact that experimental protocols and normalization procedures are adjusted to assure that the average or sum of measurements from each experiment are approximately the same. Since a tissue sample will have millions of cells, we consider
is a normalizing constant needed to account for the fact that experimental protocols and normalization procedures are adjusted to assure that the average or sum of measurements from each experiment are approximately the same. Since a tissue sample will have millions of cells, we consider 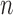 to be large enough to be treated as infinity. Note that some of these
to be large enough to be treated as infinity. Note that some of these 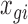 can be zero even when
can be zero even when 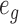 is a large number. In fact, this is part of the biological explanation for why single-cell measurements have more zeros: a gene appearing expressed in bulk RNA-seq need not be expressed in every single cell at the time the cells were isolated and measured.
is a large number. In fact, this is part of the biological explanation for why single-cell measurements have more zeros: a gene appearing expressed in bulk RNA-seq need not be expressed in every single cell at the time the cells were isolated and measured.
In a single-cell experiment, we take a random sample of 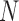 cells from the population. We denote the expression values for these as
cells from the population. We denote the expression values for these as 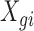 where
where  . Using scRNA-seq technology, we obtain measurements:
. Using scRNA-seq technology, we obtain measurements:
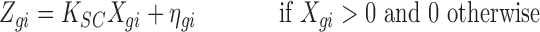 |
Here  is the normalizing constant and
is the normalizing constant and  is measurement error. Because the single-cell data is a random sample, it follows that
is measurement error. Because the single-cell data is a random sample, it follows that
 |
and therefore, if there is no bias induced from dropouts,
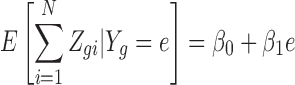 |
is a linear function with  and
and  determined by the normalization constants and the variance of the measurement error, which has been reported to be relatively low. While in a typical scRNA-seq experiment, bulk RNA-seq measurements from the same tissue is not available, the three studies described in Section 2.2 with both bulk RNA-seq and scRNA-Seq from the same biological specimens permits us to check if this relationship holds.
determined by the normalization constants and the variance of the measurement error, which has been reported to be relatively low. While in a typical scRNA-seq experiment, bulk RNA-seq measurements from the same tissue is not available, the three studies described in Section 2.2 with both bulk RNA-seq and scRNA-Seq from the same biological specimens permits us to check if this relationship holds.
As evidence that scRNA-seq technology is working as expected, previous groups (Shalek and others, 2013; Wu and others, 2014; Trapnell and others, 2014; Marinov and others, 2014; Piras and Selvarajoo, 2015) plot 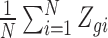 versus
versus 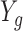 for each gene to show it generally follows a linear relationship with reported correlations around 0.80 (see e.g. Figure 1C in Shalek and others (2013) which we reproduced in Figure 2A). However, a closer look at this plot reveals a problem: the linear relationship does not appear to hold for lowly expressed genes (Figure 2B). This same pattern is observed in the other two studies with bulk and scRNA-seq data (Figure S11 of supplementary material available at Biostatistics online).
for each gene to show it generally follows a linear relationship with reported correlations around 0.80 (see e.g. Figure 1C in Shalek and others (2013) which we reproduced in Figure 2A). However, a closer look at this plot reveals a problem: the linear relationship does not appear to hold for lowly expressed genes (Figure 2B). This same pattern is observed in the other two studies with bulk and scRNA-seq data (Figure S11 of supplementary material available at Biostatistics online).
Fig. 2.

RNA-seq profiles compared to averaged scRNA-seq profile. (A) Scatter plot comparing a bulk RNA-seq profile and an averaged scRNA-seq profile, which we reproduced from Figure 1C in Shalek and others (2013). (B) The MA plot demonstrates there is a bias between the bulk profile and the single-cell profile averaged across cells as the single-cell profile averaged across cells is smaller than the bulk profile for low expressed genes. The dotted line is calculated by binning the values along the x-axis (the average between bulk and single-cell profile averaged across cells on log scale) and calculating the mean of values on the y-axis (difference between the bulk and single-cell profile averaged across cells on log scale) within each bin.
To further explore this apparent bias, we stratified the values of 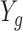 and estimated the conditional expectations of
and estimated the conditional expectations of  by averaging the scRNA-seq data in each stratum. Plotting these against each other revealed a bias that increases as
by averaging the scRNA-seq data in each stratum. Plotting these against each other revealed a bias that increases as  becomes closer to zero (Figure 3). These results are very much consistent with the theory that some of the observed zeros are due to technical and not biological differences with the actual relationship being:
becomes closer to zero (Figure 3). These results are very much consistent with the theory that some of the observed zeros are due to technical and not biological differences with the actual relationship being:
Fig. 3.

Plots comparing bulk and averaged scRNA-seq profiles that demonstrate evidence of more zeros in in scRNA-seq data for low expressed genes than what is expected. Data was obtained from three publicly available scRNA-seq studies that included a matched bulk RNA-seq sample measured on the same population of cells (Shalek and others, 2013; Wu and others, 2014; Trapnell and others, 2014). The red triangles are averages of the single-cell profiles computed in strata defined by the bulk RNA-seq values. The black solid line is what we expect if there is no bias.
 |
with 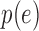 the probability of a gene with expression
the probability of a gene with expression 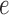 being detected. A crude estimate of
being detected. A crude estimate of 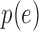 can be obtained by calculating the ratio of
can be obtained by calculating the ratio of
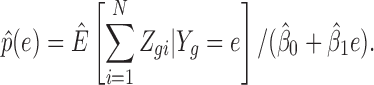 |
This estimate suggests 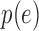 follows a logistic function (Figure S12 of supplementary material available at Biostatistics online) as others have previously noted (Kharchenko and others, 2014). In other words, genes with a lower expression
follows a logistic function (Figure S12 of supplementary material available at Biostatistics online) as others have previously noted (Kharchenko and others, 2014). In other words, genes with a lower expression 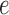 are less likely to be detected, which suggests the zeros can be considered missing not at random as the probability of the missing value depends on the level of expression.
are less likely to be detected, which suggests the zeros can be considered missing not at random as the probability of the missing value depends on the level of expression.
Motivated by Figure S12 of supplementary material available at Biostatistics online, we fit a logistic curve to determine the relationship between 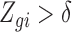 and
and 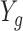 for each cell
for each cell  . We found that the biases induced by this missing data problem are exacerbated by the fact that the probability of a gene being detected varies cell to cell, as the estimates for the logistic curves intercept parameter are highly related to the cell-specific detection rate (Figure S13 of supplementary material available at Biostatistics online). We also note that the slope estimates are between 0.53 and 1.31. For example, the slopes estimated using the Trapnell and others (2014) data has an average of 0.82 and a standard deviation of 0.6 demonstrating the strong effect overall expression has on detection: note for example that a slope of 0.82 means that if the expression level is cut in half, the detection odds decrease by more than 2-fold since e
. We found that the biases induced by this missing data problem are exacerbated by the fact that the probability of a gene being detected varies cell to cell, as the estimates for the logistic curves intercept parameter are highly related to the cell-specific detection rate (Figure S13 of supplementary material available at Biostatistics online). We also note that the slope estimates are between 0.53 and 1.31. For example, the slopes estimated using the Trapnell and others (2014) data has an average of 0.82 and a standard deviation of 0.6 demonstrating the strong effect overall expression has on detection: note for example that a slope of 0.82 means that if the expression level is cut in half, the detection odds decrease by more than 2-fold since e = 2.27.
= 2.27.
3.3. Detection rate is a major source of cell-to-cell variation
Finak and others (2015) showed that detection rates correlate with the first two principal components (PCs) in two scRNA-seq data sets (Shalek and others, 2014; Finak and others, 2015). We confirmed this relationship on the 15 publicly available scRNA-seq data sets we studied (Figures S14 and S15 of supplementary material available at Biostatistics online). From this strong correlation it follows that estimated distances between cells are affected by differences in detection rate. We note that for five of these studies (Zeisel and others, 2015; Deng and others, 2014; Guo and others, 2015; Kumar and others, 2014; Burns and others, 2015), the primary variation along the first two PCs was correlated strongly with the biological groups known to have different gene expression patterns. For these studies, we stratified the data into these groups and found the same strong relationship between detection rates and first PC. In this section, we present results that demonstrate that (1) this variability is partly driven by a mathematical artifact related to scaling the original data but computing distances in the log transformed data and (2) differences in detection rate can be completely driven by technical reasons which can in turn result in false discoveries.
Currently, the most widely used unit for reporting expression values is the TPM unit. Using this unit guarantees that the sum of gene expression measurements is constant. This is also true for CPM and approximately true with the RPKM (Mortazavi and others, 2008) and FPKM (Trapnell and others, 2010) units. However, distance calculations are performed after log transformations and cell expression profiles are not always reported as being centered (centered by removing overall mean expression from the  th cell) in published analyses (Macosko and others, 2015; Treutlein and others, 2014; Burns and others, 2015; Kowalczyk and others, 2015). We can show, mathematically, that if we normalize expression profiles to have the same mean across cells, the mean after the
th cell) in published analyses (Macosko and others, 2015; Treutlein and others, 2014; Burns and others, 2015; Kowalczyk and others, 2015). We can show, mathematically, that if we normalize expression profiles to have the same mean across cells, the mean after the 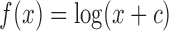 transformation used for RNA-Seq data will not be the same, and it will depend on the detection rate (Figure S16 of supplementary material available at Biostatistics online).
transformation used for RNA-Seq data will not be the same, and it will depend on the detection rate (Figure S16 of supplementary material available at Biostatistics online).
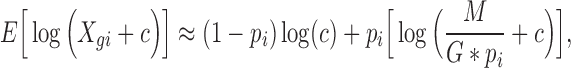 |
(3.1) |
where 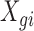 is the expression value for the
is the expression value for the 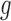 th gene and
th gene and 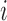 th cell,
th cell,  is a pseudo count,
is a pseudo count,  is the number of genes (or features),
is the number of genes (or features), 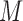 is sequencing depth, and
is sequencing depth, and  is the marginal probability of detection for the
is the marginal probability of detection for the 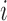 th cell (mathematical details are provided in the Supplementary material available at Biostatistics online). The implication of this result is that although the means are constant across cells, the means of the log-transformed data depend on the detection rate. In fact, when the sequencing depth is large, the mathematical relationship above is approximately a linear function of the detection rate. Because these mean values affect the entire vector, they can result in large overall variability and therefore be correlated with the first PC. Therefore, the result in Figures S14 and S15 of supplementary material available at Biostatistics online can be explained by differences in mean values that correlate with the detection rate.
th cell (mathematical details are provided in the Supplementary material available at Biostatistics online). The implication of this result is that although the means are constant across cells, the means of the log-transformed data depend on the detection rate. In fact, when the sequencing depth is large, the mathematical relationship above is approximately a linear function of the detection rate. Because these mean values affect the entire vector, they can result in large overall variability and therefore be correlated with the first PC. Therefore, the result in Figures S14 and S15 of supplementary material available at Biostatistics online can be explained by differences in mean values that correlate with the detection rate.
If this mathematical artifact was the primary reason driving the association between the detection rate and the first PC of each study, then we would expect the relationship should be greatly reduced after we center the cell-specific means before computing the PCs. We found that the correlation between the detection rate and first PC does decrease some if we center the data before computing the PCs. However, despite the decrease, the correlation is still strong (Figures S17 and S18 of supplementary material available at Biostatistics online). This suggests it is not just the proportion of observed zeros in each cell that drives this relationship. If we plot the relationship between the detection rate and the 25th, 50th, and 75th percentile of non-zero expressed genes in each cell, we found the entire distribution of the non-zero measurements depends on the detection rate (Figure 4).
Fig. 4.

The distribution of gene expression changes with the detection rate using processed scRNA-seq data available on GEO. Failure to account for differences of the proportion of detected genes between cells over-inflates the gene expression estimates of cells with a low detection rate. The curves were obtained by fitting a locally weighted scatter plot smoothing (loess) with a degree of 1. Because the range of detection rate varied from study to study, the range of the x-axis differs across plots.
To demonstrate that technical variability can lead to differences in detection rates, which in turn can lead to false discoveries, we used a data set composed of a group of cells from the same biological specimen, but processed at different times for which we know that cells from the same biological sample were randomly split into two groups differing in a technical variable: the sequencing instrument. Specifically, we used a subset of 118 single cells from Patel and others (2014), which were isolated from one tumor, but processed in two different sequencing instruments. For these data, there are no biological reasons for the two groups, defined by the sequencing instrument used, to be different since the cells were randomly selected from the same tumor. If we apply an unsupervised clustering algorithm to these data, two strong clusters appear (Figure S19 of supplementary material available at Biostatistics online) even after removing the cell mean before computing distances. A PCA plot shows that a batch effect drives the clustering (Figure 5A). We then note that the first PC strongly correlates with the detection rate (Figure 5B), which is substantially different between the two batches (Figure 5C). In addition, the differences in detection rates are highly related to the logistic curves intercept parameter when estimating the probability of a gene being detected, which varies cell to cell (Figure S20 of supplementary material available at Biostatistics online). Therefore, we see how a batch effect can produce differences in detection rate that drive distances between transcription profiles and leads to false discoveries.
Fig. 5.

Illustration of how technical variation can lead to differences in detection rates, which in turn can lead to false differences. Data from Patel and others (2014). (A) Using PCs analysis, scRNA-seq samples cluster by sequencing instrument. (B) The first PC is strongly associated the detection rate. (C) Boxplots of detection rates stratified by sequencing instrument used to sequence cells.
3.4. The impact of the detection rate in applications of unsupervised learning methods
In the previous sections, we demonstrated how the detection rate is a major source of observed cell-to-cell variation, which can be driven by technical variation. For example, we considered cells from the same biological specimen for which we knew no differences should be discovered, but we found a batch effect that had differences in the detection rate and could drive the variation between the cells. In this section, we examine detection rates in data used in published studies as evidence for the discovery of new cell types.
In the first case-study (Burns and others, 2015), we found differences in the detection rate between two groups of discovered cell types (Figure S21A of supplementary material available at Biostatistics online), which was associated with how the cells were processed in different batches (Figure S21B of supplementary material available at Biostatistics online) (Fisher’s exact test: 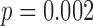 ). For example, 12 of 13 cells from one discovered cell type were processed in the same batch (Figure S21C of supplementary material available at Biostatistics online), which had a smaller median detection rate than the other batches. Furthermore, the detection rate was associated with the first PC (Figure S21D of supplementary material available at Biostatistics online), which could be partly driving the variation across the two groups of discovered cell types. Similarly, we found differences in the detection rate between groups of discovered cell types in another other study (Zeisel and others, 2015) (Figure S22 of supplementary material available at Biostatistics online), which was associated with how the cells were processed in different batches (Chi-squared test:
). For example, 12 of 13 cells from one discovered cell type were processed in the same batch (Figure S21C of supplementary material available at Biostatistics online), which had a smaller median detection rate than the other batches. Furthermore, the detection rate was associated with the first PC (Figure S21D of supplementary material available at Biostatistics online), which could be partly driving the variation across the two groups of discovered cell types. Similarly, we found differences in the detection rate between groups of discovered cell types in another other study (Zeisel and others, 2015) (Figure S22 of supplementary material available at Biostatistics online), which was associated with how the cells were processed in different batches (Chi-squared test:  ).
).
4. Discussion
We have demonstrated how detection varies substantially across scRNA-seq experiments. We presented evidence that part of this variability is technically driven. Given the logistics of how scRNA-Seq experiments are performed, and the fact that this technology is being used to discover new cell-types, batch effects are of particular concern. Specifically, when two groups of cells are cultured, captured, and sequenced separately from another group of cells in a second condition, correlated measurements may lead to the incorrect conclusion that these groups have different expression profiles. This experimental limitation presents a challenge in distinguishing biologically driven differences from technical ones because it is logistically difficult to avoid processing cells from different biological specimens in different batches. For the eight studies that were interested in comparing predefined biological groups, we used the standardized Pearson contingency coefficient to assess the level of confounding between the run number from the sequencing instrument and outcome of interest and found values ranging from 82.1 to 100
to 100 (perfect confounding) (Table 1; Tables S1–S8 of supplementary material available at Biostatistics online). Note that with this level of confounding, it is difficult to impossible to parse technical from biological variation.
(perfect confounding) (Table 1; Tables S1–S8 of supplementary material available at Biostatistics online). Note that with this level of confounding, it is difficult to impossible to parse technical from biological variation.
Furthermore, explicitly modeling confounding factors as in published batch correction methods (Leek, 2014; Love and others, 2014; Risso and others, 2014) is not appropriate in this context because the biological variation or signal of interest is often confounded with the unwanted technical variability. For the specific application of differential expression, a proposed solution is to account for differences in the proportion of detected genes by explicitly including it as a covariate in a linear regression model (Finak and others, 2015). However, given the current levels of confounding, this approach will not be able to distinguish biological from technical effects. For example, some studies have demonstrated cells with different biological phenotypes can express a different number of genes (Ramsköld and others, 2009).
An experimental design solution is to use biological replicates, namely independently repeating the experiment multiple times for each biological condition (Figure S1 of supplementary material available at Biostatistics online). This approach allows for multiple batches of cells to be randomized across sequencing runs, flow cells, and lanes as in bulk RNA-seq. With this design we can then model and adjust for batch effects due to systematic experimental bias. A more detailed discussion of how these factors affect the experimental design has been recently published (Bacher and Kendziorski, 2016; Stegle and others, 2015; Grün and van Oudenaarden, 2015).
5. Conclusions
Technical variability is considered to be a major challenge in the analysis of data measured on next-generation sequencing platforms. For example, amplification bias leading to batch effects has been shown to induce false positives in differential expression studies with bulk RNA-seq data (Lahens and others, 2014; Love and others, 2016). By examining three assessment experiments containing both bulk and single-cell RNA-seq data, we demonstrated that technical variability is a challenge in scRNA-seq as well, with a major problem arising due to differences in capture inefficiencies. Using public data from 15 studies, we showed that these inefficiencies lead to substantial differences in detection rates that lead to distortion in distance calculations, which in turn can lead to false discoveries when using unsupervised clustering.
6. Software
All the code for this analysis is available on GitHub at https://github.com/stephaniehicks/scBatchPaper.
Supplementary Material
Acknowledgements
We thank Bradley Bernstein who provided comments that we used to improve the manuscript. Conflict of Interest: None declared.
Funding
NIH R01 (GM083084, RR021967/GM103552, and HG005220); and NIH/NHGRI (K99HG009007).
References
- Achim, K., Pettit, J.-B., Saraiva, L. R., Gavriouchkina, D., Larsson, T., Arendt, D. and Marioni, J. C. (2015). High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nature Biotechnology 33, 503–509. [DOI] [PubMed] [Google Scholar]
- Bacher, R. and Kendziorski, C. (2016). Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biology 17, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel, C., Ferreira, P. G., Santoni, F., Delaneau, O., Fort, A., Popadin, K. Y., Garieri, M., Falconnet, E., Ribaux, P., Guipponi, M., Padioleau, I., Carninci, P., Dermitzakis, E. T.. and others (2015). Biased allelic expression in human primary fibroblast single cells. American Journal of Human Genetics 96, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, N. L., Pimentel, H., Melsted, P. and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brennecke, P., Anders, S., Kim, J. K., Kołodziejczyk, A. A., Zhang, X., Proserpio, V., Baying, B., Benes, V., Teichmann, S. A., Marioni, J. C.. and others (2013). Accounting for technical noise in single-cell RNA-seq experiments. Nature Methods 10, 1093–1095. [DOI] [PubMed] [Google Scholar]
- Buettner, F., Natarajan, K. N., Casale, F. P., Proserpio, V., Scialdone, A., Theis, F. J., Teichmann, S. A., Marioni, J. C. and Stegle, O. (2015). Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nature Biotechnology 33, 155–160. [DOI] [PubMed] [Google Scholar]
- Burns, J. C., Kelly, M. C., Hoa, M., Morell, R. J. and Kelley, M. W. (2015). Single-cell RNA-seq resolves cellular complexity in sensory organs from the neonatal inner ear. Nature Communications 6, 8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs, P. A. and Eisen, M. B. (2015). Low-cost, low-input RNA-seq protocols perform nearly as well as high-input protocols. Peer Journal 3, e869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Q., Ramsköld, D., Reinius, B. and Sandberg, R. (2014). Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196. [DOI] [PubMed] [Google Scholar]
- Edgar, R., Domrachev, M. and Lash, A. E. (2002). Gene expression omnibus: Ncbi gene expression and hybridization array data repository. Nucleic Acids Research 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak, G., McDavid, A., Chattopadhyay, P., Dominguez, M., De Rosa, S., Roederer, M. and Gottardo, R. (2014). Mixture models for single-cell assays with applications to vaccine studies. Biostatistics 15, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak, G., McDavid, A., Yajima, M., Deng, J., Gersuk, V., Shalek, A. K., Slichter, C. K., Miller, H. W., McElrath, M., Juliana, P., Martin, L., Peter S.. and others (2015). Mast: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biology 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad, Y. and Mizrahi-Man, O. (2015). A reanalysis of mouse encode comparative gene expression data. F1000Research 4, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün, D. and van Oudenaarden, A. (2015). Design and analysis of single-cell sequencing experiments. Cell 163, 799–810. [DOI] [PubMed] [Google Scholar]
- Guo, F., Yan, L., Guo, H., Li, L., Hu, B., Zhao, Y., Yong, J., Hu, Y., Wang, X., Wei, Y., Wang, W., Li, R., Yan, J., Zhi, X., Zhang, Y., Jin, H., Zhang, W., Hou, Y., Zhu, P., Li, J., Zhang, L., Liu, S., Ren, Y., Zhu, X., Wen, L., Gao, Y. Q., Tang, F.. and others (2015). The transcriptome and DNA methylome landscapes of human primordial germ cells. Cell 161, 1437–1452. [DOI] [PubMed] [Google Scholar]
- Harrow, J., Denoeud, F., Frankish, A., Reymond, A., Chen, C.-K., Chrast, J., Lagarde, J., Gilbert, J. G. R., Storey, R., Swarbreck, D., Rossier, C., Ucla, C., Hubbard, T., Antonarakis, S. E.. and others (2006). Gencode: producing a reference annotation for encode. Genome Biology 7 Suppl 1, S4.1–S4.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony, T., Wagner, F., Sher, N. and Yanai, I. (2012). Cel-seq: single-cell RNA-seq by multiplexed linear amplification. Cell Reports 2, 666–673. [DOI] [PubMed] [Google Scholar]
- Islam, S., Kjällquist, U., Moliner, A., Zajac, P., Fan, J.-B., Lönnerberg, P. and Linnarsson, S. (2011). Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Research 21, 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin, D. A., Kenigsberg, E., Keren-Shaul, H., Elefant, N., Paul, F., Zaretsky, I., Mildner, A., Cohen, N., Jung, S., Tanay, A.. and others (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko, P. V., Silberstein, L. and Scadden, D. T. (2014). Bayesian approach to single-cell differential expression analysis. Nature Methods 11, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivioja, T., Vähärautio, A., Karlsson, K., Bonke, M., Enge, M., Linnarsson, S. and Taipale, J. (2011). Counting absolute numbers of molecules using unique molecular identifiers. Nature Methods 9, 72–74. [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk, A. A., Kim, J. K., Svensson, V., Marioni, J. C. and Teichmann, S. A. (2015). The technology and biology of single-cell RNA sequencing. Molecular Cell 58, 610–620. [DOI] [PubMed] [Google Scholar]
- Korthauer, K. D, Chu, L.-F., Newton, M. A., Li, Y., Thomson, J., Stewart, R. and Kendziorski, C. (2016). A statistical approach for identifying differential distributions in single-cell RNA-seq experiments. Genome Biology 17, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk, M. S., Tirosh, I., Heckl, D., Rao, T. N., Dixit, A., Haas, B. J., Schneider, R. K., Wagers, A. J., Ebert, B. L. and Regev, A. (2015). Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Research 25, 1860–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, R. M., Cahan, P., Shalek, A. K., Satija, R., DaleyKeyser, A., Li, H., Zhang, J., Pardee, K., Gennert, D., Trombetta, J. J., Ferrante, T. C., Regev, A., Daley, G. Q.. and others (2014). Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature 516, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon, S and Lee, A. B. (2006). Diffusion maps and coarse-graining: a unified framework for dimensionality reduction, graph partitioning, and data set parameterization. IEEE Transactions on Pattern Analysis and Machine Intelligence 28, 1393–1403. [DOI] [PubMed] [Google Scholar]
- Lahens, N. F., Kavakli, I. H., Zhang, R., Hayer, K., Black, M. B., Dueck, H., Pizarro, A., Kim, J., Irizarry, R., Thomas, R. S., Grant, G. R.. and others (2014). Ivt-seq reveals extreme bias in RNA sequencing. Genome Biology 15, R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek, J. T. (2014). svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Research 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek, J. T., Scharpf, R. B., Bravo, H. C., Simcha, D., Langmead, B., Johnson, W. E., Geman, D., Baggerly, K. and Irizarry, R. A. (2010). Tackling the widespread and critical impact of batch effects in high-throughput data. Nature Reviews Genetics 11, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen, R., Sugawara, H., Shumway, M. and International Nucleotide Sequence Database Collaboration. (2011). The sequence read archive. Nucleic Acids Research 39(Database issue), D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng, N., Chu, L.-F., Barry, C., Li, Y., Choi, J., Li, X., Jiang, P., Stewart, R. M., Thomson, J. A. and Kendziorski, C. (2015). Oscope identifies oscillatory genes in unsynchronized single-cell RNA-seq experiments. Nature Methods 12, 947–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. and Dewey, C. N. (2011). Rsem: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M. I., Hogenesch, J. B. and Irizarry, R. A. (2016). Modeling of RNA-seq fragment sequence bias reduces systematic errors in transcript abundance estimation. Nature Biotechnology 34, 1287–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M. I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with deseq2. Genome Biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun, A. T. L., Bach, K. and Marioni, J. C. (2016). Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biology 17, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., Tirosh, I., Bialas, A. R., Kamitaki, N., Martersteck, E. M., Trombetta, J. J., Weitz, D. A., Sanes, J. R., Shalek, A. K., Regev, A.. and others (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinov, G. K., Williams, B. A., McCue, K., Schroth, G. P., Gertz, J., Myers, R. M. and Wold, B. J. (2014). From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Research 24, 496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by rna-seq. Nature Methods 5, 621–8. [DOI] [PubMed] [Google Scholar]
- Nadler, B., Lafon, S., Coifman, R. R. and Kevrekidis, I. G. (2006). Diffusion maps, spectral clustering and the reaction coordinates of dynamical systems. Applied and Computational Harmonic Analysis: Special Issue on Diffusion Maps and Wavelets 21, 113–127. [Google Scholar]
- O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., Rajput, B., Robbertse, B., Smith-White, B., Ako-Adjei, D., Astashyn, A., Badretdin, A., Bao, Y., Blinkova, O., Brover, V., Chetvernin, V., Choi, J., Cox, E., Ermolaeva, O., Farrell, C. M., Goldfarb, T., Gupta, T., Haft, D., Hatcher, E., Hlavina, W., Joardar, V. S., Kodali, V. K., Li, W., Maglott, D., Masterson, P., McGarvey, K. M., Murphy, M. R., O’Neill, K., Pujar, S., Rangwala, S. H., Rausch, D., Riddick, L. D., Schoch, C., Shkeda, A., Storz, S. S, Sun, H., Thibaud-Nissen, F., Tolstoy, I., Tully, R. E., Vatsan, A. R., Wallin, C., Webb, D, Wu, W., Landrum, M. J., Kimchi, A., Tatusova, T., DiCuccio, M., Kitts, P., Murphy, T. D.. and others (2016). Reference sequence (refseq) database at ncbi: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, A. P., Tirosh, I., Trombetta, J. J., Shalek, A. K., Gillespie, S. M., Wakimoto, H., Cahill, D. P., Nahed, B. V., Curry, W. T., Martuza, R. L., Louis, D. N., Rozenblatt-Rosen, O., Suvà, M. L., Regev, A.. and others (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, K. (1901). On lines and planes of closest fit to systems of points in space. Philiosphical Magazine 2, 559–572. [Google Scholar]
- Picelli, S., Björklund, Å. K., Faridani, O. R., Sagasser, S., Winberg, G. and Sandberg, R. (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Pierson, E. and Yau, C. (2015). Zifa: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biology 16, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piras, V. and Selvarajoo, K. (2015). The reduction of gene expression variability from single cells to populations follows simple statistical laws. Genomics 105, 137–144. [DOI] [PubMed] [Google Scholar]
- Ramsköld, D., Luo, S., Wang, Y.-C., Li, R., Deng, Q., Faridani, O. R., Daniels, G. A., Khrebtukova, I., Loring, J. F., Laurent, L. C., Schroth, G. P.. and others (2012). Full-length mRNA-seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotechnology 30, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld, D., Wang, E. T., Burge, C. B. and Sandberg, R. (2009). An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Computational Biology 5, e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso, D., Ngai, J., Speed, T. P. and Dudoit, S. (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nature Biotechnology 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba, A.-E., Westermann, A. J., Gorski, S. A. and Vogel, J. (2014). Single-cell RNA-seq: advances and future challenges. Nucleic Acids Research 42, 8845–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. and Regev, A. (2015). Spatial reconstruction of single-cell gene expression data. Nature Biotechnology 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek, A. K., Satija, R., Adiconis, X., Gertner, R. S., Gaublomme, J. T., Raychowdhury, R., Schwartz, S., Yosef, N., Malboeuf, C., Lu, D., Trombetta, J. J., Gennert, D., Gnirke, A., Goren, A., Hacohen, N., Levin, J. Z., Park, H.. and others (2013). Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek, A. K., Satija, R., Shuga, J., Trombetta, J. J., Gennert, D., Lu, D., Chen, P., Gertner, R. S., Gaublomme, J. T., Yosef, N., Schwartz, S., Fowler, B., Weaver, S., Wang, J., Wang, X., Ding, R., Raychowdhury, R., Friedman, N., Hacohen, N., Park, H., May, A. P.. and others (2014). Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, E., Biezuner, T. and Linnarsson, S. (2013). Single-cell sequencing-based technologies will revolutionize whole-organism science. Nature Reviews Genetics 14, 618–630. [DOI] [PubMed] [Google Scholar]
- Stegle, O., Teichmann, S. A. and Marioni, J. C. (2015). Computational and analytical challenges in single-cell transcriptomics. Nature Reviews Genetics 16, 133–145. [DOI] [PubMed] [Google Scholar]
- Svensson, V., Natarajan, K. N., Ly, L.-H., Miragaia, R. J., Labalette, C., Macaulay, I. C., Cvejic, A. and Teichmann, S. A. (2017). Power analysis of single-cell RNA-sequencing experiments. Nature Methods 14, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., Wang, X., Bodeau, J., Tuch, B. B., Siddiqui, A., Lao, K.. and others (2009). mRNA-seq whole-transcriptome analysis of a single cell. Nature Methods 6, 377–382. [DOI] [PubMed] [Google Scholar]
- Tipping, M. E. and Bishop, C. M. (1999). Probablistic principal components analysis. JR Stat Soc: Series B (Statistical Methodology) 61(611–622). [Google Scholar]
- Torgerson, W. S. (1952). Multidimensional scaling I: Theory and method. Psychometrika 17, 401–419. [DOI] [PubMed] [Google Scholar]
- Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokharel, P., Li, S., Morse, M., Lennon, N. J., Livak, K. J., Mikkelsen, T. S. and Rinn, J. L. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J. and Pachter, L. (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein, B., Brownfield, D. G., Wu, A. R., Neff, N. F., Mantalas, G. L., Espinoza, F. H., Desai, T. J., Krasnow, M. A. and Quake, S. R. (2014). Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung, P.-Y., Blischak, J. D., Hsiao, C. J., Knowles, D. A., Burnett, J. E., Pritchard, J. K. and Gilad, Y. (2017). Batch effects and the effective design of single-cell gene expression studies. Science Reports 7, 39921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner, C., Barber, G. P., Casper, J., Clawson, H., Diekhans, M., Eisenhart, C., Fischer, C. M., Gibson, D., Gonzalez, J. N., Guruvadoo, L., Haeussler, M., Heitner, S., Hinrichs, A. S., Karolchik, D., Lee, B. T., Lee, C. M., Nejad, P., Raney, B. J., Rosenbloom, K. R., Speir, M. L., Villarreal, C., Vivian, J., Zweig, A. S., Haussler, D., Kuhn, R. M.. and others (2017). The UCSC genome browser database: 2017 update. Nucleic Acids Research 45, D626–D634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usoskin, D., Furlan, A., Islam, S., Abdo, H., Lönnerberg, P., Lou, D., Hjerling-Leffler, J., Haeggström, J., Kharchenko, O., Kharchenko, P. V., Linnarsson, S.. and others (2015). Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nature Neuroscience 18, 145–153. [DOI] [PubMed] [Google Scholar]
- Vallejos, C. A., Risso, D., Scialdone, A., Dudoit, S. and Marioni, J. C. (2017). Normalizing single-cell RNA sequencing data: challenges and opportunities. Nature Methods 14, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten, L. (2008). Visualizing data using t-sne. Journal of Machine Learning Research 9, 2579–2605. [Google Scholar]
- Wilson, N. K., Kent, D. G., Buettner, F., Shehata, M., Macaulay, I. C., Calero-Nieto, F. J., Sánchez C., Manuel, O., Caroline, A., Diamanti, E., Schulte, R., Ponting, C. P., Voet, T., Caldas, C., Stingl, J., Green, A. R., Theis, F. J.. and others (2015). Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell 16, 712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, A. R., Neff, N. F., Kalisky, T., Dalerba, P., Treutlein, B., Rothenberg, M. E., Mburu, F. M., Mantalas, G. L., Sim, S., Clarke, M. F.. and others (2014). Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods 11, 41–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel, A., Muñoz-Manchado, A. B., Codeluppi, S., Lönnerberg, P., La, M., Gioele, J., Anna, M., Sueli, M., Hermany, H., Liqun, B., Christer, R., Charlotte, Castelo-Branco, G., Hjerling-Leffler, J.. and others (2015). Brain structure. cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zheng, G. X. Y., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., Ziraldo, S. B., Wheeler, T. D., McDermott, G. P., Zhu, J., Gregory, M. T., Shuga, J., Montesclaros, L., Underwood, J. G., Masquelier, D. A., Nishimura, S. Y., Schnall-Levin, M., Wyatt, P. W., Hindson, C. M., Bharadwaj, R., Wong, A., Ness, K. D., Beppu, L. W., Deeg, H. J., McFarland, C., Loeb, K. R., Valente, W. J., Ericson, N. G., Stevens, E. A., Radich, J. P., Mikkelsen, T. S., Hindson, B. J.. and others (2017). Massively parallel digital transcriptional profiling of single cells. Nature Communications 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L., Lei, J. and Roeder, K. A. (2016). A unified statistical framework for single cell and bulk RNA sequencing data. arXiv:1609.08028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegenhain, C., Vieth, B., Parekh, S., Reinius, B., Guillaumet-Adkins, A., Smets, M., Leonhardt, H., Heyn, H., Hellmann, I. and Enard, W. (2017). Comparative analysis of single-cell RNA sequencing methods. Molecular Cell 65, 631–643.e4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.