

Abstract
Mendelian susceptibility to mycobacterial disease (MSMD) is characterized by clinical disease caused by weakly virulent mycobacteria, such as environmental mycobacteria and Bacillus Calmette–Guérin vaccines, in otherwise healthy individuals. All known genetic etiologies disrupt interferon (IFN)-γ immunity. Germline bi-allelic mutations of IFNGR2 can underlie partial or complete forms of IFN-γ receptor 2 (IFN-γR2) deficiency. Patients with partial IFN-γR2 deficiency express a dysfunctional molecule on the cell surface. We studied three patients with MSMD from two unrelated kindreds from Turkey (P1, P2) and India (P3), by whole-exome sequencing. P1 and P2 are homozygous for a mutation of the initiation codon(c.1A>G) of IFNGR2, whereas P3 is homozygous for a mutation of the second codon (c.4delC). Overexpressed mutant alleles produce small amounts of full-length IFN-γR2 resulting in an impaired, but not abolished, response to IFN-γ. Moreover, SV40-fibroblasts of P1 and P2 responded weakly to IFN-γ, and Epstein Barr virus-transformed B cells had a barely detectable response to IFN-γ. Studies in patients’ primary T cells and monocyte-derived macrophages yielded similar results. The residual expression of IFN-γR2 protein of normal molecular weight and function is due to the initiation of translation between the second and ninth non-AUG codons. We thus describe mutations of the first and second codons of IFNGR2, which define a new form of partial recessive IFN-γR2 deficiency. Residual levels of IFN-γ signaling were very low, accounting for the more severe clinical phenotype of these patients with residual expression levels of normally functional surface receptors than of patients with partial recessive IFN-γR2 deficiency due to surface-expressed dysfunctional receptors, whose residual levels of IFN-γ signaling were higher.
Introduction
Mendelian susceptibility to mycobacterial disease (MSMD) is a primary immunodeficiency predisposing otherwise healthy individuals to severe clinical manifestations upon infection with weakly virulent mycobacteria, such as Bacillus Calmette–Guérin (BCG) vaccines and environmental mycobacteria (EM) (1,2). Clinical onset is generally in childhood, with a wide spectrum of clinical symptoms and signs, ranging from localized to disseminated disease, with or without recurrences, caused by one or more mycobacterial species. These patients are also vulnerable to the more virulent Mycobacterium tuberculosis, and about half of them also suffer from non-typhoidal or, more rarely, typhoidal Salmonella extra-intestinal infections (1,2). Other infections, mostly due to intra-macrophagic microorganisms, have been reported, although typically in individual patients (2,3). The first genetic etiology of MSMD was described in 1996, involving mutations of the ligand-binding chain of the interferon-γ receptor (IFN-γR1) (4,5). To date, the genetic dissection of MSMD has revealed germline mutations in nine autosomal genes [IFNGR1, IFNGR2, STAT1, interleukin (IL) 12B, IL12RB1, IRF8, ISG15, TYK2 and SPPL2A] and two X-linked genes (NEMO and CYBB), the products of which control the induction of (IL12B, IL12RB1, IRF8, ISG15, TYK2 and NEMO) and/or response to IFN-γ (IFNGR1, IFNGR2, STAT1, IRF8, CYBB and SPPL2A) (2,6,7). Allelic heterogeneity at several of these loci results in up to 20 different etiologies, depending on the mode of inheritance (dominant or recessive), the severity of the protein defect (complete or partial), the expression of the protein underlying a complete defect (abolished or maintained) and the mechanism of protein loss-of-function (LOF) (depending on the domain disrupted).
IFN-γR2 deficiency has been reported in 24 patients and has probably been diagnosed in many more (8–22). Autosomal recessive (AR) complete IFN-γR2 deficiency is characterized by a complete loss of function of the protein, and the following two forms have been described: with and without cell surface expression of the receptor (20,21). In 10 patients from seven kindreds and seven countries, a premature stop codon was found to result in a lack of receptor expression at the cell surface (9,10,13–17,19,20). Seven other patients from six kindreds and five countries have a non-functional IFN-γR2 protein molecule expressed on their cells (11,19–22). In three of these patients, from two kindreds and two countries, a missense mutation (p.T168N) has been shown to create a new N-glycosylation site, which abolishes the cellular response to IFN-γ, despite the expression of IFN-γR2 at the cell surface (21–23). In another patient, the mutation (c.382-387dup) does not cause a gain of glycosylation but instead results in misfolded proteins that can also, surprisingly, be rescued with inhibitors of glycosylation (20). Finally, it has been suggested that cells from a patient with a frameshift mutation display residual IFN-γR2 expression, although antibody (Ab) specificity was not demonstrated (11). AR complete IFN-γR2 deficiency is the most severe form of IFN-γR2 deficiency, characterized by high levels of IFN-γ in plasma (15) and an abolished response to IFN-γ in all cell types (2). The most commonly encountered microbial pathogens include BCG, Mycobacterium abscessus, Mycobacterium avium, Mycobacterium fortuitum, Mycobacterium porcinum and Mycobacterium simiae. The disease manifests in early childhood, with severe infections invariably occurring before the age of five years, and involving the development of poorly defined multibacillary granulomas. AR complete IFN-γR2 deficiency thus has a very poor prognosis, with hematopoietic stem cell transplantation (HSCT) as the only known curative treatment, although gene therapy is a potentially attractive option for the future (2,24).
Partial, as opposed to complete, IFN-γR2 deficiency can present in two forms. AR partial IFN-γR2 deficiency has been reported in six patients from six kindreds and four countries homozygous for the p.R114C (8), p.G227R (13), p.S124F, p.G141R (16) or c.958insT (15) mutation, all of which underlie modest but detectable IFN-γR2 expression and function. Indeed, these patients display similar levels of impairment of cellular responses to IFN-γ, but not a total abolition of these responses. All these mutations are hypomorphic, as opposed to LOF. Four of these mutant alleles (p.R114C, p.G227R, p.S124F and p.G141R) have been shown to encode misfolded proteins that are mostly retained in the endoplasmic reticulum (16). Cellular responses to IFN-γ can be rescued by treating the patients’ cells with glycosylation inhibitors (16). The partial form of AR IFN-γR2 deficiency is clinically milder than the complete form, with relatively mild infections, albeit with an early onset, before the age of 8 months in the small sample of known patients (16). Plasma IFN-γ levels are high, but the clinical outcome is relatively good, given that six patients with AR complete IFN-γR2 deficiency have died (14,15,19–21), versus only two patients with AR partial IFN-γR2 deficiency (13,15). Furthermore, six patients with AR complete IFN-γR2 deficiency have undergone HSCT (10–12,14,17), versus only one patient with AR partial IFN-γR2 deficiency (15). Antibiotics may be sufficient to prevent and treat infections and the response to recombinant IFN-γ is good (2). Autosomal dominant (AD) partial IFN-γR2 deficiency by haploinsufficiency has been reported in only one MSMD patient with a mild form of localized BCG disease (14). This patient carried a heterozygous frame-shift IFNGR2 mutation. IFN-γR2 levels were lower than in healthy individuals and the patient’s cells displayed mildly impaired responses to IFN-γ (14). This defect has low clinical penetrance, as only one of the 18 heterozygous relatives tested had MSMD (14). Another heterozygous IFNGR2 mutation identified in a healthy subject was shown to be dominant-negative at the cellular level (17). In this context, we studied three new patients with MSMD from India and Turkey, with molecular defects defining a novel form of AR partial IFN-γR2 deficiency.
Results
Identification of IFN-γR2 deficiency
We investigated three MSMD patients from two unrelated kindreds (Figs. 1A and S1). P1 and P2 were born to consanguineous parents in Turkey and P3 was born to consanguineous parents in India. Whole-exome sequencing (WES) was carried out on the patients’ genomic DNA (gDNA). We searched for rare variants (Minor Allele Frequency < 0.01) of known MSMD-causing genes. In kindred A (P1 and P2), we found a homozygous nucleotide substitution at position c.1A>G of IFNGR2 (Fig. 1B), leading to the replacement of the first methionine (M1)-encoding codon with a valine-encoding codon (Fig. 1C). In kindred B (P3), we identified a homozygous nucleotide deletion, c.4delC (Fig. 1B), predicted in silico to create a premature stop codon 22 amino acids after the M1 (Fig. 1C). We mined public databases (Genome Aggregation Database (GnomAD), Exome Aggregation Consortium (ExAC) and BRAVO) and found that the c.1A>G mutation was reported in the heterozygous state in one European individual in GnomAD (of 14 324 individuals, 7150 of whom were European), whereas the c.4delC mutation was not reported in any of these databases. Both the M1 residue (c.1ATG) and the arginine that follows it (R2, c.4CGA) are highly conserved in the species in which IFNGR2 has been sequenced (Fig. S2A). These mutations had a high combined annotation-dependent depletion score (25), higher than the mutation significance cutoff for IFNGR2 (26) (Fig. S2B). All mutations were confirmed by Sanger sequencing. The parents of kindreds A and B were heterozygous for the corresponding mutation (Fig. S2C). No DNA sample was available from the siblings of P3. These mutations therefore probably underlie AR MSMD by causing complete or partial IFN-γR2 deficiency.
Figure 1.
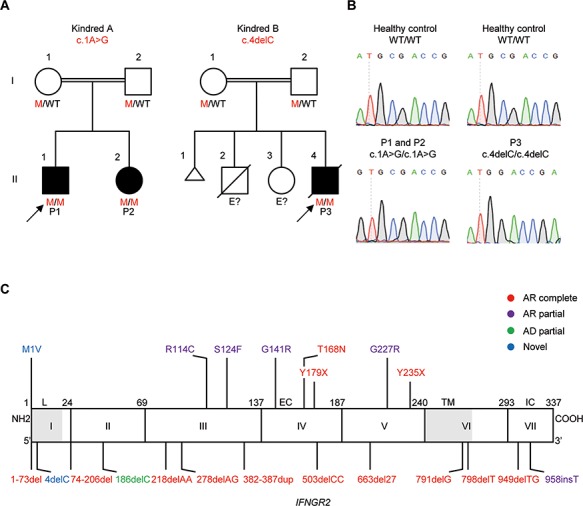
Identification of three new patients with severe AR partial IFN-γR2 deficiency and MSMD. (A) Familial segregation of the c.1A>G and c.4delC mutations (mutations marked in red). Kindred A is from Turkey and Kindred B is from India. E? indicates an ungenotyped individual. Healthy individuals are shown in white. Solid black shapes indicate patients with MSMD. The probands are indicated by arrows. Each kindred is designated by a capital letter (A and B), each generation by a roman numeral (I and II), and each individual by an Arabic numeral. (B) Electropherogram showing the ATG-GTG mutation in P1 and P2, and the CGA-GA mutation in P3. (C) Schematic diagram of the IFNGR2 gene with all previously described mutations and the c.1A>G and c.4delC mutations described here (in blue). Coding exons are numbered with roman numerals and delimited by a vertical bar. Regions corresponding to the leader sequence (L, 1–21), extracellular domain (EC, 23–248), transmembrane domain (TM, 249–272) and intracellular domain (IC, 273–337) are indicated. Mutations marked in red cause AR complete IFN-γR2 deficiency. The mutations marked in purple cause AR partial IFN-γR2 deficiency. The mutation marked in green causes AD partial IFN-γR2 deficiency. The mutations marked in blue are described in this paper.
Impaired production of IFN-γR2 protein
We first characterized the c.1A>G and c.4delC alleles in vitro by overexpressing the corresponding cDNAs in HEK293-T cells or IFN-γR2-deficient primary human fibroblasts immortalized with SV-40 T antigen [SV40-fibroblasts (SV40F)] (9) (homozygous for the c.278delAG mutation). We transiently transfected cells with wild-type (WT), c.1A>G and c.4delC IFNGR2 cDNAs, or with an empty plasmid (EV), and analyzed the corresponding products by western blotting. IFNGR2 c.278delAG, which has been reported to be a loss-of-expression and LOF mutation, was included as a negative control (9). As expected, the WT-IFNGR2 plasmid encoded a single full-length 70 kDa protein, and no protein was detected for the negative control and the EV (Fig. 2A). Interestingly, both the c.1A>G and c.4delC mutations resulted in the production of a single full-length 70 kDa protein, albeit in smaller amounts than the WT (Fig. 2A). IFN-γR2 is a type I transmembrane protein, and its glycosylation is an important post-translational modification (27). Glycosylation ensures that the protein is correctly folded and binds correctly to its ligands. It results in a range of molecular weights (MWs) for this and other membrane proteins (22). The lysates from cells transiently transfected with the WT, c.1A>G, c.4delC and c.278delAG constructs were treated with peptide N-glycosidase-F (PNGase-F) to remove N-glycans (28). The same patterns of expression were obtained, with c.1A>G and c.4delC producing products of similar MW (approximately 45 kDa) but in smaller amounts than the WT (Fig. S3). Similar results were obtained with HEK293-T cells and IFN-γR2-deficient SV40F (data not shown). These findings suggest that even though one of these mutations affects the M1 and the other creates an upstream frameshift, the encoded messenger Ribonucleic acids (mRNAs) seem to be correctly translated, resulting in the production of an apparently full-length protein. These findings also confirm that the two mutations are pathogenic, resulting in complete or partial AR IFN-γR2 deficiency in the patients.
Figure 2.
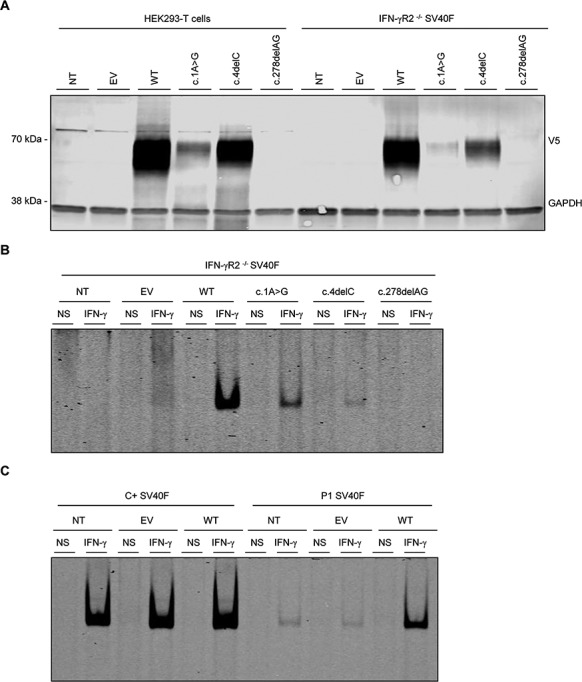
IFN-γR2 protein levels, impaired STAT1-DNA-binding activity in response to IFN-γ stimulation in vitro and complementation of the IFN-γ response with WT IFNGR2. (A) HEK293-T cells or IFN-γR2 deficient SV40F (IFN-γR2-/- SV40F) were left NT) or were transiently transfected with EV, WT, c.1A>G mutant, c.4delC mutant and c.278delAG (negative control) IFNGR2 plasmids. Total lysis and western blots were performed with anti-V5 antibody, with GAPDH as the loading control. (B) IFN-γR2-/- SV40F were left NT or were transiently transfected with EV, WT, c.1A>G mutant, c.4delC mutant and negative control IFNGR2 plasmids, and were either left non-stimulated (NS) or stimulated with 105 IU/ml IFN-γ (IFN-γ). DNA-binding activity was then analyzed by EMSA with a gamma activated sequence (GAS) probe in LI-COR, A700. (C) SV40F from a healthy control and P1 were left NT or were transiently transfected with EV and WT IFNGR2 plasmid and were either left non-stimulated (NS) or stimulated with 105 IU/ml IFN-γ (IFN-γ). DNA-binding activity was then analyzed by fluorescence EMSA with a GAS probe in LI-COR, A700. The results shown are representative of three independent experiments.
Defective IFN-γ response in IFNGR2 mutants
We then assessed the functional consequences of c.1A>G and c.4delC mutations. We transiently transfected IFN-γR2-deficient SV40F with EV, WT, c.1A>G, c.4delC and c.278delAG. We then stimulated these cells with IFN-γ (105 IU/ml) for 20 min. Nuclear proteins were extracted and gamma-activated factor (GAF)-DNA binding activity was assessed in an electrophoretic mobility shift assay (EMSA) with a gamma-activating sequence (GAS) probe. As expected, binding activity was strong in cells transfected with the WT-IFNGR2 plasmid but absent in cells transfected with EV or the negative control (Fig. 2B). Cells transfected with c.1A>G or c.4delC cDNA had impaired, but not abolished, responses to IFN-γ (Fig. 2B). These findings are consistent with the levels of the corresponding proteins, demonstrating that both the c.1A>G and c.4delC IFNGR2 mutations impair, but do not abolish, the production and function of IFN-γR2 proteins. For comparison of the observed activity with that in previously reported forms of partial AR deficiency, we tested the five corresponding hypomorphic mutations (p.G227R, p.G141R, p.S124F, p.R114C and c.958insT) (8,13,15,16). After stimulation, the mean level of binding followed a gradient WT > p.G227R > c.1A>G > p.G141R> c.958insT> p.S124F> p.R114C> c.4delC> c.278delAG (Fig. S4). Overexpressed c.1A>G showed one of the strongest bindings and c.4delC showed the lowest binding, whereas p.G227R and c.1A>G were the least deleterious on overexpression of all the alleles tested. We then transfected SV40F from P1 (cells from P3 were not available) with WT IFNGR2 and evaluated their response to IFN-γ by EMSA. GAF-binding activity was restored in the transfected cells, reaching higher levels to those observed in when compared with EV-transfected and untransfected cells (NT) (Fig. 2C). These results strongly suggest that the c.1A>G mutation of IFNGR2 is responsible for the poor response to IFN-γ observed in the overexpression system. Collectively, these data indicate that the patients from the two kindreds had a severe form of AR partial IFN-γR2 deficiency.
Impaired STAT1 phosphorylation following the stimulation with IFN-γ of SV40F from P1 and P2
A mutation of the first ATG of IFNGR1 leading to the replacement of the M1 by a lysine residue (p.M1K) has already been described in a patient with MSMD (29). In Epstein–Barr virus-transformed B (EBV-B) cells homozygous for this IFNGR1 mutation, the response to IFN-γ is impaired but not abolished. By contrast, this response is completely abolished in SV40F homozygous for this mutation. We compared the early responses to IFN-γ and IFN-α (as a control for stimulation) of SV40F from a healthy control, P1, P2 (cells from P3 being unavailable), a patient with AR complete IFN-γR2 deficiency (homozygous for the c.278delAG mutation, hereafter referred to as the negative control), a patient with AR complete IFN-γR1 deficiency (compound heterozygous for the c.104_107dupTTAC and c.200+1G>A mutations), three patients with AR partial IFN-γR2 deficiency (8,16) (corresponding to p.S124F, p.G141R and p.R114C) and the patient with AR partial IFN-γR1 deficiency due to the p.M1K mutation (hereafter referred to as M1K) (Figs. 3A and S5). We performed western blotting to assess the ability of these cells to phosphorylate the tyrosine 701 residue of STAT1 (p-Y701-STAT1) after stimulation with IFNs. The healthy control produced phosphorytlation of STAT1 (p-STAT1) upon on stimulation with IFN-γ, whereas the negative control, the cells with AR complete IFN-γR1 deficiency and cells from the M1K-IFN-γR1 patient displayed no response (Fig. 3A). Cells from the three patients with AR partial IFN-γR2 deficiency had impaired, but not abolished, responses to IFN-γ. P1 and P2 displayed equally strong decreases in response to IFN-γ (Figs. 3A and S5), but their responses were not abolished, just weaker than those of the other three patients with AR partial deficiency (Fig. 3A). However, the response was detectable, unlike that in cells from the patient with a mutation of the initiation codon of IFNGR1. Phosphorylation of STAT1 after IFN-α stimulation was found to be similar in all cell lines (Figs. 3A and S5). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels were also similar across cell lines, demonstrating that the differences in STAT1 phosphorylation were not caused by differences in protein loading. The IFN-γ signaling pathway was impaired, but not abolished in the SV40F of P1 and P2, whereas M1K-IFN-γR1 SV40F displayed a complete abolition of IFN-γ signaling in terms of STAT1 phosphorylation.
Figure 3.
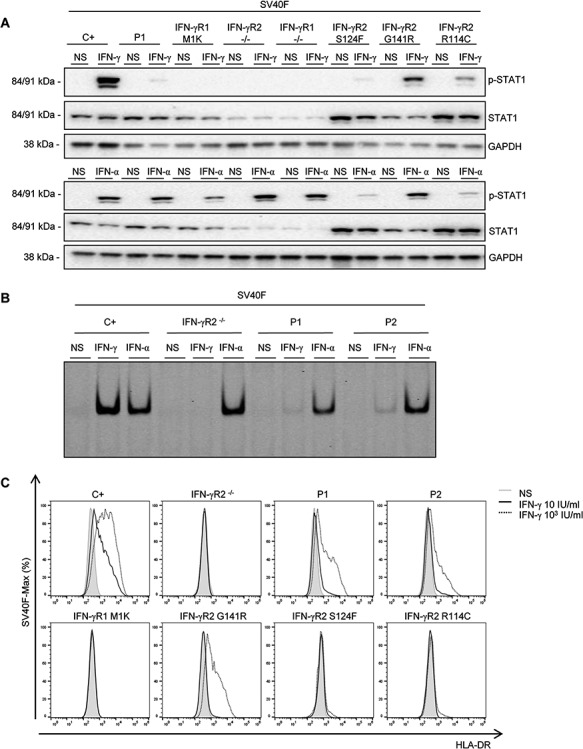
Impaired response of P1’s SV40F to IFN-γ stimulation. (A) SV40F from a healthy control (C+), an IFN-γR2-deficient patient (IFN-γR2-/-), an IFN-γR1-deficient patient (IFN-γR1-/-), P1, a patient with a mutation affecting the M1 of IFN-γR1 (IFN-γR1-M1K) and three patients with AR partial IFN-γR2 deficiency (S124F, G141R and R114C) were left unstimulated (NS) or were stimulated with 105 IU/ml IFN-γ (IFN-γ) or IFN-α (IFN-α) for 20 min. STAT1 phosphorylation on Tyr-701(p-STAT1), total STAT1 levels and levels of GAPDH (as a loading control) were measured by western blotting with specific antibodies. Upper panel: IFN-γ stimulation, lower panel: IFN-α stimulation. (B) SV40F from a healthy control (C+), an IFN-γR2-deficient patient (IFN-γR2-/-), P1 and P2 were left non-stimulated (NS) or were stimulated with 105 IU/ml IFN-γ (IFN-γ) or IFN-α (IFN-α) for 20 min and GAS probe-binding nuclear proteins were determined by fluorescence EMSA with LI-COR, A700. (C) SV40F from a healthy control (C+), an IFN-γR2-deficient patient (IFN-γR2-/-), P1, P2, the patient with a mutation affecting the M1 of IFN-γR1 (IFN-γR1-M1K) and three patients with AR partial IFN-γR2 deficiency (S124F, G141R and R114C) were stimulated with the indicated doses of IFN-γ for 48 h. HLA-DR induction was determined by flow cytometry. Overlapping histograms relative to the mode are shown. Gray area: no stimulation (NS), continuous black line: 10 IU/ml IFN-γ, discontinuous black line: 103 IU/ml IFN-γ. The results shown are representative of three independent experiments.
Impaired DNA binding upon the stimulation with IFN-γ of SV40F from P1 and P2
We next studied the ability of IFN-γ to stimulate the formation of GAS-binding nuclear complexes by EMSA. We performed the same activation as for the western blot, in SV40F. The healthy control displayed binding activity after stimulation with IFN-γ, whereas no binding was observed for the negative control (Fig. 3B). P1 and P2 displayed equally strongly diminished, but detectable, binding activity after IFN-γ stimulation. Binding after IFN-α stimulation was similar for all cell lines (Fig. 3B), confirming the western blot results. We then analyzed later events in the IFN-γ pathway and measured the induction of Human Leukocyte Antigen-antigen D Related (HLA-DR) after stimulation with two concentrations of IFN-γ (10 or 103 IU/ml) for 48 h. The cells from the healthy control displayed an increase in HLA-DR levels after stimulation with the lower concentration of IFN-γ and a larger increase after stimulation with the higher dose, whereas the negative control and M1K-IFN-γR1 cells displayed no such increase with either concentration (Fig. 3C). No HLA-DR induction was observed in S124F or R114C cells, for either of the IFN-γ concentrations tested (Fig. 3C). Low concentrations of IFN-γ did not induce HLA-DR expression in G141R cells, whereas higher concentrations did, albeit to a lesser extent than in control cells (Fig. 3C). The results obtained for these patients with AR partial IFN-γR2 deficiency are consistent with previous reports (16). Cells from P1 and P2 displayed a slight increase in HLA-DR expression in the presence of low concentrations of IFN-γ, greater than that observed for the other three patients with AR partial IFN-γR2 deficiency tested, and a stronger increase in response to higher concentrations, much smaller than that for the healthy control but similar to that for the G141R patient and greater than that for the S124F and R114C patients (Fig. 3C). Thus, both early and late events in the IFN-γ signaling pathway are impaired but not abolished in the SV40F of P1 and P2, whereas M1K-IFN-γR1 SV40F display a complete abolition of IFN-γ signaling.
Impaired STAT1 phosphorylation upon IFN-γ stimulation in Epstein Barr virus-transformed B cells from P1, as shown by western blotting
We then characterized Epstein Barr virus-transformed B (EBV-B) cells from P1 (no EBV-B cells were available for P2 and P3). We analyzed the early response to IFN-γ and IFN-α of cells from a healthy control, P1, the patient with AR complete IFN-γR2 deficiency (homozygous for c.278delAG), a patient with AR complete IFN-γR1 deficiency (homozygous for the c.202-1G>T mutation), S124F, G141R, R114C, a patient with AD partial IFN-γR2 deficiency (14) (heterozygous for the c.186delC mutation) and the patient with M1K-IFN-γR1 deficiency. We first assessed the ability of these cells to phosphorylate STAT1 after stimulation with IFNs, as measured by western blotting. For the healthy control, p-STAT1 was detected after stimulation with IFN-γ, whereas the negative control, the patient with AR complete IFN-γR1 deficiency and the M1K patient displayed no response (Fig. 4A). P1 had a severely impaired, but not abolished, response to IFN-γ (Fig. 4A). The three patients with partial IFN-γR2 deficiency described in previous studies and the patient with the AD c.186delC variant had impaired, but not abolished, responses to IFN-γ (Fig. 4A). STAT1 phosphorylation in response to IFN-α was found to be similar in all cell lines (Fig. 4A). Tubulin levels were also similar across cell lines, demonstrating that the differences in STAT1 phosphorylation were not caused by differences in protein loading. The western blot of EBV-B cells from P1 demonstrated a severe impairment of the IFN-γ signaling pathway, in terms of STAT1 phosphorylation.
Figure 4.
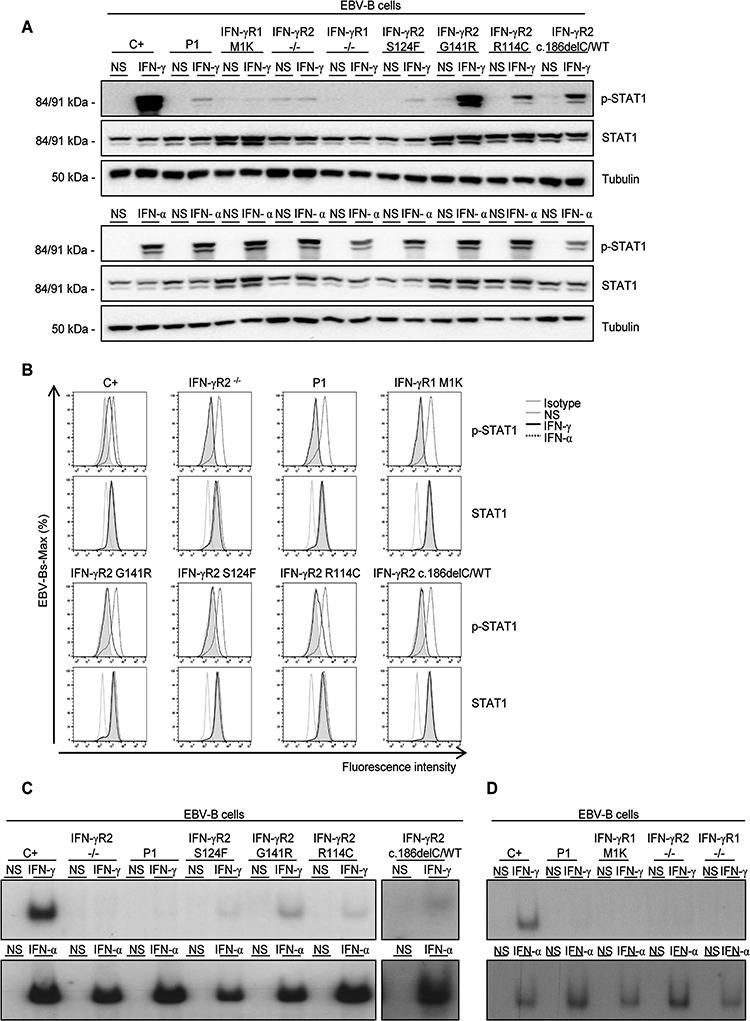
Response of P1’s EBV-B cells to IFN-γ stimulation. (A) EBV-B cells from a healthy control (C+), an IFN-γR2-deficient patient (IFN-γR2-/-), an IFN-γR1-deficient patient (IFN-γR1-/-), P1, a patient with a mutation affecting the M1 of IFN-γR1 (IFN-γR1-M1K), three patients with AR partial IFN-γR2 deficiency (S124F, G141R and R114C) and a patient with AD partial IFN-γR2 deficiency (c.186delC) were left non-stimulated (NS) or were stimulated with 105 IU/ml IFN-γ (IFN-γ) or IFN-α (IFN-α) for 20 min. Phosphorylation of STAT1 on Tyr-701 (p-STAT1), total STAT1 levels and levels of tubulin (as loading control) were determined by western blotting with specific antibodies. Upper panel: IFN-γ stimulation, lower panel: IFN-α stimulation. (B) Phosphorylation of STAT1 on Tyr-701 (p-STAT1) and total STAT1 levels were measured by flow cytometry in the same cells with and without stimulation with 105 IU/ml IFN-γ or IFN-α for 30 min. Overlapping histograms relative to the mode are shown. Gray line: isotype control or no stimulation (NS), continuous black line: 105 IU/ml IFN-γ, discontinuous black line: 105 IU/ml IFN-α(C) EBV-B cells from a healthy control, a negative control, P1, three patients with AR partial IFN-γR2 deficiency and a patient with AD partial IFN-γR2 deficiency were left non-stimulated (NS) or were stimulated with 105 IU/ml IFN-γ (IFN-γ) or IFN-α (IFN-α) for 20 min. GAS probe-binding nuclear proteins were determined by radioactive EMSA with 32P-dATP. Upper panel: IFN-γ stimulation, lower panel: IFN-α stimulation. The results shown are representative of three independent experiments. Space between images has been inserted to indicate a different gel. (D) EBV-B cells from a healthy control, P1, the IFN-γR1 M1K patient, a negative control for IFN-γR2 (IFN-γR2-/-) and a negative control for IFN-γR1 (IFN-γR1-/-) were left non-stimulated (NS) or were stimulated with 105 IU/ml IFN-γ (IFN-γ) or IFN-α (IFN-α) for 20 mi. GAS probe-binding nuclear proteins were determined by radioactive EMSA. Upper panel: IFN-γ stimulation, lower panel: IFN-α stimulation. The results shown are representative of three independent experiments.
Impaired STAT1 phosphorylation upon IFN-γ stimulation in EBV-B cells from P1, as demonstrated by flow cytometry
We also evaluated p-STAT1 levels by flow cytometry. Following the same activation procedure as for the western blot, we stained the cells for p-STAT1 and total STAT1 (as a control to check that basal levels of STAT were similar in all cells) after stimulation with IFN-γ or IFN-α. The healthy control displayed an increase in p-STAT1 levels after IFN-γ stimulation, whereas the negative control did not (Fig. 4B). The M1K-IFN-γR1 cells displayed a slight increase after stimulation with IFN-γ, as previously reported (29) (Fig. 4B), and a similar increase was observed in the cells of P1 (Fig. 4B). Moreover, the three patients with AR (R114C, G141R and S124F) and AD partial IFN-γR2 deficiency (c.186delC) displayed slight increases in p-STAT1 levels after IFN-γ stimulation, greater than those observed for M1K-IFN-γR1 cells and cells from P1, but lower than that observed for the healthy control (Fig. 4B). These results suggest that the IFNGR2 c.1A>G mutation behaves like IFNGR1 M1K in EBV-B cells.
Abolition of DNA-binding activity upon IFN-γ stimulation in EBV-B cells from P1
We investigated the ability of STAT1 homodimers to bind the GAS probe, by EMSA on EBV-B cells. We performed the same activation as for the western blot in cells from a healthy control, the negative control, the patient with AR complete IFN-γR1 deficiency, P1, patients with AR partial IFN-γR2 deficiency (S124F, G141R, R114C, c.186delC) and IFN-γR1-deficient (M1K) cells (Fig. 4C and D). As expected, the healthy control displayed binding activity after stimulation with IFN-γ, unlike the negative control and the patient with AR complete IFN-γR1 deficiency (Fig. 4C and D). Consistent with published findings, G141R, S124F, R114C and c.186delC displayed impaired but not abolished binding to GAS after IFN-γ stimulation (Fig. 4C). P1 and the M1K IFN-γR1 patient displayed no detectable GAS binding (Fig. 4D). Binding after IFN-α stimulation was essentially similar in all cell lines (Fig. 4C and D). These findings are consistent with the hypothesis that IFNGR2 c.1A>G mimics IFNGR1 M1K in EBV-B cells. However, for P1, the defect in EBV-B cells is more pronounced than that in SV40F, whereas the reverse is true for the M1K IFN-γR1 patient. The impact of the mutations affecting the M1 residues of the two chains of the IFN-γ receptor seems to depend on the chain mutated and the cell type tested.
Impairment of IFN-γ signaling in fresh leukocytes
IFN-γR2-deficient patients had high plasma IFN-γ levels, but little or no production of IL-12p40 and IL-12p70 in whole blood following stimulation with BCG and IFN-γ (2). We measured plasma IFN-γ levels in P1, P2 and P3 by ELISA, and compared with the results obtained with those for healthy controls and patients with complete or partial IFN-γR2 deficiency (Fig. 5A). The healthy controls and the patient with AD partial IFN-γR2 deficiency had no detectable IFN-γ in their plasma, whereas P1, P2 and P3 had high plasma IFN-γ levels, similar to those in patients with AR complete or partial IFN-γR2 deficiency (Fig. 5A). We also assessed IFN-γ, IL-12p40 and IL-12p70 production in whole blood from healthy controls, P1, and P2 (P3 was not available) after 48 h of stimulation with live BCG, with or without IFN-γ or IL-12, as previously described (30,31), comparing the results obtained with those for patients with complete or partial IFN-γR2 deficiency (Figs. S6A and B and 5B). Healthy controls, IFN-γR2-deficient patients, P1, and P2 displayed similar levels of IFN-γ secretion after BCG stimulation and higher levels of secretion after stimulation with BCG and IL-12 (Fig. S6A). All the individuals tested produced IL-12p40 after BCG stimulation, but no IL-12p70 (Figs. S6B and 5B). After stimulation with both BCG and IFN-γ, the healthy controls displayed larger increases in IL-12p40 production than were observed after stimulation with BCG alone (Fig. S6B), together with the induction of IL-12p70 production (Fig. 5B). By contrast, patients with AR complete IFN-γR2 deficiency produced no extra IL-12p40 in response to BCG plus IFN-γ, relative to BCG alone, and no response in terms of IL-12p70 production was observed in response to either type of stimulation (Figs. S6B and 5B). Patients with AR partial IFN-γR2 deficiency displayed an increase in IL-12p40 levels following stimulation with BCG plus IFN-γ, although this increase was smaller than that observed for controls (Fig. S6B). Patients with an AR partial defect produced no IL-12p70, whereas those with an AD partial defect displayed normal levels of IL-12p70 production (Fig. 5B). P1 and P2 also displayed increases in IL-12p40 production (Fig. S6B) in response to BCG plus IFN-γ, and no IL-12p70 production (Fig. 5B), like known patients with AR partial IFN-γR2 deficiency. Thus, the IFN-γR2 deficiency in P1 and P2 is more severe than that in previously reported patients with AR partial IFN-γR2 deficiency, but less severe than that in patients with AR complete IFN-γR2 deficiency.
Figure 5.
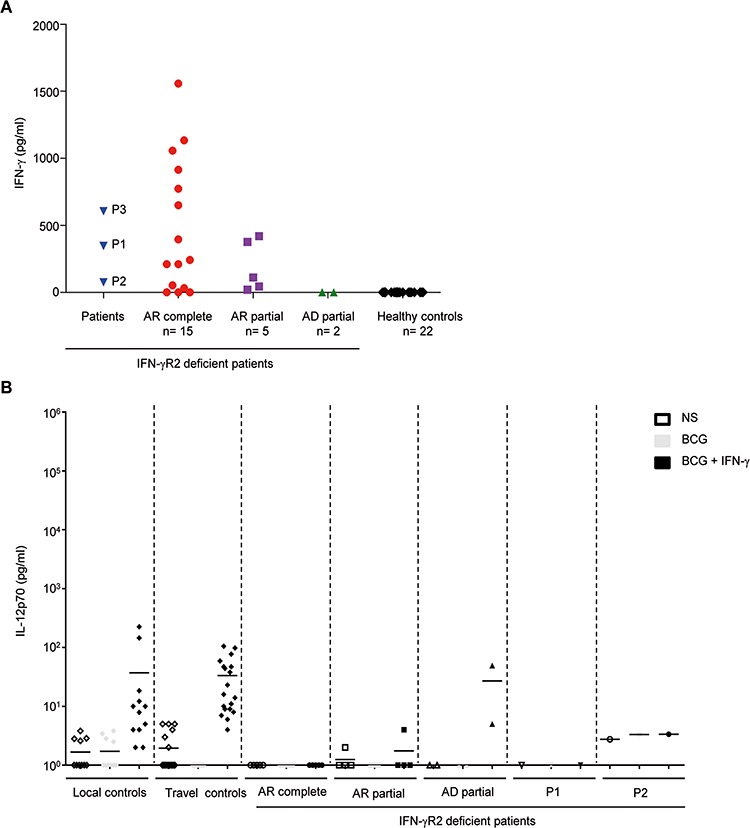
Impaired response to IFN-γ in terms of the production of IL-12p70 in the whole-blood supernatant from the patients. (A) IFN-γ levels in plasma from P1, P2, P3, AR complete IFN-γR2 deficient patients (n = 16), AR partial IFN-γR2 deficient patients (n = 5), AD partial patient (n = 2) and healthy controls (n = 22). (B) IL-12p70 in whole blood cells from local controls, travel controls, AR complete IFN-γR2 deficient patients, AR partial IFN-γR2 deficient patients, AD partial patient, P1 and P2 either not stimulated (NS) or stimulated for 48 h with live BCG alone or live BCG plus IL-12 or IFN-γ, as assessed by ELISA. Each symbol represents a value from an independent test and horizontal bars represent means.
Study of individual leukocyte subsets
We then investigated the consequences of the c.1A>G mutation in fresh leukocyte subsets from two healthy controls, P1 and P2, and a patient with AR complete IFN-γR2 deficiency (c.278delAG mutation). p-STAT1 levels in the naïve CD4+ T cells and memory IL-4 producing T cells of patients with AD partial IFN-γR2 deficiency have been reported to respond poorly to IFN-γ, whereas no such impairment has been observed in monocyte-derived macrophages (MDMs) (14). We tested naïve CD4+ T cells and MDMs generated by stimulating monocytes in vitro with macrophage colony-stimulating factor (M-CSF) plus IL-4 (no monocytes were available for the patient with AR complete IFN-γR2 deficiency). The various cell subsets were stimulated with IFN-γ and IFN-α, as previously described (14). In naïve CD4+ T cells, low levels of STAT1 phosphorylation were observed in P1 and P2 after IFN-γ stimulation, whereas p-STAT1 levels were high in healthy controls and no STAT-1 phosphorylation was detected in the patient with IFN-γR2 deficiency (Fig. 6A). MDMs from P1 and P2 displayed low levels of STAT-1 phosphorylation after IFN-γ stimulation, much lower than those in healthy controls, but phosphorylation was not completely abolished (Fig. 6B). The levels of p-STAT1 after IFN-α stimulation were similar in all cell types (T lymphocytes and MDMs), for patients and controls (Fig. 6A and B). Total STAT1 levels were similar in all situations and GAPDH or tubulin were used as loading controls (Fig. 6A and B). These results are consistent with published data (14), with the c.1A>G mutation seeming to affect cell types normally expressing low levels of IFN-γR2 at their surface most strongly. They also support the hypothesis that the c.1A>G mutation confers partial IFN-γR2 activity at levels lower than reported for the known hypomorphic mutations underlying AR partial IFN-γR2 deficiency.
Figure 6.
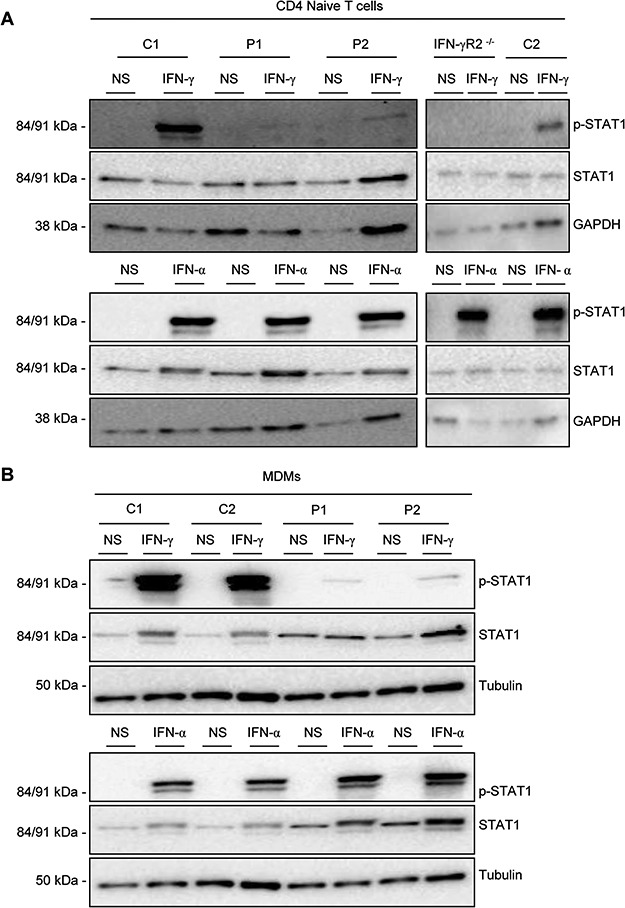
Functional study of T cells and MDMs. (A) CD4+ naïve T cells from two healthy controls (C1 and C2), an IFN-γR2-deficient patient (IFN-γR2-/-), P1 and P2 were isolated and left non-stimulated (NS) or were stimulated with 105 IU/ml IFN-γ (IFN-γ) or IFN-α (IFN-α) for 20 min. Phosphorylation of STAT1 on Tyr-701(p-STAT1), total STAT1 levels and levels of GAPDH (as loading control) were measured by western blotting with specific antibodies. Upper panel: IFN-γ stimulation, lower panel: IFN-α stimulation. Space between images has been inserted to indicate a different gel. (B) CD14+ cells from a healthy control (C1), P1 and P2 were isolated and induced to differentiate into MDMs. They were then left non-stimulated (NS) or were stimulated with 105 IU/ml IFN-γ (IFN-γ) or IFN-α (IFN-α) for 20 min. Phosphorylation of STAT1 on Tyr-701 (p-STAT1), total STAT1 levels and levels of tubulin (as a loading control) were determined by western blotting with specific antibodies. Left panel: IFN-γ stimulation, right panel: IFN-α stimulation.
Lack of translation re-initiation at a distal AUG
Several studies have shown that the most common start site for translation in mammalian mRNAs is an AUG codon in a favorable context for translation initiation −3 A or G and +4 G, collectively defined as a Kozak consensus sequence (32−34). The c.1A>G and c.4delC mutations underlie a new form of AR partial IFN-γR2 deficiency, in which a normal receptor is expressed, but in very small amounts. It is a quantitative defect, as opposed to the qualitative defects previously reported for the p.G227R, p.G141R, p.R114C, p.S124F and c.958insT mutations. We therefore investigated the mostly likely codon position for initiation of the residual IFN-γR2 expression. We searched the IFNGR2 sequence for AUG codons downstream from c.1ATG with appropriate characteristics, using the Prediction of Translation Initiation ATG Web site (http://atgpr.dbcls.jp) (35). The next AUG predicted to be a suitable site for the re-initiation of translation (and the next AUG in the sequence) is located at position c.238, corresponding to amino acid 80. The use of this initiation codon would result in the loss of the leader sequence and part of the extracellular domain (Fig. 1C). We nevertheless constructed several plasmids—‘Start’ [c.1delATG (Start 2), c.1delATGC (Start 2 + c.4delC), c.238AT>GC (p.M80A), c.1_237del (Start80) and Start 2 + p.M80A]—with the aim of identifying the translation start site after position c.4 and investigating the likelihood of IFN-γR2 protein expression following deletion of the first 237 base pairs (first 79 amino acids) or the replacement of the methionine at this position by an alanine (Fig. 7A). All the mutants except Start80 generated products with the usual MW but in much smaller amounts than WT IFNGR2, although the intensity of the band obtained was nevertheless stronger than that for c.1A>G (Fig. 7B). No product was obtained with Start80, or with c.278delAG, which was used as a negative control (Fig. 7B). As expected, these data ruled out the re-initiation of translation by the Kozak codon c.238.
Figure 7.
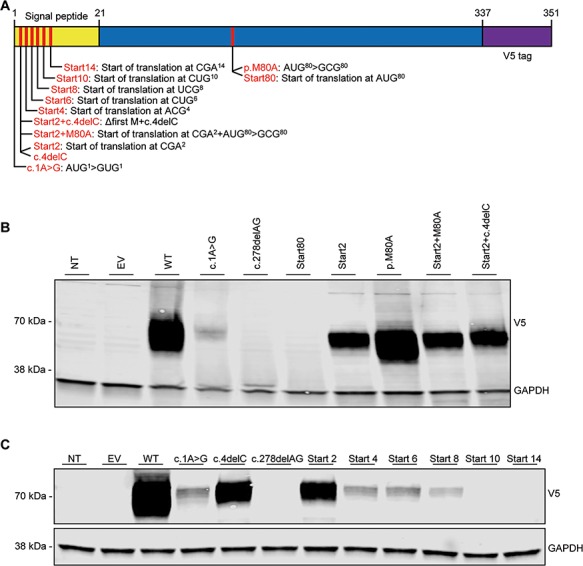
Identification of potential initiation codons. (A) Schematic representation of the IFNGR2 gene inserted into the pcDNA3.1-V5 plasmid. The positions of various translation start codon candidates in the signal peptide and performed mutagenesis are indicated. (B) HEK293-T cells were either left NT or were transiently transfected with EV, WT IFNGR2, c.1A>G, c.4delC, c.278delAG, Start80, Start2, p.M80A, Start2 combined with p.M80A or Start2 combined with c.4delC. After 48 h, the cells were lysed and total lysates were subjected to western blotting with an anti-V5 antibody, with GAPDH as the loading control. (C) HEK293-T cells were either left NT or were transiently transfected with EV, WT IFNGR2, c.1A>G, c.4delC, c.278delAG, Start2, Start4, Start6, Start8, Start10 and Start14. After 48 h, the cells were lysed and total lysate was subjected to western blotting with an anti-V5 antibody, with GAPDH as the loading control. The results shown are representative of three independent experiments.
Re-initiation of translation at a proximal non-canonical codon
Instead, these data suggest that the c.1A>G and c.4delC mutants are hypomorphic due to the initiation of translation at a non-AUG codon, probably within the leader sequence (signal peptide) of IFN-γR2, and even possibly, in the case of c.1A>G, by the first codon itself (GUG) (36). It has been shown that translation can be initiated at non-AUG codons, such as CUG, GUG and ACG (37–39). An analysis of the IFNGR2 sequence identified an ACG codon at position c.10, two consecutive CTG codons starting at position c.13 and five consecutive CTG codons starting at position c.25. Using a signal peptide predictor (http://www.predisi.de/), PrediSi (40), we deleted IFN-γR2 amino acids in silico from M1 until no signal peptide was predicted, which occurred at L10 (c.28CTG). We then created plasmids starting from different non-AUG codons until the predicted loss of the signal peptide (Start 2, 4, 6, 8, 10 and 14) (Fig. 7A). IFN-γR2 of the expected MW (70 kDa) was produced for the mutations encompassing the start at amino acids 2, 4, 6 and 8, but at lower levels than the WT (Fig. 7C). The Start2 mutation generated levels of protein similar to those obtained for the c.4delC mutation, whereas Start4 and Start6 generated protein levels similar to those obtained for the c.1A>G mutation; the amount of protein produced from Start8 was smaller than that for either of the mutations reported in this study (Fig. 7C). Neither Start10 nor Start14 generated any detectable protein, consistent with the in silico predictions (Fig. 7C). These findings suggest that several codons, all located within the signal sequence, are involved in the translation of IFNGR2, and that the c.1ATG codon is the strongest site for the start of translation. This hypothesis was confirmed by immunoprecipitation and protein sequencing of the overexpressed IFN-γR2 WT, c.1A>G and c.4delC. A full-length protein extending from positions 22 (the position after signal peptide cleavage) to 337 (last amino acid of IFN-γR2) was detected for the three lysates tested (Fig. S7). Thus, the two new mutations described here produce a full-length mature IFN-γR2 protein, at lower levels than the WT protein, due to the initiation of translation by non-AUG codons located between codons 2 and 9 within the 21 amino acid signal peptide.
Discussion
We report the following two new mutations of IFNGR2 in three MSMD patients: c.1A>G in P1 and P2 from a Turkish family, and c.4delC in P3 from an Indian family. Together, they define a novel form of AR partial IFN-γR2 deficiency that is purely quantitative, with the expression on the cell surface of very small numbers of normal receptor chains, contrasting with the patients described in previous studies, who displayed cell-surface expression of small number of receptors bearing amino acid substitutions. This report is novel in that it defines a new type of AR partial IFN-γR2 deficiency, caused by mutations decreasing the number of receptors expressed on the cell surface, without affecting their nature. These two IFN-γR2 mutants can be overexpressed, albeit to much lower levels than the WT, resulting in a severe impairment of functional activity, but not its total abolition. Both SV40F and EBV-B cells from P1 and P2, and, by inference from overexpression studies, cells from P3 as well, displayed a much more severe impairment of STAT1 and GAF activation than cells from patients with other forms of AR partial IFN-γR2 deficiency. Analyses of fresh leukocyte subsets from P1 and P2 showed that STAT1 phosphorylation was severely impaired in naïve CD4+ T cells and MDMs. The cellular phenotype in these patients is, therefore, more severe than that in patients with other known forms of AR partial IFN-γR2 deficiency, but less severe than that in patients with complete IFN-γR2 deficiency.
Like other patients with AR complete or partial IFN-γR2 deficiency, these patients had high plasma concentrations of IFN-γ. Their clinical phenotype appears to be intermediate, between that of patients with AR complete deficiency (9–11,14,15,17–21) and patients with AR partial deficiency due to missense mutations (8,13,15,16). The three patients presented clinical complications after BCG vaccination. P1 also presented multifocal osteomyelitis caused by BCG at the ages of 5 and 7 years. This condition was successfully treated and P1 is now 9 years old and healthy. P2 was treated for complications after BCG vaccination and was healthy at her last follow-up visit, at the age of 6 years. P3 died of Mycobacterium chelonei infection at the age of 5 years. The clinical phenotype of these patients, with the possible exception of P2, is thus globally more severe than that of other patients with AR partial IFN-γR2 deficiency. HSCT is therefore recommended for the two surviving patients. This treatment is generally considered for patients with complete but not partial IFN-γR2 deficiency (2,12).
Our study also shows that non-canonical initiation of IFN-γR2 translation can operate in cells lacking the canonical AUG codon upstream from the segment encoding the signal peptide. This is physiologically and clinically relevant, as these three patients have a milder phenotype than that observed in complete IFN-γR2 deficiency. During translation initiation in eukaryotic cells, the small subunit (40S) of the eukaryotic ribosome binds to the capped 5′-end of the mRNA. It then migrates, stopping at the first AUG codon in a favorable context for translation initiation (41). If the first ATG is mutated or the context is altered, translation may occur through re-initiation or context-dependent leaky scanning (42–45). In the IFNGR2 mRNA, the mutation of the first AUG to a GUG, or the loss of a C at position +4, prevents efficient translation initiation at this location. The next AUG codon downstream from the start codon is located 238 base pairs downstream. The use of this codon would lead to a loss of the leader sequence and 58 amino acids of the extracellular domain. Non-AUG codons, such as CUG, GUG and ACG, have been reported to initiate translation (29,37–39). The IFNGR2 gene has an ACG codon at position c.10, two consecutive CTG codons starting at position c.13 and five consecutive CTG codons starting at position c.25.
We have shown that the codons at amino acid positions 2, 4, 6 and 8 contribute to the residual translation of IFN-γR2 and, in the case of the c.1A>G mutation, that the GUG codon may also contribute to this process. The 12 to 19 of the 21 amino acids remaining in the signal peptide were sufficient for trafficking of the protein through the secretory pathway. The residual amounts of IFN-γR2 translated and transported to the cell surface were sufficient for a weak, but not entirely abolished, cellular response to IFN-γ. However, the shorter signal peptide was not as efficient as its full-length version, in terms of protein expression on the cell surface. Germline mutations of other signal peptides have previously been reported to underlie human genetic diseases, impairing protein secretion (46–48). Paradoxically, we found that the total expression of IFN-γR2 proteins encoded by the c.1A>G and c.4delC alleles is inversely correlated with GAS binding activity upon cell stimulation with IFN-γ (Figs. 2A and S4). Previous studies in prokaryotic cells have shown that protein secretion can be affected by the codon encoding the second amino acid position of the signal peptide (49–51). The shorter signal peptide of proteins encoded by c.4delC might thus result in intracellular accumulation of the mutant proteins, thereby accounting for lower levels of receptor expression on the cell surface, when compared with proteins encoded by c.1A>G. Unfortunately, currently available antibodies do not enable a robust detection of IFN-γR2 on the cell surface, preventing us from testing this hypothesis.
The re-initiation of translation downstream from the first AUG has been reported for other inborn errors of immunity [such as IFNGR1 (29), NEMO (45), RAG1 (52,53), FAC (54) and NBS1 (55) defects], and in other fields of human genetics (56–66). Most of these cases correspond to N-terminal frameshift mutations resulting in premature stop codons, similar to one of the IFNGR2 mutations reported here, c.4delC. In such cases, there is usually an AUG codon downstream from the frameshift, from which translation is re-initiated; this is not the case, however, for the c.4delC mutation. We are aware of only one case, for IFNGR1 (29), in which a missense mutation affecting the first AUG of the gene has been reported, as for the other IFNGR2 mutation reported here, c.1A>G. The re-initiation of translation in IFNGR1 has been reported to occur at downstream AUG codons, with additional contributions from non-AUG codons (29). By contrast, we describe here a genetic form of human disease due to the re-initiation of translation exclusively from non-AUG codons. Translation from non-AUG codons has already been shown to take place in neurodegenerative diseases, cancer and stress responses (67). This is the first example of such translation initiation to be reported for inborn errors of immunity. This form affects the function of IFN-γR2 differently in different cell types, because the efficiency of translation re-initiation at codons downstream from the first AUG is low and potentially different between cell types (41). The inefficiency of translation initiation from codons downstream from the first AUG may reflect differences in the amounts of endoplasmic reticulum and post-translational modification procedures between cell types (68) and tissue-specific differences in transfer-RNA profile (t-RNA) (37,69). These factors contribute to the observed re-initiation of translation downstream from the lost canonical AUG, highlighting the importance of this compensatory mechanism in human genetics. As translation re-initiation cannot be predicted in silico, mutations disrupting the canonical AUG should not be considered LOF until demonstrated to be so experimentally (70).
Materials and Methods
Case report
P1 (Kindred A, II.1) was born in 2009 to a consanguineous family from Turkey (Fig. 1A). He was vaccinated with BCG at 2 months of age. At 9 months of age, he developed local lymphadenitis, which was apparently resolved by antibiotics, but he had a relapse at 18 months of age. At the age of 5 years, he developed multiple cervical lymphadenopathies, deep neck abscesses and cervical bone lesions affecting movement of the neck. Serological tests for Salmonella were positive, but this bacterium was not isolated from blood cultures. At the age of 7 years, P1 was admitted to hospital for osteomyelitis in the cervix, thorax, lumbar spine and sacral bones (Fig. S1). Mycobacterium bovis-BCG was identified as the causal microorganism. The proportions of T, B and Natural Killer (NK) cells were normal, as was the results of dihydrorhodamine (DHR) assay on neutrophils for evaluation of the respiratory burst after activation with phorbol myristate acetate (PMA). A diagnosis of MSMD was suspected. P1 was treated with multiple antibiotics and subcutaneous IFN-γ, leading to clinical improvement. He is currently 9 years old and healthy.
P2 (Kindred A, II.2) was born in 2012, and she is the sister of P1 (Fig. 1A). She was vaccinated with BCG at the age of 2 months and developed vaccination-related suppurative lymphadenitis that drained spontaneously. No other infectious diseases have been detected in this patient. She was treated with multiple antibiotics and subcutaneous IFN-γ. Laboratory investigations showed normal proportions of T, B and NK cells for age; the respiratory burst, as evaluated in the DHR assay, was normal in neutrophils after PMA activation. Serum levels of IgG, IgA and IgM were normal for age. P2 is now 6 years old and is well, with no treatment.
P3 (Kindred B, II.4) was born in 2011 to second degree consanguineous parents (Fig. 1A). He was vaccinated with BCG at the age of 10 days. At the age of 3 months, he developed swelling of the left axillary lymph node, which was drained. Pathology examinations showed reactive follicular hyperplasia with no granulomas, but a blood smear tested positive for acid-fast bacilli (AFB). The treatment given at this time point was not recorded in the patient’s medical history. At the age of 11 months, P3 presented inguinal and axillary lymphadenitis and hepatosplenomegaly. Chest X-ray showed a persistent patch on the right middle lobe. Gastric AFB tests were positive and blood cultures for mycobacteria were negative. Cytological studies of fine-needle aspirates from inguinal nodes revealed ill-defined granulomas and AFB. A mycobacterium identified as M. chelonei was cultured from a lymph node. Treatment with rifampicin, ethambutol, clarithromycin and aminoglycoside was initiated. IFN-γ could not be procured and there were not good donors for HSCT. P3 died at the age of 5 years, from persistent disseminated EM disease. He had an elder brother who died of possible tuberculosis, but his sister is well.
Extraction of DNA and WES
gDNA was extracted from whole blood from a healthy control, the patients and parents, with the iPrep PureLink gDNA Blood kit and iPrep instruments from Thermo Fisher Scientific. The method used to sequence the WES has been described elsewhere (6,71,72).
Polymerase chain reaction and sequencing
Polymerase chain reaction (PCR) was carried out on gDNA with Taq polymerase (Invitrogen) and Fast Start Taq (Roche), in a GeneAmp PCR system (9700; Applied Biosystems). The PCR products were purified by centrifugation through Sephadex G-50 Superfine resin (GE Healthcare) and sequenced with the BigDye Terminator Cycle Sequencing kit (Applied Biosystems). Sequencing products were purified by centrifugation through Sephadex G-50 Superfine resin, and sequences were analyzed with an ABI Prism 3700 apparatus (Applied Biosystems). The sequences obtained were then aligned with the genomic sequence of IFNGR2 (NM_005534.3) with Genalys, version 2.0 β software.
Cell culture and stimulation
EBV-B cells, SV40F and HEK293-T cells were cultured as previously described (45,73). EBV-B cells and SV40F were stimulated with the indicated doses of IFN-γ (Imukin, Boehringer Ingelheim) and IFN-α2b (IntronA, Schering Plough) for 20 min.
Expression plasmids and cell transfection
The WT IFNGR2 allele was inserted into V5-topo-pcDNA3.1 (Invitrogen) according to the manufacturer’s instructions. The c.1A>G and c.4delC mutants were generated and various nucleotides between the first and second ATG codons were deleted by site-directed mutagenesis (Agilent Technologies, PfuUltra II Fusion HS DNA polymerase), according to the kit manufacturer’s instructions, and then digested with DpnI (New England Biolabs). HEK293-T cells, IFN-γR2-deficient SV40F (9) and the patients’ SV40F were transfected with one of the various IFNGR2 V5-tagged pcDNA3.1 plasmids or an insert-less V5-tagged pcDNA3.1 plasmid (EV), in the presence of X-treme GENE9 transfection reagent (Sigma Aldrich), according to the manufacturer’s instructions.
EMSA
EMSA was carried out as previously described (74). Briefly, cells were stimulated for 20 min with IFN-γ or IFN-α at the indicated doses. We then obtained a nuclear extract from these cells, and we incubated 5–10 μg of this nuclear extract with 32P-labeled (α-dATP) or IRDye 700 GAS probe, corresponding to the Fc-γR1 promoter, and subjected the mixture to electrophoresis in a polyacrylamide gel.
Cell lysis, immunoprecipitation and immunoblotting
Total proteins were solubilized in extraction buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl2, 1% NP-40, 1 mm EDTA, 1× proteinase inhibitor cocktail mix, 1 mm PMSF and 1 mm Na3VO4). Their concentration was then measured by the Bradford method and they were incubated with Laemmli buffer, with NuPage (Invitrogen) as a reducing agent. Samples were then subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in 10% polyacrylamide gels (Bio-Rad), and the resulting bands were transferred onto normal- or low-fluorescence PVDF membranes (Bio-Rad) with the Trans-Blot Turbo Transfer System (Bio-Rad). Immunoblotting was performed with antibodies against IFN-γRβ (sc-377291, Santa Cruz Biotechnology), GAPDH (14C10, Cell Signaling Technology), the V5 tag (Thermo Fisher Scientific), pY701 STAT1 (612133, Becton Dickinson (BD) Biosciences), STAT1 (610116, BD Biosciences) or α-tubulin (sc-23948, Santa Cruz Biotechnology). Bound antibody was detected with ECL western blotting detection reagents (Bio-Rad) on Chemidoc or with a LI-COR Clx fluorescence detector. For immunoprecipitation experiments, V5-tagged IFNGR2-transfected HEK293-T cells were lysed and subjected to immunoprecipitation with an anti-V5 tag antibody and agarose A/G beads (Santa Cruz Biotechnology), after clearing with an isotype control and agarose A/G beads (Santa Cruz Biotechnology). Immunoprecipitated proteins were then resolved by SDS-PAGE. Blots were either probed with anti-IFN-γRβ antibody or the gel was incubated with Coomassie brilliant blue stain.
Flow cytometry
The methods for detecting the cell-surface expression of HLA-DR have been described elsewhere (8,75). Briefly, 5 × 105 SV40F were incubated with 10 and 103 IU/ml IFN-γ for 48 h. HLA-DR levels on the cell surface were then evaluated by flow cytometry with an anti-human HLA-DR Fluorescein isothiocyanate (FITC) antibody (556643, BD Biosciences). STAT1 phosphorylation was assessed by activating EBV-B cells stained with the Aqua Live/Dead Cell Stain kit (Thermo Fisher Scientific) with IFN-γ or IFN-α for 30 min, permeabilizing the nuclei by incubation in BD Phosflow buffers and washing the cells in cold PBS. The cells were then washed and incubated for 1 h at 4°C with either an antibody against phosphorylated STAT1 (612597, BD Biosciences) or with the corresponding isotype control antibody (565357, BD Biosciences). Cells were washed three times and samples were acquired on a Gallios flow cytometer (Beckman Coulter), with FlowJo as the analysis software.
Whole-blood assay of the IFN-γ circuit
Whole-blood assays were performed as previously described (30,31). Heparin-treated blood samples from healthy controls, patients and parents were stimulated in vitro with BCG alone or with BCG plus IFN-γ or IL-12 (R&D). Supernatants were collected after 48 h of stimulation and ELISA was performed with specific antibodies directed against IFN-γ, IL-12p70 or IL-12p40, with the human Quantikine HS kits for IL-12p70 and IL-12p40 from R&D Systems and the human Pelipair IFN-γ kit from Sanquin, used according to the manufacturer’s instructions.
MDMs and CD4+ T cells
We isolated CD14+ monocytes from peripheral blood mononuclear cells with a positive selection kit from Miltenyi Biotec (76). Briefly, 0.5 million monocytes were incubated in six-well plates with 50 ng/ml M-CSF (216-MC, R&D Systems) and 50 ng/ml IL-4 (204-IL, R&D systems) to induce the differentiation of MDMs, as previously described (76). CD4+ naive T cells were obtained with human naive CD4+ T cell isolation kit II (Miltenyi Biotec). The cells were stimulated with IFN-γ or IFN-α, as previously described (9).
Research involving human participants
Informed consent for participation in this study was obtained from the patients and/or parents in accordance with local regulations, with approval from the Institutional Review Board (IRB). The experiments described here were performed in France, in accordance with local regulations, and with the approval of the IRB of Necker Hospital for Sick Children, France.
Supplementary Material
Acknowledgements
We would like to thank the patients and their families for their collaboration, and both branches of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions and support. We would also like to thank Dominick Papandrea, Yelena Nemirovskaya, Cécile Patissier and Céline Desvallées for administrative support. We acknowledge use of the bioresources of the DNA biobank of the Imagine Institute (BB-033-00065).
Conflict of Interest statement. None declared.
Authorship
C.O.-Q., J.-L.C. and J.B., designed the study and wrote the manuscript. C.O.-Q. and C.D. performed the experiments. A.M., I.K.R., S.K.-Y., B.G., S.M. and A.O.-P. provided material from the patients. R.P.D., S.B.-D., X.-F.K., M.M.-V., R.M.-B., A.N.-P. and A.G. provided expertise and feedback. J.-L.C., L.A. and J.B. secured funding. A.M., I.K.R., S.K.-Y., B.G., S.M. and A.O.-P. conducted the medical follow-up and provided expertise and critical reading. All the authors critically reviewed the manuscript.
Funding
National Institute of Allergy and Infectious Diseases (5R37AI095983); the National Center for Research Resources and the National Center for Advancing Sciences (NCATS) of the National Institutes of Health (UL1TR001866); The Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris Descartes University, the St. Giles Foundation and by the French National Research Agency (ANR) under the ‘Investments for the future’ program (ANR-10-IAHU-01); Laboratoire d'Excellence ‘Integrative Biology of Emerging Infectious Diseases’ (ANR-10-LABX-62-IBEID) and GENMSMD grant (ANR-16-CE17-0005-01 to J.B.). SRC2017 to J.B. ANR-HGDIFD (ANR-14-CE15-006-01 by C.O.-Q. was supported by ANR-HGDIFD (ANR-14-CE15-006-01. A.G. was supported by ANR-IFNGPHOX (ANR13-ISV3-0001-01), ANR-GENMSMD (ANR-16-CE17-0005-01) and Imagine Institute.
References
- 1. Boisson-Dupuis S., Bustamante J., El-Baghdadi J., Camcioglu Y., Parvaneh N., El Azbaoui S., Agader A., Hassani A., El Hafidi N., Mrani N.A. et al. (2015) Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol. Rev., 264, 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bustamante J., Boisson-Dupuis S., Abel L. and Casanova J.L. (2014) Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol., 26, 454–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dorman S.E., Picard C., Lammas D., Heyne K., Dissel J.T., Baretto R., Rosenzweig S.D., Newport M., Levin M., Roesler J. et al. (2004) Clinical features of dominant and recessive interferon γ receptor 1 deficiencies. Lancet, 364, 2113–2121. [DOI] [PubMed] [Google Scholar]
- 4. Jouanguy E., Altare F., Lamhamedi S., Revy P., Emile J.F., Newport M., Levin M., Blanche S., Seboun E., Fischer A. et al. (1996) Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette–Guerin infection. N. Engl. J. Med., 335, 1956–1961. [DOI] [PubMed] [Google Scholar]
- 5. Newport M.J., Huxley C.M., Huston S., Hawrylowicz C.M., Oostra B.A., Williamson R. and Levin M. (1996) A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med., 335, 1941–1949. [DOI] [PubMed] [Google Scholar]
- 6. Bogunovic D., Byun M., Durfee L.A., Abhyankar A., Sanal O., Mansouri D., Salem S., Radovanovic I., Grant A.V., Adimi P. et al. (2012) Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science, 337, 1684–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kong X.-F., Martínez-Barricarte R., Kennedy J., Mele F., Lazarov T., Deenic E.K., Ma C.S., Breton G., Lucero K.B., Langlais D. et al. (2018) Disruption of an antimycobacterial circuit between dendritic and T helper cells in humans with inherited SPPL2a deficiency. Nat. Immunol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doffinger R., Jouanguy E., Dupuis S., Fondaneche M.C., Stephan J.L., Emile J.F., Lamhamedi-Cherradi S., Altare F., Pallier A., Barcenas-Morales G. et al. (2000) Partial interferon-gamma receptor signaling chain deficiency in a patient with bacille Calmette–Guerin and Mycobacterium abscessus infection. J. Infect. Dis., 181, 379–384. [DOI] [PubMed] [Google Scholar]
- 9. Dorman S.E. and Holland S.M. (1998) Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J. Clin. Invest., 101, 2364–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holzer U., Reinhardt K., Lang P., Handgretinger R. and Fischer N. (2013) Influence of a mutation in IFN-γ receptor 2 (IFNGR2) in human cells on the generation of Th17 cells in memory T cells. Hum. Immunol., 74, 693–700. [DOI] [PubMed] [Google Scholar]
- 11. Hoyos-Bachiloglu R., Chou J., Sodroski C.N., Beano A., Bainter W., Angelova M., Al Idrissi E., Habazi M.K., Alghamdi H.A., Almanjomi F. et al. (2017) A digenic human immunodeficiency characterized by IFNAR1 and IFNGR2 mutations. J. Clin. Invest., 127, 4415–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamoun C., Morsheimer M., Sullivan K.E., Holland S.M., Rundles C.C., Bunin N. and Heimall J.R. (2016) Successful unrelated cord blood transplant for complete IFN-γ receptor 2 deficiency. J. Allergy Clin. Immunol., 138, 1489–1491. [DOI] [PubMed] [Google Scholar]
- 13. Kilic S.S., Wengen A., Paus R.A., Celebi S., Meziane B., Hafizoglu D., Dissel J.T. and Vosse E. (2012) Severe disseminated mycobacterial infection in a boy with a novel mutation leading to IFN-γR2 deficiency. J. Infect., 65, 568–572. [DOI] [PubMed] [Google Scholar]
- 14. Kong X.F., Vogt G., Itan Y., Macura-Biegun A., Szaflarska A., Kowalczyk D., Chapgier A., Abhyankar A., Furthner D., Djambas Khayat C. et al. (2013) Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum. Mol. Genet., 22, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martinez-Barricarte R., Megged O., Stepensky P., Casimir P., Moncada-Velez M., Averbuch D., Assous M.V., Abuzaitoun O., Kong X.F., Pedergnana V. et al. (2014) Mycobacterium simiae infection in two unrelated patients with different forms of inherited IFN-γR2 deficiency. J. Clin. Immunol., 34, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moncada-Velez M., Martinez-Barricarte R., Bogunovic D., Kong X.F., Blancas-Galicia L., Tirpan C., Aksu G., Vincent Q.B., Boisson B., Itan Y. et al. (2013) Partial IFN-γR2 deficiency is due to protein misfolding and can be rescued by inhibitors of glycosylation. Blood, 122, 2390–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rosenzweig S.D., Dorman S.E., Uzel G., Shaw S., Scurlock A., Brown M.R., Buckley R.H. and Holland S.M. (2004) A novel mutation in IFN-γ receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J. Immunol., 173, 4000–4008. [DOI] [PubMed] [Google Scholar]
- 18. Tesi B., Sieni E., Neves C., Romano F., Cetica V., Cordeiro A.I., Chiang S., Schlums H., Galli L., Avenali S. et al. (2015) Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN-γ receptor deficiency. J. Allergy Clin. Immunol., 135, 1638–1641. [DOI] [PubMed] [Google Scholar]
- 19. Toyoda H., Ido M., Nakanishi K., Nakano T., Kamiya H., Matsumine A., Uchida A., Mizutani H., Beaucoudrey L., Vogt G. et al. (2010) Multiple cutaneous squamous cell carcinomas in a patient with interferon γ receptor 2 (IFN γ R2) deficiency. J. Med. Genet., 47, 631–634. [DOI] [PubMed] [Google Scholar]
- 20. Vogt G., Bustamante J., Chapgier A., Feinberg J., Boisson Dupuis S., Picard C., Mahlaoui N., Gineau L., Alcais A., Lamaze C. et al. (2008) Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J. Exp. Med., 205, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vogt G., Chapgier A., Yang K., Chuzhanova N., Feinberg J., Fieschi C., Boisson-Dupuis S., Alcais A., Filipe-Santos O., Bustamante J. et al. (2005) Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat. Genet., 37, 692–700. [DOI] [PubMed] [Google Scholar]
- 22. Vogt G., Vogt B., Chuzhanova N., Julenius K., Cooper D.N. and Casanova J.L. (2007) Gain-of-glycosylation mutations. Curr. Opin. Genet. Dev., 17, 245–251. [DOI] [PubMed] [Google Scholar]
- 23. Blouin C.M., Hamon Y., Gonnord P., Boularan C., Kagan J., Viaris de Lesegno C., Ruez R., Mailfert S., Bertaux N., Loew D. et al. (2016) Glycosylation-dependent IFN-γR partitioning in lipid and actin nanodomains is critical for JAK activation. Cell, 166, 920–934. [DOI] [PubMed] [Google Scholar]
- 24. Hetzel M., Mucci A., Blank P., Nguyen A.H.H., Schiller J., Halle O., Kuhnel M.P., Billig S., Meineke R., Brand D. et al. (2018) Hematopoietic stem cell gene therapy for IFNγR1 deficiency protects mice from mycobacterial infections. Blood, 131, 533–545. [DOI] [PubMed] [Google Scholar]
- 25. Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M. and Shendure J. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Itan Y., Shang L., Boisson B., Ciancanelli M.J., Markle J.G., Martinez-Barricarte R., Scott E., Shah I., Stenson P.D., Gleeson J. et al. (2016) The mutation significance cutoff: gene-level thresholds for variant predictions. Nat. Methods, 13, 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomson A.W. and Lotze M.T. (2003) The Cytokine Handbook. Elsevier Academic Press. Cambridge, Massachusetts, United States. [Google Scholar]
- 28. Maley F., Trimble R.B., Tarentino A.L. and Plummer T.H. Jr. (1989) Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem., 180, 195–204. [DOI] [PubMed] [Google Scholar]
- 29. Kong X.F., Vogt G., Chapgier A., Lamaze C., Bustamante J., Prando C., Fortin A., Puel A., Feinberg J., Zhang X.X. et al. (2010) A novel form of cell type-specific partial IFN-γR1 deficiency caused by a germ line mutation of the IFNGR1 initiation codon. Hum. Mol. Genet., 19, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esteve-Sole A., Sologuren I., Martinez-Saavedra M.T., Deya-Martinez A., Oleaga-Quintas C., Martinez-Barricarte R., Martin-Nalda A., Juan M., Casanova J.L., Rodriguez-Gallego C. et al. (2018) Laboratory evaluation of the IFN-γ circuit for the molecular diagnosis of Mendelian susceptibility to mycobacterial disease. Crit. Rev. Clin. Lab. Sci., 55, 184–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feinberg J., Fieschi C., Doffinger R., Feinberg M., Leclerc T., Boisson-Dupuis S., Picard C., Bustamante J., Chapgier A., Filipe-Santos O. et al. (2004) Bacillus Calmette Guerin triggers the IL-12/IFN-γ axis by an IRAK-4- and NEMO-dependent, non-cognate interaction between monocytes, NK, and T lymphocytes. Eur. J. Immunol., 34, 3276–3284. [DOI] [PubMed] [Google Scholar]
- 32. Kozak M. (1986) Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell, 44, 283–292. [DOI] [PubMed] [Google Scholar]
- 33. Kozak M. (1987) An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res., 15, 8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kozak M. (1987) At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol., 196, 947–950. [DOI] [PubMed] [Google Scholar]
- 35. Salamov A.A., Nishikawa T. and Swindells M.B. (1998) Assessing protein coding region integrity in cDNA sequencing projects. Bioinformatics, 14, 384–390. [DOI] [PubMed] [Google Scholar]
- 36. Kapp K., Schrempf S., Lemberg M.K. and Dobberstein B. (2009) Post-targeting functions of signal peptides. Landes Bioscience; Chapter 1: 1–16. [Google Scholar]
- 37. Ivanov I.P., Firth A.E., Michel A.M., Atkins J.F. and Baranov P.V. (2011) Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res., 39, 4220–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mehdi H., Ono E. and Gupta K.C. (1990) Initiation of translation at CUG, GUG, and ACG codons in mammalian cells. Gene, 91, 173–178. [DOI] [PubMed] [Google Scholar]
- 39. Peabody D.S. (1989) Translation initiation at non-AUG triplets in mammalian cells. J. Biol. Chem., 264, 5031–5035. [PubMed] [Google Scholar]
- 40. Hiller K., Grote A., Scheer M., Munch R. and Jahn D. (2004) PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res., 32, W375–W379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kozak M. (2002) Pushing the limits of the scanning mechanism for initiation of translation. Gene, 299, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kozak M. (1989) Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol. Cell Biol., 9, 5073–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kozak M. (1995) Adherence to the first-AUG rule when a second AUG colon follows closely upon the first. Proc. Natl. Acad. Sci. U. S. A., 92, 7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kozak M. (2002) Emerging links between initiation of translation and human diseases. Mamm. Genome, 13, 401–410. [DOI] [PubMed] [Google Scholar]
- 45. Puel A., Reichenbach J., Bustamante J., Ku C.L., Feinberg J., Doffinger R., Bonnet M., Filipe-Santos O., Beaucoudrey L., Durandy A. et al. (2006) The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am. J. Hum. Genet., 78, 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lagerstrom-Fermer M., Nilsson M., Backman B., Salido E., Shapiro L., Pettersson U. and Landegren U. (1995) Amelogenin signal peptide mutation: correlation between mutations in the amelogenin gene (AMGX) and manifestations of X-linked amelogenesis imperfecta. Genomics, 26,159–162. [DOI] [PubMed] [Google Scholar]
- 47. Datta R., Waheed A., Shah G.N. and Sly W.S. (2007) Signal sequence mutation in autosomal dominant form of hypoparathyroidism induces apoptosis that is corrected by a chemical chaperone. Proc. Natl. Acad. Sci. USA, 104, 19989–19994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vezzoli V., Duminuco P., Vottero A., Kleinau G., Schulein R., Minari R., Bassi I., Bernasconi S., Persani L. and Bonomi M. (2015) A new variant in signal peptide of the human luteinizing hormone receptor (LHCGR) affects receptor biogenesis causing leydig cell hypoplasia. Hum. Mol. Genet., 24, 6003–6012. [DOI] [PubMed] [Google Scholar]
- 49. Zalucki Y.M., Power P.M. and Jennings M.P. (2007) Selection for efficient translation initiation biases codon usage at second amino acid position in secretory proteins. Nucleic Acids Res., 35, 5748–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zalucki Y.M., Beacham I.R. and Jennings M.P. (2009) Biased codon usage in signal peptides: a role in protein export. Trends Microbiol., 17, 146–150. [DOI] [PubMed] [Google Scholar]
- 51. Owji H., Nezafat N., Negahdaripour M., Hajiebrahimi A. and Ghasemi Y. (2018) A comprehensive review of signal peptides: structure, roles, and applications. Eur. J. Cell Biol., in press. [DOI] [PubMed] [Google Scholar]
- 52. Villartay J.P., Lim A., Al-Mousa H., Dupont S., Dechanet-Merville J., Coumau-Gatbois E., Gougeon M.L., Lemainque A., Eidenschenk C., Jouanguy E. et al. (2005) A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J. Clin. Invest., 115, 3291–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Santagata S., Gomez C.A., Sobacchi C., Bozzi F., Abinun M., Pasic S., Cortes P., Vezzoni P. and Villa A. (2000) N-terminal RAG1 frameshift mutations in Omenn's syndrome: internal methionine usage leads to partial V(D)J recombination activity and reveals a fundamental role in vivo for the N-terminal domains. Proc. Natl. Acad. Sci. USA, 97, 14572–14577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yamashita T., Wu N., Kupfer G., Corless C., Joenje H., Grompe M. and D'Andrea A.D. (1996) Clinical variability of Fanconi anemia (type C) results from expression of an amino terminal truncated Fanconi anemia complementation group C polypeptide with partial activity. Blood, 87, 4424–4432. [PubMed] [Google Scholar]
- 55. Maser R.S., Zinkel R. and Petrini J.H. (2001) An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat. Genet., 27, 417–421. [DOI] [PubMed] [Google Scholar]
- 56. Cain J.T., Kim D.I., Quast M., Shivega W.G., Patrick R.J., Moser C., Reuter S., Perez M., Myers A., Weimer J.M. et al. (2017) Nonsense pathogenic variants in exon 1 of PHOX2B lead to translational reinitiation in congenital central hypoventilation syndrome. Am. J. Med. Genet. A., 173, 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gerards J., Ritter M.M., Kaminsky E., Gal A., Hoeppner W. and Quinkler M. (2017) A novel stop mutation (p.(Gln22*)) of DAX1 (NR0B1) results in late-onset X-linked adrenal hypoplasia congenita. Endocrinol. Diabetes Metab. Case Rep., Sep 4, 2017 pii: 17-0054 doi: 10.1530/EDM-17-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gurvich O.L., Maiti B., Weiss R.B., Aggarwal G., Howard M.T. and Flanigan K.M. (2009) DMDexon 1 truncating point mutations: amelioration of phenotype by alternative translation initiation in exon 6. Hum. Mutat., 30, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moey C., Topper S., Karn M., Johnson A.K., Das S., Vidaurre J. and Shoubridge C. (2016) Reinitiation of mRNA translation in a patient with X-linked infantile spasms with a protein-truncating variant in ARX. Eur. J. Hum. Genet., 24, 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Neu-Yilik G., Amthor B., Gehring N.H., Bahri S., Paidassi H., Hentze M.W. and Kulozik A.E. (2011) Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA, 17, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ozisik G., Mantovani G., Achermann J.C., Persani L., Spada A., Weiss J., Beck-Peccoz P. and Jameson J.L. (2003) An alternate translation initiation site circumvents an amino-terminal DAX1 nonsense mutation leading to a mild form of X-linked adrenal hypoplasia congenita. J. Clin. Endocrinol. Metab., 88, 417–423. [DOI] [PubMed] [Google Scholar]
- 62. Paulsen M., Lund C., Akram Z., Winther J.R., Horn N. and Moller L.B. (2006) Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am. J. Hum. Genet., 79, 214–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Peixeiro I., Inacio A., Barbosa C., Silva A.L., Liebhaber S.A. and Romao L. (2012) Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations. Nucleic Acids Res., 40, 1160–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pittis M.G., Ricci V., Guerci V.I., Marcais C., Ciana G., Dardis A., Gerin F., Stroppiano M., Vanier M.T., Filocamo M. et al. (2004) Acid sphingomyelinase: identification of nine novel mutations among Italian Niemann Pick type B patients and characterization of in vivo functional in-frame start codon. Hum. Mutat., 24, 186–187. [DOI] [PubMed] [Google Scholar]
- 65. Stump M.R., Gong Q., Packer J.D. and Zhou Z. (2012) Early LQT2nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation. J. Mol. Cell Cardiol., 53, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stump M.R., Gong Q. and Zhou Z. (2013) LQT2nonsense mutations generate trafficking defective NH2-terminally truncated channels by the reinitiation of translation. Am. J. Physiol. Heart Circ. Physiol., 305, H1397–H1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kearse M.G. and Wilusz J.E. (2017) Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev., 31, 1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schwarz D.S. and Blower M.D. (2016) The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol. Life Sci., 73, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dittmar K.A., Goodenbour J.M. and Pan T. (2006) Tissue-specific differences in human transfer RNA expression. PLoS Genet., 2, e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Itan Y. and Casanova J.L. (2015) Can the impact of human genetic variations be predicted? Proc. Natl. Acad. Sci. USA, 112, 11426–11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bolze A., Byun M., McDonald D., Morgan N.V., Abhyankar A., Premkumar L., Puel A., Bacon C.M., Rieux-Laucat F., Pang K. et al. (2010) Whole-exome-sequencing-based discovery of human FADD deficiency. Am. J. Hum. Genet., 87, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Byun M., Abhyankar A., Lelarge V., Plancoulaine S., Palanduz A., Telhan L., Boisson B., Picard C., Dewell S., Zhao C. et al. (2010) Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med., 207, 2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jouanguy E., Dupuis S., Pallier A., Doffinger R., Fondaneche M.C., Fieschi C., Lamhamedi-Cherradi S., Altare F., Emile J.F., Lutz P. et al. (2000) In a novel form of IFN-γ receptor 1 deficiency, cell surface receptors fail to bind IFN-γ. J. Clin. Invest., 105, 1429–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dupuis S., Dargemont C., Fieschi C., Thomassin N., Rosenzweig S., Harris J., Holland S.M., Schreiber R.D. and Casanova J.L. (2001) Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science, 293, 300–303. [DOI] [PubMed] [Google Scholar]
- 75. Jouanguy E., Lamhamedi-Cherradi S., Altare F., Fondaneche M.C., Tuerlinckx D., Blanche S., Emile J.F., Gaillard J.L., Schreiber R., Levin M. et al. (1997) Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette–Guerin infection and a sibling with clinical tuberculosis. J. Clin. Invest., 100, 2658–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bustamante J., Arias A.A., Vogt G., Picard C., Galicia L.B., Prando C., Grant A.V., Marchal C.C., Hubeau M., Chapgier A. et al. (2011) Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat. Immunol., 12, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.