

Abstract
Mutations in paralogous mitochondrial proteins CHCHD2 and CHCHD10 cause autosomal dominant Parkinson Disease (PD) and Amyotrophic Lateral Sclerosis/Frontotemporal Dementia (ALS/FTD), respectively. Using newly generated CHCHD2, CHCHD10 and CHCHD2/10 double knockout cell lines, we find that the proteins are partially functionally redundant, similarly distributed throughout the mitochondrial cristae, and form heterodimers. Unexpectedly, we also find that CHCHD2/CHCHD10 heterodimerization increases in response to mitochondrial stress. This increase is driven by differences in the proteins' stability and mutual affinity: CHCHD2 is preferentially stabilized by loss of mitochondrial membrane potential, and CHCHD10 oligomerization depends on CHCHD2 expression. Exploiting the dependence of CHCHD10 oligomerization on CHCHD2, we developed a heterodimer incorporation assay and demonstrate that CHCHD2 and CHCHD10 with disease-causing mutations readily form heterodimers. As we also find that both proteins are highly expressed in human Substantia nigra and cortical pyramidal neurons, mutant CHCHD2 and CHCHD10 may directly interact with their wild-type paralogs in the context of PD and ALS/FTD pathogenesis. Together, these findings demonstrate that differences in the stability and mutual affinity of CHCHD2 and CHCHD10 regulate their heterodimerization in response to mitochondrial distress, revealing an unanticipated link between PD and ALS/FTD pathogenesis.
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disease affecting 1–2% of the population over the age of 60. Although most cases of PD are sporadic, approximately 10% of cases are familial, many with a monogenic cause (1).
An autosomal dominant missense mutation in the mitochondrial protein coiled-helix-coiled-helix domain containing 2 (CHCHD2) was recently demonstrated to cause early onset PD with a typical phenotype (2). Both gain-of-function and haploinsufficiency have been proposed as the pathogenic mechanism but neither has been established definitively (3). Likewise, the function of CHCHD2 is not fully understood, although previous work has suggested that CHCHD2 expression influences complex I and complex IV mitochondrial biogenesis and/or stability, mitochondrial cristae structure and the regulation of apoptosis (4–8).
CHCHD2 shares 58% amino acid sequence identity with CHCHD10, autosomal dominant missense mutations in which also cause neurodegenerative disorders, including amyotrophic lateral sclerosis/Frontotemporal dementia (ALS/FTD) (R15L and S59L), mitochondrial myopathy (G58R), Charcot–Marie–Tooth syndrome (G66V) and adult onset spinal muscular atrophy (G66V) (9–15). As with mutations in CHCHD2, the mechanism of pathogenesis for these mutations remains unclear and the function of CHCHD10 is not fully understood. However, recent studies propose that CHCHD10 is a structural component of the mitochondrial cristae organizing system (MICOS) that together with the sorting and assembly machinery bridges the inner and outer mitochondrial membranes and shapes inner mitochondrial membrane cristae structure (10,16).
Sharing a common ortholog in budding yeast, Mix17, human CHCHD2 and CHCHD10 are thought to have resulted from a gene duplication event (9). Two recent reports have further found that CHCHD2 and CHCHD10 form a complex, suggesting their evolutionary conservation may translate into conservation also of form and function (17,18). Assessment of the similarity and difference between the proteins, however, has been limited, as the proteins have generally been studied in isolation (the two recent reports excepted) and isogenic CHCHD2 and CHCHD10 knockouts (KOs) have not been evaluated together.
In this study, we generated cell lines null for CHCHD2, CHCHD10 and both CHCHD2 and CHCHD10 on a common cellular background. Study of these cells reveals that the proteins are partially functionally redundant, share localization in the mitochondrial cristae and form heterodimers. Unexpectedly, mitochondrial distress promotes CHCHD2/CHCHD10 heterodimerization by a mechanism that exploits differences in stability and mutual affinity between the proteins. CHCHD2 abundance increases following loss of the mitochondrial inner membrane potential and promotes the oligomerization of CHCHD10, which requires CHCHD2 for oligomerization. CHCHD2 and CHCHD10 containing patient mutations incorporate into heterodimers suggesting a mechanism by which a mutation in CHCHD2 could affect CHCHD10 function and vice versa. These findings demonstrate that CHCHD2 and CHCHD10 respond dynamically to mitochondrial distress and suggest an unanticipated link between PD and ALS pathogenesis.
Results
CHCHD2 and CHCHD10 are partially functionally redundant
CHCHD2 and CHCHD10, which exhibit 58% amino acid sequence identity, are thought to have arisen through gene duplication, as they share a common ortholog in budding yeast and other evolutionary distant organisms (Supplementary Material, Fig. S1A and B) (9). To assess whether they have retained common functions in their divergent evolution, we generated HEK293 cell lines lacking CHCHD2 (CHCHD2 KO), CHCHD10 (CHCHD10 KO) and both CHCHD2 and CHCHD10 [CHCHD2/10 double knockout (DKO] using CRISPR-Cas9 genomic editing (Fig. 1A). Interestingly, CHCHD10 protein levels elevated with an average increase of 25.7% following CHCHD2 loss (SD 23%, P-value = 0.0034). A similar significant increase of 46.3% in CHCHD10 was observed in an independent CHCHD2 KO clone (SD 26%, P-value <= 0.001). Conversely, CHCHD2 protein levels increased an average of 42.9% following loss of CHCHD10 (SD 66%, P-value = 0.013), suggesting that there are mild reciprocal compensatory responses to loss of either protein (Fig. 1A).
Figure 1.
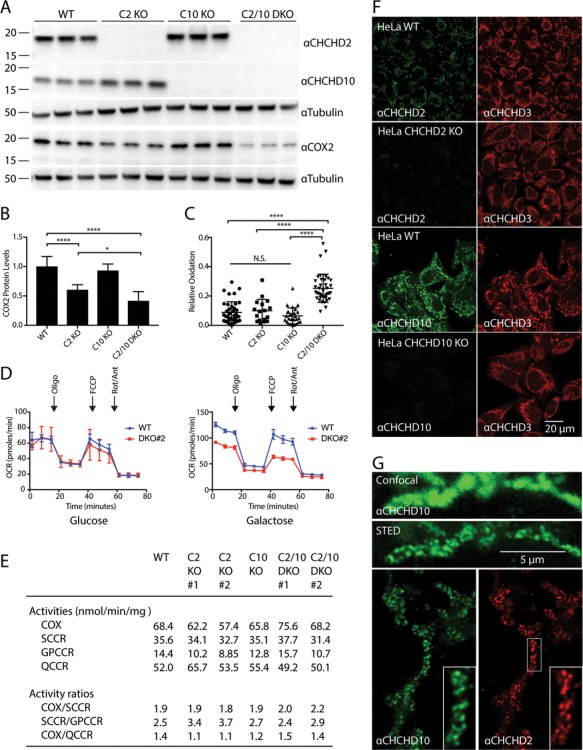
CHCHD2 and CHCHD10 are functionally redundant. (A) Lysates from wildtype (WT), CHCHD2 knockout (C2 KO), CHCHD10 knockout (C10 KO) or CHCHD2/10 DKO (C2/10 DKO) HEK293 cells were immunoblotted with CHCHD2, CHCHD10, COX2 and Tubulin antibodies. Tubulin served as a loading control. (B) Quantification of (A) from >= 3 biological replicates performed on at least 2 separate occasions. (C) Quantification of relative cytosolic oxidation of HEK293 WT, C2 KO, C10 KO and DKO cells using roGFP probe. (D) Oxygen consumption rate (OCR) of WT and C2/10 DKO HEK293 cell lines incubated in glucose (left panel) or galactose (right panel). (E) Spectrophotometric analysis of mitochondrial respiratory chain activities. COX, (complex IV); SCCR, succinate cytochrome c reductase (complex II and III); GPCCR, glycerol-3-phosphate cytochrome c reductase; QCCR, ubiquinol cytochrome c reductase (complex III). Samples were tested in duplicate and average values reported. (F) Representative confocal images of WT, C2 KO and C10 KO HeLa cells immunostained with antibodies against endogenous C2, C10 and MIC19/CHCHD3. MIC19/CHCHD3 staining was used as a mitochondrial marker. (G) Representative confocal and STED images of HeLa cells immunostained with a CHCHD10 antibody (top panels). Representative STED image of HeLa cells immunostained with CHCHD2 and CHCHD10 antibodies (bottom panels).
Steady-state levels of the cytochrome c oxidase (COX) subunit COX2 were previously reported to decrease following CHCHD2 knockdown (7). Consistently, we found that in CHCHD2 KO cells COX2 levels were decreased to an average of 60.4% of levels in wild-type (WT) cells by immunoblotting (Fig. 1A and B). We also observed a mild decrease in assembled complex IV by blue native PAGE (BN-PAGE) in two separate CHCHD2 KO clones and a mild decrease in nine COX subunits by quantitative proteomics, three of which were statistically significant after correcting for multiple comparisons (Supplementary Material, Fig. S2A and B). To test for functional redundancy of CHCHD2 and CHCHD10, we assessed COX2 levels also in the CHCHD10 KO and CHCHD2/10 DKO cells. Although COX2 levels were similar in CHCHD10 KO and WT cells, KO of both CHCHD2 and CHCHD10 leads to a more severe loss of COX2 expression (41.7% of WT for CHCHD2/10 DKO versus 60.4% of WT for CHCHD2 KO, P-value = 0.0237 corrected for multiple comparisons), consistent with partial functional redundancy between CHCHD2 and CHCHD10. These results also point to a more severe effect of CHCHD2 loss on COX2 levels compared to loss of CHCHD10 at least in HEK293 cells.
Knockdown of CHCHD2 has previously been reported to increase reactive oxygen species production in the cytosol (8). We used the ratiometric probe reduction-oxidation sensitive green fluorescent protein (roGFP), which has different excitation maxima in its reduced and oxidized forms (19), to assess whether loss of CHCHD2, CHCHD10 or of both proteins leads to a more oxidative environment within the cytosol. Cytosolic roGFP was reduced in CHCHD2 KO and CHCHD10 KO cells similar to WT cells (Fig. 1C). In contrast, the cytosolic roGFP population was partially oxidized in CHCHD2/10 DKO cells, as has been previously observed with acute knockdown of CHCHD2 (8). Interestingly, the redox potential of the mitochondrial matrix, measured with matrix-targeted roGFP, was not different between WT and CHCHD2/10 DKO cells (also, similar to what was previously observed with CHCHD2 knockdown) (Supplementary Material, Fig. S2C) (8). The source of the oxidative stress may be the mitochondrial intermembrane space (IMS), as reactive oxygen species from this compartment may be detected more readily in the cytosol than in the mitochondrial matrix. Alternatively, the source of reactive oxygen species may lie within the cytosol. In any event, these results are consistent with decompensation resulting from loss of both CHCHD2 and CHCHD10 and functional redundancy between the proteins.
Although we detected a decrease in COX2 protein expression, oxygen consumption was normal in two independent CHCHD2/10 DKO clones relative to WT HEK293 cells basally and following the sequential addition of oligomycin, Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) and antimycin/rotenone, when tested in glucose (Fig. 1D, left; Supplementary Material, Fig. S2D). Respiratory complex activities (including CII, CIII and CIV) were also found to be normal in intact CHCHD2 KO, CHCHD10 KO and CHCHD2/10 DKO cells, as measured spectrophotometrically, when the cells were tested in glucose (Fig. 1E). We next tested CHCHD2/10 DKO cells in galactose, which forces the cell to rely on oxidative phosphorylation rather than glycolysis for ATP production, and observed a mild decrease in basal and maximal oxygen consumption (Fig. 1D, right). These results are consistent with a mild respiratory defect in CHCHD2/10 DKO HEK293 cells.
CHCHD2 and CHCHD10 localize to the mitochondrial cristae and are dispensable for MICOS complex assembly and cristae structure
Having found that CHCHD2 and CHCHD10 are partially functionally redundant, we next assessed by super-resolution microscopy and immuno-electron microscopy (EM) whether the proteins have a similar localization. CHCHD2 and CHCHD10 co-localized with the mitochondrial protein MIC19/CHCHD3 at the resolution of confocal microscopy (Fig. 1F). Interestingly, both CHCHD2 and CHCHD10 exhibited a punctate pattern that was better resolved by stimulated emission depletion (STED) microscopy (Fig. 1G). Endogenous CHCHD2 and CHCHD10 puncta co-localized by two-color STED, suggesting the proteins are in the same mitochondrial subcompartment (Fig. 1G). CHCHD2 and CHCHD10 signals were absent or greatly attenuated in the CHCHD2 and CHCHD10 KO HeLa cells giving us high confidence in the specificity of staining (data not shown).
We next used immuno-EM to identify the mitochondrial subcompartment to which CHCHD2 and CHCHD10 localize. We made use of CHCHD2 and CHCHD10 KO HeLa cells to establish the specificity of our pre-embedding nanogold labeling procedure (data not shown). Consistent with the STED results, we found that endogenous CHCHD2 and CHCHD10 exhibited a similar pattern of localization with the majority of nanogold particles inside of mitochondria localizing to the cristae of the inner membrane (Fig. 2A and B).
Figure 2.
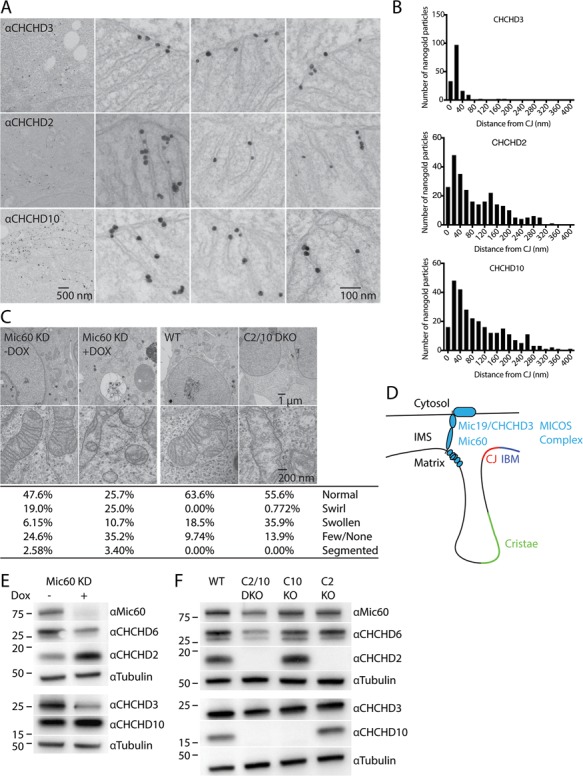
CHCHD2 and CHCHD10 localize to mitochondria cristae and are not essential for MICOS complex stability. (A) Representative Immuno-TEM images labelled pre-embedding with MIC19/CHCHD3, CHCHD2 or CHCHD10 antibodies followed by nanogold-conjugated secondary antibodies. (B) Histograms depicting distance from individual intra-mitochondrial nanogold particles to the nearest cristae junction (CJ) for MIC19/CHCHD3 (top), CHCHD2 (middle) and CHCHD10 (bottom). Particles in the cytosol or associated with the cytosolic face of the outer membrane were excluded from the analysis. (C) Representative TEM images from Doxycycline (Dox) inducible knockdown HeLa cells targeting MIC60 (MIC60 KD), WT HEK293 cells and C2/10 DKO cell lines. Dox-inducible cell lines were treated with 2 μm Dox for 7 days (Dox+) or left untreated (Dox−). Greater than 200 mitochondria were scored per condition. (D) Model of MICOS complex relative to the intermembrane space (IMS) and inner membrane composed of the inner boundary membrane (IBM), CJ and cristae. (E) Lysates from MIC60 KD HeLa cells +/− Dox were immunoblotted with MIC60, MIC25/CHCHD6, CHCHD2, MIC19/CHCHD3, CHCHD10 and Tubulin antibodies. Tubulin served as a loading control. (F) Lysates from WT, C2 KO, C10 KO, C2/10 DKO HEK293 cells were immunoblotted with the same antibodies as in (E).
As the pattern of CHCHD10 localization on the mitochondrial cristae was previously reported to be largely confined to the cristae junction (CJ) (10), we compared the localization of CHCHD2 and CHCHD10 to MIC19/CHCHD3, a stable constituent of the MICOS complex concentrated at the CJ (Fig. 2A and B). Whereas 75% of MIC19/CHCHD3 localized within 27 nm of the CJ, the majority of CHCHD2 and CHCHD10 particles were greater than 67 nm from the CJ. These results suggest that although CHCHD2 and CHCHD10 exhibit partial localization to the CJ, their distribution is more extensive along the cristae and distinct from MIC19/CHCHD3, which is structurally integral to the MICOS complex and confined to the CJ.
Next, we assessed the effect of CHCHD2 and CHCHD10 KO on cristae structure, which relies on MICOS complex integrity. Loss of critical components of the MICOS complex leads to characteristic cristae ‘swirls’ with limited or no connection to the inner boundary membrane (IBM) (20–22). As a positive control for these typical ultrastructural changes resulting from MICOS complex disruption, we examined a HeLa cell line with doxycycline-regulated expression of shRNA against MIC60/mitofilin, a central component of the MICOS complex (23). Treatment with doxycycline for 7 days leads to substantial reduction in MIC60 protein levels (Fig. 2C and E). In MIC60 knockdown cells, only a quarter of mitochondria had a normal morphology with another quarter forming the characteristic ‘swirl’ morphology and the remainder exhibiting swollen cristae, few or no cristae or an unusual segmented phenotype (Fig. 2C). In contrast, CHCHD2/10 DKO HEK293 cells had subtle ultrastructural abnormalities when compared to WT HEK293 cells with slightly fewer normal mitochondria and more organelles with swollen cristae (36.9% versus 18.5%). Less than 1% of CHCHD2/10 DKO cells possessed a ‘swirl’ pattern typically seen following disruption of an essential component of the MICOS complex. Collectively, the ultrastructural morphology of CHCHD2/10 DKO cells suggests that CHCHD2 and CHCHD10 are largely dispensable for normal cristae structure.
To further test the hypothesis that CHCHD2 and/or CHCHD10 may be an integral component of the MICOS complex (Fig. 2D), we assessed whether CHCHD2 and CHCHD10 levels are labile following knockdown of MIC60, as has been reported for integral MICOS complex components such as MIC19/CHCHD3 and MIC25/CHCHD6 (23). Consistent with previous reports, levels of MIC19/CHCHD3 and MIC25/CHCHD6 were consistently reduced upon MIC60 knockdown (Fig. 2E). In contrast, CHCHD2 levels were increased and CHCHD10 levels were stable or only mildly decreased. We next assessed the effects of single as well as DKOs of CHCHD2 and CHCHD10 on proteins integral to the MICOS complex in HEK293 cells. In contrast to their substantial decrease in response to MIC60 knockdown, MIC60, MIC19/CHCHD3 and MIC25/CHCHD6 levels were not reduced by KO of CHCHD2 or CHCHD10 alone (Fig. 2F). DKO in CHCHD2 and CHCHD10 resulted in a mild decrease in MIC60, MIC19/CHCHD3 and MIC25/CHCHD6 levels, compared to the more severe decrease following knockdown of MIC60 (Fig. 2E and F). Finally, in HeLa cell lines stably expressing CHCHD2-Flag or MIC19/CHCHD3-Flag, CHCHD2-Flag failed to pulldown MIC25, MIC27 and MIC60; whereas under the same conditions MIC19/CHCHD3-Flag pulled down all three subunits of the MICOS complex (Supplementary Material, Fig. S3). These findings suggest that CHCHD2 and CHCHD10 are not integral components of the MICOS complex, in contrast to MIC60, MIC19/CHCHD3 and MIC25/CHCHD6.
CHCHD2 is required for oligomerization of CHCHD10
Having found evidence for functional redundancy between CHCHD2 and CHCHD10 and their similar sub-mitochondrial localization, we next assessed whether CHCHD2 and CHCHD10 directly interact. We found that the addition of the crosslinker disuccinimidyl glutarate (DSG) led to the formation of two CHCHD2 immunoreactive bands that migrated near the 37 kDa marker (Fig. 3A and B; Supplementary Material, Fig. S4A). These were interpreted to represent a CHCHD2 homodimer and a more abundant heterodimer composed of CHCHD2 and the slightly smaller CHCHD10, respectively. We concluded that the lower band represents a CHCHD2/CHCHD10 heterodimer, as this CHCHD2 immunoreactive band was absent in CHCHD10 KO cells and a single band at this molecular weight was observed upon immunoblotting for CHCHD10. The slower than predicted migration (∼37 kDa versus 29–31 kDa) was thought to reflect summation of the higher apparent molecular weight (MW) of the constituents (∼18 kDa for CHCHD2 and ∼16 kDa for CHCHD10) together with alteration in migration due to crosslinking of the polypeptide chains. The homodimer and heterodimer bands were also detected with antibodies raised against the C-terminal (residues 99–148) and N-terminal regions (residues 2–29) of CHCHD2, respectively (Supplementary Material, Fig. S4B). Two non-specific bands migrating just below the 37 kDa marker (marked by asterisks) in the untreated samples (Fig. 3A, right, lane 1) were absent following DSG treatment as evidenced by the absence of bands at this level in the CHCHD2 KO cell lines (Fig. 3A, right, lane 6–7; Supplementary Material, Fig. S4C).
Figure 3.
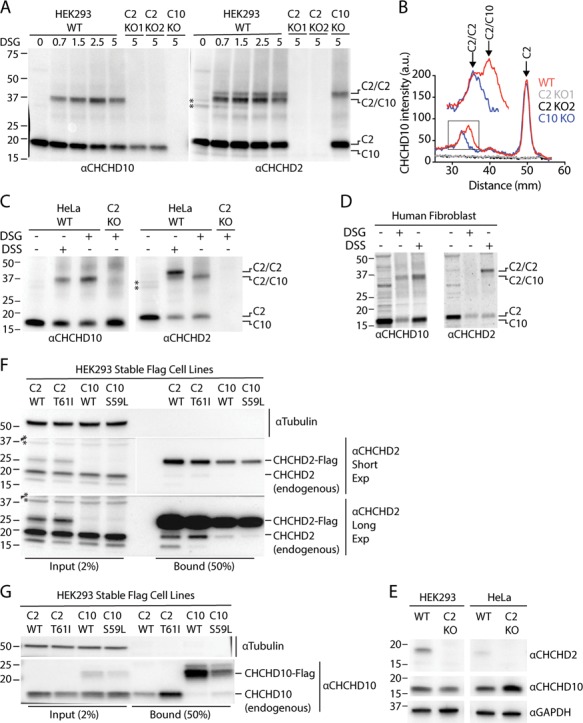
CHCHD2 promotes oligomerization of endogenous CHCHD10. (A) Lysates from WT, C2 KO and C10 KO HEK293 cells treated with the indicated DSG concentration or untreated were immunoblotted with CHCHD2 and CHCHD10 antibodies. Two independent C2 KO cell lines (C2 KO1 and C2 KO2) were tested. (B) Representative line scans of experiment described in (A). Experiment was performed >= 3 times on at least two separate occasions with similar results. (C) Lysates from untreated, DSG (5 mm) or DSS (5 mm) treated WT and C2 KO HeLa cells were immunoblotted with CHCHD2 and CHCHD10 antibodies. (D) Lysates from untreated, DSG (5 mm) or DSS (5 mm) treated primary human fibroblasts were immunoblotted with CHCHD2 and CHCHD10 antibodies. (E) Lysates from HEK293 and HeLa WT and C2 KO cells were immunoblot ted with antibodies against CHCHD2, CHCHD10 and GAPDH. (F and G) Lysates from HEK293 cells stably expressing CHCHD2 WT-Flag (C2 WT), CHCHD2 T61I-Flag (C2 T61I), CHCHD10 WT-Flag (C10 WT) and CHCHD10 S59L-Flag (C10 S59L) were immunocaptured with anti-Flag beads, followed by immunoblotting with Tubulin, CHCHD2 (F) and CHCHD10 (G) antibodies. Tubulin was used as a loading control. Experiment was performed >= 3 times on at least two separate occasions with similar results. In all panels ‘*’ indicates two non-specific bands cross-reactive with the mouse monoclonal CHCHD2 antibody that was used in all panels of the figure. These were observed in untreated but not in DSG or DSS treated samples.
In two independent CHCHD2 KO HEK293 clones and a CHCHD2 KO HeLa cell line, only CHCHD10 monomers were observed (Fig. 3A–C). This suggests that CHCHD2 is required for oligomerization of CHCHD10 at endogenous levels of expression. In contrast, CHCHD2 homodimers were readily observed in the absence of CHCHD10. Together these results demonstrate that CHCHD2 forms heterodimers with CHCHD10 but can also form homodimers in the absence of CHCHD10. CHCHD10, in contrast, requires CHCHD2 for oligomerization at endogenous levels of expression.
We obtained the same general pattern of results using the crosslinker disuccinimidyl suberate (DSS) in HeLa, HEK293 cells and human primary fibroblasts (Fig. 3C and D and Supplementary Material, Fig. S4D). Compared to DSG treatment, the majority of CHCHD2 immunoreactivity was present in the dimer and the homodimer appeared more intense, suggesting either that DSS forms the CHCHD2 homodimer more efficiently than DSG or that DSG partially blocks the CHCHD2 epitope for the antibody used. The latter interpretation is supported by the observation that the CHCHD2 homodimer stabilized by DSG is more strongly reactive with an N-term-CHCHD2 antibody than the C-term-CHCHD2 antibody (Supplementary Material, Fig. S4). As with DSG treatment, the CHCHD10 oligomers did not form in the absence of CHCHD2.
In contrast to HEK293 cells, the CHCHD2 homodimer in HeLa cells was detectable with DSS treatment but not with DSG treatment (Fig. 3C). We hypothesized that the difference may be due to a lower CHCHD2/CHCHD10 ratio in HeLa cells compared to HEK293 cells. Consistently, we found that CHCHD2 levels are ∼4-fold lower in HeLa cells compared with HEK293 cells, whereas CHCHD10 levels are similar in both cell lines (Fig. 3E and Supplementary Material, Fig. S5A).
To assess for interaction between CHCHD2 and CHCHD10 in an independent assay, we immunoprecipitated CHCHD2-Flag and CHCHD10-Flag from stable HEK293 cell lines. CHCHD2-Flag pulled down both endogenous CHCHD2 and CHCHD10 (Fig. 3F and G; Supplementary Material, Fig. S5B). CHCHD10-Flag also pulled down endogenous CHCHD2 but pulled down substantially less endogenous CHCHD10 compared to CHCHD2-Flag (Fig. 3F and G). Similar results were observed for HeLa cells stably expressing CHCHD2-Flag. CHCHD2-Flag cells but not similarly treated MIC19/CHCHD3-Flag cells pulled down endogenous CHCHD2 and CHCHD10 (Supplementary Material, Fig. S3). These results are consistent with the crosslinking experiments suggesting that CHCHD10 has relatively high affinity for CHCHD2 but low affinity for itself, whereas CHCHD2 has high affinity for both CHCHD10 and itself.
CHCHD2 carrying the PD-causing T61I point mutation and CHCHD10 carrying the S59L point mutation also interacted with CHCHD10 and CHCHD2, respectively, consistent with them also being capable of forming heterodimers, as discussed further below (Fig. 3F and G).
Disease-causing mutations cluster near GXXXGXXXG motif and promote aggregation
CHCHD2 and CHCHD10 have a predicted α-helix N-terminal to their dual CX9C domain. Interestingly, most mutations that segregate with disease in families with neurodegenerative disorders, including CHCHD2 T61I, CHCHD10 G58R, CHCHD10 S59L and CHCHD10 G66V, cluster within this predicted α-helix within or near a conserved GXXXGXXXG motif (Fig. 4A). GXXXG motifs are known to mediate homo- or hetero-dimerization of proteins in other structural contexts (24). Structurally the motif forms a tight α-helix with 3.9 amino acids per turn placing glycine residues on a common face of the α-helix. Notably, three of the four mutations in CHCHD2 and CHCHD10 lie on the glycine face of the predicted α-helix modeled with a 3.9 amino acid turn, with the remaining mutation lying on an adjacent face (Fig. 4B).
Figure 4.
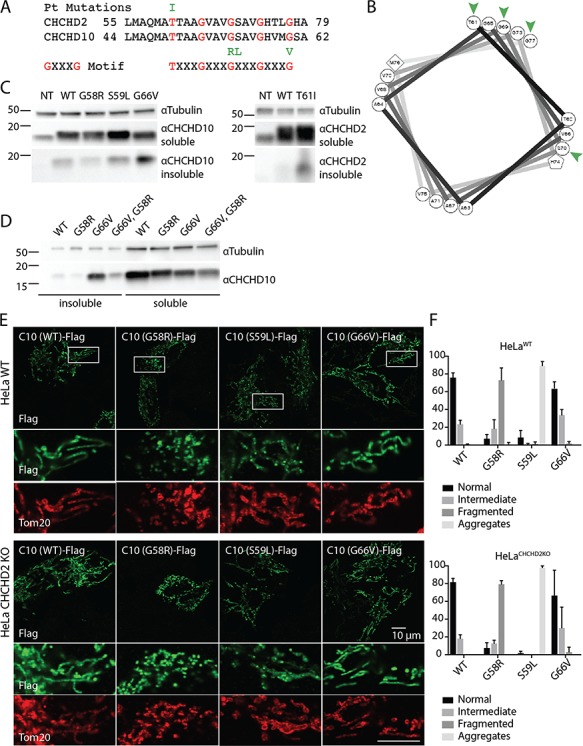
Disease-causing mutations cluster in conserved GXXXGXXXG motif and cause protein aggregation. (A) Position of disease-causing mutations (green) is indicated relative to conserved GXXXGXXXG motif (red). (B) Helical wheel representation of CHCHD2 sequence modeled with 3.9 amino acids per turn, as is classically observed for GXXXG motif containing helices. Position of disease mutations in CHCHD2 and CHCHD10 is indicated by green arrowheads. (C and D) Triton-X100 soluble and insoluble fractions from WT HEK293 cells transiently transfected with CHCHD2 WT, CHCHD2 T61I, CHCHD10 G58R, CHCHD10 S59L and CHCHD10 G66V or CHCHD10 G58R, G66V cDNA were immunoblotted for CHCHD2, CHCHD10 and Tubulin. Tubulin from the soluble fraction was used as a loading control. (E) Representative images from WT ( top panels) or CHCHD2 KO (bottom panels) HeLa cells were transiently transfected with C-terminal Flag tagged CHCHD10 WT, CHCHD10 G58R, CHCHD10 S59L, CHCHD10 G66V and immunostained with Flag and Tom20 antibodies. Tom20 was used as a mitochondrial marker. (F) Percentage of cells with indicated mitochondrial phenotypes in cells treated as described in (E) from >= 3 biological replicates performed on at least two separate occasions.
As mutations in the GXXXG motif are also associated with protein aggregation in the context of other diseases, we tested whether these disease-causing mutations formed either visible aggregates by confocal microscopy or accumulation of protein in a Triton-X100 insoluble fraction by immunoblotting, following exogenous expression (25). We found that three of the four mutations exhibited evidence of aggregation. CHCHD2 T61I, CHCHD10 G66V and (to a lesser extent) CHCHD10 S59L accumulated in the Triton-X100 insoluble fraction upon exogenous expression (Fig. 4C and D). CHCHD10 S59L formed striking visible aggregates in the mitochondria by confocal microscopy, as has been reported previously (Fig. 4E and F) (17). The fourth mutation (CHCHD10 G58R) caused severe mitochondrial fragmentation, as previously observed (12), but did not obviously accumulate in the insoluble fraction or form aggregates visible by confocal microscopy (Fig. 4E and F). That CHCHD10 G58R appears not to aggregate may relate to its predicted disruptive effect on intermolecular hydrophobic interactions; whereas the other three substitutions would be predicted to strengthen hydrophobic interactions, the G58R substitution would be predicted to weaken them. Consistently, we found that introduction of the G58R in cis with the G66V mutation partially suppressed accumulation of mutant CHCHD10 in the insoluble fraction (Fig. 4D). Mitochondrial fragmentation with CHCHD10 G58R expression and microaggregate formation with CHCHD10 S59L expression were also observed in CHCHD2 KO HeLa cells, in which CHCHD10 does not form physiological oligomers, suggesting that physiological heterodimerization is not required for these aggregation and fragmentation phenotypes (Fig. 4E and F).
As the GXXXG motif mediates homo- and/or hetero-dimerization in other proteins, we tested the hypothesis that the predicted α-helix containing the GXXXGXXXG motif mediates CHCHD2 homo-dimerization. CHCHD2 (Δ59-83), which lacks this α-helix, was exogenously expressed in CHCHD2 KO and CHCHD2/10 DKO cells. Surprisingly, CHCHD2 (Δ59–83) formed homodimers and hetero-oligomers with CHCHD10 similarly to full-length CHCHD2, suggesting that the GXXXGXXXG motif is not necessary for oligomerization of CHCHD2 (Supplementary Material, Fig. S6A and B). These findings suggest that homo- and hetero-dimerization is likely mediated at least in part by a domain separate from the GXXXGXXXG motif, the identification of which will require further study.
CHCHD2 and CHCHD10 accumulate asymmetrically in response to loss of Δψ
The observation that CHCHD2 levels and oligomerization status change in response to CHCHD10 (and MIC60) levels led us to question whether CHCHD2 is responsive to mitochondrial stressors more generally. To address this question, we assessed the effect of a variety of mitochondrial stressors on CHCHD2 protein expression. Surprisingly, the protonophore carbonyl cyanide 3-chlorophenylhydrazone (CCCP) led to a robust increase in CHCHD2 expression (Fig. 5A–C). The CCCP responsive increase was also observed for CHCHD2 T61I in a stable cell line, as well as for endogenous CHCHD2 and CHCH10 in primary newborn mouse fibroblasts (Fig. 5C; Supplementary Material, Fig. S7A).
Figure 5.
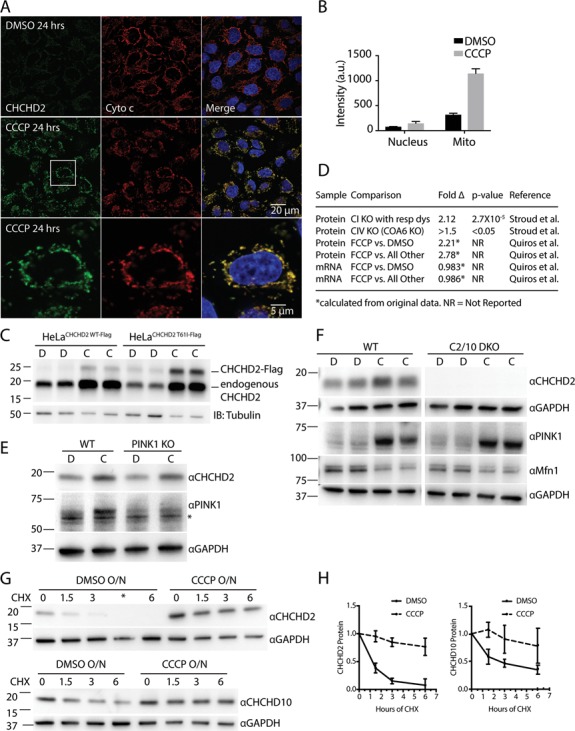
CHCHD2 and CHCHD10 are stabilized in mitochondria post-translationally following loss of Δψ. (A) Representative confocal images of WT HeLa cells treated for 24 h with dimethyl sulfoxide (DMSO) or CCCP (10 μM) and immunostained with CHCHD2 (green) and cytochrome c (red) antibodies and incubated with 4',6-diamidino-2-phenylindole (DAPI) to label nuclei (blue). Cytochrome c was used as a mitochondrial marker. Retention of cytochrome c in mitochondria also indicates that the cells analyzed have not undergone apoptosis after 24 h CCCP treatment, as cytochrome c is released from mitochondria during apoptosis. (B) CHCHD2 intensity in regions overlapping with DAPI and cytochrome c, representing nuclear and mitochondrial compartments, respectively, was measured in 11 fields from three biological replicates for each condition in cells treated as described in (A). (C) Lysates from WT HeLa cells stably expressing CHCHD2 WT-Flag or CHCHD2 T61I-Flag and treated with DMSO or CCCP (10 μm) for 24 h were immunoblotted (IB) using CHCHD2 and Tubulin antibodies. Tubulin was used as a loading control. (D) Table summarizes changes in CHCHD2 protein and mRNA expression in response to mitochondrial stressors from previously published datasets. CI KO with resp dys, average fold change resulting from KO of complex I supernumerary subunits that cause respiratory dysfunction; all other, fold change of CHCHD2 protein level following FCCP treatment versus average of control, actinonin treatment, doxycycline treatment and MitoBloCK-6 treatment. All treatments were for 24 h (E) Lysates from WT and PINK1 KO HeLa cells treated with CCCP (C) or DMSO (D) for 6 h were immunoblotted for GAPDH, CHCHD2 and PINK1. The * indicates non-specific band. (F) Lysates from WT and C2/10 DKO HEK293 cells were immunoblotted with GAPDH, CHCHD2, PINK1 and Mfn1. (G) Lysates from WT HeLa cells treated overnight with DMSO (D) or CCCP (C) followed by the addition of CHX for 0–6 h were immunoblotted with CHCHD2, CHCHD10 and GAPDH antibodies. The * indicates misloaded well. (H) Quantification of CHCHD2 and CHCHD10 protein levels from experiment shown in (G) from >= 3 biological replicates performed on at least two separate occasions CHCHD2 and CHCHD10 protein levels at each time point were normalized to their value at time point 0 hr.
By immunocytochemistry, the accumulated CHCHD2 localized to mitochondria with a roughly 4-fold increase in CHCHD2 immunostaining intensity in regions overlapping with cytochrome c immunostaining following treatment with CCCP (Fig. 5A). CHCHD2 and CHCHD10 were also observed to increase specifically in the mitochondria-enriched heavy membrane fraction (HMF) by immunoblotting following treatment with CCCP (Supplementary Material, Fig. S7B). Most of CHCHD2 was found to be mitochondrial by immuno-EM, where they localized to the mitochondrial cristae following CCCP treatment in a pattern similar to that observed in vehicle-treated cells (Supplementary Material, Fig. S7C). Similarly, protease protection assays demonstrated that the majority of CHCHD2 localized to following treatment with vehicle alone (Supplementary Material, Fig. S7D). Together these results demonstrate that CHCHD2 accumulates within the IMS of mitochondria following treatment with the mitochondrial uncoupler CCCP.
CCCP dissipates the voltage and pH component of the electrochemical gradient across the inner mitochondrial membrane while increasing electron transport and decreasing ATP production. To further evaluate the stimulus for the CHCHD2 increase we assessed other uncouplers and inhibitors of the respiratory chain for their ability to regulate CHCHD2 levels (Supplementary Material, Fig. S7E). Complex I inhibition with rotenone and complex III inhibition with antimycin (which tend to reduce membrane potential similar to CCCP but have an opposite effect on electron flux and superoxide production) caused a similar (but more modest) increase in CHCHD2 levels after 6 h, suggesting that reduction of the electrochemical potential and not a change in electron flux or superoxide production drives the CHCHD2 increase. The complex V inhibitor, oligomycin, which increases membrane potential while decreasing ATP production, led to a slight decrease or did not affect CHCHD2 levels, whereas concurrent treatment with oligomycin and antimycin led to an increase in CHCHD2 levels. These observations suggest that the CHCHD2 increase is sensitive to membrane potential and not mitochondrial ATP production. Additionally, the potassium ionophore valinomycin caused a similar increase in CHCHD2 levels as CCCP treatment suggesting that loss of Δψ component (as opposed to the ΔpH component) of the electrochemical potential causes the CHCHD2 increase. Finally, as CCCP can induce cell death we tested whether CCCP accumulation occurred to a similar extent in the presence and absence of the pan-caspase inhibitor, z-vad, which inhibits apoptosis (Supplementary Material, Fig. S7F). We found that the CHCHD2 increase is unaffected by caspase inhibition. Together with the observations that CHCHD2 can increase after a relatively short treatment with CCCP (6 h) and that CHCHD2 increases in cells that have retained cytochrome c (a protein that is released during apoptosis), we conclude that the CHCHD2 increase in response to CCCP is independent of cell death (Fig. 5A and B; Supplementary Material, Fig. S7E).
Consistent with the observation that CHCHD2 increases in response to dissipation of Δψ, mining of high quality proteomics and transcriptomics data sets from recent publications, identified CHCHD2 as one of the proteins most responsive to genetic or pharmacological manipulations affecting Δψ (26,27). In data analyzed from Quirós et al., CHCHD2 was among a small group of proteins (0.35%) that increased greater than 2-fold in response to FCCP treatment (26). This was not a class effect of twin CX9C proteins as CHCHD2 was the only protein in this class observed to increase in response to FCCP among the 17 detected (CHCHD10 was not detected in the dataset). Analysis of data from this study also indicates that CHCHD2 mRNA levels measured by RNAseq are unchanged by FCCP treatment, suggesting that FCCP regulates CHCHD2 expression post-transcriptionally (Fig. 5D). Consistently, we found that exogenously expressed CHCHD2, which lacks CHCHD2-specific transcriptional elements, also increases in response to mitochondrial uncoupling (Fig. 5C). In another dataset published by Stroud et al., CHCHD2 increased an average of 2.12-fold in response to KO of the 8 supernumerary subunits of complex I that lead to respiratory dysfunction, consistent with our finding that complex I inhibition increases CHCHD2 protein expression (Fig. 5D). CHCHD2 was also found to be significantly upregulated following KO of the complex IV assembly factor COA6 in the same dataset, which, similar to inhibition of complex III with antimycin, leads to a severe reduction in electron transport and is predicted to decrease Δψ. Considered together, these data suggest that CHCHD2 expression responds robustly to manipulations that cause loss of Δψ and that the mechanism underlying increased CHCHD2 protein abundance is likely post-transcriptional.
As we and others previously found that the Parkinson-related PINK1/Parkin pathway is also regulated by Δψ (28–31), we tested whether CHCHD2 accumulation and PINK1/Parkin activation are interdependent. We found that CHCHD2 accumulation is independent of PINK1, as it accumulated to equal extent in WT and PINK1 KO HeLa cells (Fig. 5E). Likewise, PINK1 accumulation was found to be independent of CHCHD2 and CHCHD10, as PINK1 accumulated to a similar extent in WT and CHCHD2/10 DKO cells following CCCP treatment (Fig. 5F). Additionally, the presence of CHCHD2/10 had no effect on degradation of the Parkin substrate Mitofusin-1 (Mfn1), indicating that the PINK1/Parkin pathway is intact in the absence of CHCHD2/10. Similarly, we found that steady-state levels of the CHCHD2/CHCHD10 heterodimer were unaffected by PINK1 loss (data not shown). In sum, these results indicate that the CHCHD2/CHCHD10 and PINK1/Parkin pathways, while responsive to similar mitochondrial stressors, are independently regulated.
To further assess the mechanism of CHCHD2 increase, we tested whether CCCP treatment affects protein stability. Consistent with a previous report (17), CHCHD2 was found to have a very short half-life under basal conditions as measured by protein stability following translation inhibition with cycloheximide (CHX) (Fig. 5F and G). Pretreatment with CCCP overnight, however, significantly increased the half-life of CHCHD2, presumably by reducing its degradation rate. CHCHD10 had a longer half-life than CHCHD2 under basal conditions, as reported previously, but CHCHD10 was similarly stabilized by CCCP pretreatment. The longer half-life of CHCHD10 may account for the more modest effect of depolarization on CHCHD10 levels. Together these results suggest that loss of Δψ leads to CHCHD2 and CHCHD10 increase by blocking the degradation of these otherwise short-lived proteins. The precise mechanism of CHCHD2 degradation basally and its block by CCCP will be important to explore in future studies.
CHCHD2 levels have previously been reported to increase under hypoxic conditions at 4% O2 by a transcriptional mechanism (8). To test whether a hypoxia-like effect accounts for the increase in CHCHD2 and CHCHD10 observed with CCCP treatment, we assessed the effect of CCCP treatment under hypoxic (4% O2) and normoxic (21% O2) conditions. If CCCP and hypoxia increase CHCHD2 through the same mechanism, then CCCP should not increase CHCHD2 levels above what occurs with hypoxia alone. We found that even under hypoxic conditions CCCP increased CHCHD2 levels, suggesting the effects of hypoxia and CCCP treatment on CHCHD2 levels may be mediated by different mechanisms (Supplementary Material, Fig. S8A). Furthermore, the increase in CHCHD2 and CHCHD10 appears to be post-translational both under hypoxic and normoxic conditions, as CHCHD2 and CHCHD10 protein levels pretreated with CCCP are relatively stable following translation shutoff with CHX. In contrast, CHCHD2 and CHCHD10 were unstable in hypoxia-treated cells in the absence of CCCP, consistent with distinct mechanisms underlying the increase in CHCHD2 in response to CCCP and hypoxia.
Post-translational regulation of CHCHD2 levels has a potential ‘advantage’ of regulating CHCHD2 levels in a mitochondrion-autonomous manner. This might allow for higher CHCHD2 levels within impaired mitochondria and lower levels in healthy mitochondria within a single cell. We previously described such a mitochondrion-autonomous mechanism of regulation for products of the PD-related genes Parkin and PINK1 (28,29). To test whether CHCHD2 levels might be regulated in a mitochondrion-autonomous manner, we examined endogenous CHCHD2 expression in Mfn1/Mitofusin-2 (Mfn2) DKO mouse embryonic fibroblasts (Mfn1/2 DKO MEFs), which have a bioenergetically heterogeneous mitochondrial pool due to defective mitochondrial fusion (32). Bioenergetically active mitochondria in Mfn1/2 DKO MEFs were labeled with Mitotracker Red (MTR), a Δψ-dependent mitochondrial dye. Cells were then fixed and immunostained for CHCHD2 as well as cytochrome c to label all mitochondria. Within single cells, we found that mitochondria with high CHCHD2 expression frequently had low or absent MTR intensity (indicating reduced Δψ) whereas mitochondria with high MTR intensity typically had low CHCHD2 expression (Supplementary Material, Fig. S8B). These findings suggest that CHCHD2 levels are bioenergetically regulated within individual mitochondria.
Loss of Δψ promotes CHCHD2/CHCHD10 heterodimerization, and disease-causing CHCHD2/CHCHD10 mutants incorporate into heterodimers
We next assessed the effect of mitochondrial uncoupling on oligomerization of CHCHD2 and CHCHD10. Monomeric and oligomeric CHCHD2 increased roughly proportionally in response to CCCP treatment (Fig. 6A, bottom; Supplementary Material, Fig. S8C). Oligomeric CHCHD10 levels, in contrast, increased to a much greater extent than monomeric CHCHD10 (Fig. 6A, top; Supplementary Material, Fig. S8C). The large increase in oligomeric CHCHD10 is presumably due to the increase in CHCHD2 which is required for CHCHD10 oligomerization. The presence of CHCHD2 and CHCH10 in these high molecular weight bands was confirmed by the absence of these immunoreactive bands, in CHCHD2 KO and CHCHD10 KO cells, respectively, which were processed in parallel. Importantly, these findings suggest that under basal conditions levels of CHCHD2 may be limiting for CHCHD10 oligomerization, allowing CHCHD2 stabilization to shift the monomer/oligomer ratio of CHCHD10.
Figure 6.
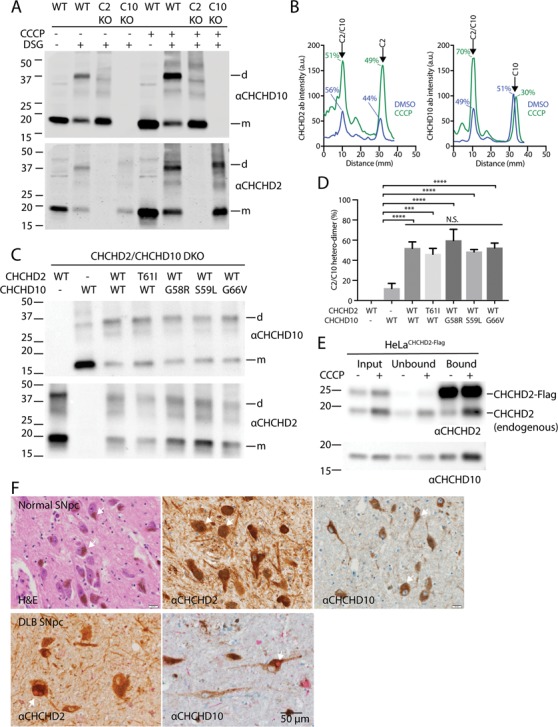
Loss of Δψ promotes CHCHD2/10 heterodimerization, and CHCHD2/10 with disease-causing mutations readily incorporate into heterodimers. (A) Lysates from HeLa cells treated with DMSO or CCCP (10μM) for 24 h immunoblotted for CHCHD2 and CHCHD10. (B) Line scan of immunoblot in (A). (C) Lysates from CHCHD2/10 DKO HEK293 cells co-transfected with the indicated CHCHD2 and CHCHD10 WT or missense mutation-containing cDNAs were treated with DSG (5mM). In control experiments (lanes 1–2), CHCHD2 or CHCHD10 was co-transfected with YFP cDNA. (D) Quantification of experiment described in (C). Intensity of CHCHD10 antibody signal was determined by line-scanning in three biological replicates. The percent dimer represented the ratio of the peak corresponding to the dimer divided by the sum of the dimer and monomer peaks. One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test was used to determine statistical significance. ‘n.s.’, not significant; ‘****’, P-value < 0.0001; and ‘***, P-value = 0.0001. (E) Lysates from HeLa cells stably expressing CHCHD2 WT-Flag (C2 WT) and treated with CCCP or DMSO overnight were immunocaptured with anti-Flag beads, followed by immunoblotting (IB) with CHCHD2 (top) and CHCHD10 (bottom) antibodies. Experiment was performed >= 3 times on at least two separate occasions with similar results. (F) Representative images from normal human substantia nigra pars compacta (SNpc) stained with hematoxylin and eosin (left panel), CHCHD2 antibody (middle panel) and CHCHD10 antibody (right panel). CHCHD2 and CHCHD10 stain are lighter brown than the neuromelanin pigment (arrows) characteristic of dopaminergic neurons in the adult human SNpc. Representative images of SNpc from a DLB case co-immunostained for CHCHD2 (brown) and α-synuclein (red) (left panel) or CHCHD10 (brown) and α-synuclein (red) (right panel). Arrows indicate characteristic brainstem Lewy bodies.
To further test whether relative CHCHD2 levels can change the monomer/dimer ratio of CHCHD10, CHCHD10 was co-expressed with CHCHD2 or a yellow fluorescent protein (YFP) control, in the presence of DSG. Consistent with CHCHD2 promoting oligomerization of CHCHD10, exogenous CHCHD10 was predominately monomeric upon co-expression with YFP and predominately oligomeric upon co-expression with CHCHD2 (Fig. 6C and D). This suggests that a change in CHCHD2 expression has a profound impact on the oligomerization status of CHCHD10.
Additionally, in HeLa cells stably expressing CHCHD2-Flag, CHCHD2-Flag pulled down more endogenous CHCHD10 and more CHCHD2 following CCCP treatment (Fig. 6E). Together these data are consistent with a model in which accumulating CHCHD2 binds more endogenous CHCHD10 following mitochondrial stress.
The heterodimerization of CHCHD2 and CHCHD10 suggests a mechanism by which a missense mutation in either CHCHD2 or CHCHD10 could directly affect its WT paralog. To test whether disease-causing mutations incorporate into the heterodimer, CHCHD2 T61I was co-expressed with WT CHCHD10; and CHCHD10 G58R, CHCHD10 S59L and CHCHD10 G66V were co-expressed with WT CHCHD2 (Fig. 6C and D). In each case the mutant protein was found to promote heterodimerization of CHCHD10 to a similar extent as its WT counterpart, consistent with their incorporating into the heterodimer. Thus, these disease-causing mutations do not seem to alter the heterodimerization dynamics of CHCHD2 and CHCHD10. As a corollary, these findings demonstrate that CHCHD2 and CHCHD10 with disease-causing mutations can directly interact with their WT paralogs and suggest a mechanism for a possible dominant negative effect.
We next addressed whether CHCHD2 and CHCHD10 tend to be expressed in the same neuronal cell populations in the human brain. We reasoned that heterodimerization is most likely of physiologic relevance in cell types that express both CHCHD2 and CHCHD10. We found that CHCHD2 and CHCHD10 were strongly expressed within Substantia nigra pars compacta (SNpc) neurons affected in PD (Fig. 6F). Similarly, CHCHD2 and CHCHD10 were highly expressed in pyramidal neurons within the cortex and the hippocampus affected in ALS/FTD (Supplementary Material, Fig. S9). As the gene products of some but not all familial PD genes accumulate in Lewy bodies, we also assessed CHCHD2 and CHCHD10 immunostaining in these characteristic protein inclusions. In a PD case and a case of dementia with Lewy bodies (DLB), α-synuclein immunostaining was prominent in Lewy bodies, as expected (Fig. 6F and data not shown). CHCHD2 and CHCHD10 immunoreactivity, in contrast, was excluded from Lewy bodies in the same sections, indicating that CHCHD2 and CHCHD10 do not accumulate in Lewy bodies. Together these findings suggest that CHCHD2 and CHCHD10 are highly co-expressed in cell types affected in PD and ALS/FTD, underscoring the potential pathophysiological significance of CHCHD2/10 heterodimerization.
Discussion
CHCHD2 and CHCHD10 are paralogous mitochondrial proteins, autosomal dominant mutations in which were recently found to cause PD and ALS/FTD, respectively (2,10). Despite a shared ontology, studies to date have tended to focus on either CHCHD2 or CHCHD10 alone (with the notable exception of two very recent publications) (17,18). By evaluating novel single KO cell lines of CHCHD2 and CHCHD10 and, to our knowledge, the first CHCHD2/CHCHD10 DKO cell lines, we identify similarities in the function and localization of CHCHD2 and CHCHD10. We also reveal that unanticipated differences in their stability and mutual affinity govern the dynamic heterodimerization of CHCHD2 and CHCHD10 in response to bioenergetic stress (Fig. 7).
Figure 7.
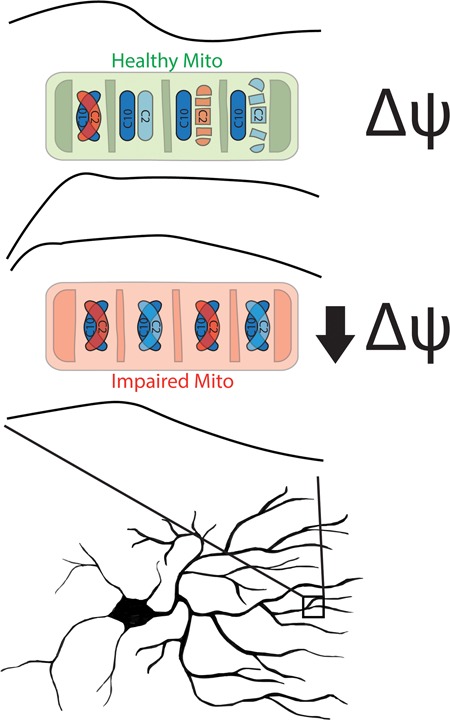
Model of CHCHD2/10 heterodimerization in response to loss of Δψ. In well-coupled mitochondria (green) with high Δψ CHCHD2 (C2) is unstable and CHCHD10 (C10) is primarily monomeric. In contrast, in impaired mitochondria with low Δψ, CHCHD2 is stabilized and CHCHD2/10 heterodimerization promoted. CHCHD2 containing the T61I patient mutation (red) incorporates into the heterodimer similarly to WT protein, suggesting a possible mechanism for a dominant effect on CHCHD10 function.
Similar to prior reports examining CHCHD2 knockdown, we find that KO of CHCHD2 leads to lower steady-state levels of the complex IV subunit COX2, which is labile outside of the assembled complex (7). Assembled complex IV and several other subunits detected by quantitative mass spectrometry were also mildly reduced. Levels of COX2 are further decreased with DKO of CHCHD2 and CHCH10. Despite decreased COX2 levels, we observe only a mild effect on oxygen consumption, which required incubation in galactose to elicit, suggesting that although reduced, COX2 levels are sufficient in the CHCHD2/CHCHD10 DKO HEK293 cells to maintain near normal respiration. The mild effect of CHCHD2/10 loss on oxidative phosphorylation is largely in-keeping with what has been observed for CHCHD2 and CHCHD10 ortholog in other model systems. Notably, KO of Mix17, the single budding yeast ortholog of CHCHD2 and CHCHD10, had relatively minor effects on oxygen consumption, respiratory complex formation, complex III and IV activities and growth on non-fermentable carbon sources, in contrast to the vast majority of twin CX9C domain containing proteins (33). Similarly, KO of CHCHD2 in the fly and KO of CHCHD10 in human fibroblasts and mice had no effect or relatively mild effects, respectively, on oxygen consumption (4,17,18). Together, these results seem to be consistent with the idea that CHCHD2 and CHCHD10 impinge on but are largely dispensable for respiratory complex function in many cell types and tissues. This is in contrast to, e.g. the twin CX9C domain-containing protein COA6, which serves as an assembly factor for complex IV and was recently found to be essential for respiratory function in HEK293T cells (34).
We found that CHCHD2 and CHCHD10 localize primarily to the mitochondrial cristae by immuno-EM. This likely accounts for their co-localizing punctate pattern observed by two-color STED microscopy. Our results agree at least partially with a prior report using the same CHCHD10 antibody for immuno-EM in that we detected a substantial portion of CHCHD2 and CHCHD10 near the CJ (10). However, differing from the prior report we conclude that the distribution of CHCHD2 and CHCHD10 is not limited to the CJ and therefore argue against CHCHD2 and CHCHD10 being a structural component of the MICOS complex. Our differing interpretation is supported by our inclusion of MIC19/CHCHD3 as a positive control for the MICOS complex, which we observed to have a pattern of localization that is distinct from CHCHD2 and CHCHD10.
Additional observations made in this study also argue against CHCHD2 and CHCHD10 being essential structural components of the MICOS complex. First, as was shown in a previous report and replicated here, CHCHD2 and CHCHD10 have short half-lives, contrary to expectations for a structural protein (17). Second, the characteristic cristae ‘swirls’ observed with loss of MIC60 were extremely rare in CHCHD2/10 null cells in this study. Instead, almost half of the mitochondria were normal, and among the abnormal mitochondria a milder and less specific phenotype was observed. Third, CHCHD2 was not observed to be labile and CHCHD10 levels were only mildly affected following MIC60 knockdown in contrast to structural components of MICOS complex, such as MIC19/CHCHD3 and MIC25/CHCHD6. Conversely, levels of MICOS complex proteins MIC19/CHCHD3, MIC25/CHCHD6 and MIC60 were not affected by KO of either CHCHD2 or CHCHD10 alone and only mildly affected by loss of both CHCHD2 and CHCHD10, in contrast with what is observed with loss of some (but not all) MICOS complex subunits. Finally, whereas MIC19/CHCHD3-Flag was found to pull down three subunits of the MICOS complex, CHCHD2-Flag failed to pull down these subunits under the same conditions. Whereas these results could be compatible with CHCHD2 and CHCHD10 having a peripheral association with the MICOS complex, together they suggest that CHCHD2 and CHCHD10 are not essential, structural components of the MICOS complex. Nonetheless, it is possible that a peripheral interaction with the MICOS complex could form the basis for a gain-of-function disruption of the MICOS complex by mutant CHCHD2 or CHCHD10, as has been suggested previously for CHCHD10 (16).
In addition to localizing CHCHD2 and CHCHD10 within the same submitochondrial compartment and showing evidence for partial functional redundancy, we find that CHCHD2 and CHCHD10 form heterodimers, using cross-linking and co-immunoprecipitation methods. This observation is consistent with two recent reports that demonstrated CHCHD2 and CHCHD10 are in the same complex by co-immunoprecipitation, co-sedimentation in sucrose gradients and co-migration in BN-PAGE gels (17,18). They are also consistent with the previously reported observation that exogenously expressed CHCHD2 can form homodimers following crosslinking with DSS (4). Our study using novel CHCHD2 and CHCHD10 null cells extends previous findings in a number of important ways. Crosslinking with DSG (which has a short 7.5 Å crosslinker arm) suggests that CHCHD2 and CHCHD10 directly interact. We were able to detect the endogenous DSS or DSG crosslinked CHCHD2/CHCHD2 homodimer and the CHCHD2/CHCHD10 heterodimer in intact cells and measure changes in their abundance in response genetic and pharmacological manipulations. In conjunction with our novel CHCHD2 and CHCHD10 KO cells lines, this allowed us to establish that endogenous CHCHD10 requires CHCHD2 to oligomerize; and that CHCHD2, in contrast, does not require CHCHD10 to oligomerize. These findings suggest that CHCHD2 and CHCHD10 have evolved a stark asymmetry in their mutual affinities that may be important to their regulation in response to changes in mitochondrial membrane potential.
Our results suggest that CHCHD2 and CHCHD10 directly interact as they lie within the 7.5 Å crosslinker arm of DSG. It is notable that CHCHD2 has also been reported to interact with a number of other mitochondrial proteins, including P32/C1QBP, complex IV, cytochrome c and BCL-XL, suggesting that they may lie within a larger complex with one or more of these proteins (6,17,18,35). Supporting the notion that CHCHD2 and CHCHD10 may reside in a larger complex, the proteins were previously observed to comigrate in a complex of approximately 220 kDa by BN-PAGE (18). Additionally, although we stabilize dimeric CHCHD2 and CHCHD10 by crosslinking, it is possible that CHCHD2 and CHCH10 form larger homo- and hetero-oligomeric complexes with reactive groups outside of the crosslinking distance of DSG and DSS. It will be interesting to investigate whether any of these interactions are strengthened following mitochondrial uncoupling as CHCHD2 levels and its association with CHCHD10 increase. In particular, as CHCHD10 was also recently found to depend on CHCHD2 for its interaction with complex IV, it will be interesting to investigate whether the indirect interaction between CHCHD10 and COX increases as CHCHD2 accumulates in response to mitochondrial uncoupling (35).
In addition to the asymmetry in their affinities, we were surprised to find CHCHD2 and CHCHD10 respond differently to loss of Δψ. CHCHD2 abundance dramatically increases in response to loss of Δψ, whereas the increase in CHCHD10 is more modest. Consistent with our observation that CHCHD2 increases in response to loss of the Δψ, Drosophila CHCHD2 was recently found to be more abundant in a number of Drosophila lines that exhibit mitochondrial dysfunction (4). It is also consistent with data from a number of high quality proteomics studies in which CHCHD2 was found to be among the proteins that increase substantially in response to a pharmacological or genetic manipulation that would be predicted to result in loss of Δψ (26,27). Using a panel of respiratory complex inhibitors and ionophores, our results point specifically to the loss of the Δψ or a highly correlated event, such as loss of mitochondrial import, as the trigger for CHCHD2 increase within mitochondria. The more modest increase in CHCHD10 levels in response to loss of Δψ is likely due to its longer half-life. The decreased stability of CHCHD2 under basal conditions may account for our observation that most of CHCHD10 is monomeric under basal conditions, as CHCHD2 is limiting for heterodimer formation. When CHCHD2 levels increase in response to loss of Δψ, we observe most of CHCHD10 is pulled into heterodimers with CHCHD2.
Using several complementary lines of evidence, we establish that Δψ regulates CHCHD2 levels post-translationally by decreasing the rate of its degradation. This post-translational mechanism of regulation is reminiscent of the regulation of PD-linked proteins PINK1 and Parkin by Δψ (28–31). The mitochondrion-autonomous regulation of CHCHD2/10 and PINK1/Parkin may be particularly important in the projection neurons affected in PD, which have mitochondria distributed in an extensive axonal arbor, remote from the cell nucleus (36). The post-translational regulation of these proteins may allow individual mitochondria to respond to local disruptions in optimal mitochondrial function, without affecting other mitochondria in one of the other axonal branches. The coincidence of CHCHD2 and PINK1/Parkin both being upregulated by loss of Δψ also highlights the importance of adaptive quality control responses to mitochondrial distress in PD. Finally, our observations suggest that CHCHD2 protein levels might be used as marker of mitochondrial stress particularly in PD models.
That both CHCHD2 and PINK1 are stabilized in response to loss of the Δψ raises the possibility that both proteins employ a similar mechanism for the Δψ ‘sensing’. It is notable that CHCHD2 and CHCHD10, like PINK1, are predicted to have two mitochondrial targeting sequences, one of which is Δψ-sensitive (37). CHCHD2/10 have a predicted mitochondrial targeting signal at their N-terminus, which would be expected to mediate Δψ-dependent import to the inner membrane or the matrix via the Tim23 pathway. Additionally, they have a predicted internal mitochondrial targeting signal in their twin CX9C domain that should target the proteins to the IMS via the Mia40/Erv1 pathway, which is not directly Δψ-dependent. It is interesting to speculate that the Δψ ‘sensing’ of CHCHD2 and CHCHD10 may result from an interplay between these targeting sequences, similar to an interplay between inner and outer mitochondrial membrane targeting sequences of PINK1. Whether Δψ ‘sensing’ by CHCHD2/10 is connected to mitochondrial import and the precise mechanism responsible for CHCHD2/10 degradation under basal conditions are potentially rich areas for future study.
The mechanism by which CHCHD2 and CHCHD10 are constitutively degraded is also unclear at present. CHCHD2 and CHCHD10 are unstable under basal conditions as reported previously and confirmed here, suggesting that they normally undergo rapid degradation after synthesis (17). Most proteins within the IMS are degraded by proteases within this compartment, which include the iAAA protease (composed of a YMEL1 homo-oligomer), HTRA2/OMI, OMA1 and ATP23 (38). The constitutive degradation of CHCHD2 and CHCHD10 may be mediated by one or more of these IMS-facing proteases or, alternatively, by the proteasome, which has been shown to mediate degradation of other CX9C motif containing proteins including Mix17, the ortholog of CHCHD2 and CHCHD10 in budding yeast (39). Similarly, the block in CHCHD2/CHCHD10 degradation following loss of the inner membrane potential could result from a change in the activity of these proteases. Notably, OMA1 is activated and degraded in response to loss of membrane potential, and YME1 is degraded in the setting of low membrane potential and ATP depletion (40,41). Alternatively, the cleavage site of CHCHD2 and CHCHD10 may no longer be available to a protease that retains its activity following Δψ loss due to a change in CHCHD2/CHCHD10 conformation, association with other proteins or localization within the mitochondrial IMS.
Heterodimerization of CHCHD2 and CHCHD10 suggests a mechanism by which mutant CHCHD2 or CHCHD10 could affect function of its WT paralog in a dominant negative manner. Consistent with this possibility we found that CHCHD2 and CHCHD10 with disease-causing mutations efficiently heterodimerize with WT CHCHD10 or CHCHD2, respectively. Both CHCHD2 and CHCHD10 were found to be expressed in the neuronal populations affected in PD and ALS/FTD, suggesting that heterodimerization could contribute to disease pathogenesis. It is notable that most of the mutations with clear segregation in families cluster around a conserved GXXXGXXXG motif in a predicted α-helix N-terminal to the twin CX9C domain. We find these mutations alter CHCHD2 or CHCHD10 behavior when exogenously expressed, with most promoting aggregation and one causing severe mitochondrial fragmentation. Together these findings provide some support for a possible dominant negative or gain-of-function mechanism for disease. However, definitive evidence for the mechanism of disease is lacking with recent reports also finding evidence for both haploinsufficiency and gain-of-function (4,10,16,17,23,42–44). Comparison of knock-in mice carrying disease-causing mutations and KO mice holds promise for providing more definitive answers. As the phenotypes resulting from CHCHD10 mutations, in particular, are diverse, ranging from myopathy to motor neuron disease to frontotemporal dementia, it is also possible that different pathogenic mechanisms are at play in different diseases.
The association of PD, motor neuron disease and other neuromuscular disorders with mutations in CHCHD2 and CHCHD10 highlight their clinical importance. Two very recent studies suggest that CHCHD2 and CHCHD10 form a complex, suggesting that the two proteins may be structurally and functionally related (17,18). Our results with novel CHCHD2, CHCHD10 and CHCHD2 and CHCHD10 null cells highlight the functional redundancy between these proteins and their similar distribution along mitochondrial cristae. Surprisingly, we uncover important asymmetries in their affinities for each other and in their regulation by the bioenergetic status of mitochondria. We demonstrate how these asymmetries drive CHCHD2/CHCHD10 heterodimerization in response to mitochondrial distress and establish that CHCHD2 and CHCHD10 with disease-causing mutations readily incorporate with WT protein into heterodimers. Collectively, these findings suggest that CHCHD2/CHCHD10 are parts of a bioenergetically responsive pathway and reveal unexpected links between the pathogenesis of PD and motor neuron disease.
Materials and Methods
Antibodies
The following commercially available primary antibodies were used: ATP5A (Novus Biologicals, Cat#NB600-1171), CHCHD2 (Proteintech, Cat#63302-1), CHCHD2 N-term (Sigma, Cat#HPA052510), CHCHD2 C-term (Sigma, Cat#HPA027407), CHCHD10 (Sigma, Cat#HPA003440), COX2 (abcam, Cat#ab110258), Cytochrome c (BD Biosciences, Cat#556432), anti-FLAG M2 (Sigma cat# F1804-1MG), GAPDH (Proteintech, Cat#60004-1-lg), HSP60 (Enzo Life Science, Cat#ADI-SPA-806-F), HSP90 (Santa Cruz, Cat#SC-7947), MIC19/CHCHD3 (Sigma, Cat#HPA042935), MIC25/CHCHD6 (Proteintech, Cat#20693-1-AP), MIC27/APOOL (Sigma, Cat#HPA000612), MIC60/Mitofilin (Abcam, Cat#ab48139), Mfn-1 (Abcam, Cat#ab57602), MTCO1 (life technology, Cat#459600), NDUFS1 (Abcam, Cat#AB169540), PINK1 (Cell Signaling Technology, Cat#6946), SDHA (Abcam, Cat#ab14715), TIMM50 (abcam, Cat#ab109436), Tom20 antibody (Santa Cruz Biotechnologies, FL-175), Tubulin (Sigma, Cat#T8328) and UQCRC2 (Abcam, Cat#ab14745). CHCHD2 (Proteintech, Cat#63302-1) was used for all immunoblotting and CHCHD2 C-term (Sigma, Cat#HPA027407) for all immuno-EM and immunocytochemistry for STED and confocal imaging, except where stated otherwise.
Construction of Plasmids
CHCHD2 from PCR amplified from complementary DNA (cDNA) (Transgenomic) and cloned into the YFP-N1 vector (Clontech), replacing the YFP sequence. The C-terminal Flag tag was introduced by PCR. CHCHD10-Flag and CHCHD2 (Δ59-83)-Flag were synthesized as a codon-optimized gBlock by Integrated DNA Technologies (IDT) and cloned into the YFP-N1 vector (Clontech), replacing the YFP sequence. CHCHD2 and CHCHD10 coding regions with a C-terminal Flag-GFP11 tag were also cloned into a targeting vector for the CLYBL locus (pZT-C12-R1), a gift from Jizhong Zou (Addgene, plasmid#66578), which had been modified to express mApple from a second promoter to aid selection, a gift from Michael Ward National Institutes of Health (NIH). Point mutations were introduced into the CHCHD2-Flag and CHCHD10-Flag plasmids using the QuikChange II Site-Directed Mutagenesis protocol (Stratagene).
Cell culture, transfection
HeLa and HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium with high glucose, L-glutamine and pyruvate (Gibco), with 10% fetal bovine serum, 1X pen/strep (Gibco) and uridine 50 μg/ml (Sigma) added. Cells were transfected with plasmids using FugeneHD (Promega), XtremeGENE9 (Roche) or Polyethylenimine, Linear (MW 25 000) (Polysciences, Inc.). Primary human fibroblasts were obtained from ATCC. Primary neonatal mouse fibroblasts were generated from mice less than 3 days after birth. Mouse and human fibroblasts were cultured in an incubator set to 5% O2, 5% CO2.
CRISPR/Cas9-mediated gene editing and generation of stable cell lines
PINK1-/- HeLa cells were described previously and were provided as a gift from Richard Youle (45). Mfn1/2 DKO MEFs were described previously and were provided as a gift from David Chan (32). MIC60/Mitofilin Dox-inducible shRNA cell lines were described previously (23). HEK293 and HeLa cells lacking CHCHD2 and/or CHCHD10 were generated using CRISPR/Cas9 gene editing. Briefly, complementary oligos encoding guide sequences were synthesized by IDT, annealed and cloned into the pX458 vector (Addgene, plasmid#48138), which was a gift from Feng Zhang. Plasmids were introduced into cells by transient transfection. Single GFP positive cells were sorted into single wells of 96 well plates. Resulting colonies were tested for CHCHD2 and CHCH10 protein expression by immunocytochemistry followed by immunoblotting to confirm KO. CHCHD2/CHCHD10 DKO cells were generated in a single round of targeting with guides against CHCHD2 and CHCHD10 co-transfected. CHCHD2-Flag and CHCHD10-Flag were stably integrated into the CLBYL ‘safe harbor’ locus of HEK293 or HeLa cells. Cells were co-transfected with targeting vectors containing CHCHD2 or CHCHD10 together with pZT-C13-R1 and pZT-C13-L1, expressing TALENs directed against the CLYBL locus (Addgene, Plasmid#62897 and Plasmid#62196), which were gifts from Jizhong Zou. mApple positive cells were selected by multiple (typically 3–4) rounds of fluorescence-activated cell sorting (FACS).
Immunoblot analysis, subcellular fractionation, crosslinking experiments and immunoprecipitation experiments
HEK293, HeLa and fibroblast cells were lysed in a buffer containing 1% sodium dodecyl sulfate (SDS) and 63 mm Tris (pH 6.95). DNA was sheared using a bath sonicator. Insoluble material was separated by centrifugation at 21 130 g at 4°C for 3 min with the soluble fraction retained. Protein concentration was measured using the BCA assay. Bromophenol blue (final concentration 0.0008%), glycerol (11%) and β-mercaptoethanol (0.284 mm) were added to the supernatant. Sample was heated at 80°C for 10 min. Insoluble material was separated by centrifugation at 21 130 g at room temperature for 3 min with the soluble fraction retained. Soluble fraction was subsequently analyzed by SDS-PAGE and immunoblotting. Densitometry measurements were made using ImageJ (NIH).
For the subcellular fractionation experiment, cells were washed with phosphate buffered saline (PBS) and then scrapped into subcellular fractional buffer (SF buffer) [250 mm sucrose, 20 mm HEPES (pH 7.4), 10 mm KCl, 1.5 mm MgCl2,1 mm EDTA and 1 mm EGTA, 1mm DTT, cOmplete protease Mini inhibitor (Roche)] and incubated at 4°C for 30 min with end-over-end rotation. Lysates were separated by centrifugation at 720 g at 4°C for 5 min. The supernatant was transferred to a new tube and separated by centrifugation at 10 000 g at 4°C for 10 min. The pellet (designated the HMF) was washed once in SF buffer, re-pelleted and then lysed in RIPA buffer. The supernatant was cleared by ultracentrifugation at 100 000 g at 4°C for 1 h. The supernatant was saved as the cytosolic fraction.
For cross-linking experiments, HEK293 or HeLa cells were trypsinized, washed three times with PBS. Cells in PBS (25 × 106 cells/ml) were incubated with 5 mm of DSG or DSS (or dose indicated) in PBS for 30 min at room temperature with continuous end-over-end rotation. Tris (pH 8.0) was added for a final concentration of 5 mm to quench remaining crosslinker and the cells were incubated for a further 15 min at room temperature. Cells were subsequently lysed and subjected to SDS-PAGE separation and immunoblotting as described above. Blots were imaged for chemiluminescent using the ChemiDoc Imaging system (Biorad) or the Odyssey Imaging System (LiCor).
For immunoprecipitation experiments, cells were incubated with isolation buffer [20 mm HEPES (pH 7.4), 10% glycerol, HEPES and miniComplete protease inhibitor cocktail (Roche)] containing 1% w/v digitonin (Sigma) for 10 min on ice with periodic mixing by pipetting. Insoluble material was separated by centrifugation at 16 000 g at 4°C. The soluble fraction was retained and diluted to 0.2% digitonin with the same buffer concentration. A total of 10% was retained as input and the remainder was incubated with 40 μL anti-Flag M2 magnetic beads (Sigma) with end-over-end mixing at 4°C for 2–3 h. Beads were washed three times with isolation buffer containing 0.05% digitonin. The bound fraction was eluted by heating beads in 2X SDS-sample buffer (Thermo Fisher) at 80°C for 3–5 min. The input (2%) and the bound fraction (50%) were analyzed by SDS-PAGE and immunoblotting.
For protein solubility assays, HEK293 cells were transfected using polyethylenimine (PEI) linear (Polysciences) and harvested 24 h later by scraping into PBS. An aliquot was used for protein concentration determination using the BCA assay. Cell numbers between conditions were normalized based on protein concentration. The cells were pelleted at 400 g and resuspended in Triton X-100 lysis buffer (20 mm Tris-HCl pH 7.5, 150 mm NaCl, 10 mm EDTA pH 8.0, 1% Na-Doc, 1% Triton X-100). After lysing in Triton X-100 lysis buffer, the sample was centrifuged at 21 130 g to obtain a soluble (supernatant) and insoluble fraction (pellet). Laemmli buffer was added to both fractions to obtain equal volume of 1x buffer with 2.5% (v/v) 2-mercaptoethanol. Lysates were boiled at 98°C, separated by SDS-PAGE and analyzed by immunoblotting.
Mitochondrial isolation and BN-PAGE
Mitochondrial isolation was performed as previously described (46). Mitochondria were isolated from HEK293 cells and resuspended in 10 mm of Hepes pH 7.6 and 0.5 M of sucrose. Protein concentration was measured using the BCA assay. Mitochondria were solubilized in 1% n-Dodecyl-β-D-Maltoside (DDM) and 1X NativePAGE sample buffer. The NativePAGE Novex Bis-Tris Gel System (Thermo Fisher Scientific) was used following the manufacturer’s instructions with the following modifications: 20 μg of mitochondria were loaded, only Light Blue Cathode Buffer was used and the electrophoresis was performed at 150 V for 1 h then 250 V for 2 h. For immunoblotting, polyvinylidene fluoride (PVDF) was used as the membrane, and transfer was performed at 130 V for 1 h at 4°C. The membrane was then washed with 10% acetic acid for 20 min and air-dried. After air-drying, the membrane was washed five times with methanol to remove residual Coomassie Blue dye, blocked with 5% milk in TBST for 1 h at room temperature and probed for proteins of interests.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
HEK293 cells were cultured in DMEM media for SILAC (stable isotope labeling with amino acids in cell culture) (Cambridge Isotope Laboratories) supplemented with 10% (v/v) dialyzed FBS (Sigma), penicillin/streptomycin, 50 μg/ml uridine and either ‘light’ amino acids (73 mg/L L-lysine · HCl and 42 mg/L L-arginine · HCl), ‘medium’ amino acids (42.9 mg/L 2H4-L-lysine · HCl and 52.2 mg/L 13C6-L-arginine · HCl) or ‘heavy’ amino acids (52.2 mg/L 13C615N2-L-lysine · 2 HCl and 53.2 mg/L 13C615N4-L-arginine · HCl) at 37°C under 5% CO2. After full amino acid incorporation, equal numbers of labeled WT, CHCHD2 KO and CHCHD10 KO HEK293 cell lines were mixed for the following comparisons: WT and CHCHD2 KO; WT and CHCHD10 KO; and WT, CHCHD2 KO; and CHCHD10 KO. Mitochondria were then isolated as described above, and protein concentration was measured using the BCA assay. One hundred μg of each mitochondrial mixture was mixed with 1x Laemmli sample buffer and β-mercaptoethanol (0.355 mm) and then boiled at 80°C for 10 min. Samples were then separated by SDS-PAGE. Five to ten SDS-PAGE bands were excised for each sample and digested with trypsin overnight. Extracted peptides were desalted and used for LC-MS/MS data acquisition on an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) coupled with a 3000 Ultimate high pressure liquid chromatography instrument (Thermo Fisher Scientific). Peptides were separated on an ES802 column (Thermo Fisher Scientific) with mobile phase B increased from 2 to 27% over 50 min (for 12–2017 samples) or 60 min (for 08–2107 samples). The LC-MS/MS data were acquired in data-dependent mode. The resolution of the survey scan (300–1600 m/z) was set at 60k at m/z 400 with a target value of 10 × 106 ions. Collision-induced dissociation was performed on the top ten most abundant precursor ions with an isolation window of 2.0 Da. Database search and H/L ratio calculation were performed using Mascot Distiller (2.5.1) (Matrix Science) against Sprot Human database. The error tolerances for precursor and product ions were ±5 ppm and ±0.3 Da, respectively. Oxidation (M), Label:2H(4) (K), Label:13C(6) (R), Label:13C(6)15N(2) (K) and Label:13C(6)15N(4) (R) were included as variable modifications in the database search. SILAC ratios were log2-transformed and median normalized for proteins quantified in all three biological replicates followed by filtering for proteins present in MitoCarta2. Statistical testing (multiple one-sample t-testing) was performed in the Perseus software package (version 1.6.1.1) using standard settings and correcting P-value for multiple comparisons.
EM
HeLa cells were fixed with 2% glutaraldehyde (Electron Microscopy Services) in EM buffer (0.1 N sodium cacodylate at pH 7.4 with 2 mm calcium chloride) for at least 24 h. Samples were washed with buffer and treated with 1% osmium tetroxide in 0.1 N cacodylate buffer at pH 7.4 for 1 h on ice, washed and en bloc stained with 0.25–1% uranyl acetate in 0.1 N acetate buffer at pH 5.0 overnight at 4°C, dehydrated with a series of graded ethanol and finally embedded in epoxy resins. Ultrathin sections (70 nm) were stained with lead citrate and imaged with a JEOL 1200 EXII Transmission Electron Microscope. Images were blinded for a separate rater, who scored mitochondria as possessing swirls (defined as changing direction more than 180 degrees in its course), swollen, few/none (defined as less than or equal to 2), segmented (in which cristae appear to segment the mitochondria at areas of constriction) or, if not possessing any of these features, as normal.
For immuno-EM studies, cells were fixed with 4% paraformaldehyde for 30 min in PBS. Samples were then blocked and made permeable 0.1% saponin in with 5% bovine serum albumin (BSA) for 40–60 min. Samples were incubated with primary and secondary antibodies (Nanogold, Nanoprobes, Yaphand, NY) for 1–2 h, fixed with 2% glutaraldehyde in PBS for 30 min overnight, silver enhanced (HQ kit, Nanoprobes), en block mordanted with 0.25–0.5% uranyl acetate in acetate buffer at pH 5.0 for 1 h, treated with 0.2% osmium tetroxide in 0.1 M phosphate buffer at pH 7.4 for 30 min, dehydrated with a series of graded ethanol and embedded in epoxy resin. The primary antibodies used were CHCHD2 C-term, CHCHD10 and MIC19/CHCHD3. The distance from individual nanogold particles associated with the inner membrane to the nearest CJ were measured using ImageJ (NIH). Measurements were only taken for nanogold particles in which a continuous inner membrane was apparent between the nanogold particle and the nearest CJ.
Light Microscopy
For confocal imaging of fixed cells, HeLa cells grown on chambered coverglass (LabTek II or Ibidi) were fixed for 10 min with 4% paraformaldehyde in PBS, permeabilized with 0.25% Triton X-100, blocked with 1% BSA for 30 min and then incubated with primary antibody in 1% BSA for 1–2 h at room temperature. Cells were washed with PBS three times and then incubated with Alexa Fluro-labeled goat secondary antibodies (488, 555 or 647) at 1:1 000 dilution in 1% BSA for 45 min. Confocal fluorescence microscopy was performed on a LSM 880 Spectral (Zeiss) or a Fluoview3000 (Olympus). For two-color STED microscopy experiments, a sequential labeling protocol was used as the highest quality antibodies for CHCHD2, CHCHD10 and MIC19/CHCHD3 had all been raised in rabbit. Samples were fixed as above and then blocked in 5% BSA prior to incubation with the first primary antibody (anti-CHCHD10) in 5% BSA, washed in PBS and the incubated with the first secondary antibody (anti-rabbit Atto 488) in 5% BSA. Samples were subsequently washed and then incubated with fab anti-rabbit IgG (1:30, Jackson cat#111-007-003) in 5% BSA followed by a brief fixation with 4% paraformaldehyde (PFA) in PBS. This step blocks remaining free Fc epitopes of the first primary antibody. Samples were then incubated with the second primary (anti-CHCHD2 or anti-Mic19/CHCHD3), washed in PBS and incubated with the second secondary (anti-rabbit Alexa 555), prior to the final washes in PBS. To ensure specificity of antibody signal was maintained throughout the sequential labeling procedure CHCHD2 and CHCHD10 KO HeLa cells were processed in parallel with the WT HeLa cells. After immunostaining, samples were embedded in ProLong™ Diamond Antifade Mountant (Invitrogen cat# P36965) and three-image z-stacks were obtained using a TCS SP8 STED (Leica). STED images were then deconvolved using Huygens Professional software. Maximum projection images of the resulting deconvolved z-stacks were generated using ImageJ (NIH).
For roGFP experiments, HEK293 cells were transfected with a plasmid expressing cytosolic roGFP or matrix roGFP, both of which were gifts from Paul Schumacker (Addgene plasmid #49435 and #49437). Confocal z-stacks were obtained live at baseline, following 100 μm 2-aldrithiol (Sigma), and following 2 mm DTT (Sigma) to fully oxidize and fully reduce roGFP, respectively. Three z-stacks at ∼1 min intervals were obtained for each of the three conditions. Imaging was performed on a LSM 880 Spectral confocal microscope (Zeiss) at 37°C. roGFP was excited sequentially by a 405 nm laser and a 488 nm laser to preferentially excite oxidized and reduced roGFP, respectively. 405/488 fluorescence ratio (roGFP405/488) was measured in individual cells for each time point and the three time points averaged for each condition using ImageJ (NIH). Cells that did not exhibit a higher roGFP405/488 with 2-aldrithiol compared to DTT were excluded from analysis. Relative oxidation was calculated as (roGFP405/488baseline - roGFP405/488DTT )/ (roGFP405/488aldrithiol – roGFP405/488DTT).
For measurement of CHCHD2 intensity following treatment with DMSO (vehicle) or CCCP, fields from DMSO and CCCP treated samples immunostained with CHCHD2 and cytochrome c were obtained using constant microscope settings on a Fluoview3000 confocal microscope (Olympus) with 60X oil immersion lens. In ImageJ (NIH), DAPI and cytochrome c channels were made binary using default settings. Regions of interest corresponding to DAPI (representing the nucleus) and cytochrome c (representing mitochondria) were created using the ‘create selection’ feature and transferred to the CHCHD2 channel for measurement of CHCHD2 intensity in the nuclear ROIs and mitochondrial ROIs, respectively. Measurements were made in 11 fields for each sample from three biological replicates.
The histological analysis of all brain specimens was authorized by the ethics committees on human studies of the cantons of Basel. Explicit consent was obtained for all cases. The guidelines of the ethics committees of Basel (www.eknz.ch) were strictly followed for the analysis of all human samples. Specimens were collected and archived as formalin-fixed paraffin blocks by the Institute for Medical Genetics and Pathology, University Hospital Basel. Four μm paraffin sections were mounted on Superfrost slides (Menzel Glas, Germany) and air-dried. Sections were either deparaffinized and stained with eosin & haematoxylin or immunostained on a Ventana Benchmark Ultra® autostainer using a 3,3'-diaminobenzidine (DAB)-based detection system with enzyme-linked anti-mouse/anti-rabbit protocols provided by Ventana. The following antibodies were used (manufacturer, catalog number, dilution, pretreatment protocol and chromogen in brackets): anti-CHCHD2 (Sigma, Cat#HPA027407, 1:800, CC1 16, UltraView DAB), anti-CHCHD10 (Sigma-Aldrich, HPA003440, 1:100, CC1 16, UltraView DAB) and anti-alpha-Synuclein (Zymed/Thermo Fisher, 180215 clone LB509, 1:500, CC1 20+CC1 36, UltraView Red). Immunostains were performed either separately or sequentially (DAB/Red) as indicated in the respective figure legends. Counterstaining was performed on the Benchmark Ultra® instrument with hematoxylin.
Analysis of mitochondrial function
Oxygen consumption rates (OCR) were measured using the Seahorse Extracellular Flux Analyzer XFp (Agilent). 15 000 HEK293 cells were plated into each poly-L-ornithine (Sigma) coated well the day before the assay. The standard ‘mitochondria stress test’ was performed consisting of basal measurements followed by measurements after sequential addition of 1 μm oligomycin, 2 μm FCCP and 0.5 μm of rotenone and antimycin. Data are presented as means ± standard error of the mean for three replicates. Protein concentrations determined by BCA assay were used to normalize the OCRs. HEK293 cells were tested either in glucose (4.5 g/L) or galactose (25 mm) containing DMEM lacking bicarbonate. Samples tested in galactose were switched from glucose to galactose DMEM 48 h before testing. Spectrophotometric analysis of complex II, III and IV activities were performed in duplicate on HEK293 cell pellets as described previously using a Cary 50 UV-visible spectrophotometer (Varian Inc, Les Ulis, France) (47).
Statistical analyses
Quantitative data are presented as means ± SD if not indicated otherwise. The statistical significance was assessed using one-way or two-way ANOVA with post-hoc testing that was corrected for multiple comparisons. Prism 7.0 (Graphpad) and Excel 16.9 (Microsoft) were used for data analysis.
Supplementary Material
Acknowledgements
We thank Maric Dragan PhD and the National Institute of Neurological Disorders and Stroke (NINDS) Flow Cytometry Core Facility for technical assistance with FACS experiments. We thank Virginia Crocker and the NINDS EM Facility for technical assistance with transmission electron microscopy (TEM). We thank Carolyn Smith PhD and the NINDS Light Microscopy Facility for technical assistance with confocal and stimulated depletion emission microscopy. We thank Yan Li at the NINDS Protein/peptide Sequencing Facility for the SILAC-based proteomics analysis. We thank Nunziata Maio PhD for help with BN-PAGE. We thank Craig Blackstone MD PhD and Richard Youle PhD for critical reading of the manuscript and insightful comments.
Conflict of Interest statement. None declared.
Funding
Intramural Research Program of the NINDS, National Institutes of Health. National Institute of Neurological Disorders and Stroke, (Grant /Award Number: ``Intramural Program'').
References
- 1. Elbaz A., Grigoletto F., Baldereschi M., Breteler M.M., Manubens-Bertran J.M., Lopez-Pousa S., Dartigues J.F., Alpérovitch A., Tzourio C. and Rocca W.A. (1999) Familial aggregation of Parkinson’s disease: a population-based case-control study in Europe EUROPARKINSON Study Group. Neurology, 52, 1876–1882. [DOI] [PubMed] [Google Scholar]
- 2. Funayama M., Ohe K., Amo T., Furuya N., Yamaguchi J., Saiki S., Li Y., Ogaki K., Ando M., Yoshino H. et al. (2015) CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: a genome-wide linkage and sequencing study. Lancet Neurol., 14, 274–282. [DOI] [PubMed] [Google Scholar]
- 3. Koschmidder E., Weissbach A., Brüggemann N., Kasten M., Klein C. and Lohmann K. (2016) A nonsense mutation in CHCHD2 in a patient with Parkinson disease. Neurology, 86, 577–579. [DOI] [PubMed] [Google Scholar]
- 4. Meng H., Yamashita C., Shiba-Fukushima K., Inoshita T., Funayama M., Sato S., Hatta T., Natsume T., Umitsu M., Takagi J. et al. (2017) Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun., 8, 15500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wei Y., Vellanki R.N., Coyaud É., Ignatchenko V., Li L., Krieger J.R., Taylor P., Tong J., Pham N.-A., Liu G. et al. (2015) CHCHD2 is coamplified with EGFR in NSCLC and regulates mitochondrial function and cell migration. Mol. Cancer Res. MCR, 13, 1119–1129. [DOI] [PubMed] [Google Scholar]
- 6. Liu Y., Clegg H.V., Leslie P.L., Di J., Tollini L.A., He Y., Kim T.-H., Jin A., Graves L.M., Zheng J. et al. (2015) CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation. Cell Death Differ., 22, 1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baughman J.M., Nilsson R., Gohil V.M., Arlow D.H., Gauhar Z. and Mootha V.K. (2009) A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet., 5, e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aras S., Bai M., Lee I., Springett R., Hüttemann M. and Grossman L.I. (2015) MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion, 20, 43–51. [DOI] [PubMed] [Google Scholar]
- 9. Cavallaro G. (2010) Genome-wide analysis of eukaryotic twin CX 9 C proteins. Mol. Biosyst., 6, 2459–2470. [DOI] [PubMed] [Google Scholar]
- 10. Bannwarth S., Ait-El-Mkadem S., Chaussenot A., Genin E.C., Lacas-Gervais S., Fragaki K., Berg-Alonso L., Kageyama Y., Serre V., Moore D.G. et al. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain J. Neurol., 137, 2329–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Müller K., Andersen P.M., Hübers A., Marroquin N., Volk A.E., Danzer K.M., Meitinger T., Ludolph A.C., Strom T.M. and Weishaupt J.H. (2014) Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain J. Neurol., 137, e309. [DOI] [PubMed] [Google Scholar]
- 12. Ajroud-Driss S., Fecto F., Ajroud K., Lalani I., Calvo S.E., Mootha V.K., Deng H.-X., Siddique N., Tahmoush A.J., Heiman-Patterson T.D. et al. (2015) Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics, 16, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson J.O., Glynn S.M., Gibbs J.R., Nalls M.A., Sabatelli M., Restagno G., Drory V.E., Chiò A., Rogaeva E. and Traynor B.J. (2014) Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain J. Neurol., 137, e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Penttilä S., Jokela M., Bouquin H., Saukkonen A.M., Toivanen J. and Udd B. (2015) Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol., 77, 163–172. [DOI] [PubMed] [Google Scholar]
- 15. Auranen M., Ylikallio E., Shcherbii M., Paetau A., Kiuru-Enari S., Toppila J.P. and Tyynismaa H. (2015) CHCHD10 variant p.(Gly66Val) causes axonal Charcot-Marie-Tooth disease. Neurol. Genet., 1, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Genin E.C., Plutino M., Bannwarth S., Villa E., Cisneros-Barroso E., Roy M., Ortega-Vila B., Fragaki K., Lespinasse F., Pinero-Martos E. et al. (2016) CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med., 8, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burstein S.R., Valsecchi F., Kawamata H., Bourens M., Zeng R., Zuberi A., Milner T.A., Cloonan S.M., Lutz C., Barrientos A. et al. (2018) In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet., 27, 160–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Straub I.R., Janer A., Weraarpachai W., Zinman L., Robertson J., Rogaeva E. and Shoubridge E.A. (2018) Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet., 27, 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hanson G.T., Aggeler R., Oglesbee D., Cannon M., Capaldi R.A., Tsien R.Y. and Remington S.J. (2004) Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem., 279, 13044–13053. [DOI] [PubMed] [Google Scholar]
- 20. Malsburg K., Müller J.M., Bohnert M., Oeljeklaus S., Kwiatkowska P., Becker T., Loniewska-Lwowska A., Wiese S., Rao S., Milenkovic D. et al. (2011) Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell, 21, 694–707. [DOI] [PubMed] [Google Scholar]
- 21. Hoppins S., Collins S.R., Cassidy-Stone A., Hummel E., DeVay R.M., Lackner L.L., Westermann B., Schuldiner M., Weissman J.S. and Nunnari J. (2011) A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol, 195, 323–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harner M., Körner C., Walther D., Mokranjac D., Kaesmacher J., Welsch U., Griffith J., Mann M., Reggiori F. and Neupert W. (2011) The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J., 30, 4356–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ott C., Dorsch E., Fraunholz M., Straub S. and Kozjak-Pavlovic V. (2015) Detailed analysis of the human mitochondrial contact site complex indicate a hierarchy of subunits. PLOS ONE, 10, e0120213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kleiger G., Grothe R., Mallick P. and Eisenberg D. (2002) GXXXG and AXXXA: common alpha-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry (Mosc.), 41, 5990–5997. [DOI] [PubMed] [Google Scholar]
- 25. Hung L.W., Ciccotosto G.D., Giannakis E., Tew D.J., Perez K., Masters C.L., Cappai R., Wade J.D. and Barnham K.J. (2008) Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J. Neurosci., 28, 11950–11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quirós P.M., Prado M.A., Zamboni N., D’Amico D., Williams R.W., Finley D., Gygi S.P. and Auwerx J. (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol., 216, 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stroud D.A., Surgenor E.E., Formosa L.E., Reljic B., Frazier A.E., Dibley M.G., Osellame L.D., Stait T., Beilharz T.H., Thorburn D.R. et al. (2016) Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature, 538, 123–126. [DOI] [PubMed] [Google Scholar]
- 28. Narendra D., Tanaka A., Suen D.-F. and Youle R.J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol., 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Narendra D.P., Jin S.M., Tanaka A., Suen D.-F., Gautier C.A., Shen J., Cookson M.R. and Youle R.J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol., 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vives-Bauza C., Zhou C., Huang Y., Cui M., Vries R.L.A., Kim J., May J., Tocilescu M.A., Liu W., Ko H.S. et al. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA, 107, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geisler S., Holmström K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J. and Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol., 12, 119–131. [DOI] [PubMed] [Google Scholar]
- 32. Chen H., Chomyn A. and Chan D.C. (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem., 280, 26185–26192. [DOI] [PubMed] [Google Scholar]
- 33. Longen S., Bien M., Bihlmaier K., Kloeppel C., Kauff F., Hammermeister M., Westermann B., Herrmann J.M. and Riemer J. (2009) Systematic analysis of the twin Cx9C protein family. J. Mol. Biol., 393, 356–368. [DOI] [PubMed] [Google Scholar]
- 34. Stroud D.A., Maher M.J., Lindau C., Vögtle F.-N., Frazier A.E., Surgenor E., Mountford H., Singh A.P., Bonas M., Oeljeklaus S. et al. (2015) COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet., 24, 5404–5415. [DOI] [PubMed] [Google Scholar]
- 35. Purandare N., Somayajulu M., Hüttemann M., Grossman L.I. and Aras S. (2018) The cellular stress proteins CHCHD10 and MNRR1 (CHCHD2): partners in mitochondrial and nuclear function and dysfunction. J. Biol. Chem., 293, 6517–6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsuda W., Furuta T., Nakamura K.C., Hioki H., Fujiyama F., Arai R. and Kaneko T. (2009) Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci., 29, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Modjtahedi N., Tokatlidis K., Dessen P. and Kroemer G. (2016) Mitochondrial proteins containing coiled-coil-helix-coiled-coil-helix (CHCH) domains in health and disease. Trends Biochem. Sci., 41, 245–260. [DOI] [PubMed] [Google Scholar]
- 38. Quirós P.M., Langer T. and López-Otín C. (2015) New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol., 16, 345–359. [DOI] [PubMed] [Google Scholar]
- 39. Bragoszewski P., Gornicka A., Sztolsztener M.E. and Chacinska A. (2013) The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol. Cell. Biol., 33, 2136–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rainbolt T.K., Lebeau J., Puchades C. and Wiseman R.L. (2016) Reciprocal degradation of YME1L and OMA1 adapts mitochondrial proteolytic activity during stress. Cell Rep., 14, 2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baker M.J., Lampe P.A., Stojanovski D., Korwitz A., Anand R., Tatsuta T. and Langer T. (2014) Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J., 33, 578–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brockmann S.J., Freischmidt A., Oeckl P., Müller K., Ponna S.K., Helferich A.M., Paone C., Reinders J., Kojer K., Orth M. et al. (2018) CHCHD10 mutations p.R15L and p.G66V cause motoneuron disease by haploinsufficiency. Hum. Mol. Genet., 27, 706–715. [DOI] [PubMed] [Google Scholar]
- 43. Woo J.-A.A., Liu T., Trotter C., Fang C.C., De Narvaez E., LePochat P., Maslar D., Bukhari A., Zhao X., Deonarine A. et al. (2017) Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun., 8, 15558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tio M., Wen R., Lim Y.L., Zukifli Z.H.B., Xie S., Ho P., Zhou Z., Koh T.-W., Zhao Y. and Tan E.-K. (2017) Varied pathological and therapeutic response effects associated with CHCHD2 mutant and risk variants. Hum. Mutat., 38, 978–987. [DOI] [PubMed] [Google Scholar]
- 45. Lazarou M., Sliter D.A., Kane L.A., Sarraf S.A., Wang C., Burman J.L., Sideris D.P., Fogel A.I. and Youle R.J. (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature, 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lazarou M., Jin S.M., Kane L.A. and Youle R.J. (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell, 22, 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bénit P., Goncalves S., Philippe Dassa E., Brière J.-J., Martin G. and Rustin P. (2006) Three spectrophotometric assays for the measurement of the five respiratory chain complexes in minuscule biological samples. Clin. Chim. Acta, 374, 81–86. [DOI] [PubMed] [Google Scholar]
- 48. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.-F., Guindon S., Lefort V., Lescot M. et al. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res., 36, W465–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Calvo S.E., Clauser K.R. and Mootha V.K. (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res., 44, D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.