

Abstract
Infections are estimated to contribute to 20% of all human tumours. These are mainly caused by viruses, which explains why a direct bacterial contribution to cancer formation has been largely ignored. While epidemiological data link bacterial infections to particular cancers, tumour formation is generally assumed to be solely caused by the ensuing inflammation responses. Yet, many bacteria directly manipulate their host cell in various phases of their infection cycle. Such manipulations can affect host cell integrity and can contribute to cancer formation. We here describe how bacterial surface moieties, bacterial protein toxins and bacterial effector proteins can induce host cell DNA damage, and thereby can interfere with essential host cell signalling pathways involved in cell proliferation, apoptosis, differentiation and immune signalling.
Keywords: bacteria, cancer, effectors, infection, signalling
Subject Categories: Cancer; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
Glossary
- Apc
Adenomatous polyposis coli
- BFT
Bacteroides fragilis toxin
- CagA
Cytotoxin‐associated gene A
- CCL5
Chemokine (C‐C motif) ligand 5
- CDK1
Cyclin‐dependent kinase 1
- CDT
Cytolethal distending toxin
- DDR
DNA damage responses
- DSBs
Double‐strand DNA breaks
- EF‐2
Elongation factor 2
- EGFR
Epidermal growth factor receptor
- ER
Endoplasmic reticulum
- ERK
Extracellular signal‐regulated kinase
- FadA
Fusobacterium adhesion A
- FCP
Francisella‐containing phagosome
- IKK
Iκβ kinase
- IL
Interleukin
- JNK
C‐Jun N‐terminal kinase
- LPS
Lipopolysaccharides
- MALT
Mucosa‐associated lymphoid tissue
- MAPK
Mitogen‐activated protein kinase
- MEK
Mitogen‐activated protein kinase kinase
- MyD88
Myeloid differentiation primary response 88
- NET1
Neuroepithelial cell‐transforming gene 1 protein
- NF‐κβ
Nuclear factor‐κβ
- OipA
Outer inflammatory protein A
- PAK
p21‑activated kinase
- Pks
Polyketide synthetase
- Raf
Rapidly Accelerated Fibrosarcoma
- SCV
Salmonella‐containing Vacuole
- Tcf
T‐cell factor
- TLR
Toll‐like receptor
- VacA
Vacuolating cytotoxin A
Introduction
Cancer development is the result of a series of genetic modifications that alter the normal control of cell growth and survival. These genetic alterations can be induced by a wide variety of external factors 1, including smoking, alcohol 2 and sunlight 3, 4. At least 75% of the head and neck cancers are caused by tobacco and alcohol 5 and 65–86% of the skin cancer risk can be attributed to sun exposure 4. In addition to these external factors, viral genomes have been retrieved from a variety of tumour samples 6 and this link has been further substantiated by many epidemiological studies (Table 1). For example, viral infections such as human papillomavirus and hepatitis B virus and hepatitis C virus have been associated with ~90% of cervical cancer cases 7 and ~80% of hepatocellular carcinoma cases 8, respectively.
Table 1.
Epidemiological and experimental evidence for a link between microbial infections and cancer
| Infectious agent | Type of micro‐organism | Cancer type |
|---|---|---|
| Epstein–Barr virus | Virus | Nasopharyngeal carcinoma, Burkitt lymphoma, immune suppression‐related non‐Hodgkin lymphoma, Hodgkin lymphoma, extranodal natural killer/T‐cell lymphoma (nasal type) 102 |
| Hepatitis B virus | Virus | Hepatocellular carcinoma 102 |
| Hepatitis C virus | Virus | Hepatocellular carcinoma, non‐Hodgkin lymphoma 102 |
| Kaposi sarcoma herpesvirus | Virus | Kaposi sarcoma, primary effusion lymphoma 102 |
| Human immunodeficiency virus 1 | Virus | Kaposi sarcoma, non‐Hodgkin lymphoma, Hodgkin lymphoma, carcinoma of the cervix, anus, conjunctiva 102 |
| Human papillomavirus type 16 | Virus | Carcinoma of the cervix, vulva, vagina, penis, anus, oral cavity, and oropharynx and tonsil 102 |
| Human T‐cell lymphotropic virus type 1 | Virus | Adult T‐cell leukaemia and lymphoma 102 |
| Merkel cell polyomavirus | Virus | Merkel cell carcinoma 103 |
| Opisthorchis viverrini | Trematode | Cholangiocarcinoma 102 |
| Clonorchis sinensis | Helminth | Cholangiocarcinoma 102 |
| Schistosoma heamatobium | Trematode | Urinary bladder cancer 102 |
| Helicobacter pylori | Bacterium | Non‐cardia gastric carcinoma, low‐grade B‐cell MALT gastric lymphoma 102 |
| Alfatoxin (B1) | Mould (Aspergillus flavus) | Liver cancer 102 |
| Salmonella Typhi | Bacterium | Gallbladder carcinoma 13 |
| Salmonella Enteritidis | Bacterium | Colon carcinoma in the ascending and transverse parts of the colon 14 |
| Chlamydia trachomatis | Bacterium | Carcinoma of the cervix and ovaries 104, 105 |
An even more compelling case for the link between viral infections and cancer arose from experiments showing that viruses exploit the host cell niche for their infection cycle and as a result stimulate mammalian growth‐inducing genes, leaving the cells in a cancerous state of uncontrolled cell division. It is now understood how viruses such as hepatitis B virus and human papillomavirus types 5 and 8 cause cellular transformation by inducing genetic instability through viral integration and through the activation of a large number of signalling pathways and cellular genes involved in oncogenesis, proliferation, inflammation and immune responses 9, 10.
Viruses do, however, represent only one segment of the microbiome that exploits the mammalian host during its infection cycle. Pathogenic moulds, helminths and bacteria intensively interact with mammalian host cells to ensure their survival. Although these microorganisms usually do not leave a genetically recognizable trait or piggyback on mammalian genes, such as illustrated by viral infections, strong epidemiological links exist between various microbiological infections and cancers (Table 1). Examples include connections between Schistosoma haematobium infections and bladder cancer 11, Helicobacter pylori (H. pylori) infections and gastric cancer 12, chronic Salmonella Typhi (S. Typhi) infections and gallbladder carcinoma 13, and Salmonella Enteritidis (S. Enteritidis) infections and colon carcinoma 14. Moreover, studies in germ‐free and antibiotic‐treated animals have indicated cancer‐promoting effects of microbiota in various experimental systems, varying from gastric 15, 16, colon 17, 18 and liver 19 cancers.
However, since microbiome–host interactions are extremely diverse, their exact contributions to cancer development are hard to pinpoint. Especially, pathogenic bacteria have been shown to manipulate and exploit the human host cell niche in various ways throughout various stages of their infection cycle. In this review, we will discuss how bacterial surface moieties, bacterial protein toxins and bacterial effector proteins interact with host cells, and how such encounters can result in the modification of essential host cell signalling pathways involved in cancer formation.
Bacterial cell‐surface components and cancer development
The bacterial outer surface directly contacts host cells and consists of complex structures that include various antigenic moieties that activate host innate and adaptive immune responses. As a consequence, pathogenic bacteria have evolved a wide variety of outer‐surface modifications that ensure immune escape to afford significant survival opportunities. To abolish immune recognition and clearance, Gram‐negative bacteria cover their complex outer‐surface macromolecules with a polysaccharide‐rich capsule. These capsules limit complement activation by shielding deeper structures on the membranes of pathogenic variants of Escherichia coli (E. coli), Streptococcus pneumoniae, Haemophilus influenzae type b, Neisseria meningitidis and others, and prevent engulfment by professional phagocytes 20, 21, 22, 23. Unencapsulated mutants of these bacteria rarely cause an invasive infection and are highly attenuated in various infection models due to better opsonophagocytic clearance 22, 24, 25.
In addition to their shielding capsules, many bacterial pathogens have modified their surface‐exposed molecules, including lipopolysaccharides (LPS), flagella and peptidoglycans, to limit immune recognition. For example, H. pylori has LPS surface molecules that harbour “underacylated” lipid A molecules that are a poor substrate for host Toll‐like receptor (TLR)4 and as such evade innate immune sensing 26, 27. Helicobacter pylori also produces modified flagellin molecules that are not recognized by TLR5 to prevent TLR5‐mediated interleukin (IL)‐8 secretion and subsequent immune signalling 28. Salmonella typhimurium (S. typhimurium) expresses lipid A deacetylase PagL and a lipid A palmitoyltransferase PagP to modify lipid A, resulting in a 100‐fold decrease in lipid A‐mediated TLR4 activation and nuclear factor‐κβ (NF‐κβ) activation 29. These examples illustrate how bacterial pathogens modify their outer surface to escape immune recognition.
Pathogenic bacteria that favour an intracellular lifestyle express surface proteins that promote both host cell attachment and internalization. For example, pathogenic species of the Neisseria family express a variety of surface adhesins that mediate selective interaction with certain cell types, thereby allowing the exploitation of specialized host cell niches 30. In a similar fashion, fibronectin‐binding proteins of Staphylococcus aureus and Borrelia burgdorferi mediate the interaction between bacterium and host cell through the formation of tandem β‐zippers that stimulate bacterial engulfment by non‐phagocytic cells 31, 32.
In general, these surface‐mediated assault strategies are aimed at facilitating bacterial survival within the host through both immune evasion and host invasion. However, to further control the host cell machinery, bacterial surface molecules also manipulate host cell signalling cascades and affect host cell integrity, which can coincidentally induce cellular malignancies. CagL is a type IV pilus adhesin of H. pylori that ensures the adherence of H. pylori to gastric epithelial cells and then controls a signalling cascade that induces upregulation of gastrin secretion. This results in hypergastrinemia, a major risk factor for the development of gastric adenocarcinoma. CagL binds β5‐integrin thus manipulating integrin‐linked kinase complexes and the downstream rapidly accelerated fibrosarcoma (Raf) kinase, the mitogen‐activated protein kinase kinase (MEK) and the extracellular signal‐regulated kinase (ERK) pathways (Fig 1A) 33. The outer inflammatory protein A (OipA) of H. pylori activates EGFR (epidermal growth factor receptor) and stimulates Akt and β‐catenin signalling, a phenotype observed in a number of different cancers, including gastric cancer (Fig 1B) 34, 35. OipA inactivation decreases β‐catenin nuclear localization in vitro and reduces the incidence of cancer in animal models 36. In addition, the blood group antigen‐binding adhesin BabA of H. pylori can bind human Lewis(b) surface epitopes which indirectly increases mRNA levels of proinflammatory cytokines chemokine (C‐C motif) ligand 5 (CCL5) and IL‐8, and the precancer‐related factors CDX2 and MUC2 (Fig 1C) 37. The fusobacterium adhesion A (FadA) of Fusobacterium nucleatum (F. nucleatum) can bind the extracellular domain of E‐cadherin, thereby inducing phosphorylation and internalization of E‐cadherin. This then releases β‐catenin to activate β‐catenin–T‐cell factor (Tcf)/LEF, downstream in the Wnt signalling pathway to control transcription of genes involved in apoptosis, cell proliferation and transformation (Fig 1D) 38. In patients with colon adenomas or adenocarcinomas, high expression levels of F. nucleatum fadA have been associated with upregulated expression of oncogenic and inflammatory genes associated with the Wnt signalling pathway 39, 40.
Figure 1. Bacterial outer‐surface components that manipulate host cell signalling cascades involved in cellular malignancy.
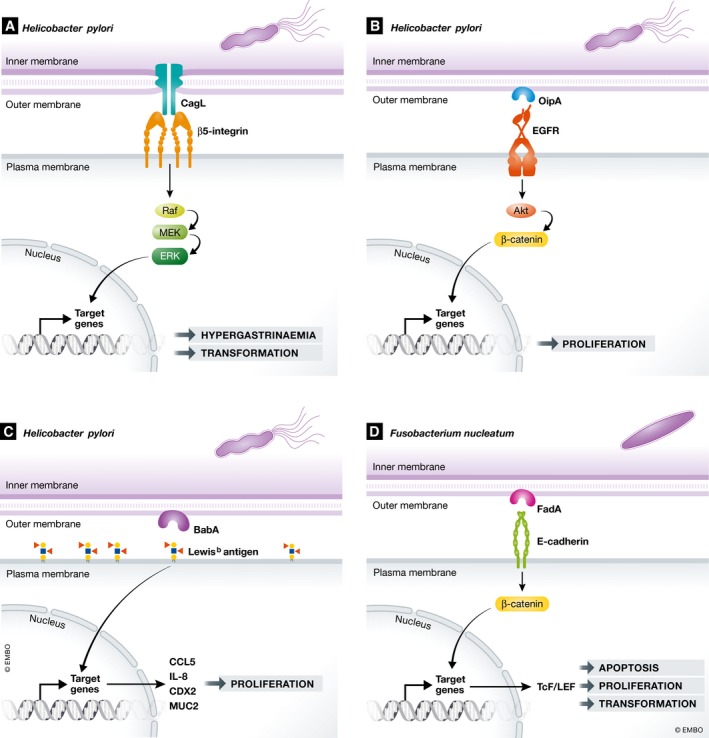
(A) Helicobacter pylori CagL binds β5‐integrin and induces downstream signalling of Raf, MEK and ERK pathways that play a central role in H. pylori‐induced gastrin production and cellular transformation. (B) H. pylori OipA activates EGFR and stimulates Akt and β‐catenin signalling, causing cell proliferation. (C) H. pylori BabA binds human Lewis(b) surface epitopes which increases levels of CCL5, IL‐8, CDX2 and MUC2, causing cell proliferation. (D) Fusobacterium nucleatum FadA binds to E‐cadherin, which releases β‐catenin that activates transcription factor Tcf/LEF which controls the transcription of genes involved in apoptosis, cell proliferation and transformation.
The major surface‐exposed component of Gram‐negative bacteria, LPS additionally activates signalling cascades that promote cancer development. LPS is present in both pathogenic and commensal bacteria and plays a central role in the activation of TLR4. TLR4‐mediated signalling is critical for the downstream activation of numerous signalling pathways that underlie a variety of inflammatory and immune responses, and can promote the development of adenomatous polyposis coli (Apc)‐dependent colorectal cancers and inflammation‐associated colorectal cancers in mice. The role of TLR signalling in intestinal tumorigenesis has been studied through the crossing of myeloid differentiation primary response 88 (MyD88)‐deficient mice that have impaired TLR4 signalling, with Apc (Apc Min/+) mice that mimic sporadic cancer and familial adenomatous polyposis. These MYD88‐deficient × Apc Min/+ mice showed a reduction in both tumour number and size compared to the Apc Min/+ control mice, suggesting that TLR4 signalling further promotes tumour growth 41, 42. Tumour tissues of mice lacking MyD88 showed lower expression of the Cox2 gene that is involved in inflammation, indicating a role of this gene in reduced tumour formation 43. It has furthermore been shown that Cox2 inhibitors, such as aspirin, reduce colorectal cancer risk in people that overexpress the 15‐PGDH gene which encodes for an enzyme that disrupts Cox2 activity 44. Studies with germ‐free and wild‐type mice showed that TLR4 activation by LPS from the intestinal microbiota pool contributes to the promotion of injury‐ and inflammation‐driven hepatocellular carcinoma by activating proliferative and anti‐apoptotic signals 19. Findings from these animal studies were further corroborated by human studies in which enhanced expression of the TLR4/MyD88 complex was detected in 20% of colorectal patient samples 45.
Bacterial toxin‐mediated host cell transformation
To ensure immune escape, rapid replication and spreading, pathogenic bacteria do not only use immune‐evasion strategies to avoid host cell clearance, but are also capable of immune cell elimination. One of the strategies employed by bacteria is the secretion of protein toxins that have cytolytic properties. Bacteria can express protein toxins from their pathogenicity islands and secrete them through specialized secretion systems for transport across bacterial outer membranes 46. The interaction of proteins toxins with the host generally occurs in an ordered series of events and can be illustrated by the mode of action of the diphtheria toxin that inhibits the synthesis of host cell proteins through the inactivation of the host elongation factor 2 (EF‐2) protein. The diphtheria toxin consists of three subunits and is secreted by Corynebacterium diphtheriae as a single polypeptide chain. Diphtheria toxin then binds to the host's heparin‐binding epidermal growth factor‐like surface receptor that then is internalized in the endosomal system. Here, the transmembrane domain of the toxin is unfolded, which translocates the toxin to the cytosolic side of the endosomal membrane. This is followed by a reduction in the disulphide bond between toxin fragments A and B and release of the C‐domain into the cytoplasm. The C‐domain is then refolded into an enzymatically active conformation that catalyses NAD+‐dependent ADP‐ribosylation of EF‐2. This then inhibits protein synthesis, ultimately resulting in cell death of the targeted cells 47.
Although pathogenic bacteria primarily use their toxin‐mediated assault strategies to create a favourable host cell environment, their toxins, likely as a side effect of their mode of action, can also contribute to carcinogenesis. Toxin‐mediated carcinogenesis can occur in multiple ways, including the induction of genomic instability, the induction of cell death resistance cell signalling and the induction of proliferative signalling 48. Genome instability is most readily caused by bacterial protein toxins that induce host cell double‐stranded DNA breaks, including the cytolethal distending toxin (CDT), the colibactin, the Shiga toxin and endonucleases. CDT is secreted by various Gram‐negative bacteria that belong to the Gamma and Epsilon class of Proteobacteria, including S. Typhi, E. coli, Shigella dysenteriae and Campylobacter jejuni. CDT is comprised of three subunits, CdtA, CdtB and CdtC. CdtA and CdtC ensure the uptake and cellular delivery of CdtB, which harbours the catalytic activity of CDT and causes double‐strand DNA breaks (DSBs) in host cells. After host cell binding and internalization by subunits CdtA and CdtC, CdtB undergoes retrograde transport via the endosomes and Golgi to the endoplasmic reticulum (ER), where it undergoes ER‐associated protein degradation‐mediated translocation into the cytosol. The CtdB subunit is then imported in the nucleus where it induces DSBs 49. These DSBs result in DNA damage responses (DDR) that cause G1‐S cell cycle arrest in endothelial and epithelial cells, and both G1‐S and G2‐M cell cycle arrest in fibroblasts and apoptosis in haematopoietic cells that are particularly sensitive to these toxins. As a result, this toxin can locally eliminate immune cells, providing an obvious advantage for the bacteria. However, prolonged exposure to sublethal doses of CDT can impair DDR sensor functionality, resulting in impaired detection of DNA damage and the accumulation of mutations. At the same time, mitogen‐activated protein kinase (MAPK) activity is upregulated by activation of the neuroepithelial cell‐transforming gene 1 protein (NET1) and the GTPase RhoA, which supports survival of the toxin‐exposed cells (Fig 2A) 50. As a consequence, these cells can propagate with DNA mutations and deletions that arise during the repair process, thus inducing genomic errors that underlie cancer formation.
Figure 2. Host cell signalling pathways involved in cell growth and transformation manipulated by bacterial toxins.
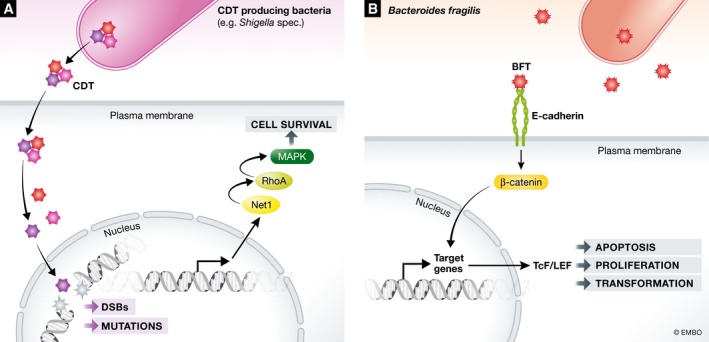
(A) The CdtB subunit of the CDT toxin is delivered to the nucleus where it causes DSBs and impairs DNA DDR sensor functionality. At the same time, NET1 and RhoA are activated which ensure upregulation of MAPK and cellular survival. (B) Bacteroides fragilis BFT binds to E‐cadherin and involved in cellular signalling, proliferation and transformation via activation of the β‐catenin/Wnt and NF‐κβ signalling pathways.
In addition to the CDT toxins, the DNA interacting colibactin toxin has also been associated with the formation of DSBs and the introduction of genomic instability. Colibactin is secreted by E. coli strains of the phylogenetic group B2 that harbours the polyketide synthetase (pks) island 51. Bacteria that harbour the pks genomic island are able to induce DSBs in eukaryotic cells, which results in the activation of the DNA damage checkpoint pathways ATM, CHK1 and CHK2. This then results in CDC25 and cyclin‐dependent kinase 1 (CDK1)‐mediated G2‐ to M‐phase cell cycle arrest and finally in apoptotic cell death. As a side effect of their mode of action, colibactin‐producing bacteria also induce incomplete DNA repair, chromosomal instability and anchorage‐dependent colony formation, phenotypes that can promote cancer formation 52, 53. This is further substantiated by epidemiological studies showing that colibactin‐producing E. coli bacteria appear with high prevalence in biopsies of patients with human colorectal tumours 54, 55. Moreover, colitis‐susceptible IL‐10‐deficient mice showed increased formation of invasive carcinoma when colonized with E. coli secreting colibactin, whereas deletion of the pks genotoxic island from these E. coli strains decreased tumour multiplicity and invasion 56.
Besides toxins that contribute to carcinogenesis by introducing DSBs and genomic instability, toxins have been reported that promote carcinogenesis by inducing resistance to cell death signalling and by promoting proliferative signalling. These toxins are generally secreted by pathogenic bacteria that favour an intracellular host cell life as part of their infectious cycle and thus directly benefit from host cell survival. An example of such a toxin is the Bacteroides fragilis (B. fragilis) toxin (BFT) that binds to intestinal epithelial cell receptors and stimulates cell proliferation by cleavage of the tumour suppressor protein E‐cadherin 57, 58. E‐cadherin is involved in the formation of intercellular adhesion junctions in the intestinal epithelium and is involved in cellular signalling, proliferation and differentiation via activation of the β‐catenin/Wnt and NF‐κβ signalling pathways (Fig 2B) 59, 60, 61. BFT induced acute and chronic colitis in C57BL/six mice, and colon tumours in the multiple intestinal neoplasia (Apc Min/+) mouse model for human colon carcinoma. This is the same mouse model where H. pylori triggers a pro‐carcinogenic multi‐step inflammatory cascade that requires IL‐17R, NF‐κβ and STAT3 signalling in colonic epithelial cells 62, 63. These mouse experiments are further substantiated by epidemiology, indicating that infections with enterotoxigenic variants of B. fragilis, as opposed to non‐toxigenic variants, are more prevalent in people with colorectal cancers. More specifically, the enterotoxigenic variant is present in only 10–20% of the healthy population, whereas 40% of CRC patients present enterotoxigenic B. fragilis in their faeces 64. In addition to BFT, multiple biologically plausible mechanisms have been reported that explain how the vacuolating cytotoxin A (VacA) of H. pylori enhances gastric cancer risk. Similar as the H. pylori outer membrane protein OipA, VacA activates the EGFR receptor that triggers PI3K–Akt signalling, and inactivates glycogen synthase kinase 3β 34, 65. As a result, β‐catenin degradation is abolished, which promotes Tcf/LEF‐controlled transcription that promotes cell growth and transformation 34, 65, 66. Another H. pylori virulence factor, cytotoxin‐associated gene A (CagA), which depends on the type IV pilus cell‐surface adhesion CagL for its host cell targeting, interacts with the c‐Met receptor to activate epithelial proliferation, as shown in human gastric organoids 67. Phosphorylated and unphosphorylated CagA can also interact with a variety of host proteins involved in the MEK, ERK, NF‐κβ and β‐catenin pathways that are all involved in host cell proliferation and cancer formation 68, 69.
Bacterial effector proteins that mediate host cell transformation
Various intracellular bacterial pathogens have developed molecular mechanisms to ensure a persistent infection within the protective environment of the host cell's interior. This requires host cell control at various steps of the intracellular infection cycle, including host cell internalization through receptor‐mediated endocytosis or phagocytosis, intracellular survival and growth, and release from the infected host cell.
After host cell internalization bacterial‐cargo generally routes across the endosomal system that usually terminates in a highly degradative organelle, the phagolysosome. To avoid phagolysosomal degradation, intracellular bacterial pathogens have evolved various mechanisms that can be broadly grouped into pathways where pathogenic bacteria either escape the phagosome or enter in the cytosol, and pathways where the phagosome is hijacked and tailored to the preferences of the bacteria. Cytosolic pathogens like Listeria, Shigella flexneri (S. flexneri), Rickettsia and Francisella are known to rapidly escape the phagosome to enter the host cytosol and thereby avoid lysosomal fusion and degradation 70. This generally involves secretion of bacterial effector proteins that induce pore formation of the endolysosomal vacuole and ensure its subsequent rupture. It has, for example, been shown that S. flexneri secretes the effector protein Invasion plasmid antigen B that forms ion channels in eukaryotic membranes and can mediate potassium influx and subsequent endolysosomal leakage 71. In addition, Listeria can secrete the listeriolysin‐O protein that induces small‐membrane perforations, which causes Ca2+ leakage from vacuoles and an increase in the vacuolar pH. Subsequently, vacuolar maturation is prevented 72, 73. Francisella tularensis (F. tularensis) also escapes into the host cytoplasm. After phagocytic uptake by macrophages, F. tularensis resides in the Francisella‐containing phagosome (FCP) that over time matures from a phagosome with an early endosomal character into a more acidic late endosomal phagosome. Since inhibition of FCP acidification delays the escape of F. tularensis into the cytosol, further acidification during phagosome maturation apparently stimulates F. tularensis to express unique, as‐yet‐undefined factors to disrupt the phagosomal membrane 74, 75, 76.
In contrast to bacteria that escape the phagosome, pathogenic bacteria have been reported that hijack the phagosome to ensure a favourable replication niche. An example of such a pathogen is Legionella pneumophila that redirects the Legionella‐containing phagosome to the ER via the secretion of bacterial proteins through the Dot‐Icm secretion system. This rearrangement prevents lysosomal degradation and ensures Legionella replication within the phagosome 77, 78. Bacterial control of phagosomal maturation has also been reported for the intracellular pathogen Salmonella. After its host cell internalization, Salmonella ends up in a membrane‐bound phagosome‐like vacuolar compartment called the Salmonella‐containing vacuole (SCV). The SCV then matures and acquires characteristics of late endocytic compartments including acidification. It does, however, not become bactericidal. Under control of the Salmonella effectors, SifA, SseJ, SseG, SseF, SopD2, and PipB2, cellular host processes are manipulated to turn the SCV into a compartment that facilitates Salmonella replication 79. SifA, which is critical in this process 80, interacts with the host cell effector of the GTPase Arl8b, the SifA and kinesin‐interacting protein SKIP. This interaction results in the formation of a tubular membrane network, known as Salmonella‐induced filaments, that is essential for the supply of nutrients to the SCV and prevents endosomal antimicrobial activities due to constant mixing of antimicrobial agents with late endosomes and lysosomes 81, 82.
Intracellular pathogenic bacteria that engage effector proteins during their intracellular life cycle manipulate host cell integrity in a major way. To this end, some of these infections have been epidemiologically linked to particular cancer types. Infections by two food‐borne Salmonella serovars, S. Typhi and S. Enteritidis, are linked to gallbladder carcinoma and colon cancer, respectively 13, 14. These bacteria introduce a series of effector proteins in the host cell to take over host cell biology and—depending on host pathway affected—can contribute to cancer formation. A Salmonella effector protein that has been linked to colon cancer formation is the acetyltransferase AvrA that alters a variety of host‐signalling pathways and modulates immune responses, apoptosis and proliferation 83, 84. AvrA modifies and stabilizes β‐catenin, thereby enhancing signalling and promoting epithelial cell proliferation (Fig 3A) 85, 86, 87. AvrA also suppresses the host immune system and its apoptotic defences via the inhibition of the c‐Jun N‐terminal kinase (JNK) and NF‐κβ signalling pathways (Fig 3A) 88. In addition to AvrA, three AvrA orthologues have been reported that similarly interact with essential host cell signalling pathways. However, in contrast to AvrA these orthologues have primarily only inhibitory effects on the host immune system. YopJ is expressed by Yersinia pestis and attenuates the ERK, p38, JNK and Iκβ kinase (IKK) pathways involved in the synthesis of cytokines as well as anti‐apoptotic factors 89. VopA of Vibrio parahaemolyticus can similarly inhibit host ERK, p38 and JNK signalling, but not the IKK pathway 90, 91, and AopP of Aeromonas salmonicida interacts with the IKK pathway 92.
Figure 3. Examples of bacterial effector proteins involved in cellular transformation.

(A) Salmonella Enteritidis AvrA stabilizes β‐catenin, which results in proliferative Wnt signalling. At the same time, AvrA inhibits JNK and NF‐κβ signalling pathways involved in inflammation and apoptosis. (B) The effector proteins SopB, SopE and SopE2 of Salmonella typhimurium activate the small GTPases RhoG, Rac1 and Cdc42 and activate members of the PAK that phosphorylate members of the Abl kinase family, leading to the activation the cytoplasmic transcription factor STAT3, which contributes to cellular transformation. The effector proteins SopB, SopE, SopE2 and SptP of S. typhimurium additionally mediate activation of the MAPK and Akt pathways, which transforms premutated fibroblasts and gallbladder organoids.
In epithelial cells infected with S. typhimurium, the effector proteins SopE, SopE2 and SopB can manipulate host Rho‐family GTPases, p21‑activated kinase (PAK) and ABL tyrosine kinase to activate STAT3 and alter transcription regulation, which 93 can mediate transformation of cells (Fig 3B). In addition, cellular transformation can occur through Salmonella effector SopE, SopE2, SopB and SptP‐mediated activation of the MAPK and AKT pathways (Fig 3B) 94. The activation of these signalling pathways enables the transformation of fibroblasts and gallbladder organoids that harbour a pre‐transformed phenotype whereby the tumour suppressor gene p53 is inactivated and the MYC oncogene is overexpressed. These findings are supported by pathology on gallbladder carcinoma samples from Indian patients that contain both S. Typhi DNA and the pre‐transformed modifications also observed in the laboratory experiments, and by an Apc Min/+ mouse model in which oral infection with S. typhimurium results in the development colorectal adenocarcinomas in a Salmonella effector‐dependent manner 13.
Conclusions
Although bacterially induced host cell manipulation can promote cancer formation, it is unlikely that bacterial pathogens themselves experience any evolutionary benefit from their carcinogenic actions. Bacterially induced cancer formation is more likely an unfortunate consequence of the bacterial infection cycle since cancer usually occurs long after the bacterium and its effectors have left the host 13, 14. Moreover, bacterial host cell manipulations involved in the induction of cancer formation usually account for only one step in the multi‐step process required for actual cellular transformation and cancer formation. This can be illustrated by Salmonella infections that only in combination with pre‐mutations allow cellular transformation in tissue culture fibroblasts and gallbladder organoids and is supported by observations of Indian gallbladder cancer patients who showed the corresponding pre‐mutations in the p53 gene, c‐MYC amplification in their tumours and had a history of S. Typhi infection. 11 In other words, Salmonella will only induce cancer when the cell has made already one or multiple pretransforming steps. This would explain why chronic bacterial infections have a higher statistical chance of initiating tumorigenesis as the likelihood of encountering a pre‐transformed cell would then be markedly increased. This may also explain the correlations of persistent Mycobacterium tuberculosis infections and pulmonary cancers 95 and chronic Coxiella burnetii infections and B‐cell non‐Hodgkin lymphoma 96.
Since many epidemiological studies reveal a link between bacterial infections and cancer incidence, and the number of bacterial mechanisms that can contribute to cellular transformation are most likely considerable larger than reported to date, we expect that the number of examples illustrating the role of bacterial infections in cancer formation will increase the coming years. It is also known that bacterial effectors from different species can act synergistically during host cell manipulation and then act in a symbiotic interspecies manner 55, 97. These combined mechanisms can induce cell transformation and cancer in an even more complex manner and further contribute to the complexity of bacterial contributions to cancer.
While the first examples of bacterial mechanisms contributing to cancer are uncovered, it is likely that bacteria will provide many new and surprising mechanisms for host cell manipulation, some of which may participate in cell transformation. These may include an expansion of mechanisms involved in immune evasion, DNA damage and signalling pathways, but may also include more indirect routes, as, for example, via the formation of carcinogenic metabolites 98. When the role of defined bacterial mechanisms in cancer formation will become more apparent and accepted (see also Box 1), studies on their prevention or control can help reduce cancer formation. On this note, antibiotic therapy during cancer treatment, which is already a standard of care in patients with gastric mucosa‐associated lymphoid tissue (MALT) lymphoma 99, might become a valuable addition to current tumour‐targeting therapies. This, however, may only help when the presence of a bacterial species is required to continuously provide signals to maintain the transformed state. Otherwise, patients diagnosed with a bacterial pathogen known to participate in cancer formation—but not necessarily maintenance—may be incorporated in cancer screening programs.
Box 1: In need of answers.
Do bacterial infections only decrease the threshold for cellular transformation or can they also initiate tumour formation?
How is transformation by activation of host signalling pathways imprinted in host cells?
How can correlations from microbiome studies be translated to causalities?
Does transformed tissue cause microbial dysbiosis 100?
It has been shown that there is a distal oncogenic effect of the gut microbiome 101. How does the gut microbiome affect tumour formation at a distance?
What is the total contribution of bacteria to cancer formation?
How to translate the collective knowledge on bacteria and cancer formation into treatment or prevention measures?
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work is supported by an ERC Advanced grant and a grant from the Dutch Cancer Society KWF to JN.
EMBO Reports (2018) 19: e46632
See the Glossary for abbreviations used in this article.
References
- 1. Wu S, Powers S, Zhu W, Hannun YA (2015) Substantial contribution of extrinsic risk factors to cancer development. Nature 529: 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siegel RL, Jacobs EJ, Newton CC, Feskanich D, Freedman ND, Prentice RL, Jemal A (2015) Deaths due to cigarette smoking for 12 smoking‐related cancers in the United States. JAMA Intern Med 175: 1574–1576 [DOI] [PubMed] [Google Scholar]
- 3. Koh HK, Geller AC, Miller DR, Grossbart TA, Lew RA (1996) Prevention and early detection strategies for melanoma and skin cancer. Current status. Arch Dermatol 132: 436–443 [PubMed] [Google Scholar]
- 4. Parkin DM, Mesher D, Sasieni P (2011) 13. Cancers attributable to solar (ultraviolet) radiation exposure in the UK in 2010. Br J Cancer 105(Suppl 2): S66–S69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blot WJ, McLaughlin JK, Winn DM, Austin DF, Greenberg RS, Preston‐Martin S, Bernstein L, Schoenberg JB, Stemhagen A, Fraumeni JF Jr (1988) Smoking and drinking in relation to oral and pharyngeal cancer. Can Res 48: 3282–3287 [PubMed] [Google Scholar]
- 6. Khoury JD, Tannir NM, Williams MD, Chen Y, Yao H, Zhang J, Thompson EJ, TCGA Network , Meric‐Bernstam F, Medeiros LJ et al (2013) Landscape of DNA virus associations across human malignant cancers: analysis of 3,775 cases using RNA‐seq. J Virol 87: 8916–8926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bosch FX, Manos MM, Munoz N, Sherman M, Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R, Shah KV (1995) Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J Natl Cancer Inst 87: 796–802 [DOI] [PubMed] [Google Scholar]
- 8. El‐Serag HB (2012) Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142: 1264–1273.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neuveut C, Wei Y, Buendia MA (2010) Mechanisms of HBV‐related hepatocarcinogenesis. J Hepatol 52: 594–604 [DOI] [PubMed] [Google Scholar]
- 10. Münger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K (2004) Mechanisms of human papillomavirus‐induced oncogenesis. J Virol 78: 11451–11460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mostafa MH, Sheweita SA, O'Connor PJ (1999) Relationship between schistosomiasis and bladder cancer. Clin Microbiol Rev 12: 97–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kikuchi S (2002) Epidemiology of Helicobacter pylori and gastric cancer. Gastric Cancer 5: 6–15 [DOI] [PubMed] [Google Scholar]
- 13. Scanu T, Spaapen RM, Bakker JM, Pratap CB, Wu LE, Hofland I, Broeks A, Shukla VK, Kumar M, Janssen H et al (2015) Salmonella manipulation of host signaling pathways provokes cellular transformation associated with gallbladder carcinoma. Cell Host Microbe 17: 763–774 [DOI] [PubMed] [Google Scholar]
- 14. Mughini‐Gras L, Schaapveld M, Kramers J, Mooij S, Neefjes‐Borst EA, Pelt WV, Neefjes J (2018) Increased colon cancer risk after severe Salmonella infection. PLoS One 13: e0189721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S et al (2011) Lack of commensal flora in Helicobacter pylori‐infected INS‐GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 140: 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee CW, Rickman B, Rogers AB, Ge Z, Wang TC, Fox JG (2008) Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS‐GAS mice. Can Res 68: 3540–3548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vannucci L, Stepankova R, Kozakova H, Fiserova A, Rossmann P, Tlaskalova‐Hogenova H (2008) Colorectal carcinogenesis in germ‐free and conventionally reared rats: different intestinal environments affect the systemic immunity. Int J Oncol 32: 609–617 [PubMed] [Google Scholar]
- 18. Li Y, Kundu P, Seow SW, de Matos CT, Aronsson L, Chin KC, Karre K, Pettersson S, Greicius G (2012) Gut microbiota accelerate tumor growth via c‐jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis 33: 1231–1238 [DOI] [PubMed] [Google Scholar]
- 19. Dapito Dianne H, Mencin A, Gwak G‐Y, Pradere J‐P, Jang M‐K, Mederacke I, Caviglia Jorge M, Khiabanian H, Adeyemi A, Bataller R et al (2012) Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21: 504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pluschke G, Mayden J, Achtman M, Levine RP (1983) Role of the capsule and the O antigen in resistance of O18:K1 Escherichia coli to complement‐mediated killing. Infect Immun 42: 907–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abeyta M, Hardy GG, Yother J (2003) Genetic alteration of capsule type but not PspA type affects accessibility of surface‐bound complement and surface antigens of Streptococcus pneumoniae . Infect Immun 71: 218–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown EJ, Hosea SW, Frank MM (1983) The role of antibody and complement in the reticuloendothelial clearance of pneumococci from the bloodstream. Rev Infect Dis 5(Suppl 4): S797–S805 [DOI] [PubMed] [Google Scholar]
- 23. Winkelstein JA, Tomasz A (1978) Activation of the alternative complement pathway by pneumococcal cell wall teichoic acid. J Immunol 120: 174–178 [PubMed] [Google Scholar]
- 24. Watson DA, Musher DM (1990) Interruption of capsule production in Streptococcus pneumonia serotype 3 by insertion of transposon Tn916. Infect Immun 58: 3135–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geno KA, Gilbert GL, Song JY, Skovsted IC, Klugman KP, Jones C, Konradsen HB, Nahm MH (2015) Pneumococcal capsules and their types: past, present, and future. Clin Microbiol Rev 28: 871–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mattsby‐Baltzer I, Mielniczuk Z, Larsson L, Lindgren K, Goodwin S (1992) Lipid A in Helicobacter pylori . Infect Immun 60: 4383–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tran AX, Stead CM, Trent MS (2005) Remodeling of Helicobacter pylori lipopolysaccharide. J Endotoxin Res 11: 161–166 [DOI] [PubMed] [Google Scholar]
- 28. Gewirtz AT, Yu Y, Krishna US, Israel DA, Lyons SL, Peek RM Jr (2004) Helicobacter pylori flagellin evades toll‐like receptor 5‐mediated innate immunity. J Infect Dis 189: 1914–1920 [DOI] [PubMed] [Google Scholar]
- 29. Kawasaki K, Ernst RK, Miller SI (2004) 3‐O‐deacylation of lipid A by PagL, a PhoP/PhoQ‐regulated deacylase of Salmonella typhimurium, modulates signaling through toll‐like receptor 4. J Biol Chem 279: 20044–20048 [DOI] [PubMed] [Google Scholar]
- 30. Popp A, Billker O, Rudel T (2001) Signal transduction pathways induced by virulence factors of Neisseria gonorrhoeae . Int J Med Microbiol 291: 307–314 [DOI] [PubMed] [Google Scholar]
- 31. Meenan NA, Visai L, Valtulina V, Schwarz‐Linek U, Norris NC, Gurusiddappa S, Hook M, Speziale P, Potts JR (2007) The tandem beta‐zipper model defines high affinity fibronectin‐binding repeats within Staphylococcus aureus FnBPA. J Biol Chem 282: 25893–25902 [DOI] [PubMed] [Google Scholar]
- 32. Raibaud S, Schwarz‐Linek U, Kim JH, Jenkins HT, Baines ER, Gurusiddappa S, Hook M, Potts JR (2005) Borrelia burgdorferi binds fibronectin through a tandem beta‐zipper, a common mechanism of fibronectin binding in staphylococci, streptococci, and spirochetes. J Biol Chem 280: 18803–18809 [DOI] [PubMed] [Google Scholar]
- 33. Wiedemann T, Hofbaur S, Tegtmeyer N, Huber S, Sewald N, Wessler S, Backert S, Rieder G (2012) Helicobacter pylori CagL dependent induction of gastrin expression via a novel alphavbeta5‐integrin‐integrin linked kinase signalling complex. Gut 61: 986–996 [DOI] [PubMed] [Google Scholar]
- 34. Tabassam FH, Graham DY, Yamaoka Y (2009) Helicobacter pylori activate epidermal growth factor receptor‐ and phosphatidylinositol 3‐OH kinase‐dependent Akt and glycogen synthase kinase 3beta phosphorylation. Cell Microbiol 11: 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Polk DB, Peek RM Jr (2010) Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 10: 403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, Wroblewski LE, Piazuelo MB, Correa P, Peek RM Jr (2008) Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Can Res 68: 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ishijima N, Suzuki M, Ashida H, Ichikawa Y, Kanegae Y, Saito I, Boren T, Haas R, Sasakawa C, Mimuro H (2011) BabA‐mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J Biol Chem 286: 25256–25264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E‐cadherin/beta‐catenin signaling via its FadA adhesin. Cell Host Microbe 14: 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J et al (2012) Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22: 292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen‐Vercoe E, Moore RA et al (2012) Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22: 299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coleman OI, Haller D (2017) Bacterial signaling at the intestinal epithelial interface in inflammation and cancer. Front Immunol 8: 1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rakoff‐Nahoum S, Medzhitov R (2007) Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 317: 124–127 [DOI] [PubMed] [Google Scholar]
- 43. Abreu MT (2010) Toll‐like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol 10: 131–144 [DOI] [PubMed] [Google Scholar]
- 44. Fink SP, Yamauchi M, Nishihara R, Jung S, Kuchiba A, Wu K, Cho E, Giovannucci E, Fuchs CS, Ogino S et al (2014) Aspirin and the risk of colorectal cancer in relation to the expression of 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH, HPGD). Sci Transl Med 6: 233re2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, Kudo E, Shimada M, Sano T (2010) High expression of Toll‐like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer 102: 908–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Costa TRD, Felisberto‐Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, Waksman G (2015) Secretion systems in Gram‐negative bacteria: structural and mechanistic insights. Nat Rev Microbiol 13: 343 [DOI] [PubMed] [Google Scholar]
- 47. Murphy JR (2011) Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 3: 294–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosadi F, Fiorentini C, Fabbri A (2016) Bacterial protein toxins in human cancers. Pathog Dis 74: ftv105 [DOI] [PubMed] [Google Scholar]
- 49. Cortes‐Bratti X, Chaves‐Olarte E, Lagergard T, Thelestam M (2000) Cellular internalization of cytolethal distending toxin from Haemophilus ducreyi . Infect Immun 68: 6903–6911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guerra L, Carr HS, Richter‐Dahlfors A, Masucci MG, Thelestam M, Frost JA, Frisan T (2008) A bacterial cytotoxin identifies the RhoA exchange factor Net1 as a key effector in the response to DNA damage. PLoS One 3: e2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Putze J, Hennequin C, Nougayrede JP, Zhang W, Homburg S, Karch H, Bringer MA, Fayolle C, Carniel E, Rabsch W et al (2009) Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun 77: 4696–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E (2006) Escherichia coli induces DNA double‐strand breaks in eukaryotic cells. Science 313: 848–851 [DOI] [PubMed] [Google Scholar]
- 53. Cuevas‐Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP (2010) Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA 107: 11537–11542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille‐Michaud A, Pezet D, Bonnet R (2013) High prevalence of mucosa‐associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One 8: e56964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, Wu X, DeStefano Shields CE, Hechenbleikner EM, Huso DL et al (2018) Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359: 592–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Arthur JC, Perez‐Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB et al (2012) Intestinal inflammation targets cancer‐inducing activity of the microbiota. Science 338: 120–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu S, Rhee KJ, Zhang M, Franco A, Sears CL (2007) Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and gamma‐secretase‐dependent E‐cadherin cleavage. J Cell Sci 120: 1944–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sears CL (2009) Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev 22: 349–369, Table of Contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu S, Lim KC, Huang J, Saidi RF, Sears CL (1998) Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E‐cadherin. Proc Natl Acad Sci USA 95: 14979–14984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu S, Powell J, Mathioudakis N, Kane S, Fernandez E, Sears CL (2004) Bacteroides fragilis enterotoxin induces intestinal epithelial cell secretion of interleukin‐8 through mitogen‐activated protein kinases and a tyrosine kinase‐regulated nuclear factor‐kappaB pathway. Infect Immun 72: 5832–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nelson WJ, Nusse R (2004) Convergence of Wnt, beta‐catenin, and cadherin pathways. Science 303: 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F et al (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15: 1016–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, Rabizadeh S, Golub JE, Mathews LE, Shin J et al (2009) Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild‐type C57BL/6 mice. Infect Immun 77: 1708–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Toprak NU, Yagci A, Gulluoglu BM, Akin ML, Demirkalem P, Celenk T, Soyletir G (2006) A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect 12: 782–786 [DOI] [PubMed] [Google Scholar]
- 65. Nakayama M, Hisatsune J, Yamasaki E, Isomoto H, Kurazono H, Hatakeyama M, Azuma T, Yamaoka Y, Yahiro K, Moss J et al (2009) Helicobacter pylori VacA‐induced inhibition of GSK3 through the PI3K/Akt signaling pathway. J Biol Chem 284: 1612–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sokolova O, Bozko PM, Naumann M (2008) Helicobacter pylori suppresses glycogen synthase kinase 3beta to promote beta‐catenin activity. J Biol Chem 283: 29367–29374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y et al (2014) Modelling human development and disease in pluripotent stem‐cell‐derived gastric organoids. Nature 516: 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu X, Liu Z, Fang M, Yu H, Liang X, Li X, Liu X, Chen C, Jia J (2012) Helicobacter pylori CagA induces ornithine decarboxylase upregulation via Src/MEK/ERK/c‐Myc pathway: implication for progression of gastric diseases. Exp Biol Med (Maywood) 237: 435–441 [DOI] [PubMed] [Google Scholar]
- 69. Wang F, Meng W, Wang B, Qiao L (2014) Helicobacter pylori‐induced gastric inflammation and gastric cancer. Cancer Lett 345: 196–202 [DOI] [PubMed] [Google Scholar]
- 70. Fredlund J, Enninga J (2014) Cytoplasmic access by intracellular bacterial pathogens. Trends Microbiol 22: 128–137 [DOI] [PubMed] [Google Scholar]
- 71. Senerovic L, Tsunoda SP, Goosmann C, Brinkmann V, Zychlinsky A, Meissner F, Kolbe M (2012) Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis 3: e384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Henry R, Shaughnessy L, Loessner MJ, Alberti‐Segui C, Higgins DE, Swanson JA (2006) Cytolysin‐dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes . Cell Microbiol 8: 107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shaughnessy LM, Lipp P, Lee K‐D, Swanson JA (2007) Localization of protein kinase C ε to macrophage vacuoles perforated by Listeria monocytogenes cytolysin. Cell Microbiol 9: 1695–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Santic M, Asare R, Skrobonja I, Jones S, Abu Kwaik Y (2008) Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect Immun 76: 2671–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ozanic M, Marecic V, Abu Kwaik Y, Santic M (2015) The divergent intracellular lifestyle of Francisella tularensis in evolutionarily distinct host cells. PLoS Pathog 11: e1005208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, Klose KE, Celli J (2008) The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun 76: 5488–5499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR (2002) A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295: 679–682 [DOI] [PubMed] [Google Scholar]
- 78. Tilney LG, Harb OS, Connelly PS, Robinson CG, Roy CR (2001) How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J Cell Sci 114: 4637–4650 [DOI] [PubMed] [Google Scholar]
- 79. Rajashekar R, Liebl D, Chikkaballi D, Liss V, Hensel M (2014) Live cell imaging reveals novel functions of Salmonella enterica SPI2‐T3SS effector proteins in remodeling of the host cell endosomal system. PLoS One 9: e115423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Beuzon CR, Meresse S, Unsworth KE, Ruiz‐Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW (2000) Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 19: 3235–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sindhwani A, Arya SB, Kaur H, Jagga D, Tuli A, Sharma M (2017) Salmonella exploits the host endolysosomal tethering factor HOPS complex to promote its intravacuolar replication. PLoS Pathog 13: e1006700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Stein MA, Leung KY, Zwick M, Garcia‐del Portillo F, Finlay BB (1996) Identification of a Salmonella virulence gene required for formation of filamentous structures containing lysosomal membrane glycoproteins within epithelial cells. Mol Microbiol 20: 151–164 [DOI] [PubMed] [Google Scholar]
- 83. Lu R, Wu S, Zhang YG, Xia Y, Zhou Z, Kato I, Dong H, Bissonnette M, Sun J (2016) Salmonella protein AvrA activates the STAT3 signaling pathway in colon cancer. Neoplasia 18: 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lu R, Bosland M, Xia Y, Zhang YG, Kato I, Sun J (2017) Presence of Salmonella AvrA in colorectal tumor and its precursor lesions in mouse intestine and human specimens. Oncotarget 8: 55104–55115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lu R, Wu S, Zhang YG, Xia Y, Liu X, Zheng Y, Chen H, Schaefer KL, Zhou Z, Bissonnette M et al (2014) Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic beta‐catenin signaling pathway. Oncogenesis 3: e105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ye Z, Petrof EO, Boone D, Claud EC, Sun J (2007) Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am J Pathol 171: 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sun J, Hobert ME, Rao AS, Neish AS, Madara JL (2004) Bacterial activation of beta‐catenin signaling in human epithelia. Am J Physiol Gastrointest Liver Physiol 287: G220–G227 [DOI] [PubMed] [Google Scholar]
- 88. Jones RM, Wu H, Wentworth C, Luo L, Collier‐Hyams L, Neish AS (2008) Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 3: 233–244 [DOI] [PubMed] [Google Scholar]
- 89. Orth K, Palmer LE, Bao ZQ, Stewart S, Rudolph AE, Bliska JB, Dixon JE (1999) Inhibition of the mitogen‐activated protein kinase kinase superfamily by a Yersinia effector. Science 285: 1920–1923 [DOI] [PubMed] [Google Scholar]
- 90. Trosky JE, Mukherjee S, Burdette DL, Roberts M, McCarter L, Siegel RM, Orth K (2004) Inhibition of MAPK signaling pathways by VopA from Vibrio parahaemolyticus . J Biol Chem 279: 51953–51957 [DOI] [PubMed] [Google Scholar]
- 91. Trosky JE, Li Y, Mukherjee S, Keitany G, Ball H, Orth K (2007) VopA inhibits ATP binding by acetylating the catalytic loop of MAPK kinases. J Biol Chem 282: 34299–34305 [DOI] [PubMed] [Google Scholar]
- 92. Fehr D, Casanova C, Liverman A, Blazkova H, Orth K, Dobbelaere D, Frey J, Burr SE (2006) AopP, a type III effector protein of Aeromonas salmonicida, inhibits the NF‐kappaB signalling pathway. Microbiology 152: 2809–2818 [DOI] [PubMed] [Google Scholar]
- 93. Hannemann S, Gao B, Galán JE (2013) Salmonella modulation of host cell gene expression promotes its intracellular growth. PLoS Pathog 9: e1003668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kuijl C, Savage ND, Marsman M, Tuin AW, Janssen L, Egan DA, Ketema M, van den Nieuwendijk R, van den Eeden SJ, Geluk A et al (2007) Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 450: 725–730 [DOI] [PubMed] [Google Scholar]
- 95. Kuo SC, Hu YW, Liu CJ, Lee YT, Chen YT, Chen TL, Chen TJ, Fung CP (2013) Association between tuberculosis infections and non‐pulmonary malignancies: a nationwide population‐based study. Br J Cancer 109: 229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Melenotte C, Million M, Audoly G, Gorse A, Dutronc H, Roland G, Dekel M, Moreno A, Cammilleri S, Carrieri MP et al (2016) B‐cell non‐Hodgkin lymphoma linked to Coxiella burnetii . Blood 127: 113–121 [DOI] [PubMed] [Google Scholar]
- 97. Fulbright LE, Ellermann M, Arthur JC (2017) The microbiome and the hallmarks of cancer. PLoS Pathog 13: e1006480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12: 661 [DOI] [PubMed] [Google Scholar]
- 99. Wundisch T, Thiede C, Morgner A, Dempfle A, Gunther A, Liu H, Ye H, Du MQ, Kim TD, Bayerdorffer E et al (2005) Long‐term follow‐up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 23: 8018–8024 [DOI] [PubMed] [Google Scholar]
- 100. Dmitrieva O, Grivennikov SI (2017) Microbiota and cancer: a complex equation with a lot of exciting unknowns. Semin Immunol 32: 1–2 [DOI] [PubMed] [Google Scholar]
- 101. Erdman SE, Poutahidis T (2015) Gut bacteria and cancer. Biochem Biophys Acta 1856: 86–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans (2012) Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr Eval Carcinog Risks Hum 100: 1–441 [PMC free article] [PubMed] [Google Scholar]
- 103. Liu W, MacDonald M, You J (2016) Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr Opin Virol 20: 20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zhu H, Shen Z, Luo H, Zhang W, Zhu X (2016) Chlamydia trachomatis infection‐associated risk of cervical cancer: a meta‐analysis. Medicine (Baltimore) 95: e3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Trabert B, Waterboer T, Idahl A, Brenner N, Brinton LA, Butt J, Coburn SB, Hartge P, Hufnagel K, Inturrisi F et al (2018) Antibodies against Chlamydia trachomatis and ovarian cancer risk in two independent populations. J Natl Cancer Inst 111: djy084 [DOI] [PMC free article] [PubMed] [Google Scholar]