

Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by dopaminergic neuron loss. At present, there are no drugs that stop the progression of PD. As with other multifactorial genetic disorders, genome-wide association studies (GWASs) found multiple risk loci for PD, although their clinical significance remains uncertain. Here, we report the identification of candidate drugs for PD by a method using GWAS data and in silico databases. We identified 57 Food and Drug Administration-approved drug families as candidate neuroprotective drugs for PD. Among them, dabrafenib, which is known as a B-Raf kinase inhibitor and is approved for the treatment of malignant melanoma, showed remarkable cytoprotective effects in neurotoxin-treated SH-SY5Y cells and mice. Dabrafenib was found to inhibit apoptosis, and to enhance the phosphorylation of extracellular signal-regulated kinase (ERK), and inhibit the phosphorylation of c-Jun NH2-terminal kinase. Dabrafenib targets B-Raf, and we confirmed a protein–protein interaction between B-Raf and Rit2, which is coded by RIT2, a PD risk gene in Asians and Caucasians. In RIT2-knockout cells, the phosphorylation of ERK was reduced, and dabrafenib treatment improved the ERK phosphorylation. These data indicated that dabrafenib exerts protective effects against neurotoxicity associated with PD. By using animal model, we confirmed the effectiveness of this in silico screening method. Furthermore, our results suggest that this in silico drug screening system is useful in not only neurodegenerative diseases but also other common diseases such as diabetes mellitus and hypertension.
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder, and is characterized by the loss of dopaminergic neurons in the substantia nigra and the formation of Lewy bodies that are primarily composed of aggregated α-synuclein in the neurons (1). Despite extensive investigation, there are currently no disease-modifying drugs available that can halt the progression of PD. The discovery of new drugs is an expensive and time-consuming process. It takes ∼15 years and >$1 billion to develop and bring a new drug to market (2). Furthermore, <5% of the new molecules that enter Phase 1 clinical trials are approved by the US Food and Drug Administration (FDA) (3). Under such circumstances, drug repurposing, which is the identification of new indications for existing drugs, is thought to be a promising strategy for intractable diseases such as PD.
Genome-wide association study (GWAS) results have now been reported for many common adult diseases (metabolic, auto-immune and psychiatric and so on). The common form of PD is also a multifactorial disorder, and previous GWASs have identified several genetic loci as genetic risks for sporadic PD (4,5). In 2014, 24 risk loci for sporadic PD were reported from a meta-analysis of Caucasian GWASs (6). Although GWAS data have provided valuable biological insight into the molecular mechanisms of PD, translation of the genetic findings from GWAS into the clinic has remained limited. Recently, a new method of drug discovery utilizing risk genes from GWAS and computational databases were developed for rheumatoid arthritis (7). This in silico screening method was subsequently used to search for drugs for colorectal cancer and type 2 diabetes, and some drugs that have been approved for other diseases were identified as candidate drugs (8,9), although their biological effects in vitro or in vivo were uncertain.
In the present study, we applied this method to search for disease-modifying drugs for sporadic PD, and identified some candidate drugs. Then, we analyzed their neuroprotective effects in in vitro and in vivo PD models, and demonstrated that dabrafenib is a promising neuroprotective drug for PD.
Results
In silico identification of potential disease-modifying drugs
We applied the in silico screening method (7) to identify disease-modifying drugs for PD. We first defined 32 PD risk-genes within PD-risk loci that were detected in the previous meta-GWAS (6). Using protein–protein interaction (PPI) databases, InWeb (10) and PINA (11), we obtained 834 protein products showing direct PPI with protein products of the PD-risk genes. We considered that a total of 866 protein products from the 32 PD-risk genes and 834 genes in direct PPI have the possibility of involvement in PD pathogenesis. We further identified 871 drug target genes from the drug databases DrugBank (12) and Therapeutic Target Database (13). Among the 866 PD-risk/direct PPI genes, we found that 48 genes were targeted by 57 FDA-approved drug families for other diseases, and considered these to be candidate disease-modifying drugs for PD (Supplementary Material, Fig. S1). Neuroprotective effects in in vitro or in vivo PD model have already been reported in 17 of the 57 FDA-approved drug families (∼30%) (14–30) (Fig. 1). Therefore, our results suggest that this combinational analysis of GWAS-data and in silico database can efficiently identify drugs with neuroprotective effects.
Figure 1.
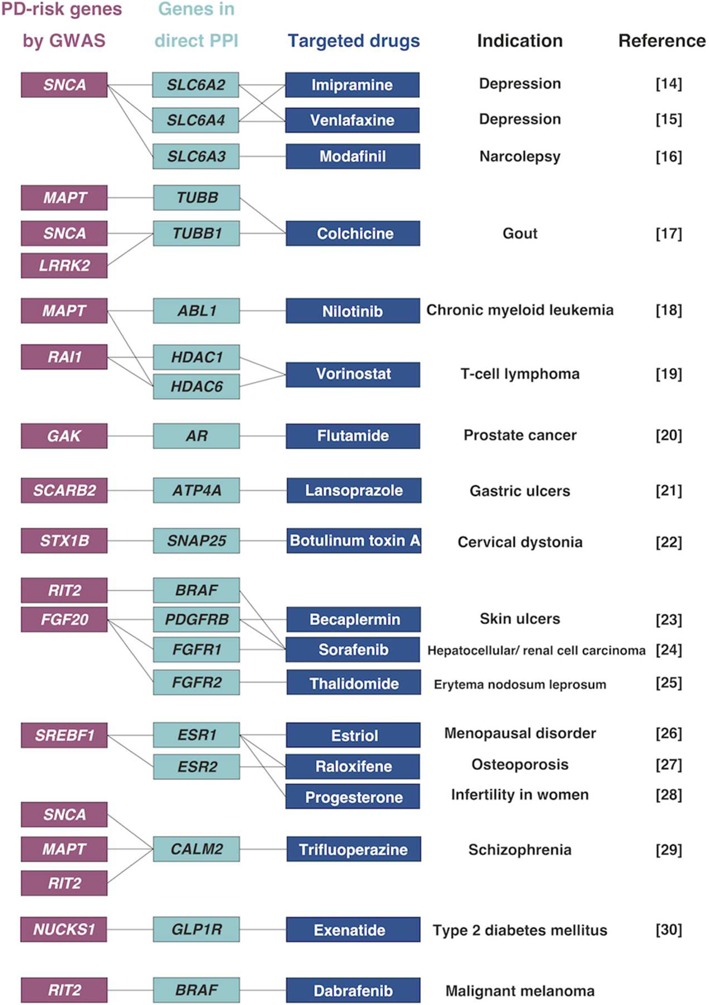
Examples of candidate drugs for PD identified by in silico drug screening
‘PD-risk genes’ were listed from the data of meta-GWAS for PD, and ‘genes in direct PPI’ were extracted using PPI databases. Using drug databases, we identified FDA-approved drugs that targeted ‘PD-risk genes’ or ‘genes in direct PPI’. These drugs are all approved for other diseases, and have already been reported as being neuroprotective for PD. We found dabrafenib as a new candidate drug for PD.
Neuroprotective effects of dabrafenib in vitro and in vivo
We then searched the previous literature for information on these candidates, focusing on the following five drugs that were expected to have neuroprotective effects from the data of other reports: dasatinib, duloxetine, furosemide, regorafenib and dabrafenib. Dasatinib was reported to show neuroprotective effects in in vitro and in vivo amyotrophic lateral sclerosis model (31). Duloxetine inhibited oxidative stress and apoptosis in rat pheochromocytoma-derived PC12 cells (32). Furosemide was reported to rescue the inhibitory effects of midazolam (benzodiazepine) on neural stem cell proliferation (33). Regorafenib was synthesized as a more potent multi-kinase inhibitor than sorafenib, which has already been reported to be neuroprotective in PD (24,34). Dabrafenib targets B-Raf, which was reported to be associated with neuronal survival (35). To examine whether these candidate drugs actually exert neuroprotective effects, we treated human neuroblastoma SH-SY5Y cells with each candidate drug together with 1-methyl-4-phenylpyridinium ion (MPP+), and performed the lactate dehydrogenase (LDH) assay and cell viability assay. MPP+ is a toxic molecule that is known to increase LDH and decrease cell viability as a result of mitochondrial dysfunction and apoptosis, and is hence thought to mimic the pathogenesis of PD (36).
Among these five drugs, we found that dabrafenib, a drug approved for melanoma, most robustly decreased LDH release (P-value < 0.01) and increased cell viability (P-value < 0.01) in cells exposed to MPP+ (Fig. 2A and Supplementary Material, Fig. S2).
Figure 2.
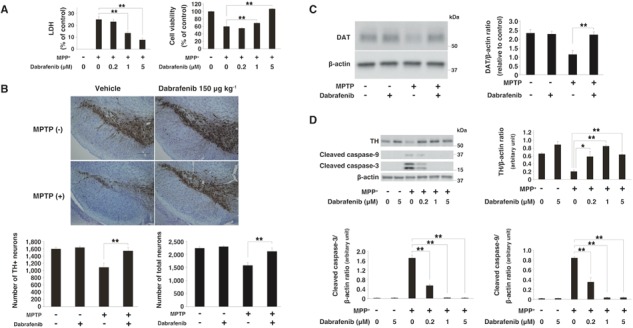
Neuroprotective effects of dabrafenib both in vitro and in vivo
(A) LDH/cell viability assay under the condition of MPP+. Dabrafenib inhibited LDH release and the loss of cell viability at 24 h after MPP+ (3 mM) treatment in a concentration-dependent manner. Data represent the mean ± s.e.m. **, P-value < 0.01 compared with MPP+ alone (one-way ANOVA with the Tukey post-hoc test; n = 4 per group). (B) Immunohistochemistry staining of TH. Dabrafenib (150 μg kg−1) prevented the MPTP-induced loss of TH-positive and total neurons in the substantia nigra at 3 weeks after MPTP injection. Scale bar represents 200 μm. Data represent the mean ± s.e.m. **, P-value < 0.01 compared with vehicle + MPTP group (one-way ANOVA with the Tukey post-hoc test; n = 5–9 per group). (C) Immunoblotting showed that dabrafenib rescued the loss of DAT in the substantia nigra of mice 3 weeks after MPTP injection. Data represent the mean ± s.e.m. **, P-value < 0.01 compared with vehicle + MPTP group (one-way ANOVA with the Tukey post-hoc test; n = 3–4 per group). (D) Western blot analysis showed that dabrafenib reduced the levels of cleaved caspase-3 and 9 at 48 h after MPP+ (3 mM) treatment. Data are represented as the mean ± s.e.m. **, P-value < 0.01 compared with MPP+ alone (one-way ANOVA with the Tukey post-hoc test; n = 3 per group).
To assess the effects of these drugs in vivo, we analyzed their neuroprotective effects in a subacute 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) mice model. MPTP is a neurotoxin, which is known to be converted to MPP+ and reproduce most of the biochemical and pathological hallmarks of PD, including the extensive degeneration of dopaminergic neurons (36).
Intracerebroventricular (icv) injection of dabrafenib 3 days before MPTP administration significantly prevented the loss of tyrosine hydroxylase (TH)-positive and total neurons (P-value < 0.01) at 3 weeks after MPTP injection (Fig. 2B). We also investigated the expression of dopamine transporter (DAT), which mediates the active reuptake of dopamine from the synapse and is a principal regulator of dopaminergic neurotransmission. At 3 weeks after the administration of MPTP, the DAT expression was also rescued by dabrafenib (Fig. 2C). Icv injection of dabrafenib 3 days before MPTP administration also significantly inhibited TH-positive neuronal death in a dose-dependent manner, assessed 1 week after MPTP injection (Supplementary Material, Fig. S3A and B). These data further confirmed the neuroprotective effects of dabrafenib in an in vivo PD model.
Next, we analyzed the anti-apoptotic effects of dabrafenib, because vemurafenib, a selective inhibitor of B-Raf containing the melanoma-causing V600E pathogenic mutation (BRAFV600E), was reported to suppress apoptosis in non-BRAFV600E-mutated cells (37). Dabrafenib significantly reduced the expression of cleaved caspase-3 and caspase-9 under MPP+-treated conditions (Fig. 2D). Furthermore, dabrafenib rescued the expression of TH in the presence of MPP+ (Fig. 2D). Dabrafenib also tended to reduce the expression of cleaved caspase-3, induced by MPTP in vivo, although the effect was not significant (Supplementary Material, Fig. S3C). These results indicated the anti-apoptotic effects of dabrafenib. In this subacute MPTP mice model, dabrafenib did not affect the expression of other proteins such as α-synuclein, p62, glial fibrillary acidic protein (GFAP) and ionized calcium-binding adapter molecule 1 (IBA-1), which are known to be associated with the pathophysiology of PD (Supplementary Material, Fig. S3C).
Expression of BRAF and RIT2 and interaction of their encoded proteins
Dabrafenib targets B-Raf, which directly interacts with the protein product of the Ras-like without CAAX 2 (RIT2) gene. A previous report (38) and our co-immunoprecipitation experiment confirmed the interaction between Rit2 and B-Raf (Supplementary Material, Fig. S4A). We confirmed the expression of RIT2 and BRAF transcripts in the mouse substantia nigra pars compacta (SNpc) by reverse transcription–polymerase chain reaction (RT-PCR) (Fig. 3A). The expression and phosphorylation of B-Raf were also observed in other brain regions, such as the cortex, hippocampus, striatum and cerebellum (Supplementary Material, Fig. S4B). The phosphorylation of B-Raf was not affected by MPTP or dabrafenib at 3 weeks after treatment (Supplementary Material, Fig. S4C). It has been reported that RIT2 mRNA was specifically expressed in TH-positive neurons (39) and that BRAF mRNA was expressed in several brain regions, including SNpc, as shown by in situ hybridization (40), which is consistent with our results.
Figure 3.
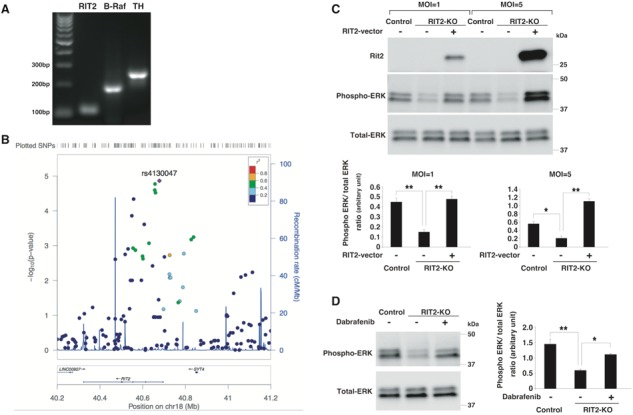
Significant association between RIT2 and PD, and the effect of dabrafenib on RIT2-knockout (KO) cells
(A) RT-PCR data for the RIT2, BRAF and TH expression in the mouse substantia nigra. (B) Regional association plot of the RIT2 locus. (C) Immunoblot of phosphorylated ERK and Rit2. Phospho-ERK was reduced in RIT2-KO (clone 42) compared to control cells (clone 7), and overexpression of RIT2 in RIT2-KO cells rescued the phosphorylation of ERK. (Upper) representative immunoblots and (bottom) summarized data of phospho-ERK/total ERK levels in each cell line. (D) Dabrafenib upregulated the expression of phospho-ERK in RIT2-KO cells (clone 42). (Left) representative immunoblots and (right) summarized data of phospho-ERK/total ERK levels in each cell line. Data in (C) and (D) represent the mean ± s.e.m. (*, P-value < 0.05 and **, P-value < 0.01; one-way ANOVA with the Tukey post-hoc test; n = 3 per group).
PD-susceptibility of East Asians and Caucasians with RIT2
A significant association between PD and RIT2 was reported in two meta-GWASs of Caucasian samples (6,41), but it remains unclear as to whether there is an association between PD and RIT2 in other racial populations (42,43). To assess the association between RIT2 and PD in the Asian population, we investigated the association between PD and single nucleotide polymorphisms (SNPs) within this locus identified from our GWAS data, which we previously conducted using genotype data from 435 470 SNPs in 988 PD cases and 2521 controls of Japanese origin (4). We found a positive association between RIT2 and PD (rs4130047, P trend = 1.37 × 10−5 by the Cochran–Armitage trend test and 6.8 × 10−5 by a 2 × 3 chi-squared test) in the East Asian population (Fig. 3B and Table 1). Furthermore, we identified rs4130047 as the most significant SNP in this locus. This SNP is located within intron 1 of RIT2, and is located 4.8 kb away from rs12456492, the peak SNP reported in the Caucasian meta-GWASs (6,41). We did not have direct genotype data of rs12456492, because our genotyping array did not include this SNP, but HaploReg (44) indicated that rs4130047 is in perfect linkage disequilibrium with rs12456492 (r2 = 1) (Table 1). The detection of a positive association between RIT2 and PD in the East Asian, as well as Caucasian population, demonstrates the definite PD susceptibility of this locus. We also performed a meta-analysis for the SNP rs4130047 using the MyMeta function implemented in the PDGene database (http://www.pdgene.org/my_meta). A forest plot indicated that in our study (4), SNP rs4130047 had an odds ratio of 1.27 (C versus T; 95% confidence interval = 1.14 – 1.41), and the association was in the same direction as in the original study (Supplementary Material, Table S1).
Table 1.
Summary of association results for representative SNPs within the RIT2 locus
| SNP | hg19 | Allele | MAF | Genotype count | Ptrend | SNP call | SNP call | PHWD | r 2 with | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | position | Minor | Major | Case | Control | Case | Control | Case | Control | Control | rs12456492 | ||
| rs879215 | 18 | 40557661 | T | C | 0.124 | 0.1626 | 19/207/762 | 58/704/1759 | 4.59 × 10−5 | 1.0000 | 1.0000 | 0.2146 | < 0.2 |
| rs4243267 | 18 | 40656531 | G | T | 0.3163 | 0.3697 | 99/427/462 | 340/1184/997 | 2.53 × 10−5 | 1.0000 | 1.0000 | 0.7324 | 0.42 |
| rs4441358 | 18 | 40657643 | G | A | 0.3148 | 0.3693 | 97/428/463 | 340/1182/999 | 1.69 × 10−5 | 1.0000 | 1.0000 | 0.7648 | 0.44 |
| rs4536548 | 18 | 40659924 | A | G | 0.3168 | 0.3697 | 99/428/461 | 340/1184/997 | 3.01 × 10−5 | 1.0000 | 1.0000 | 0.7324 | 0.42 |
| rs4130047 | 18 | 40678235 | C | T | 0.4195 | 0.3635 | 176/477/335 | 330/1172/1018 | 1.37 × 10−5 | 1.0000 | 0.9996 | 0.8296 | 1 |
Nucleotide positions refer to hg19. Ptrend values were obtained using the Cochran–Armitage trend test (1 d.f.). MAF, minor allele frequency. PHWD, P-value of deviation from the Hardy–Weinberg equilibrium
Although we also explored two eQTL databases, namely, the GTEx database (https://www.gtexportal.org/) and The Brain eQTL Almanac (www.Braineac.org), to examine potential influence of SNPs on the expression of RIT2, no convincing conclusion could be reached because of inconsistent reports on our target SNPs in the two databases.
Rescue of the effects of RIT2-KO by dabrafenib
A reduced expression of RIT2 was reported in the substantia nigra of patients with PD (45). To determine the effect of RIT2 deficiency in neuronal cells, we created RIT2-KO SH-SY5Y cells by using CRISPR/Cas9 genome editing (Supplementary Material, Fig. S5A). RIT2 is known to increase the expression of phosphorylated extracellular signal-regulated kinase (ERK) by activating B-Raf (38), and consistently, we observed a reduced expression of phospho-ERK in our RIT2-KO cells (Fig. 3C and Supplementary Material, Fig. S5B).
In RIT2-KO cells, rescue experiments with lentiviral overexpression of RIT2 upregulated the phosphorylation of ERK in an expression level-dependent manner (Fig. 3C and Supplementary Material, Fig. S5B). These data confirmed that the reduction in phospho-ERK levels was caused by a loss of RIT2.
Previous studies demonstrated that adenosine 5′-triphospate (ATP)-competitive RAF inhibitors, such as dabrafenib, inhibit ERK signaling in cells with BRAFV600E, but enhance ERK signaling in cells with wild-type BRAF (37,46). Furthermore, we confirmed ERK activation by dabrafenib in SH-SY5Y cells (Supplementary Material, Fig. S6A). We found that treatment with dabrafenib improved the phosphorylation level of ERK in RIT2-KO cells (Fig. 3D and Supplementary Material, Fig. S5C).
Inhibition of phosphorylated JNK/c-jun by dabrafenib
To examine whether the neuroprotective effects of dabrafenib result from the activation of ERK, we inhibited ERK activity in cells using the ERK inhibitors PD98059 or U0126, and performed the LDH and cell viability assay. We found that the neuroprotective effects of dabrafenib (1 μM) were cancelled by ERK inhibition. On the other hand, these inhibitors could not block the neuroprotective effects of dabrafenib (5 μM), although ERK inhibitors suppressed ERK phosphorylation (Fig. 4A and Supplementary Material, Fig. S6B).
Figure 4.
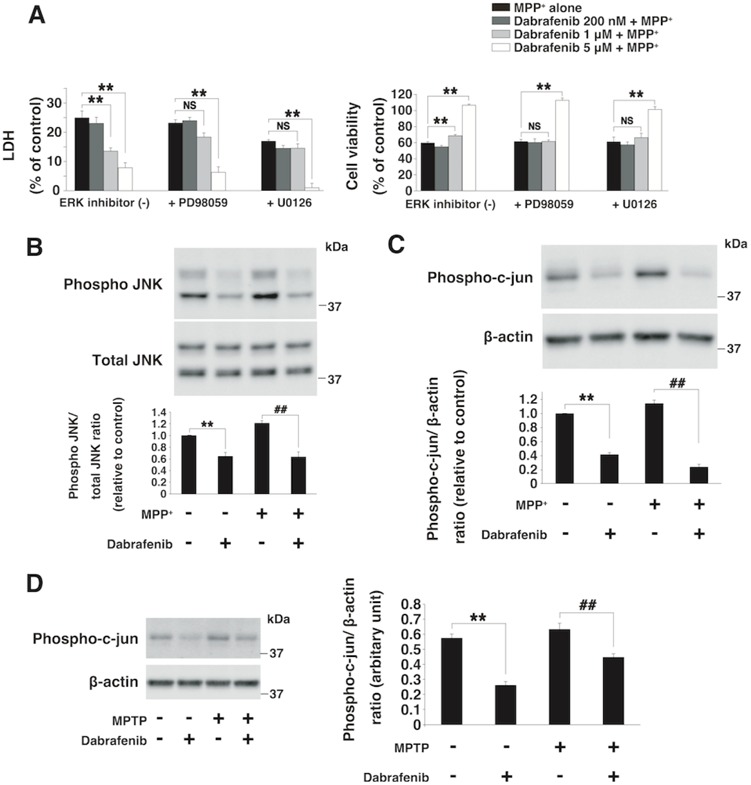
Dabrafenib reduced the expression of phosphorylated JNK and c-jun in both SH-SY5Y cells and mice
(A) LDH/cell viability assay under the condition of ERK inhibitor. Neuroprotective effects of dabrafenib (5 μM) were not dependent on the downregulation of activated ERK expression by PD98059 (30 μM) or U0126 (3 μM). Neuroprotective effects were assessed by the LDH and cell viability assay in SH-SY5Y cells at 24 h after MPP+ (3 mM) exposure. **, P-value < 0.01 compared with the MPP+ alone group; NS, not significant (n = 3 per group). (B) Immunoblot of phosphorylated JNK in SH-SY5Y cells at 30 min after drug treatment. Dabrafenib (5 μM) inhibited the phosphorylation of JNK with or without MPP+ (3 mM). **, P-value < 0.01 compared with control; ##, P-value < 0.01 compared with MPP+ alone (n = 3 per group). (C) Immunoblot for phospho-c-jun in SH-SY5Y cells at 24 h after drug treatment. Dabrafenib (5 μM) decreased the expression of phospho-c-jun with or without MPP+ 3 mM. **, P-value < 0.01 compared with control; ##, P-value < 0.01 compared with MPP+ alone (n = 3 per group). (D) Protein expression of phospho-c-jun in the substantia nigra of mice at 1 week after MPTP-injection. Dabrafenib significantly decreased the expression of phospho-c-jun with or without MPTP. **, P-value < 0.01 compared with the normal control group; ##, P-value < 0.01 compared with the vehicle + MPTP group (n = 4–5 per group). Data represent the mean ± s.e.m. All statistical analyses in this figure were performed using one-way ANOVA with the Tukey post-hoc test.
These results indicated that although the neuroprotective effects of 1 μM of dabrafenib were dependent on ERK activation, the neuroprotective effects of 5 μM of dabrafenib were not completely dependent on ERK activation. Then, we investigated whether the c-Jun NH2-terminal kinase (JNK)/c-jun cascade was involved in the neuroprotective effects of dabrafenib, because a previous study reported that vemurafenib, a BRAFV600E inhibitor similar to dabrafenib, also inhibited the phosphorylation of JNK (37). Furthermore, we found that dabrafenib downregulated the activation of JNK and c-jun independently of MPP+ exposure (Fig. 4B and C). These effects continued for at least 24 h (Supplementary Material, Fig. S6C). In MPTP-treated mice, dabrafenib also significantly decreased the expression of phospho-c-jun at 1 week after MPTP injection (Fig. 4D). These data suggested that dabrafenib demonstrated neuroprotective effects through the inhibition of JNK/c-jun, as well as the activation of ERK.
Discussion
By utilizing GWAS data and in silico databases, we identified dabrafenib as a neuroprotective drug for sporadic PD, demonstrating the usefulness of this screening method practically. Dabrafenib exerted anti-apoptotic effects, activated ERK and inhibited JNK/c-jun phosphorylation. We showed the PD susceptibility of the RIT2 gene in the Asian population, in addition to Caucasians, and showed that dabrafenib rescued the effects of RIT2-KO cells.
Dabrafenib (TafinlarⓇ) was approved by the FDA in 2013 for the treatment of BRAFV600E mutation-positive melanoma. Dabrafenib is known as a selective B-Raf kinase inhibitor (47), and induces cytotoxic effects specifically against cells with the BRAFV600E mutation, which is observed in 40% of all melanoma patients (48). On the other hand, dabrafenib is reported to activate B-Raf in wild-type cells that do not have the BRAFV600E mutation. B-Raf is a member of the Raf family, which comprises A-Raf, B-Raf and C-Raf. Cells of the central nervous system mainly express B-Raf and C-Raf, and the cytoplasmic concentration of B-Raf in neuronal cells appears higher than that of C-Raf (49). Previous reports demonstrated that B-Raf plays a role in neuronal survival (35) and maturation (50). These data indicated the close association between B-Raf and the nervous system.
Our data confirmed that dabrafenib activated ERK in SH-SY5Y cells, and the cytoprotective effects of dabrafenib (1 μM) were dependent on ERK activation (Fig. 4A). It was reported that activation of ERK showed neuroprotective effects in a PD model (14,51). We also investigated the effect of RIT2 deficiency in SH-SY5Y cells because the expression of RIT2 was decreased in the substantia nigra of PD patients. In RIT2-KO cells, phosphorylated ERK was suppressed, and this suppression was ameliorated by dabrafenib treatment. Other candidate of neuroprotective drug for PD, nilotinib (Abl-inhibitor), was identified on the basis of the fact that Abl levels were increased in the nigrostriatal region of PD patients (18). Our data demonstrated that dabrafenib improved the phosphorylation level of ERK in RIT2-KO cells. The viability of RIT2-KO cells was lower than that of control cells, but lentiviral overexpression of RIT2 (multiplicity of infection of 10) increased the cell viability (Supplementary Material, Fig. S5D). However, treatment of RIT2-KO cells with dabrafenib failed to improve the cell viability (Supplementary Material, Fig. S5E). It is possible that the influence of the complete deletion of RIT2 was severe and thus negated the effect of dabrafenib on cell viability. It is also possible that the complete deletion of RIT2 may have changed several cellular pathways, thus suppressing the improvement in cell viability.
On the other hand, our data indicated that the neuroprotective effects of dabrafenib (5 μM) were not completely dependent on ERK activation. Dabrafenib at this concentration also inhibited JNK/c-jun phosphorylation. Some reports suggested that the inhibition of JNK had neuroprotective effects in both in vitro and in vivo PD models (52). Thus, we concluded that dabrafenib (5 μM) showed neuroprotective effects by the inactivation of JNK/c-jun, in addition to the activation of ERK.
We further found that dabrafenib exerted anti-apoptotic effects. Apoptosis is known to be one of the mechanisms contributing to the etiology of PD (53). An association between the overexpression of B-Raf and anti-apoptotic effects was reported (54). Furthermore, the activation of ERK (55) and the suppression of JNK (55) were found to inhibit apoptosis. These data suggested the possibility that dabrafenib exerted anti-apoptotic effects through the activation of ERK and the inactivation of JNK.
It is interesting that in silico drug screening using GWAS data of PD identified the anti-melanoma drug dabrafenib, because many epidemiological studies have demonstrated associations between PD and melanoma (56,57). Our present study is the first to demonstrate the neuroprotective effects of an anti-melanoma drug in a PD model. Peak serum concentrations of dabrafenib at the clinically used dose (150 mg twice daily) are equivalent to 1.55 μM in vitro (58), which is similar to the concentrations in our experiment.
Previous studies have reported that oral administration of dabrafenib could reduce the size of brain metastases in patients with malignant melanoma (58,59), which suggested a good penetration ability of dabrafenib to the central nervous system. It is also possible that dabrafenib may be injected to PD patients intrathecally, similar to the administration of nusinersen (Spinraza®) to patients with spinal muscular atrophy (60).
Several studies have suggested that L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesia is associated with the activation of ERK (61). Dabrafenib also activates ERK in cells with wild-type BRAF and thus may potentially exacerbate dyskinesia. However, a previous report has demonstrated that the severity of L-DOPA-induced dyskinesia correlated with the extent of long-term ERK activation and that no persistent ERK activation was detected in the non-dyskinetic group (62). Our experiment with SH-SY5Y cells showed that the activation of ERK by dabrafenib was transient (< 24 h), suggesting a relatively low risk of dyskinesia (Supplementary Material, Fig. S6A). However, in real patients, the duration of ERK activation by dabrafenib will depend on the dosing regimen. Therefore, the best schedule of its administration should be considered to avoid persistent activation of ERK and to minimize the risk of dyskinesia.
Drug repurposing is a promising strategy for drug development because the safety data of these drugs in human patients are already available (63). However, it is inefficient to examine all FDA-approved drugs, because of their vast number. This in silico drug screening, which links the data of GWAS to drug/PPI databases, may narrow down candidate drugs for many polygenic diseases and save cost and time. Our present study is the first to demonstrate the efficacy of this drug screening method by using in vitro and in vivo PD model. In this study, we used the LDH/cell viability assay to verify the neuroprotective effects of candidate drugs. This enabled the identification of drugs with anti-apoptotic effects. Further studies are required to identify drugs with other neuroprotective effects, such as those that inhibit the aggregation or propagation of α-synuclein, by performing other assays. This screening method is also a promising strategy to identify fundamental therapeutic agents for not only other neurodegenerative diseases, such as Alzheimer’s disease and amyotrophic lateral sclerosis, but also many multifactorial genetic disorders, like diabetes mellitus and hypertension.
Materials and Methods
Materials
Dabrafenib was obtained from Cayman Chemical Company (Ann Arbor, MI, USA) and Selleck Chemicals (Houston, TX, USA). MPP+, MPTP-HCl and duloxetine were from Sigma-Aldrich (St. Louis, MO, USA). PD98059 and U0126 were from Cayman Chemical Company. Regorafenib was obtained from Adooq Bioscience (Irvine, CA, USA). Dasatinib was from BioVision (Milpitas, CA, USA). Furosemide was obtained from Abcam (Cambridge, MA, USA).
Antibodies against caspase-3 (#9665, 1:1000), cleaved caspase-9 (#7237, 1:1000), ERK 1/2 (#4695, 1:1000), phospho-ERK 1/2 (#4370, 1:2000), JNK (#9252, 1:1000), phospho-JNK (#4668, 1:1000), phospho-c-jun (#3270, 1:1000) and phospho-B-Raf (Ser445; #2696, 1:1000) were all purchased from Cell Signaling Technology (Beverly, MA, USA). The antibody against β-actin (A1978, 1:2000) was from Sigma-Aldrich. An anti-DAT antibody (MAB369, 1:1,000) was from Merck Millipore (Darmstadt, Germany). An anti-p62/SQSTM1 antibody (18420–1-AP, 1:1000) was from Proteintech (Chicago, IL, USA). An anti-α-synuclein antibody (610787, 1:1000) was from BD Transduction Laboratories (San Diego, CA, USA). An anti-IBA-1 antibody (016–20001, 1:1000) was from Wako (Osaka, Japan). An anti-GFAP antibody (Z0334, 1:2000) was from Dako (Copenhagen, Denmark). An anti-TH antibody (657012, 1:5000) was from Calbiochem (San Diego, CA, USA). Antibodies against Rit2 (sc-58474, 1:200), B-Raf (sc-5284, 1:200) and c-Myc (sc-40, 1:200) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). An anti-V5 antibody (04434–36, 1:1000) was from Nacalai Tesque (Kyoto, Japan).
In silico drug screening
We focused on the candidate PD-risk genes that were identified by a meta-analysis of GWASs (6); GBA, SYT11, RAB7L1, NUCKS1, SIPA1L2, ACMSD, TMEM163, STK39, MCCC1, TMEM175, GAK, DGKQ, BST1, FAM47E, SCARB2, SNCA, HLA-DQB1, GPNMB, INPP5F, MIR4697, LRRK2, CCDC62, GCH1, VPS13C, BCKDK, STX1B, MAPT, RIT2, DDRGK1, FGF20, SREBF1 and RAI1. We performed an interaction simulation of these PD-risk genes using InWeb (10) and PINA (11) databases, and identified genes in direct PPI with the PD-risk genes. PD-risk genes by GWAS and genes in direct PPI were combined as PD-associated genes. Next, we obtained all genes that were targeted by drugs from DrugBank (12) and Therapeutic Targets Database (13). We identified the overlapping genes between the PD-associated genes and all drug target genes. Drugs that targeted these overlapping genes were identified as candidate drugs for PD.
Cell culture
Human neuroblastoma SH-SY5Y cells (a kind gift from Dr Miho Murata, National Center Hospital, National Center of Neurology and Psychiatry) and HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum, 1% penicillin/streptomycin at 37°C in a humidified incubator with a 5% CO2 environment. All experiments were carried out 24–48 h after the cells were seeded.
Measurement of LDH release and cell viability
SH-SY5Y cells (2 × 104) were grown on collagen-coated 96-well plates in serum-free medium. Twenty-four h later, cells were treated with candidate drugs, followed by MPP+ (3 mM) for another 24 h. After incubation, the release of LDH was measured with a cytotoxicity detection kit (Roche Applied Science, Indianapolis, IN, USA). For measurement of cell viability, the CellTiter 96 AQueous One Solution cell proliferation assay (Promega, Madison, WI, USA) was used. We performed these assays according to the manufacturer’s instructions, and measured absorbance at 490 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA). To compare cell viability between control and RIT2-KO cells, absorbance was measured 48 h after the administration of dabrafenib or lentiviral overexpression of RIT2.
Animals
C57BL/6J mice (8–12 weeks old; 20–25 g; Charles River Laboratories, Kingston, NY, USA) were used. Animals were housed five per cage in a temperature-controlled room under a 12 h light/dark cycle with ad libitum access to food and water. All animal experiments were conducted according to the Osaka University Medical School Guidelines for the Care and Use of Laboratory Animals.
All animal procedures were approved by the Osaka University Medical School Animal Care and Use Committee.
Drug treatment
Mice were anesthetized using sodium pentobarbital (Kyoritsu Seiyaku, Tokyo, Japan), injected intraperitoneally and placed in a stereotaxic instrument (Narishige, Tokyo, Japan). Using a 10 μL Hamilton syringe, vehicle (5% dimethylsulfoxide and polyethylene glycol 300) or dabrafenib (150 μg kg−1, 15 μg kg−1 or 1.5 μg kg−1 per mouse) was injected into the lateral ventricle of mice at the following coordinates: 0.2 mm posterior to bregma, 1 mm lateral to the midline and 3 mm ventral to the skull surface.
Three days after the treatment, the mice were intraperitoneally injected with MPTP-HCl (30 mg kg−1; Sigma) in saline daily for five consecutive days. The mice were subsequently sacrificed 7 days or 21 days after the last administration of MPTP.
Immunohistochemistry
After anesthetization with sodium pentobarbital, mice were perfused transcardially with phosphate-buffered saline (PBS), followed by 4% paraformaldehyde in PBS. Then, the brains were removed and immersed for 48 h in 4% paraformaldehyde at 4°C and in 30% sucrose containing 0.01% sodium azide for another 48 h at 4°C. Nigral coronal sections (20 μm thick) were prepared with a cryostat. The sections were first rinsed with PBS, and then they were immersed in a solution of PBS containing 3% hydrogen peroxidase for 10 min. After washing with PBS, the sections were incubated overnight at 4°C with primary antibodies diluted with blocking solution (PBS-T containing 10% Block-Ace; DS Pharma Biomedical, Osaka, Japan). The sections were washed with PBS, and incubated with biotinylated secondary antibodies (VECTOR Laboratories, Burlingame, CA, USA, 1:500) in PBS-T for 1 h and then with VECTASTAIN ABC reagent (VECTOR Laboratories) for 1 h at room temperature. Finally, the sections were incubated for 5 min with 3,3′-diaminobenzidine (Sigma) for visualization.
Stereological cell counting
The number of dopaminergic neurons in the SNpc was analyzed by unbiased stereological counting method (64). TH- and Nissl-double-positive neurons and total (i.e. Nissl-positive) neurons were counted in the right and left SNpc of every fourth 20 μm section throughout the entire extent of the SNpc. To avoid double counting of neurons with unusual shapes, TH- and Nissl-double-positive neurons were counted only when their nuclei and nucleoli were optimally visualized.
Western blot analysis
SH-SY5Y cells were seeded at a density of 0.5–1 × 106 cells in six-well plates and treated with dabrafenib and/or MPP+ for various times. Cells were lysed with RIPA buffer (50 mM Tris–HCl, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate and 0.1% sodium dodecyl sulfate [SDS]), or SDS buffer (62.5 mM Tris–HCl, 2% SDS and 10% glycerol) and ultrasonicated for ∼20 s. The substantia nigra were isolated from each mouse brain, and then lysed in CelLytic MT Cell Lysis Reagent (#C3228, Sigma) or SDS buffer followed by ultrasonication. Protein concentrations were estimated using the BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Proteins (10–20 μg) were separated by 4–15% SDS-polyacrylamide gradient gel electrophoresis (BioRad, Hercules, CA, USA), followed by transfer to a polyvinylidene difluoride membrane (Merck Millipore). Membranes were blocked with 5% skim milk in TBS-T for 1 h, and then incubated with a primary antibody overnight at 4°C. After incubation with an appropriate secondary antibody for 1 h at room temperature, protein bands were visualized by the ECL western blotting detection system (GE Healthcare, Madison, WI, USA). The bands were semi-quantified by densitometric analysis with Image J software.
RT-PCR
Total RNA was isolated from the SNpc of a control mouse using TRIzol (Invitrogen, Carlsbad, CA, USA) and converted to cDNA using SuperScript III reverse transcriptase (Invitrogen) in accordance with the manufacturer’s protocols. The primers for mouse RIT2 were 5′-GATGATGCTTTTCAAGGCTT-3′ and 5′-GGCTTTTATCTTCTTCCACA-3′; primers for mouse BRAF were 5′-GCTCTGTTCAATGGCGACATGG-3′ and 5′-AGGCCTCCAGGTATATTGATGGTG-3′ and primers for mouse TH were 5′-TACGCCACGCTGAAGGGCCTCTAT-3′ and 5′-AGGTGAGGAGGCATGACGGATGTA-3′.
Association study and genomic analysis
We used summary statistics of the Cochran–Armitage trend test from our previous GWAS, which consisted of 435 470 SNPs in 988 PD and 2521 control Japanese samples (4). We generated regional plots presenting the P-values of SNPs across the RIT2 locus (40.2–41.2 Mb on chromosome 18) using LocusZoom (65). We used linkage disequilibrium information from the Asian population of 1000 Genomes. We examined r2 values between rs12456492 and each SNP using HaploReg (Asian population of 1000 Genomes) (44). The MyMeta function implemented in the PDGene database (http://www.pdgene.org/my_meta) was used to perform a meta-analysis for the SNP rs4130047.
Co-immunoprecipitation
Mammalian expression constructs containing the human c-Myc-tagged RIT2 or V5-tagged BRAF in the pcDNA3.1/myc-His A vector (Invitrogen) were generated by PCR amplification. To examine the interaction between the Rit2 and B-Raf proteins, HEK293T cells were transiently transfected with the plasmids encoding c-Myc-Rit2 or V5-B-Raf using Effectene (Qiagen, Hilden, Germany). After 48 h, the cells were lysed with RIPA buffer containing protease and phosphatase inhibitors. The cell lysates were incubated with anti-c-Myc antibody-conjugated agarose beads (M047–8; MBL, Woburn, MA, USA) or anti-V5 antibody-conjugated agarose beads (PM003–8; MBL). After the incubation, the beads were washed five times and subjected to immunoblot analysis with the indicated antibodies.
Preparation of RIT2-KO cells with CRISPR/Cas9 gene editing
We used the Cas9 Smart Nuclease All-in-One vector (System Biosciences, Mountain View, CA, USA). The CRISPR/Cas9 targeting sequence for RIT2 was GATTATCATGACCCTACTAT. The All-in-One vector was transfected into SH-SY5Y cells together with the zeocin-resistant gene in pcDNA3.1/myc-His A (20:1 ratio) using Lipofectamine 2000 reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. The resulting zeocin-resistant colonies were dissociated into single cells and plated at 5000 cells per 10 cm dish. Mutations in the cell clones were verified by DNA sequencing of RIT2. The mutations in each clone were as follows: clone 13 (homozygous 1 bp insertion in exon 2); clone 21 (homozygous 1 bp deletion in exon 2) and clone 42 (homozygous 2 bp deletion in exon 2). We used clones that underwent genome editing but revealed no mutation in RIT2 as controls (clones 7, 8 and 9).
Lentiviral infection
The lentivirus vector CSII-CMV-MCS-IRES2-Venus, and the two packaging vectors pCAG-HIVgp and pCMV-VSV-G-RSV-Rev, were kindly provided by Dr Hiroyuki Miyoshi (Riken BioResource Center, Tsukuba, Japan). To generate the lentiviruses, the recombinant RIT2 vector was transiently transfected into Lenti-X 293 T cells (Clontech, Mountain View, CA, USA) together with two packaging vectors. Forty-eight h after transfection, the viral supernatants were concentrated using a LentiX concentrator (Clontech). Viral particles were resuspended into DMEM. Analysis of lentiviral experiments was performed 2 days post infection at a multiplicity of infection of 1, 5 and 10.
Statistical analysis
Data are expressed as the mean ± s.e.m. All data were acquired from at least three independent experiments. Statistical analyses were performed by one-way ANOVA followed by the Tukey’s post-hoc test. A P-value of >0.05 was considered to indicate a statistically significant difference between two groups.
Supplementary Material
Funding
Platform Program for Promotion of Genome Medicine, Advanced Genome Research and Bioinformatics Study to Facilitate Medical Innovation from the Japan Agency for Medical Research and Development (AMED; JP17km0405206 to T.T.); the Practical Research Project for Rare/Intractable Diseases from AMED (JP16ek0109029 to T.T., JP17ek0109207 to W.S.); a Grant-in-Aid for Scientific Research (B) from Japan Society for the Promotion of Science (JSPS; JP17H04056 to W.S.); the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) from AMED (to H.M.); and the Practical Research Project for Allergic Diseases and Immunology from AMED (JP17ek0410041 to Y.O.).
Acknowledgements
We would like to thank the members of Dr Toda’s laboratory for fruitful discussions/scientific contributions and Chi-Jing Choong for experimental advice and mouse maintenance.
Conflict of Interest statement. None declared.
References
- 1. Volta M., Milnerwood A.J. and Farrer M.J. (2015) Insights from late-onset familial parkinsonism on the pathogenesis of idiopathic Parkinson's disease. Lancet Neurol., 14, 1054–1064. [DOI] [PubMed] [Google Scholar]
- 2. Sirota M., Dudley J.T., Kim J., Chiang A.P., Morgan A.A., Sweet-Cordero A., Sage J. and Butte A.J. (2011) Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med., 3, 96ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Plenge R.M., Scolnick E.M. and Altshuler D. (2013) Validating therapeutic targets through human genetics. Nat. Rev. Drug Dis., 12, 581–594. [DOI] [PubMed] [Google Scholar]
- 4. Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A. et al. (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet., 41, 1303–1307. [DOI] [PubMed] [Google Scholar]
- 5. Lill C.M., Roehr J.T., McQueen M.B., Kavvoura F.K., Bagade S., Schjeide B.M., Schjeide L.M., Meissner E., Zauft U., Allen N.C. et al. (2012) Comprehensive research synopsis and systematic meta-analyses in Parkinson's disease genetics: the PDGene database. PLoS Genet., 8, e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nalls M.A., Pankratz N., Lill C.M., Do C.B., Hernandez D.G., Saad M., DeStefano A.L., Kara E., Bras J., Sharma M. et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet., 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Okada Y., Wu D., Trynka G., Raj T., Terao C., Ikari K., Kochi Y., Ohmura K., Suzuki A., Yoshida S. et al. (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature, 506, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J., Jiang K., Lv L., Wang H., Shen Z., Gao Z., Wang B., Yang Y., Ye Y. and Wang S. (2015) Use of genome-wide association studies for cancer research and drug repositioning. PLoS One, 10, e0116477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Imamura M., Takahashi A., Yamauchi T., Hara K., Yasuda K., Grarup N., Zhao W., Wang X., Huerta-Chagoya A., Hu C. et al. (2016) Genome-wide association studies in the Japanese population identify seven novel loci for type 2 diabetes. Nat. Commun., 7, 10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rossin E.J., Lage K., Raychaudhuri S., Xavier R.J., Tatar D., Benita Y., International Inflammatory Bowel Disease Genetics Constortium, Cotsapas C. and Daly M.J. (2011) Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet., 7, e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu J., Vallenius T., Ovaska K., Westermarck J., Makela T.P. and Hautaniemi S. (2009) Integrated network analysis platform for protein-protein interactions. Nat. Methods, 6, 75–77. [DOI] [PubMed] [Google Scholar]
- 12. Knox C., Law V., Jewison T., Liu P., Ly S., Frolkis A., Pon A., Banco K., Mak C., Neveu V. et al. (2011) DrugBank 3.0: a comprehensive resource for 'omics' research on drugs. Nucleic Acids Res., 39, D1035–D1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu F., Shi Z., Qin C., Tao L., Liu X., Xu F., Zhang L., Song Y., Liu X., Zhang J. et al. (2012) Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res., 40, D1128–D1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shadfar S., Kim Y.G., Katila N., Neupane S., Ojha U., Bhurtel S., Srivastav S., Jeong G.S., Park P.H., Hong J.T. et al. (2018) Neuroprotective effects of antidepressants via upregulation of neurotrophic factors in the MPTP model of Parkinson's disease. Mol. Neurobiol., 55, 554–566. [DOI] [PubMed] [Google Scholar]
- 15. Sharma N., Jamwal S. and Kumar P. (2016) Beneficial effect of antidepressants against rotenone induced Parkinsonism like symptoms in rats. Pathophysiology, 23, 123–134. [DOI] [PubMed] [Google Scholar]
- 16. Jenner P., Zeng B.Y., Smith L.A., Pearce R.K.B., Tel B., Chancharme L. and Moachon G. (2000) Antiparkinsonian and neuroprotective effects of modafinil in the mptp-treated common marmoset. Exp. Brain Res., 133, 178–188. [DOI] [PubMed] [Google Scholar]
- 17. Salama M., Ellaithy A., Helmy B., El-Gamal M., Tantawy D., Mohamed M., Sheashaa H., Sobh M. and Arias-Carrion O. (2012) Colchicine protects dopaminergic neurons in a rat model of Parkinson's disease. CNS Neurol. Disord. Drug Targets, 11, 836–843. [DOI] [PubMed] [Google Scholar]
- 18. Hebron M.L., Lonskaya I. and Moussa C.E. (2013) Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson's disease models. Hum. Mol. Genet., 22, 3315–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen S.H., Wu H.M., Ossola B., Schendzielorz N., Wilson B.C., Chu C.H., Chen S.L., Wang Q., Zhang D., Qian L. et al. (2012) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. Bri. J. Pharmacol., 165, 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cunningham R.L., Giuffrida A. and Roberts J.L. (2009) Androgens induce dopaminergic neurotoxicity via caspase-3-dependent activation of protein kinase Cdelta. Endocrinology, 150, 5539–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hashioka S., Klegeris A. and McGeer P.L. (2009) Proton pump inhibitors exert anti-inflammatory effects and decrease human microglial and monocytic THP-1 cell neurotoxicity. Exp. Neurol., 217, 177–183. [DOI] [PubMed] [Google Scholar]
- 22. Antipova V., Hawlitschka A., Mix E., Schmitt O., Drager D., Benecke R. and Wree A. (2013) Behavioral and structural effects of unilateral intrastriatal injections of botulinum neurotoxin a in the rat model of Parkinson's disease. J. Neurosci. Res., 91, 838–847. [DOI] [PubMed] [Google Scholar]
- 23. Zachrisson O., Zhao M., Andersson A., Dannaeus K., Haggblad J., Isacson R., Nielsen E., Patrone C., Ronnholm H., Wikstrom L. et al. (2011) Restorative effects of platelet derived growth factor-BB in rodent models of Parkinson's disease. J. Parkinsons Dis., 1, 49–63. [DOI] [PubMed] [Google Scholar]
- 24. Liu Z., Hamamichi S., Lee B.D., Yang D., Ray A., Caldwell G.A., Caldwell K.A., Dawson T.M., Smith W.W. and Dawson V.L. (2011) Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson's disease models. Hum. Mol. Genet., 20, 3933–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boireau A., Bordier F., Dubedat P., Peny C. and Imperato A. (1997) Thalidomide reduces MPTP-induced decrease in striatal dopamine levels in mice. Neurosci. Lett., 234, 123–126. [DOI] [PubMed] [Google Scholar]
- 26. Hirohata M., Ono K., Morinaga A., Ikeda T. and Yamada M. (2009) Anti-aggregation and fibril-destabilizing effects of sex hormones on alpha-synuclein fibrils in vitro. Exp. Neurol., 217, 434–439. [DOI] [PubMed] [Google Scholar]
- 27. Baraka A.M., Korish A.A., Soliman G.A. and Kamal H. (2011) The possible role of estrogen and selective estrogen receptor modulators in a rat model of Parkinson's disease. Life Sci., 88, 879–885. [DOI] [PubMed] [Google Scholar]
- 28. Bourque M., Morissette M., Al Sweidi S., Caruso D., Melcangi R.C. and Di Paolo T. (2016) Neuroprotective effect of progesterone in MPTP-treated male mice. Neuroendocrinology, 103, 300–314. [DOI] [PubMed] [Google Scholar]
- 29. Hollerhage M., Goebel J.N., Andrade A., Hildebrandt T., Dolga A., Culmsee C., Oertel W.H., Hengerer B. and Hoglinger G.U. (2014) Trifluoperazine rescues human dopaminergic cells from wild-type alpha-synuclein-induced toxicity. Neurobiol. Aging, 35, 1700–1711. [DOI] [PubMed] [Google Scholar]
- 30. Aviles-Olmos I., Dickson J., Kefalopoulou Z., Djamshidian A., Ell P., Soderlund T., Whitton P., Wyse R., Isaacs T., Lees A. et al. (2013) Exenatide and the treatment of patients with Parkinson's disease. J. Clin. Invest., 123, 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Katsumata R., Ishigaki S., Katsuno M., Kawai K., Sone J., Huang Z., Adachi H., Tanaka F., Urano F. and Sobue G. (2012) c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PLoS One, 7, e46185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akpinar A., Uguz A.C. and Naziroglu M. (2014) Agomelatine and duloxetine synergistically modulates apoptotic pathway by inhibiting oxidative stress triggered intracellular calcium entry in neuronal PC12 cells: role of TRPM2 and voltage-gated calcium channels. J. Membr. Biol., 247, 451–459. [DOI] [PubMed] [Google Scholar]
- 33. Zhao S., Zhu Y., Xue R., Li Y., Lu H. and Mi W. (2012) Effect of midazolam on the proliferation of neural stem cells isolated from rat hippocampus. Neural Regen. Res., 7, 1475–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carr B.I., Cavallini A., Lippolis C., D'Alessandro R., Messa C., Refolo M.G. and Tafaro A. (2013) Fluoro-Sorafenib (Regorafenib) effects on hepatoma cells: growth inhibition, quiescence, and recovery. J. Cell. Physiol., 228, 292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wiese S., Pei G., Karch C., Troppmair J., Holtmann B., Rapp U.R. and Sendtner M. (2001) Specific function of B-Raf in mediating survival of embryonic motoneurons and sensory neurons. Nat. Neurosci., 4, 137–142. [DOI] [PubMed] [Google Scholar]
- 36. Beal M.F. (2001) Experimental models of Parkinson's disease. Nat. Rev. Neurosci., 2, 325–334. [DOI] [PubMed] [Google Scholar]
- 37. Vin H., Ojeda S.S., Ching G., Leung M.L., Chitsazzadeh V., Dwyer D.W., Adelmann C.H., Restrepo M., Richards K.N., Stewart L.R. et al. (2013) BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. ELife, 2, e00969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi G.X., Han J. and Andres D.A. (2005) Rin GTPase couples nerve growth factor signaling to p38 and b-Raf/ERK pathways to promote neuronal differentiation. J. Biol. Chem., 280, 37599–37609. [DOI] [PubMed] [Google Scholar]
- 39. Zhou Q., Li J., Wang H., Yin Y. and Zhou J. (2011) Identification of nigral dopaminergic neuron-enriched genes in adult rats. Neurobiol. Aging, 32, 313–326. [DOI] [PubMed] [Google Scholar]
- 40. Di Benedetto B., Hitz C., Holter S.M., Kuhn R., Vogt Weisenhorn D.M. and Wurst W. (2007) Differential mRNA distribution of components of the ERK/MAPK signalling cascade in the adult mouse brain. J. Comp. Neurol., 500, 542–556. [DOI] [PubMed] [Google Scholar]
- 41. Pankratz N., Beecham G.W., DeStefano A.L., Dawson T.M., Doheny K.F., Factor S.A., Hamza T.H., Hung A.Y., Hyman B.T., Ivinson A.J. et al. (2012) Meta-analysis of Parkinson's disease: identification of a novel locus, RIT2. Ann. Neurol., 71, 370–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li J.Y., Zhang J.H., Li N.N., Wang L., Lu Z.J., Cheng L., Sun X.Y. and Peng R. (2017) Genetic association study between RIT2 and Parkinson's disease in a Han Chinese population. Neurol. Sci., 38, 343–347. [DOI] [PubMed] [Google Scholar]
- 43. Nie K., Feng S.J., Tang H.M., Ma G.X., Gan R., Zhao X., Zhao J.H., Wang L.M., Huang Z.H., Huang J. et al. (2015) RIT2 polymorphism is associated with Parkinson's disease in a Han Chinese population. Neurobiol. Aging, 36, 1603e1615–1607. [DOI] [PubMed] [Google Scholar]
- 44. Ward L.D. and Kellis M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bossers K., Meerhoff G., Balesar R., Dongen J.W., Kruse C.G., Swaab D.F. and Verhaagen J. (2009) Analysis of gene expression in Parkinson's disease: possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain Pathol., 19, 91–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poulikakos P.I., Zhang C., Bollag G., Shokat K.M. and Rosen N. (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature, 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rheault T.R., Stellwagen J.C., Adjabeng G.M., Hornberger K.R., Petrov K.G., Waterson A.G., Dickerson S.H., Mook R.A. Jr., Laquerre S.G., King A.J. et al. (2013) Discovery of dabrafenib: a selective inhibitor of Raf kinases with antitumor activity against B-Raf-driven tumors. ACS Med. Chem. Lett., 4, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hauschild A., Grob J.J., Demidov L.V., Jouary T., Gutzmer R., Millward M., Rutkowski P., Blank C.U., Miller W.H. Jr., Kaempgen E. et al. (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet, 380, 358–365. [DOI] [PubMed] [Google Scholar]
- 49. Frebel K. and Wiese S. (2006) Signalling molecules essential for neuronal survival and differentiation. Biochem. Soc. Trans., 34, 1287–1290. [DOI] [PubMed] [Google Scholar]
- 50. Zhong J., Li X., McNamee C., Chen A.P., Baccarini M. and Snider W.D. (2007) Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo. Nat. Neurosci., 10, 598–607. [DOI] [PubMed] [Google Scholar]
- 51. Gordon R., Neal M.L., Luo J., Langley M.R., Harischandra D.S., Panicker N., Charli A., Jin H., Anantharam V., Woodruff T.M. et al. (2016) Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nat. Commun., 7, 12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xia X.G., Harding T., Weller M., Bieneman A., Uney J.B. and Schulz J.B. (2001) Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc. Natl. Acad. Sci. USA, 98, 10433–10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yasuda T. and Mochizuki H. (2010) The regulatory role of alpha-synuclein and parkin in neuronal cell apoptosis; possible implications for the pathogenesis of Parkinson's disease. Apoptosis, 15, 1312–1321. [DOI] [PubMed] [Google Scholar]
- 54. Erhardt P., Schremser E.J. and Cooper G.M. (1999) B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol. Cell. Biol., 19, 5308–5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xia Z., Dickens M., Raingeaud J., Davis R.J. and Greenberg M.E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science, 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- 56. Pan T., Li X. and Jankovic J. (2011) The association between Parkinson's disease and melanoma. Int. J. Cancer, 128, 2251–2260. [DOI] [PubMed] [Google Scholar]
- 57. Bertoni J.M., Arlette J.P., Fernandez H.H., Fitzer-Attas C., Frei K., Hassan M.N., Isaacson S.H., Lew M.F., Molho E., Ondo W.G. et al. (2010) Increased melanoma risk in Parkinson disease: a prospective clinicopathological study. Arch. Neurol., 67, 347–352. [DOI] [PubMed] [Google Scholar]
- 58. Falchook G.S., Long G.V., Kurzrock R., Kim K.B., Arkenau T.H., Brown M.P., Hamid O., Infante J.R., Millward M., Pavlick A.C. et al. (2012) Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet, 379, 1893–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Long G.V., Trefzer U., Davies M.A., Kefford R.F., Ascierto P.A., Chapman P.B., Puzanov I., Hauschild A., Robert C., Algazi A. et al. (2012) Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol., 13, 1087–1095. [DOI] [PubMed] [Google Scholar]
- 60. Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E. et al. (2017) Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med., 377, 1723–1732. [DOI] [PubMed] [Google Scholar]
- 61. Spigolon G. and Fisone G. (2018) Signal transduction in L-DOPA-induced dyskinesia: from receptor sensitization to abnormal gene expression. J. Neural Trans. (Vienna), 125, 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fiorentini C., Savoia P., Savoldi D., Barbon A. and Missale C. (2013) Persistent activation of the D1R/Shp-2/Erk1/2 pathway in l-DOPA-induced dyskinesia in the 6-hydroxy-dopamine rat model of Parkinson's disease. Neurobiol. Dis., 54, 339–348. [DOI] [PubMed] [Google Scholar]
- 63. Hurle M.R., Yang L., Xie Q., Rajpal D.K., Sanseau P. and Agarwal P. (2013) Computational drug repositioning: from data to therapeutics. Clin. Pharmacol. Ther., 93, 335–341. [DOI] [PubMed] [Google Scholar]
- 64. Hasegawa K., Yasuda T., Shiraishi C., Fujiwara K., Przedborski S., Mochizuki H. and Yoshikawa K. (2016) Promotion of mitochondrial biogenesis by necdin protects neurons against mitochondrial insults. Nat. Commun., 7, 10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R. and Willer C.J. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics, 26, 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.