

Abstract
Previous work from our group showed that tamoxifen, an oral drug that has been in use for the treatment of breast cancer for over 40 years, is active both in vitro and in vivo against several species of Leishmania, the etiological agent of leishmaniasis. Using a combination of metabolic labeling with [3H]-sphingosine and myo-[3H]-inositol, alkaline hydrolysis, HPTLC fractionations and mass spectrometry analyses, we observed a perturbation in the metabolism of inositolphosphorylceramides (IPCs) and phosphatidylinositols (PIs) after treatment of L. amazonensis promastigotes with tamoxifen, with a significant reduction in the biosynthesis of the major IPCs (composed of d16:1/18:0-IPC, t16:0/C18:0-IPC, d18:1/18:0-IPC and t16:0/20:0-IPC) and PIs (sn-1-O-(C18:0)alkyl -2-O-(C18:1)acylglycerol-3-HPO4-inositol and sn-1-O-(C18:0)acyl-2-O-(C18:1)acylglycerol-3-HPO4-inositol) species. Substrate saturation kinetics of myo-inositol uptake analyses indicated that inhibition of inositol transport or availability were not the main reasons for the reduced biosynthesis of IPC and PI observed in tamoxifen treated parasites. An in vitro enzymatic assay was used to show that tamoxifen was able to inhibit the Leishmania IPC synthase with an IC50 value of 8.48 μM (95% CI 7.68–9.37), suggesting that this enzyme is most likely one of the targets for this compound in the parasites.
Keywords: Leishmania amazonensis, Tamoxifen, Sphingolipids, Inositolphosphorylceramide, Phosphatidylinositols, IPC synthase
Graphical abstract
Highlights
-
•
Tamoxifen alters the sphingolipid metabolism of L. amazonensis.
-
•
Tamoxifen treated parasites show a significant reduction of IPC and PI species.
-
•
Tamoxifen-treated parasites present a reduction of inositol transport.
-
•
Tamoxifen is an inhibitor of L. major's IPC synthase in a micromolar range.
1. Introduction
Leishmania spp. are the etiological agent of leishmaniasis, a group of vector-borne neglected disease affecting approximately 12 million people worldwide with 1.2 million new cases per year (Reithinger et al., 2007; Alvar et al., 2012). Taken together they show a spectrum of clinical manifestations, ranging from self-healing cutaneous forms to fatal visceral leishmaniasis in endemic areas. This clinical diversity depends on parasite species, host immunity and genetics, amongst other factors (Reithinger et al., 2007; WHO, 2010). Leishmania (Leishmania) amazonensis is one of the most prevalent species causing human cutaneous leishmaniasis (CL) and the main etiological agent responsible for diffuse cutaneous leishmaniasis (DCL) in South America. DCL is characterized by multiple lesions with uncontrolled progression of infection and poor or absent response to chemotherapy due to host defective parasite-specific cell mediated immunity (Convit and Ulrich, 1993).
Only a few drugs are available for leishmaniasis treatment. These drugs are in general expensive, toxic and of systemic administration, and therapeutic failure is a problem in endemic areas (Croft and Coombs, 2003; Alvar et al., 2006). Against this background, drug repurposing is an attractive option for the discovery for new antileishmanials (Charlton et al., 2018).
Tamoxifen, an oral drug that has been in use for the treatment of breast cancer for over 40 years (Jordan, 2003), has been shown to be active against several species of Leishmania in vitro (Miguel et al., 2007) and in vivo (Miguel et al., 2008, 2009). It has also been shown to be a good partner when used in combination with amphotericin B (Trinconi et al., 2014), miltefosine (Trinconi et al., 2016) and meglumine antimoniate (Trinconi et al., 2017) in an established CL animal model. In many different lineages of human cancer cells tamoxifen has been proven to be a multi-target drug interfering in distinct cell pathways, such as sphingolipid (SL) metabolism (Cabot et al., 1996). SLs are essential cell membrane components in eukaryotic organisms (Mina and Denny, 2018), including protozoa of the Trypanosomatidae family such as Leishmania (Kaneshiro et al., 1986; Denny et al., 2004; Sutterwala et al., 2008). SLs act as important mediators of cell signaling and control several critical and important cell biology processes, including endocytosis, cell growth, differentiation, apoptosis, and oncogenesis (Shayman, 2000). The most abundant SL in Leishmania is inositolphosphorylceramide (IPC), corresponding to 5–10% of membrane total lipids (Kaneshiro et al., 1986) and abundantly present in membrane fractions known as lipid rafts (Yoneyama et al., 2006). IPC is also abundant in yeast (Shayman, 2000), Trypanosoma cruzi and Trypanosoma brucei (Figueiredo et al., 2005; Sutterwala et al., 2008). IPC synthase activity has been shown to be essential for S. cerevisiae survival (Nagiec et al., 1997) and T. brucei blood forms (Sutterwala et al., 2008; Mina et al., 2009). Mammalians do not synthetize IPC, with predominance of sphingomyelin (SM) instead (Merrill, 2011). IPC abundance in Leishmania and its absence in mammalian cells (Denny and Smith, 2004) suggest that the enzyme responsible for its synthesis, IPC synthase (Denny et al., 2006), might be a good target for therapeutic intervention. Remarkably, tamoxifen's activity over sphingolipid (SL) metabolism in cancer cells (Cabot et al., 1996) has been already demonstrated. These information prompted us to investigate whether this could be part of its mechanism of action against Leishmania.
In the present work, we show that L. amazonensis promastigotes treated with tamoxifen display a perturbation in SL metabolism with a significant reduction of IPCs/PIs species, increased abundance of acyl ceramide and direct inhibition of IPC synthase.
2. Material and methods
2.1. Parasites
L. amazonensis (MHOM/BR/73/M2269) promastigotes were cultivated in M-199 medium supplemented with 10% heat inactivated-fetal calf serum (FCS), 25 mM HEPES (pH 6.9), 12 mM NaHCO3, 7.6 mM hemin, 50 U/mL penicillin, 50 μg/mL streptomycin at 25 °C.
2.2. Drug and lipid standards
Tamoxifen (T5648) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Stock solutions of tamoxifen (10 mM) were prepared in DMSO and kept at −20 °C. Subsequent dilutions were done in culture media. d18:1/16:0 C16-ceramide (N-palmitoyl-D-erythro-sphingosine, Avanti®Polar Lipids, Inc.), 18:1 PI [1,2-dioleoyl-sn-glycero-3-phosphoinositol (ammonium salt), Avanti®Polar Lipids, Inc.] and acyl-ceramide (1-oleoyl-N-heptadecanoyl-D-erythro-sphingosine, Avanti®Polar Lipids, Inc.) were used as standards.
2.3. Metabolic labeling and lipid extraction
Metabolic labeling assays were performed with 8 × 108 L. amazonensis stationary phase promastigotes. After washing twice with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4), parasites were suspended in PBS-glucose (1 g/L) at a cell density of 5 × 107 cells/mL and treated with 10 μM tamoxifen during 4 h. Ninety minutes after the start of the treatment, cells were labeled with 2–6 μCi [3H]-sphingosine (Sphingosine, D-erythro-[3-3H], specific activity 18.4 Ci/mmol, PerkinElmer®, Boston, MA, USA) or 6 μCi myo-[3H]-inositol (Inositol, myo-[2–3H(N)]-, specific activity 22.5 Ci/mmol, PerkinElmer®, Boston, MA, USA) for 150 min. Then, the cells were washed three times with Hank's solution (137 mM NaCl, 5.3 mM KCl, 0.4 mM KH2PO4, 10 mM glucose, 4.2 mM NaHCO3 and 0.4 mM Na2HPO4, pH 7.2) to remove unincorporated radiolabeled precursors. Lipid extraction from 4 × 108 promastigotes was performed as described by Boath and coworkers with some modifications (Boath et al., 2008). Briefly, parasites were mixed at 1100 rpm with 1.24 mL of 1:1:0.75 (v/v/v) chloroform: methanol: ultra-pure water for 30 min. After centrifugation at 2×g during 10 min, the organic phase was reserved and the aqueous phase was re-extracted with 600 μL of chloroform. This procedure was repeated twice. The three organic phases obtained were joined, dried under N2 gas and stored at −20 °C.
2.4. High performance thin layer chromatography (HPTLC)
The lipid fractions of parasites treated or not with tamoxifen were analyzed by HPTLC. Lipid extracts were dissolved in 50 μL 1:1 (v/v) chloroform: methanol and 10 μL were spotted onto HPTLC Silica gel 60 plate (Merck, Darmastadt, Germany). Extracts were run in four different running systems: (1) 60:35:8 (v/v/v) chloroform:methanol:water during the initial run covering 20% of the plate and 90:2:8 (v/v/v) chloroform:methanol:acetic acid for the remaining of the plate (running system 1) (Ichikawa et al., 1994); (2) 60:35:8 (v/v/v) chloroform:methanol:water for 50% of the plate and 90:2:8 (v/v/v) chloroform:methanol:acetic acid for the remaining of the plate (running system 2) (Ichikawa et al., 1994 modified); (3) 58:32:9.3 (v/v/v) chloroform:methanol: methylamine 40% (running system 3) (Castro et al., 2013) and (4) 40:40:12 (v/v/v) chloroform:methanol:water (running system 4) (Martin and Smith, 2006). The migration patterns of standards were analyzed after staining in iodine vapor. Labeled lipids were visualized after spraying the plate with En3Hance® (PerkinElmer®) followed by exposure to autoradiography film (Hyperfilm™ Amersham ECL 18 × 24 cm) (GE Healthcare) at – 80 °C for 3–30 days. Autoradiography images were scanned and analyzed using the software ImageJ® (http://imagej.nih.gov/ij/). Intensity change was calculated as: % Change = 100 – [(Number of pixels treated sample area – number of pixels blank area) x 100]/(Number of pixels control sample area – number of pixels blank area). The percentage of intensity change on abundance of lipids of interest is presented as mean and standard deviation of 3–5 independent performed experiments.
2.5. Lipid extraction from silica
Labeled and unlabeled lipid fractions of parasites treated or not with tamoxifen were ran in parallel by HPTLC using running system 2. Guided by the migration of labeled lipids of interest, the unlabeled lipids were scraped from the silica plate and extracted three times with 3 mL 3:3:0.8 (v/v/v) chloroform:methanol:water followed by ultrasound treatment for 60 s in a water bath (VWR B2.500A Ultrasonic Cleaner - DTH, North American VWR), homogenization in a magnetic stirrer for 90 min at room temperature and a second ultrasound treatment for 60 s. The samples were centrifuged at 2600×g for 10 min and the organic phase transferred into a new tube. The three organic layers were dried under N2 gas and stored at - 20 °C.
2.6. Mass spectrometry analysis
Unlabeled lipids of interest, re-extracted from silica plates were analyzed by Electrospray Ionization Mass Spectrometry (ESI-MS). Total lipid extracted from silica was dissolved in 100 μL of 1:1 (v/v) chloroform:methanol containing 1 mM LiCl. Prior to injection, samples were diluted 1:10 and analyzed in the same solvent at an AmaZon SL Ion Trap ESI Mass Spectrometer (Bruker) with injection rate of 3 μL/min in positive and negative modes. The analysis was performed using the following parameters: nitrogen gas nebulizers at 10 psi, drying gas at 5 L/min, source temperature at 200 °C, ionization source at 4.5 kV, and collision gas with Helium. The analysis was carried out in the range of 50–900 m/z.
2.7. Alkaline hydrolysis
Purified lipid products, extracted from silica as described above, were incubated with 200 μL of 100 mM NaOH in 95% (v/v) aqueous methanol or 100 mM NaCl in 95% (v/v) aqueous methanol for 1 h at 37 °C. The treatment was terminated with 800 μL of 20% (v/v) acetic acid and 200 μL 0.5 M Tris-HCl pH 7.5. The products were desalted by two extractions with 2 vol of butan-1-ol saturated with water followed by 1 vol water saturated with butan-1-ol. The samples were dried under N2 gas and analyzed by HPTLC using running system 2.
2.8. Inositol uptake assay
For the uptake assay, promastigotes from exponential growth phase (day 3 of culture) were washed three times in PBS pH 7.4 through centrifugations at 3000×g for 10 min at 4 °C, suspended at a density of 3 × 108 promastigotes/mL in PBS and distributed in 100 μL aliquots (3 × 107 cells each). The uptake of the radiolabeled substrate was initiated by the addition of 100 μL of the uptake solution consisting of myo-inositol (50, 100, 200, 500, 1000 and 2000 μM) traced with 0.3 μCi myo-[3H]-inositol (Inositol, myo-[2–3H(N)]-, specific activity 22.5 Ci/mmol, PerkinElmer®, Boston, MA, USA) in PBS, pH 7.4] to the cell suspension. At different times (between 1 and 60 min), uptake was stopped by adding 800 μL of cold stop solution (50 mM unlabeled myo-inositol in PBS, pH 7.4) and immediate transfer of samples to ice baths. The parasite suspensions were washed three times in cold PBS (centrifugation at 13,000×g for 50 s) to remove all free myo-[3H]-inositol. Subsequently, cells were lysed in 100 μL of 1% SDS and the incorporated radioactivity was measured by liquid scintillation counting (Beckman LS 5000 TD scintillation photometer). Radioactivity corresponding to the experimental background was measured from parasites that were simultaneously incubated with the uptake solution and stop solution followed by the washing procedure and measurement analysis.
2.9. Effect of tamoxifen on inositol uptake
In order to evaluate the effect of tamoxifen on inositol uptake activity in L. amazonensis promastigotes, cells were washed in PBS, and then pre-incubated with 10, 30 and 50 μM tamoxifen for 10 min at 25 °C. The uptake assays were conducted with increasing substrate concentrations (50, 100, 200, 500, 1000 and 2000 μM myo-inositol plus 0.3 μCi myo-[3H]-inositol) during a fixed time of 2 min. As inhibitor control, 2 mM unlabeled myo- and scyllo-inositol were used at the Km condition (200 μM myo-inositol plus 0.3 μCi myo-[3H]-inositol).
2.10. Inositol uptake data analysis
The incorporated radioactivity, expressed as counts per minute (c.p.m.) of each experimental point was determined by subtracting the average c.p.m. of background samples from the average c.p.m. of triplicates after each time point. Curves were obtained from nonlinear regressions to the expected exponential decay function (in the case of time-course measurements) or hyperbolic function corresponding to Michaelis-Menten equation (in the case of measurements of V0 as a function of substrate concentrations). All data analysis and fittings were performed with GraphPad Prism software 5. All data came from at least three independent experiments performed with triplicate samples.
2.11. Cell viability
Parasite viability under the assay conditions was evaluated by propidium iodide staining. Briefly, after the addition of stop solution, an aliquot of parasites (10 μL) was removed, diluted 10 times in PBS and incubated with 5 μg/mL propidium iodide for 5 min at room temperature. The propidium iodide fluorescence was observed by fluorescence microscopy under a 540 nm filter. The percentage of propidium iodide positive cells was determined over the total cell count in a Neubauer chamber.
2.12. IPC synthase assay
CHAPS-washed membranes containing Leishmania IPC synthase were prepared and quantified from transgenic Saccharomyces cerevisiae reliant on expression of the protozoan enzyme (α ade−.lys−.leu−.Δaur1−.pESC-LEU_LmjFIPCS) as previously described (Norcliffe et al., 2018), and 0.75U of enzyme used in a reaction mix containing 100 μM PI (Avanti®Polar Lipids), 5 μM NBD-C6-ceramide (ThermoFisher Scientific), 50 mM PO4− pH7 and 600 μM CHAPS as described previously (Mina et al., 2010, 2011) in the presence or absence of tamoxifen at the appropriate concentration in DMSO (0.8 μL). Following incubation at 30 °C for 25 min, the reaction product, NBD-C6-IPC, was separated from the substrate, NBD-C6-ceramide, via anion exchange chromatography on protonated AG4-X4 resin (Bio-Rad) in 96-well MultiScreen® Solvinert filter plates (Merck Millipore), as previously described (Mina et al., 2010, 2011). After methanol washes, NBD-C6-IPC was eluted from the resin in black OptiPlate-96 (PerkinElmer) plates using 1M potassium formate in methanol and quantified using a Synergy HT microplate reader (Ex480/18; Em520/18) with the Gen5™ version 2 package (Biotek). IC50 values were calculated using sigmoidal regression analysis (GraphPad Prism7).
2.13. Statistical analysis
Data were analyzed for statistical significance by unpaired, two-tailed T test calculated using GraphPad Prism 5 software.
3. Results
3.1. L. amazonensis promastigotes treated with tamoxifen displayed altered sphingolipid biosynthesis
Metabolic labeling with [3H]-sphingosine or [3H]-inositol was employed to study the effect of tamoxifen on promastigotes' sphingolipid metabolism. Total lipids were purified from control and treated parasites and analyzed by HPTLC. In extracts from control parasites labeled with inositol, three well-defined bands were observed in the slower migrating region. These products were named A, B and C (Fig. 1). Metabolic labeling with sphingosine led to a more complex band pattern including bands with the same retention factor (Rf) as bands A, B and C. The detection of these 3 products after metabolic labeling with the two precursors suggested that these lipids contained inositol and sphingosine or sphingosine-derivatives in their composition. The pattern observed after sphingosine labeling included other lipids migrating with similar or higher Rfs. One of these, only slightly faster than lipid A was named X (Fig. 1). One of the faster migrating and therefore less polar molecules migrated with an Rf similar to the standard for C16 ceramide. Other labeled products were present, migrating with Rfs that were not compatible with available standards. The fastest migrating product among them (Rf = 0.97), which was consistent with a nonpolar molecule, was called band D (Fig. 1).
Fig. 1.

Effect of tamoxifen treatment on L. amazonensis sphingolipid biosynthesis. Metabolic labeling of 1 × 108L. amazonensis promastigotes with 6 μCi of [3H]-sphingosine (lanes 2 and 3) or 6 μCi of [3H]-inositol (lanes 5 and 6). Parasites were treated with 10 μM tamoxifen for 4 h and purified total lipids were analyzed by HPTLC using running system 1. ST(S): [3H]-sphingosine standard without incubation with cells, ST(I): [3H]-inositol standard without incubation with cells, Sph*: lipid extract of [3H]-sphingosine labeled parasites, Ino*: lipid extract of [3H]-inositol labeled parasites, Ct: lipid extract of untreated parasites, Tam: lipid extract of parasites treated with tamoxifen, C16-cer: C16-ceramide standard, PI 18:1: phosphatidylinositol 18:1 standard, O: origin, F: front.
Parasites treated with 10 μM tamoxifen for 4 h exhibited differences in the lipid profile compared to control extracts with a decrease in the abundance of products migrating as bands A, B and C and an increase in the abundance of lipid D (Fig. 1). The abundance of lipids A, B and C from tamoxifen-treated and [3H]-inositol-labeled parasites was reduced by 69 ± 14%, 77 ± 17% and 32 ± 13%, respectively, as compared with the control untreated sample. In [3H]-sphingosine labeled cells, the abundance of lipids B and C was 66 ± 19% and 68 ± 24% lower in tamoxifen treated parasites in relation to control cells (Fig. Suppl. 1). Quantification of lipid A after labeling with [3H]-sphingosine was not performed due to the presence of another labeled lipid (X) migrating with a similar Rf. The abundance of lipid D in treated parasites corresponded to about twice (263 ± 62%) the signal in control parasites (Fig. 1).
In summary, tamoxifen treated parasites decreased the abundance of products presenting the same Rf when labeled with [3H]-sphingosine (source for ceramide synthesis) or [3H]-inositol (source for the synthesis of PI and IPC) (Fig. 1). Therefore, lipids A, B and C were potentially molecules containing both sphingosine (or its derivatives) and inositol in their structure.
3.2. Characterization of lipids A and B
The nature of the products with altered biosynthesis in tamoxifen-treated parasites was investigated through the analysis of the products of their alkaline hydrolysis, by migration in different HPTLC running systems and/or mass spectrometry.
Lipid B, derived from parasites labeled with [3H]-sphingosine and eluted from the silica plate, showed resistance to alkaline hydrolysis (Fig. 2), as expected for the IPC structure. Mass spectrometry (MS) analysis of lipid B identified two predominant ions with m/z 778 and 806, previously described as IPC in L. major (Zhang et al., 2005; Hsu et al., 2007) (Fig. 3A and B). The tandem mass spectrometry (MS2) spectrum of m/z 778 indicated the presence of the following predominant ions: m/z 241 corresponding to cyclic inositol-1,2-phosphate, m/z 259 corresponding to inositol monophosphate and m/z 223 that is generated from 241 ions by the loss of water (Fig. 3C and D). These ions are characteristic of IPC or PI molecules. The spectrum also showed ions m/z 616 (m/z 778 − 162) and 598 (778 − 180) originated by inositol dehydration and inositol loss, respectively (Hsu et al., 2007). Furthermore, there is the emergence of the ion m/z 512 (with weak strength) which corresponds to the loss of a C18:0 acyl-bound fatty acid derived ketone substituent (778 − 266). Similarly, the MS2 fragmentation of the ion m/z 806 (Fig. 3E and F) showed characteristic ions of IPCs with m/z 223, 241, 259, 644 (806 − 162) and 626 (806 − 180) and ion m/z 540 (weakly detected) which also corresponds to the loss of a C18:0 acyl-bound fatty acid derived ketone substituent (806 − 266). These data suggest that the m/z 778 IPC is composed of d16:1/18:0 (Fig. 3G). Similarly, MS2 fragmentation of the m/z 806 ion revealed a characteristic IPC composed of d18:1/18:0 (Fig. 3H).
Fig. 2.
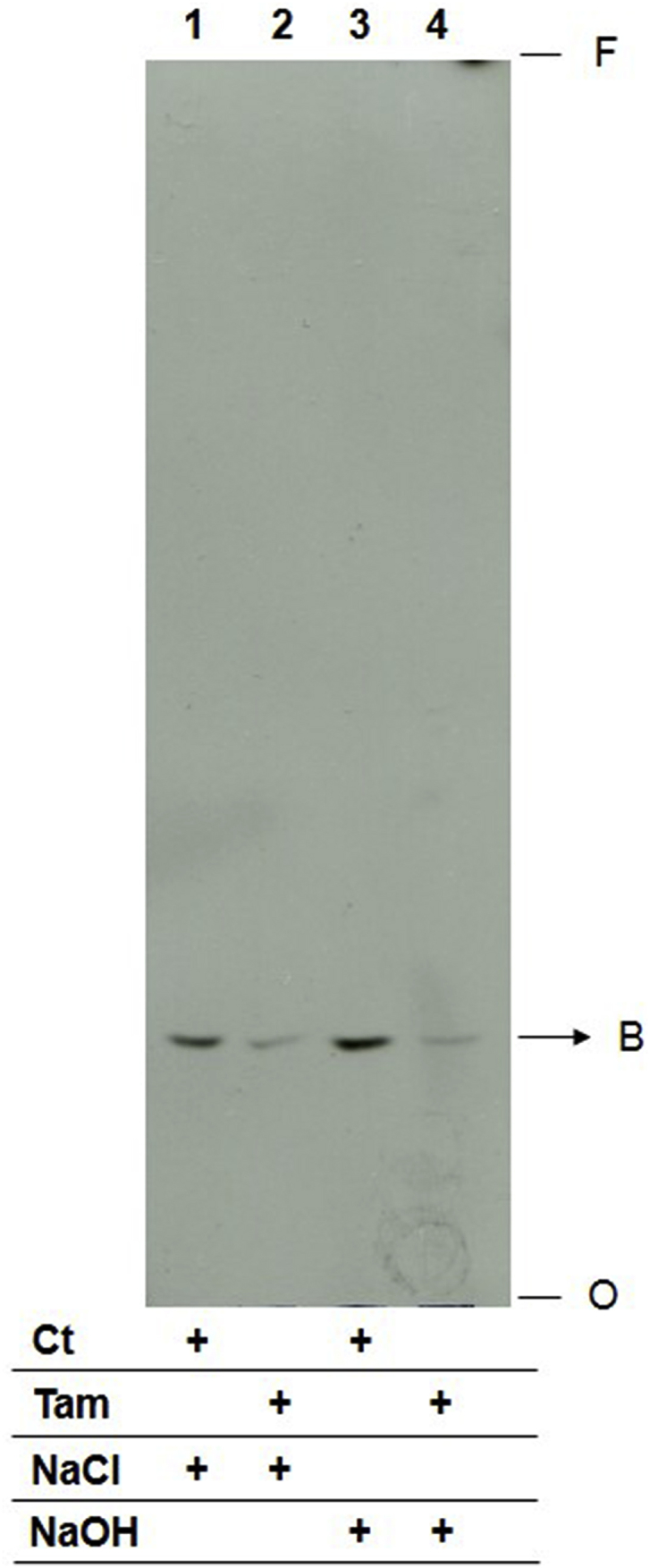
Lipid B characterization by alkaline hydrolysis. Lipid B fractions purified from HPTLC plates after separation of total lipids from L. amazonensis promastigotes metabolic labeled with 2 μCi [3H]-sphingosine. Initial lipid extracts were obtained from parasites treated with 10 μM tamoxifen or left untreated. Eluted lipids were incubated in 100 mM NaCl (lanes 1 and 2) or in 100 mM NaOH (lanes 3 and 4) in 95% methanol aqueous solution for 1 h. Extracts from 3 × 108 cells were applied to each lane and analyzed by HPTLC using running system 2. Ct: lipid extract of untreated parasites, Tam: lipid extract of parasites treated with tamoxifen, NaCl: extract incubated with 100 mM NaCl, NaOH: extract incubated with 100 mM NaOH, O: origin, F: front.
Fig. 3.

ESI-MS analysis of lipid B. Lipid B fractions of L. amazonensis promastigotes untreated (A) or treated with 10 μM tamoxifen (B) were analyzed by ESI-MS in the negative mode. MS2 spectra of ions m/z 778 and 806 (C/D and E/F, respectively) revealed characteristic ions of IPC molecules that were identified as IPC-(d16:1/18:0) (G) and IPC-(d18:1/18:0) (H), respectively.
The analysis of lipid A by ESI-MS identified the prevalent ions m/z 598, 778, 796, 806 and 824 (Fig. 4A and B). The ion m/z 806 was detected only in tamoxifen treated samples (Fig. 4A and B). Other peaks detected did not match phospholipids containing inositol phosphate. These ions can indicate the presence of possible glycerophospholipid species (m/z 621, 653 and 671) that were not characterized further.
Fig. 4.

ESI-MS analysis of lipid A. Lipid A fractions of L. amazonensis promastigotes untreated (A) or treated with 10 μM tamoxifen (B) were analyzed by ESI-MS in the negative mode. Ions m/z 598, 778, 796, 806 and 824 were identified as lyso-PI (18:1) (C), IPC (d16:1/18:0) (D), IPC (t16:0/18:0) (E), IPC (d18:1/18:0) (F) and IPC (t16:0/20:0) (G), respectively.
Based on the analysis of the MS2 spectra generated from each of these species (not shown), it was possible to identify the ion m/z 598 as a C18:1 lyso-acyl PI (Pulfer and Murphy, 2003) (Fig. 4C). Although the structure presented proposes the fatty acid bound to sn-1 of the glycerol molecule (Fig. 4C), it is not possible to exclude a structure with a C18:1 fatty acid occupying the sn-2 position (Murphy and Axelsen, 2011). The other ions detected corresponded to different IPCs composed of d16:1/18:0-IPC (m/z 778, Fig. 4D), t16:0/C18:0-IPC (m/z 796, Fig. 4E), d18:1/18:0-IPC (m/z 806, Fig. 4F) and t16:0/20:0-IPC (m/z 824, Fig. 4G). The latter species have also been detected in small amounts in ESI-MS analyses of IPCs isolated from of Leishmania major promastigotes (Hsu et al., 2007).
Together, these data confirm that IPC was present in bands B and A and suggested that tamoxifen decreased the biosynthesis of IPC in L. amazonensis promastigotes.
3.3. Characterization of lipid C
Lipid C recovered from [3H]-sphingosine-labeled parasites was sensitive to alkaline hydrolysis (data not shown) and migrated in running system 1 with Rfs compatible with an 18:1 PI standard (Fig. 1). Two other running systems were used to confirm the properties of lipid C. Total lipid extracts of [3H]-sphingosine labeled L. amazonensis were subjected to chromatography in two running systems previously used for the study of PI (Castro et al., 2013; Martin and Smith, 2006) in parallel with system 1, but with a longer run with the first solvent (named as running system 2) to obtain better separation of bands in the slower migration region (Fig. 5). In all three HPTLC analysis systems, a labeled product migrating with the Rf of the 18: 1 PI standard was observed (Fig. 5).
Fig. 5.

Separation of total lipid extracts from L. amazonensis promastigotes in different HPTLC systems. Parasites (7 × 108L. amazonensis promastigotes) were metabolic labeled with 2 μCi [3H]-sphingosine. Total lipid extract corresponding to 1 × 108 cells was added to each lane and separated by HPTLC using running systems 2 (A), 3 (B) and 4 (C). Sph*: [3H]-sphingosine standard, 18:1 PI: 18:1 phosphatidylinositol standard, O: origin, F: front.
To confirm the molecular nature of lipid C, unlabeled products migrating with the PI standard's Rf from a plate ran in the running system 2, were scraped from the silica and analyzed by mass spectrometry (Fig. 6A and B). The analysis identified ions with m/z 835, 849 and 863, consistent with L. major PI ions described previously (Fig. 6A and B) (Zhang et al., 2005; Hsu et al., 2007). The MS2 spectra of ion m/z 849 (Fig. 6C and D) demonstrated the presence of an ion with m/z 241 which, as observed on the analysis of the product identified as IPC (lipid B), corresponded to the cyclic inositol-1,2-phosphate ion. Another preponderant ion is the m/z 405, which corresponded to [HPO4—CH CH—CH2—O—(CH2)17—CH3]- (Serrano et al., 1995). Other ions observed on the MS2 analysis included m/z 281, corresponding to the carboxylated ion [CH3-(CH2)7—CH CH—(CH2)7—CO2]- and probably arising from a C18:1 fatty chain originally acyl-bound to glycerol sn-2 carbon. Finally, other two ions with m/z 585 and 567 were observed and corresponded to C18:0 lyso-alkyl PI before and after losing water (Pulfer and Murphy, 2003). These data suggested that the PI with m/z 849 was composed by a sn-1-O-(C18:0) alkyl-2-O-(C18:1)acylglycerol-3-HPO4-inositol (Fig. 6G). Similarly, the MS2 fragmentation of ion m/z 863 (Fig. 6E and F) originated PI characteristic ions such as m/z 241 and 281. However, a high intensity ion at m/z 283 was also apparent suggesting the presence of a carboxylated ion [CH3-(CH2)16—CO2]- probably derived from a C18:0 fatty chain that was originally acyl-linked to the sn-1 glycerol carbon. The m/z ∼296 ion (296.879) is most likely the signal for glycerophosphoinositol – 2H2O. Ions at m/z 437/419 and 599/581 were also noted, corresponding respectively to C18:0 lyso-acyl phosphatidic acid and C18 lyso-acyl PI before and after water loss (Pulfer and Murphy, 2003). These data suggest that PI with m/z 863 is composed by sn-1-O-(C18:0)acyl-2-O-(C18:1)acylglycerol-3-HPO4-inositol (Fig. 6H). Therefore, lipid C, reduced in abundance in tamoxifen treated parasites was identified as PI.
Fig. 6.

ESI-MS analysis of lipid C. Lipid C fractions of L. amazonensis promastigotes untreated (A) or treated with 10 μM tamoxifen (B) were analyzed by ESI-MS in the negative mode. MS2 spectra of ions m/z 849 and 863 (C/D and E/F, respectively) revealed characteristic ions of PI molecules that were identified as PI-(O-18:0/18:1) (G) and PI-(18:0/18:1) (H), respectively.
3.4. Characterization of lipid D
Total lipids re-extracted from lipid D area were submitted to alkaline hydrolysis and analyzed by HPTLC (Fig. 7). Prior to alkaline hydrolysis, extracts of tamoxifen-treated parasites presented an increased abundance of lipid D in comparison to untreated parasites (Fig. 7, lanes 1 and 2). Lipid D migrated with an Rf similar to the acyl ceramide standard. Upon alkaline treatment, a decrease in band D's intensity and an increase in the abundance of a lipid with a retention profile similar to the C16-ceramide standard was observed (Fig. 7, lane 4). Since alkaline hydrolysis characteristically results in the breakage of ester bonds, this data suggested that lipid D is a ceramide with an additional group, probably with non-polar features, that is linked to the molecule by an ester bond. If we take into account the similar retention profile to the standards, these results suggested that tamoxifen induced the increase of acyl ceramide synthesis in L. amazonensis promastigotes.
Fig. 7.

Lipid D characterization by alkaline hydrolysis. (A) Acyl ceramide structure. (B) Total lipid extracts from lipid D fractions of [3H]-sphingosine metabolic labeled L. amazonensis promastigotes treated or not with 10 μM tamoxifen were incubated in 100 mM NaOH (lane 3 and 4) or NaCl (lane 1 and 2) in 95% methanol aqueous solution for 1 h. Total lipid extracts from 3 × 108 cells were applied to each lane and analyzed by HPTLC using running system 2. D: lipid D, Acyl-cer: Acyl-ceramide standard, C16-cer: C16-ceramide standard, Ct: lipid extract of untreated parasites, Tam: lipid extract of parasites treated with tamoxifen, NaCl: extract incubated with 100 mM NaCl, NaOH: extract incubated with 100 mM NaOH, O: origin, F: front.
3.5. Inositol availability in L. amazonensis promastigotes treated with tamoxifen
Since the synthesis of IPC and PI depends on an intact pool of inositol, we evaluated whether tamoxifen altered the activity of the inositol transmembrane transporter in L. amazonensis promastigotes. An initial evaluation of inositol uptake in these parasites was performed in time-course incorporation curves during 60 min in the presence of a substrate concentration presumably close to the Km (0.5 mM) according to previous characterization of the myo-inositol transport and/or uptake in L. donovani (Drew et al., 1995; Seyfang et al., 1997; Seyfang and Landfear, 2000). A non-linear regression analysis showed that data fitted to an exponential decay function (R2 = 0.9932) which, as expected, is compatible with transporter facilitated uptake (Fig. Supl. 2A). The fact that the uptake was not saturated during the first 60 min strongly indicated that during this time the myo-inositol is metabolized while it is transported into the cells. Additionally, the myo-inositol uptake could be considered approximately linear during the first 10 min (R2 = 0.9980) (Inset, Fig Supl. 2A), allowing us to calculate V0 inside this range of time (Fig. Supl. 2A). From this analysis, we chose to calculate V0 based on the substrate uptake during 2 min of incubation of the parasites with myo-inositol.
The substrate saturation curve with substrate concentrations ranging from 0.05 to 2 mM inositol exhibited a classical Michaelis-Menten kinetics with values of Km and Vmax of 163.5 (131.8–195.1) μM and 62.5 (58.9–66.0) pmol min−1 3 × 107 cells−1 (mean ± 95% confidence interval), respectively (Fig Supl. 2B).
Under the hypothesis that inositol uptake by L. amazonensis promastigotes could be diminished by tamoxifen, this process was evaluated at substrate concentrations equivalent to the Km (0.2 mM myo-[3H]-inositol) and to concentrations allowing Vmax transport (2 mM myo-[3H]-inositol) in parasites previously incubated with 10, 30 and 50 μM tamoxifen for 10 min (Fig. 8A). In all treated samples, the number and viability of parasites were preserved as compared to the untreated group (Table 1), showing that the reduction on inositol uptake observed in parasites treated with tamoxifen was not due to parasites' death. Tamoxifen induced a dose-dependent decrease on inositol incorporation in L. amazonensis promastigotes (Fig. 8A). When assayed at the Km conditions, parasites treated with 10, 30 and 50 μM tamoxifen showed 11%, 21% and 41% reductions on myo-[3H]-inositol uptake, respectively. When assayed at Vmax conditions, inositol incorporation was decreased by 10.9%, 11.8% and 33.1%, respectively. As a control for these drug inhibition assays, 2 mM of myo- and scyllo-inositol were used as competitive inhibitors. When assayed at Km conditions, myo- and scyllo-inositol inhibited inositol uptake by 81% and 84%, respectively (Fig. 8A). Such inhibition was greater than that observed in parasites treated with the highest dose of tamoxifen.
Fig. 8.

Interference of tamoxifen on inositol uptake of L. amazonensis promastigotes. (A) Effect of tamoxifen (10, 30, and 50 μM) on myo-inositol uptake at Km (200 μM myo-inositol plus 0.3 μCi myo-[3H]-inositol) and Vmax (2000 μM myo-inositol plus 0.3 μCi myo-[3H]-inositol) conditions. Unlabeled 2 mM myo- and scyllo-inositol were used as inhibitors at the Km condition. (B) myo-inositol uptake kinetics in L. amazonensis promastigotes treated with different concentrations of tamoxifen. Inset: Double reciprocal plot of the linear inhibition of inositol uptake at the different concentrations of tamoxifen compared to untreated control. The uptake assays were conducted with increasing substrate concentrations (50, 100, 200, 500, 1000 and 2000 μM myo-inositol plus 0.3 μCi myo-[3H]-inositol) during a fixed time of 2 min. Results are representative of three experiments performed in each case. The bars represent the standard deviation of duplicates or triplicates. The myo-inositol incorporations is shown as picomoles per minute per 3 × 107 promastigotes.
Table 1.
Cell concentration and viability of L. amazonensis promastigotes treated with different tamoxifen concentrations for 10 min at 25 °C.
| Tamoxifen (μM) | Promastigotes (cells/mL) | Cell viability (%)a |
|---|---|---|
| 0 | 2.0 × 107 | 91 |
| 10 | 2.0 × 107 | 91 |
| 30 | 2.2 × 107 | 95 |
| 50 | 2.1 × 107 | 93 |
Percentage of propidium iodide unlabeled cells.
Substrate saturation curves were calculated from parasites treated with 0, 10, 30 and 50 μM tamoxifen, allowing the determination of the Km and Vmax for each condition (Fig. 8B, Table 2). Assuming that the myo-inositol transporter can be a target for tamoxifen, the observed changes on Vmax values in the presence of tamoxifen are compatible with a non-competitive inhibition is taking place, with tamoxifen binding at a distinct site from the substrate active site. The double-reciprocal plot (Inset, Fig. 8B) constructed from the obtained saturation curve supported this possibility.
Table 2.
L. amazonensis inositol transporter Km and Vmax after treatment with tamoxifen for 10 min at 25 °C.
| Tamoxifen (μM) | Kma [μM (CI95%)] | Vmaxb [pmol min−1 3 × 107 cells−1(CI95%)] |
|---|---|---|
| 0 | 182.9 (155.0–210.9) | 76.5 (72.9–80.2) |
| 10 | 193.9 (137.2–250.6) | 66.8 (60.8–72.7) |
| 30 | 203.2 (146.4–260.0) | 63.7 (58.0–69.4) |
| 50 | 197.7 (141.1–254.4) | 50.9 (46.6–55.2) |
Michaelis-Menten constant expressed in micromolar and 95% confidence interval.
Maximum velocity of inositol uptake expressed in picomoles per minute per 3 × 107 cells and 95% confidence interval.
Therefore, while 10 μM tamoxifen led to clear disturbances on lipid metabolism, its effect was only mild on inositol uptake, suggesting that the latter may not be the relevant target. Notwithstanding, to verify whether inositol availability had an impact on PI and IPC biosynthesis in tamoxifen-treated cells, a metabolic labeling assay was performed to evaluate PI and IPC synthesis in the presence of high concentrations of inositol. Metabolic labeling of L. amazonensis promastigotes previously treated with 10 μM tamoxifen was performed with [3H]-sphingosine or [3H]-inositol in the absence or in the presence of 0.2 or 2 mM unlabeled inositol (Fig. 9). In parasites labeled with [3H]-inositol (Fig. 9, lines 1 to 6), the presence of increasing concentrations of unlabeled inositol led to a gradual reduction on label incorporation on PI (Lipid C) and IPC (Lipids A and B) (Fig. 9). This effect occurred in both control and treated samples, and is probably related to the dilution of the radioactive labeling. However, tamoxifen-treated parasites metabolically labeled with [3H]-sphingosine presented reduced PI and IPC synthesis despite the increased concentrations of unlabeled inositol. These data confirmed that the reduced synthesis of PI and IPC in parasites treated with tamoxifen is not due to the lack of inositol availability to the cells.
Fig. 9.

Sphingolipid biosynthesis in L. amazonensis promastigotes treated with tamoxifen.L. amazonensis promastigotes (4 × 108) were treated with tamoxifen and metabolic labeled with 6 μCi [3H]-inositol (1–6) or 2 μCi [3H]-sphingosine (7–10). Parasites were supplemented with 0.2 (2, 5, 9) or 2 mM (3, 6, 10) myo-inositol. Total lipid extracts from 1 × 108 cells were spotted on each lane and analyzed by HPTLC using running system 1. Tam: 10 μM tamoxifen for 4 h; Sph*: labeling with [3H]-sphingosine; Ino*: labeling with [3H]-inositol; Ino: unlabeled inositol; O: origin; F: front.
3.6. Effect of tamoxifen on the Leishmania IPC synthase
Since the availability of inositol did not explain the reduced biosynthesis of PI and IPC in tamoxifen treated promastigotes, we investigated whether tamoxifen was an IPC synthase inhibitor. Tamoxifen's effect on Leishmania IPC synthase was investigated using the fluorescent based cell-free assay of L. major IPC synthase developed by Mina and co-workers (Mina et al., 2010). In this system, the drug is incubated with C6-NBD-ceramide (receiving substrate) and PI (donor substrate) in the presence of CHAPS washed S. cerevisiae microsomal extracts containing the L. major IPC synthase. Tamoxifen inhibited L. major IPC synthase with an IC50 value of 8.48 μM (95% CI 7.68–9.37) (Fig. 10).
Fig. 10.

Effect of tamoxifen on the turnover of Leishmania IPC synthase. IC50 of tamoxifen (2-fold serial dilution from 200 μM top concentration) against the L. major IPC synthase in the in vitro assay was calculated using GraphPad Prism 7 (log (inhibitor) vs. normalized response - Variable slope) as 8.48 μM (95% CI 7.68–9.37). Seven replicate data set, error bars represent standard deviation (not visible where very small), and R2 = 0.98.
4. Discussion
Current therapeutic options for leishmaniasis are of limited efficacy and safety and new alternatives are clearly needed. Due the promising therapeutic profile observed for tamoxifen in different animal models of leishmaniasis, we undertook to evaluate its mechanism of action against the parasite. [3H]-sphingosine and myo-[3H]-inositol metabolic labeling of L. amazonensis promastigotes treated with tamoxifen showed a meaningful perturbation of SL metabolism, leading to a significant reduction of IPCs and PIs, and accumulation of acyl ceramide.
IPC, the most abundant SL in Leishmania, is present in lipid rafts (Kaneshiro et al., 1986; McConville and Bacic, 1989; Yoneyama et al., 2006) that are involved in cell signaling and macrophage invasion as well as in the process of promastigote migration in the vector's gut during metacyclogenesis (Denny and Smith, 2004; Denny et al., 2004; Yoneyama et al., 2006). IPC is essential for fungi and for the bloodstream forms of Trypanosoma brucei (Nagiec et al., 1997; Sutterwala et al., 2008; Mina et al., 2009).
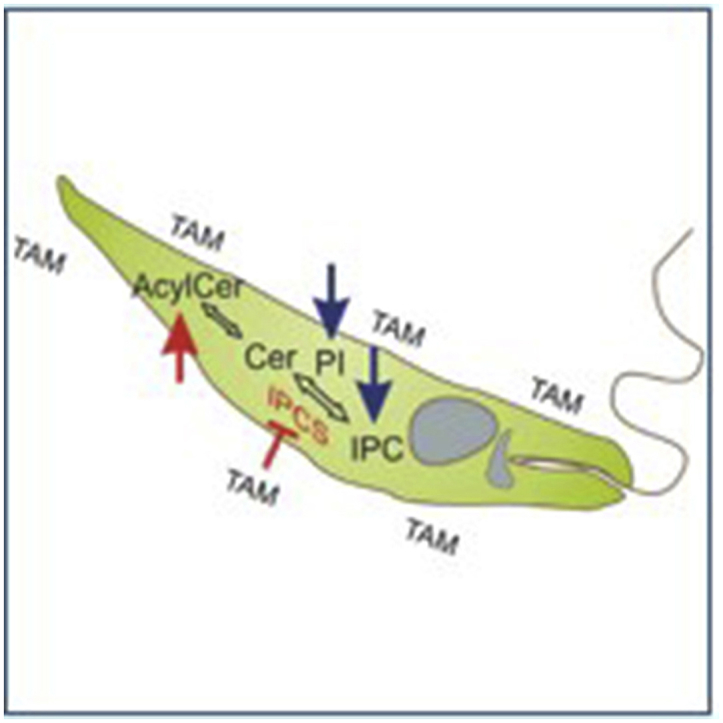
PI is an essential phospholipid in eukaryotes that, together with its metabolites, regulate a wide range of cellular processes. In addition, it is a substrate for PI kinases (PIKs), key enzymes of one of the major intracellular signaling pathways of eukaryotic cells (reviewed in Martin and Smith, 2006; Sasaki et al., 2009). This pathway consists in one of the main targets for cancer chemotherapy and may represent a very interesting focus for the treatment of parasitic diseases (Braga and de Souza, 2006; Fernandes et al., 2006). PI3k3 has been shown to mediate invasion of macrophages and neutrophils by L. mexicana in vitro and the recruitment of phagocytes and regulatory T cells into the site of infection in in vivo studies (Cummings et al., 2012; Oghumu and Satoskar, 2013). Consequently, the reduction of IPC and PI synthesis in L. amazonensis promastigotes treated with tamoxifen may have important repercussions in the establishment of Leishmania infection, as well as cell viability. Two hypotheses were formulated to explain the reduction of PI and IPC abundance in tamoxifen's treated parasites: (1) tamoxifen interferes in inositol and/or ceramide availability, both being critical elements for IPC synthesis; (2) tamoxifen inhibits IPC and/or PI synthase (Fig. 11).
Fig. 11.

Schematic diagram summarizing potential tamoxifen targets related to sphingolipid biosynthesis in Leishmania. Reduced IPC levels (blue arrows) in L. amazonensis promastigotes treated with tamoxifen are related to IPC synthase inhibition (red bars). Reduced PI and IPC intracellular levels (blue arrows) could also be related to reduced incorporation of inositol into the cell or to direct or indirect inhibition of PI and/or IPC synthase (purple bars). The increased levels of acylated ceramide may be a result from intracellular ceramide accumulation (green arrow) due to its reduced use as a source for IPC synthesis. Labeled PI observed in [3H]-Sphingosine-labeled parasites may be generated by hexadecenal pathway from sphingosine-1-phosphate degradation. DAG: diacylglycerol; CDP-DAG: cytidine diphosphate diacylglycerol; PIS: phosphatidylinositol synthase; IPC: inositolphophorylceramide; EtN-P: phosphoetalonamine; Alkyl-G3P: 1-alkylglycerol-3-phosphate; CDase: ceramidase; CDP-DAG: cytidine diphosphate diacylglycerol; CerS: ceramide synthase; DAG: diacylglycerol; EtN-P: phosphoetalonamine; IPC: inositolphophorylceramide; IPCS: inositolphosphorylceramide synthase; ISCL: inositolsphingolipid phospholipase C-like; PI: phosphatidylinositol; PIS: phosphatidylinositol synthase; S1PL: sphingosine-1-phosphate lyase; Sph-1-P: sphingosine-1-phosphate; TAM: tamoxifen. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
In Leishmania, inositol can be acquired by de novo synthesis or uptake through specific membrane transporters (Drew et al., 1995; Ilg, 2002; Majumder et al., 1997; Michell, 2008; Reynolds, 2009). Inositol de novo synthesis occurs in the cell cytoplasm by cytosolic enzymes (Donahue and Henry, 1981; Michell, 2008), which are unlikely to be a target for tamoxifen, a lipophilic molecule (Log P = 5.93) that is known to interact and incorporate into biomembranes (Custodio et al., 1998).
A myo-inositol transporter has been described in L. donovani (Drew et al., 1995). A single copy gene encodes this protein, which behaves as a myo-inositol/proton electrogenic symporter (Drew et al., 1995; Seyfang et al., 1997). Regulation of inositol uptake activity has been correlated with the availability of substrate with upregulated expression and activity when the substrate is depleted from the external environment (Seyfang and Landfear, 1999). The myo-inositol transporter, localized in the plasma membrane of L. donovani promastigotes, was shown to be redirected to the multivesicular tubule in stationary phase promastigotes and to the lysosome following transformation to the intracellular amastigote form (Vince et al., 2011), suggesting that transport activity is more important during the insect stage. The transport kinetics of the L. amazonensis myo-inositol transporter characterized here were similar to the ones described previously for L. donovani (Km 250 ± 0.05 μM and Vmax: 55,5 ± 8.8 pmol min−1 5 × 107 células−1) (Drew et al., 1995).
The characterization of inositol uptake in tamoxifen-treated parasites indicated the occurrence of a non-competitive inhibition (Fig. 8), suggesting that tamoxifen does not bind at the substrate active site. It is important to stress that even in the presence of 50 μM tamoxifen (equivalent to 5x the IC50) the maximal uptake inhibition observed was 40%. Considering that the metabolic labeling of parasites was performed after treatment with 10 μM tamoxifen (for longer periods), it seemed unlikely that inhibition of inositol transport was the sole reason for reduced synthesis of IPC and PI.
The confirmation that inositol availability had no impact on PI and IPC biosynthesis was validated by metabolic labeling of parasites with [3H]-sphingosine or [3H]-inositol in presence of high concentrations of unlabeled inositol (Fig. 9).
The reduction of PI and IPC synthesis in tamoxifen-treated parasites despite the increased concentrations of unlabeled inositol suggested that the enzymes IPC and/or PI synthase could be targets of the drug. A well-established cell-free assay of L. major IPC synthase was used to demonstrate the inhibitory activity of tamoxifen on Leishmania IPC synthase with an IC50 in the lower micromolar range [IC50 = 8.48 μM (95% CI 7.68–9.37)] (Fig. 10). Under IPC synthase inhibition, an increase in PI concentration could be expected but is not observed, suggesting that PI synthesis may also be impaired. However, a possible direct drug inhibition of PI synthase activity was not shown here and still needs to be investigated.
Both PI and IPC synthase, as well as other enzymes involved in the SLs biosynthesis, are transmembrane proteins with several transmembrane domains (Martin and Smith, 2006; Sutterwala et al., 2008). Since tamoxifen in known to interact with the cell membrane, it is possible that, in doing so, it could promote an alteration in the three-dimensional structure of IPC/PI synthase resulting in impairment of enzymatic activity. Aside from its proposed inhibitory effect on PI and IPC synthases, tamoxifen could also interfere with other enzymes of the metabolism of sphingolipids in Leishmania like the phosphorylation of sphingosine to form S1P and its degradation by S1PL that give rise to hexadecenal (that can be used to the biosynthesis of glycerophospholipids) and ethanolamine-phosphate (that can be used to the biosynthesis of phosphatidylethanolamine) (Nakahara et al., 2012). The latter product could not be traced in our metabolic labelling experiments using [3H]-sphingosine, once the [3H] was located at the sphingosine C-2. On the other hand, the resulting hexadecenal would be labeled and could be incorporated into PI molecules (more specifically to the PI with m/z 849), a process that was apparently inhibited by tamoxifen (Fig. 1). In this pathway, the labeled hexadecenal is used as a substrate for the alkyl-dihydroxyacetone phosphate (DHAP) synthase to generate alkyl-DHAP which will subsequently be reduced into alkyl-glycerol-3-phosphate that can be used in the biosynthesis of ether glycerophospholipids.
The mass spectrometry analysis also disclosed the presence of some other lipid species (m/z 621, 653, 671 and 806) that were not characterized but may be of interest to better understand the biochemistry of tamoxifen activity over these lipids in Leishmania.
The deletion of the IPC synthase gene in S. cerevisiae leads to accumulation of intracellular ceramide followed by cell death (Nagiec et al., 1997). In addition, several studies have indicated the accumulation of intracellular ceramide in tumor cells submitted to treatment with tamoxifen (Morad et al., 2013; Morad and Cabot, 2015). The anti-proliferative and cell death triggering effects of high intracellular concentrations of ceramide are well characterized in eukaryotic cells (Jayadev et al., 1995; Wang et al., 2003; Chapman et al., 2010; Tirodkar and Voelkel-Johnson, 2012). Instead, the increase of sphingosine and sphingosine-1-phosphate is related to cell survival and proliferation (Smith et al., 2000; Rodriguez et al., 2015). The balance between these and other active SLs and enzymes of SL metabolism plays a very important role in the pathogenesis and progression of cancer in humans (Ogretmen and Hannun, 2004).
Tamoxifen has been shown to inhibit the activity of acid ceramidases in human tumoral cells (Morad et al., 2013; Morad and Cabot, 2015), resulting in accumulation of ceramide. An increase in ceramide levels was not observed in tamoxifen-treated parasites (Fig. 1). On the other hand, the present data show an increased synthesis of acylated ceramide in L. amazonensis promastigotes treated with tamoxifen. Acyl-ceramide is an uncommon form of ceramide not yet described in Leishmania spp. In humans, 1-O-acylceramide has been identified as a natural component of the human and murine epidermis, which contributes to the homeostasis of the skin hydration barrier (Rabionet et al., 2013). Since intracellular ceramide level is tightly regulated in eukaryotic cells, the ceramide acylation could occur as a protective mechanism.
Considering that tamoxifen-treated parasites showed a reduction of IPC (lipid A and B), the most abundant membrane phosphosphingolipid in Leishmania, it is reasonable to assume that inhibition of IPC synthase could result in accumulation of intracellular ceramide. The similar intensity of the product with comparable Rf to C16-ceramide pattern in [3H]-sphingosine-labeled parasites treated or not with tamoxifen (Fig. 1, Fig. 7), even though in conditions of IPC synthase inhibition, supports the hypothesis that intracellular ceramides were acylated to maintain ceramide physiological levels and cell homeostasis.
In summary, we have shown that tamoxifen alters the profile of incorporation of [3H]-sphingosine and [3H]-inositol precursors in the sphingolipid biosynthesis pathway in L. amazonensis promastigotes, leading to the reduction of PI and IPC synthesis, with accumulation of acylated ceramide. The reduction in IPC, with the corollary increase in ceramide, seen in Leishmania treated with tamoxifen, coupled with the inhibitory action on parasite IPC synthase activity in vitro, indicated that inhibition of IPC synthesis may represent a key leishmanicidal mechanism of action of this drug. IPC synthase is a very interesting target since it represents an essential enzyme responsible for the synthesis of the most abundant SL in Leishmania (Kaneshiro et al., 1986; McConville and Bacic, 1989) which is absent in mammals (Denny et al., 2004). Recent work has seen a yeast-based assay employed to discover new, selective inhibitors with activity against intramacrophage Leishmania (Norcliffe et al., 2018). In addition, we have shown that tamoxifen treatment leads to the reduction of PI abundance, which could have a wide range of effects on cellular function, since numerous biological functions of PI are known in eukaryotic cells (Divecha and Irvine, 1995; Henry and Patton-Vogt, 1998; Odom et al., 2000; Martin, 2001; Sasaki et al., 2009).
We have shown previously that methods normally used to select parasites resistant to drugs failed when tamoxifen resistance is pursued (Coelho et al., 2015), making it likely that several targets in the cell are capable of justifying tamoxifen's leishmanicidal effect, just as it occurs in human tumor cells.
5. Conclusions
Promastigotes of L. amazonensis treated with tamoxifen presented reduced IPC and PI biosynthesis. The reduction in IPC biosynthesis cannot be attributed to reduction in inositol transport but is probably related to inhibition of the IPC synthase.
Funding
This work was supported by grant 2015/09080-2, São Paulo Research Foundation (FAPESP), grant 473343/2012-6, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenacão de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and the research leading to these results has, in part, received funding from the Research Councils UK Grand Challenges Research Funder under grant agreement ‘A Global Network for Neglected Tropical Diseases’ grant number MR/P027989/1. C. T. T. (2011/18858-6) was supported by a FAPESP fellowship. S. R. B. U. is the recipient of a senior researcher scholarship from CNPq.
Acknowledgments
We thank Alejandro Katzin for contributing with discussions and equipment availability. We would also like to thank Caroline Ricce Espada, Victor de Sousa Agostino, Mariana K. Galuppo e Jenicer K. U. Yokoyama-Yasunaka for support and helpful discussions.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijpddr.2018.10.007.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Alvar J., Croft S., Olliaro P. Chemotherapy in the treatment and control of leishmaniasis. Adv. Parasitol. 2006;61:223–274. doi: 10.1016/S0065-308X(05)61006-8. [DOI] [PubMed] [Google Scholar]
- Alvar J., Velez I.D., Bern C., Herrero M., Desjeux P., Cano J., Jannin J., den Boer M. Leishmaniasis worldwide and global estimates of its incidence. PloS One. 2012;7 doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boath A., Graf C., Lidome E., Ullrich T., Nussbaumer P., Bornancin F. Regulation and traffic of ceramide 1-phosphate produced by ceramide kinase: comparative analysis to glucosylceramide and sphingomyelin. J. Biol. Chem. 2008;283:8517–8526. doi: 10.1074/jbc.M707107200. [DOI] [PubMed] [Google Scholar]
- Braga M.V., de Souza W. Effects of protein kinase and phosphatidylinositol-3 kinase inhibitors on growth and ultrastructure of Trypanosoma cruzi. FEMS Microbiol. Lett. 2006;256:209–216. doi: 10.1111/j.1574-6968.2006.00125.x. [DOI] [PubMed] [Google Scholar]
- Cabot M.C., Giuliano A.E., Volner A., Han T.Y. Tamoxifen retards glycosphingolipid metabolism in human cancer cells. FEBS Lett. 1996;394:129–131. doi: 10.1016/0014-5793(96)00942-8. [DOI] [PubMed] [Google Scholar]
- Castro E.V., Yoneyama K.G., Haapalainen E.F., Toledo M.S., Takahashi H.K., Straus A.H. Myriocin, a serine palmitoyltransferase inhibitor, blocks cytokinesis in Leishmania (Viannia) braziliensis promastigotes. J. Eukaryot. Microbiol. 2013;60:377–387. doi: 10.1111/jeu.12043. [DOI] [PubMed] [Google Scholar]
- Chapman J.V., Gouaze-Andersson V., Messner M.C., Flowers M., Karimi R., Kester M., Barth B.M., Liu X., Liu Y.Y., Giuliano A.E., Cabot M.C. Metabolism of short-chain ceramide by human cancer cells--implications for therapeutic approaches. Biochem. Pharmacol. 2010;80:308–315. doi: 10.1016/j.bcp.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton R., Rossi-Bergmann B., Denny P., Steel P. Repurposing as a strategy for the discovery of new anti-leishmanials: the-state-of-the-art. Parasitology. 2018;145:219–236. doi: 10.1017/S0031182017000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho A.C., Trinconi C.T., Senra L., Yokoyama-Yasunaka J.K., Uliana S.R. Leishmania is not prone to develop resistance to tamoxifen. Int. J. Parasitol. Drugs Drug Resist. 2015;5:77–83. doi: 10.1016/j.ijpddr.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convit J., Ulrich M. Antigen-specific immunodeficiency and its relation to the spectrum of american cutaneous leishmaniasis. Biol. Res. 1993;26:159–166. [PubMed] [Google Scholar]
- Croft S.L., Coombs G.H. Leishmaniasis- current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003;19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Cummings H.E., Barbi J., Reville P., Oghumu S., Zorko N., Sarkar A., Keiser T.L., Lu B., Ruckle T., Varikuti S., Lezama-Davila C., Wewers M.D., Whitacre C., Radzioch D., Rommel C., Seveau S., Satoskar A.R. Critical role for phosphoinositide 3-kinase gamma in parasite invasion and disease progression of cutaneous leishmaniasis. Proc. Natl. Acad. Sci. U. S. A. 2012;109:1251–1256. doi: 10.1073/pnas.1110339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custodio J.B., Moreno A.J., Wallace K.B. Tamoxifen inhibits induction of the mitochondrial permeability transition by Ca2+ and inorganic phosphate. Toxicol. Appl. Pharmacol. 1998;152:10–17. doi: 10.1006/taap.1998.8510. [DOI] [PubMed] [Google Scholar]
- Denny P.W., Smith D.F. Rafts and sphingolipid biosynthesis in the kinetoplastid parasitic protozoa. Mol. Microbiol. 2004;53:725–733. doi: 10.1111/j.1365-2958.2004.04208.x. [DOI] [PubMed] [Google Scholar]
- Denny P.W., Goulding D., Ferguson M.A., Smith D.F. Sphingolipid-free Leishmania are defective in membrane trafficking, differentiation and infectivity. Mol. Microbiol. 2004;52:313–327. doi: 10.1111/j.1365-2958.2003.03975.x. [DOI] [PubMed] [Google Scholar]
- Denny P.W., Shams-Eldin H., Price H.P., Smith D.F., Schwarz R.T. The protozoan inositol phosphorylceramide synthase: a novel drug target that defines a new class of sphingolipid synthase. J. Biol. Chem. 2006;281:28200–28209. doi: 10.1074/jbc.M600796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divecha N., Irvine R.F. Phospholipid signaling. Cell. 1995;80:269–278. doi: 10.1016/0092-8674(95)90409-3. [DOI] [PubMed] [Google Scholar]
- Donahue T.F., Henry S.A. myo-Inositol-1-phosphate synthase. Characteristics of the enzyme and identification of its structural gene in yeast. J. Biol. Chem. 1981;256:7077–7085. [PubMed] [Google Scholar]
- Drew M.E., Langford C.K., Klamo E.M., Russell D.G., Kavanaugh M.P., Landfear S.M. Functional expression of a myo-inositol/H+ symporter from Leishmania donovani. Mol. Cell Biol. 1995;15:5508–5515. doi: 10.1128/mcb.15.10.5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes A.B., Neira I., Ferreira A.T., Mortara R.A. Cell invasion by Trypanosoma cruzi amastigotes of distinct infectivities: studies on signaling pathways. Parasitol. Res. 2006;100:59–68. doi: 10.1007/s00436-006-0236-6. [DOI] [PubMed] [Google Scholar]
- Figueiredo J.M., Dias W.B., Mendonca-Previato L., Previato J.O., Heise N. Characterization of the inositol phosphorylceramide synthase activity from Trypanosoma cruzi. Biochem. J. 2005;387:519–529. doi: 10.1042/BJ20041842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry S.A., Patton-Vogt J.L. Genetic regulation of phospholipid metabolism: yeast as a model eukaryote. Prog. Nucleic Acid Res. Mol. Biol. 1998;61:133–179. doi: 10.1016/s0079-6603(08)60826-0. [DOI] [PubMed] [Google Scholar]
- Hsu F.F., Turk J., Zhang K., Beverley S.M. Characterization of inositol phosphorylceramides from Leishmania major by tandem mass spectrometry with electrospray ionization. J. Am. Soc. Mass Spectrom. 2007;18:1591–1604. doi: 10.1016/j.jasms.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S., Nakajo N., Sakiyama H., Hirabayashi Y. A mouse B16 melanoma mutant deficient in glycolipids. Proc. Natl. Acad. Sci. U. S. A. 1994;91:2703–2707. doi: 10.1073/pnas.91.7.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilg T. Generation of myo-inositol-auxotrophic Leishmania mexicana mutants by targeted replacement of the myo-inositol-1-phosphate synthase gene. Mol. Biochem. Parasitol. 2002;120:151–156. doi: 10.1016/s0166-6851(01)00435-2. [DOI] [PubMed] [Google Scholar]
- Jayadev S., Liu B., Bielawska A.E., Lee J.Y., Nazaire F., Pushkareva M., Obeid L.M., Hannun Y.A. Role for ceramide in cell cycle arrest. J. Biol. Chem. 1995;270:2047–2052. doi: 10.1074/jbc.270.5.2047. [DOI] [PubMed] [Google Scholar]
- Jordan V.C. Tamoxifen: a most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- Kaneshiro E.S., Jayasimhulu K., Lester R.L. Characterization of inositol lipids from Leishmania donovani promastigotes: identification of an inositol sphingophospholipid. J. Lipid Res. 1986;27:1294–1303. [PubMed] [Google Scholar]
- Majumder A.L., Johnson M.D., Henry S.A. 1L-myo-inositol-1-phosphate synthase. Biochim. Biophys. Acta. 1997;1348:245–256. doi: 10.1016/s0005-2760(97)00122-7. [DOI] [PubMed] [Google Scholar]
- Martin K.L., Smith T.K. Phosphatidylinositol synthesis is essential in bloodstream form Trypanosoma brucei. Biochem. J. 2006;396:287–295. doi: 10.1042/BJ20051825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T.F. PI(4,5)P(2) regulation of surface membrane traffic. Curr. Opin. Cell Biol. 2001;13:493–499. doi: 10.1016/s0955-0674(00)00241-6. [DOI] [PubMed] [Google Scholar]
- McConville M.J., Bacic A. A family of glycoinositol phospholipids from Leishmania major. Isolation, characterization, and antigenicity. J. Biol. Chem. 1989;264:757–766. [PubMed] [Google Scholar]
- Merrill A.H., Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 2011;111:6387–6422. doi: 10.1021/cr2002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michell R.H. Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell Biol. 2008;9:151–161. doi: 10.1038/nrm2334. [DOI] [PubMed] [Google Scholar]
- Miguel D.C., Yokoyama-Yasunaka J.K., Andreoli W.K., Mortara R.A., Uliana S.R. Tamoxifen is effective against Leishmania and induces a rapid alkalinization of parasitophorous vacuoles harbouring Leishmania (Leishmania) amazonensis amastigotes. J. Antimicrob. Chemother. 2007;60:526–534. doi: 10.1093/jac/dkm219. [DOI] [PubMed] [Google Scholar]
- Miguel D.C., Yokoyama-Yasunaka J.K., Uliana S.R. Tamoxifen is effective in the treatment of Leishmania amazonensis infections in mice. PLoS Neglected Trop. Dis. 2008;2:e249. doi: 10.1371/journal.pntd.0000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel D.C., Zauli-Nascimento R.C., Yokoyama-Yasunaka J.K., Katz S., Barbieri C.L., Uliana S.R. Tamoxifen as a potential antileishmanial agent: efficacy in the treatment of Leishmania braziliensis and Leishmania chagasi infections. J. Antimicrob. Chemother. 2009;63:365–368. doi: 10.1093/jac/dkn509. [DOI] [PubMed] [Google Scholar]
- Mina J., Denny P. Everybody needs sphingolipids, right! Mining for new drug targets in protozoan sphingolipid biosynthesis. Parasitology. 2018;145:134–147. doi: 10.1017/S0031182017001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina J.G., Mosely J.A., Ali H.Z., Denny P.W., Steel P.G. Exploring Leishmania major inositol phosphorylceramide synthase (LmjIPCS): insights into the ceramide binding domain. Org. Biomol. Chem. 2011;9:1823–1830. doi: 10.1039/c0ob00871k. [DOI] [PubMed] [Google Scholar]
- Mina J.G., Mosely J.A., Ali H.Z., Shams-Eldin H., Schwarz R.T., Steel P.G., Denny P.W. A plate-based assay system for analyses and screening of the Leishmania major inositol phosphorylceramide synthase. Int. J. Biochem. Cell Biol. 2010;42:1553–1561. doi: 10.1016/j.biocel.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Mina J.G., Pan S.Y., Wansadhipathi N.K., Bruce C.R., Shams-Eldin H., Schwarz R.T., Steel P.G., Denny P.W. The Trypanosoma brucei sphingolipid synthase, an essential enzyme and drug target. Mol. Biochem. Parasitol. 2009;168:16–23. doi: 10.1016/j.molbiopara.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Morad S.A., Cabot M.C. Tamoxifen regulation of sphingolipid metabolism--Therapeutic implications. Biochim. Biophys. Acta. 2015;1851:1134–1145. doi: 10.1016/j.bbalip.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morad S.A., Levin J.C., Tan S.F., Fox T.E., Feith D.J., Cabot M.C. Novel off-target effect of tamoxifen--inhibition of acid ceramidase activity in cancer cells. Biochim. Biophys. Acta. 2013;1831:1657–1664. doi: 10.1016/j.bbalip.2013.07.016. [DOI] [PubMed] [Google Scholar]
- Murphy R.C., Axelsen P.H. Mass spectrometric analysis of long-chain lipids. Mass Spectrom. Rev. 2011;30:579–599. doi: 10.1002/mas.20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagiec M.M., Nagiec E.E., Baltisberger J.A., Wells G.B., Lester R.L., Dickson R.C. Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphorylceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the AUR1 gene. J. Biol. Chem. 1997;272:9809–9817. doi: 10.1074/jbc.272.15.9809. [DOI] [PubMed] [Google Scholar]
- Nakahara K., Ohkuni A., Kitamura T., Abe K., Naganuma T., Ohno Y., Zoeller R.A., Kihara A. The Sjogren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol. Cell. 2012;46:461–471. doi: 10.1016/j.molcel.2012.04.033. [DOI] [PubMed] [Google Scholar]
- Norcliffe J.L., Mina J.G., Alvarez E., Cantizani J., de Dios-Anton F., Colmenarejo G., Valle S.G., Marco M., Fiandor J.M., Martin J.J., Steel P.G., Denny P.W. Identifying inhibitors of the Leishmania inositol phosphorylceramide synthase with antiprotozoal activity using a yeast-based assay and ultra-high throughput screening platform. Sci. Rep. 2018;8:3938. doi: 10.1038/s41598-018-22063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom A.R., Stahlberg A., Wente S.R., York J.D. A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science. 2000;287:2026–2029. doi: 10.1126/science.287.5460.2026. [DOI] [PubMed] [Google Scholar]
- Oghumu S., Satoskar A.R. PI3K-gamma inhibitors in the therapeutic intervention of diseases caused by obligate intracellular pathogens. Commun. Integr. Biol. 2013;6 doi: 10.4161/cib.23360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogretmen B., Hannun Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Canc. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- Pulfer M., Murphy R.C. Electrospray mass spectrometry of phospholipids. Mass Spectrom. Rev. 2003;22:332–364. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- Rabionet M., Bayerle A., Marsching C., Jennemann R., Grone H.J., Yildiz Y., Wachten D., Shaw W., Shayman J.A., Sandhoff R. 1-O-acylceramides are natural components of human and mouse epidermis. J. Lipid Res. 2013;54:3312–3321. doi: 10.1194/jlr.M040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reithinger R., Dujardin J.C., Louzir H., Pirmez C., Alexander B., Brooker S. Cutaneous leishmaniasis. Lancet Infect. Dis. 2007;7:581–596. doi: 10.1016/S1473-3099(07)70209-8. [DOI] [PubMed] [Google Scholar]
- Reynolds T.B. Strategies for acquiring the phospholipid metabolite inositol in pathogenic bacteria, fungi and protozoa: making it and taking it. Microbiology. 2009;155:1386–1396. doi: 10.1099/mic.0.025718-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A.M., Graef A.J., LeVine D.N., Cohen I.R., Modiano J.F., Kim J.H. Association of sphingosine-1-phosphate (S1P)/S1P receptor-1 pathway with cell proliferation and survival in canine hemangiosarcoma. J. Vet. Intern. Med. 2015;29:1088–1097. doi: 10.1111/jvim.13570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T., Takasuga S., Sasaki J., Kofuji S., Eguchi S., Yamazaki M., Suzuki A. Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 2009;48:307–343. doi: 10.1016/j.plipres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Serrano A.A., Schenkman S., Yoshida N., Mehlert A., Richardson J.M., Ferguson M.A. The lipid structure of the glycosylphosphatidylinositol-anchored mucin-like sialic acid acceptors of Trypanosoma cruzi changes during parasite differentiation from epimastigotes to infective metacyclic trypomastigote forms. J. Biol. Chem. 1995;270:27244–27253. doi: 10.1074/jbc.270.45.27244. [DOI] [PubMed] [Google Scholar]
- Seyfang A., Kavanaugh M.P., Landfear S.M. Aspartate 19 and glutamate 121 are critical for transport function of the myo-inositol/H+ symporter from Leishmania donovani. J. Biol. Chem. 1997;272:24210–24215. doi: 10.1074/jbc.272.39.24210. [DOI] [PubMed] [Google Scholar]
- Seyfang A., Landfear S.M. Substrate depletion upregulates uptake of myo-inositol, glucose and adenosine in Leishmania. Mol. Biochem. Parasitol. 1999;104:121–130. doi: 10.1016/s0166-6851(99)00138-3. [DOI] [PubMed] [Google Scholar]
- Seyfang A., Landfear S.M. Four conserved cytoplasmic sequence motifs are important for transport function of the Leishmania inositol/H(+) symporter. J. Biol. Chem. 2000;275:5687–5693. doi: 10.1074/jbc.275.8.5687. [DOI] [PubMed] [Google Scholar]
- Shayman J.A. Sphingolipids. Kidney Int. 2000;58:11–26. doi: 10.1046/j.1523-1755.2000.00136.x. [DOI] [PubMed] [Google Scholar]
- Smith E.R., Merrill A.H., Obeid L.M., Hannun Y.A. Effects of sphingosine and other sphingolipids on protein kinase C. Methods Enzymol. 2000;312:361–373. doi: 10.1016/s0076-6879(00)12921-0. [DOI] [PubMed] [Google Scholar]
- Sutterwala S.S., Hsu F.F., Sevova E.S., Schwartz K.J., Zhang K., Key P., Turk J., Beverley S.M., Bangs J.D. Developmentally regulated sphingolipid synthesis in African trypanosomes. Mol. Microbiol. 2008;70:281–296. doi: 10.1111/j.1365-2958.2008.06393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirodkar T.S., Voelkel-Johnson C. Sphingolipids in apoptosis. Exp. Oncol. 2012;34:231–242. [PubMed] [Google Scholar]
- Trinconi C.T., Reimao J.Q., Bonano V.I., Espada C.R., Miguel D.C., Yokoyama-Yasunaka J.K., Uliana S.R. Topical tamoxifen in the therapy of cutaneous leishmaniasis. Parasitology. 2017:1–7. doi: 10.1017/S0031182017000130. [DOI] [PubMed] [Google Scholar]
- Trinconi C.T., Reimao J.Q., Coelho A.C., Uliana S.R. Efficacy of tamoxifen and miltefosine combined therapy for cutaneous leishmaniasis in the murine model of infection with Leishmania amazonensis. J. Antimicrob. Chemother. 2016;71:1314–1322. doi: 10.1093/jac/dkv495. [DOI] [PubMed] [Google Scholar]
- Trinconi C.T., Reimao J.Q., Yokoyama-Yasunaka J.K., Miguel D.C., Uliana S.R. Combination therapy with tamoxifen and amphotericin B in experimental cutaneous leishmaniasis. Antimicrob. Agents Chemother. 2014;58:2608–2613. doi: 10.1128/AAC.01315-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince J.E., Tull D., Landfear S., McConville M.J. Lysosomal degradation of Leishmania hexose and inositol transporters is regulated in a stage-, nutrient- and ubiquitin-dependent manner. Int. J. Parasitol. 2011;41:791–800. doi: 10.1016/j.ijpara.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Charles A.G., Frankel A.J., Cabot M.C. Increasing intracellular ceramide: an approach that enhances the cytotoxic response in prostate cancer cells. Urology. 2003;61:1047–1052. doi: 10.1016/s0090-4295(02)02511-6. [DOI] [PubMed] [Google Scholar]
- WHO . WHO; Geneva: 2010. Control of the Leishmaniasis: Report of a Meeting of the World Health Organization Expert Committee on the Control of Leishmaniases, WHO Technical Report Series. [Google Scholar]
- Yoneyama K.A., Tanaka A.K., Silveira T.G., Takahashi H.K., Straus A.H. Characterization of Leishmania (Viannia) braziliensis membrane microdomains, and their role in macrophage infectivity. J. Lipid Res. 2006;47:2171–2178. doi: 10.1194/jlr.M600285-JLR200. [DOI] [PubMed] [Google Scholar]
- Zhang K., Hsu F.F., Scott D.A., Docampo R., Turk J., Beverley S.M. Leishmania salvage and remodelling of host sphingolipids in amastigote survival and acidocalcisome biogenesis. Mol. Microbiol. 2005;55:1566–1578. doi: 10.1111/j.1365-2958.2005.04493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.