

Abstract
Microglia are specialized parenchymal‐resident phagocytes of the central nervous system (CNS) that actively support, defend and modulate the neural environment. Dysfunctional microglial responses are thought to worsen CNS diseases; nevertheless, their impact during neuroinflammatory processes remains largely obscure. Here, using a combination of single‐cell RNA sequencing and multicolour flow cytometry, we comprehensively profile microglia in the brain of lipopolysaccharide (LPS)‐injected mice. By excluding the contribution of other immune CNS‐resident and peripheral cells, we show that microglia isolated from LPS‐injected mice display a global downregulation of their homeostatic signature together with an upregulation of inflammatory genes. Notably, we identify distinct microglial activated profiles under inflammatory conditions, which greatly differ from neurodegenerative disease‐associated profiles. These results provide insights into microglial heterogeneity and establish a resource for the identification of specific phenotypes in CNS disorders, such as neuroinflammatory and neurodegenerative diseases.
Keywords: heterogeneity, lipopolysaccharide, microglia, neuroinflammation, single‐cell RNA‐seq
Subject Categories: Immunology, Methods & Resources, Neuroscience
Introduction
The healthy brain hosts distinct and specialized populations of tissue‐resident macrophages strategically placed in the parenchyma, perivascular spaces, meninges and choroid plexus where they coordinate homeostatic and immune surveillance functions 1. As the only parenchymal‐resident immune cells of the central nervous system (CNS), microglia act as critical effectors and regulators of changes in the CNS during development and adult homeostasis. Their ontogeny, together with the absence of turnover from the periphery and the exceptional environment of the CNS, makes microglia a unique immune cell population 2. By sensing any disruption of CNS homeostasis, microglia rapidly change their gene expression programmes and functional profiles. Recent genome‐wide transcriptional studies revealed a unique molecular signature selectively expressed in homeostatic microglia 3, 4, 5, 6 that is lost in disease and during ageing 4, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17. Microglia coordinate immune responses between the periphery and the CNS as they perceive and propagate inflammatory signals initiated outside the CNS 18. A multitude of signals received from the CNS environment as well as from the periphery induce microglial responses towards phenotypes that ultimately may support or harm neuronal health 2, 19. Although neuroinflammation and its associated immune responses are often linked to neurodegeneration, the inflammatory response per se provides a primary, transient and self‐limiting defence mechanism, by which harmful stimuli are resolved and tissue damage is repaired 20. Disruption of CNS homeostasis, neuronal deterioration and inflammation are common pathophysiological features of several neurodegenerative diseases. In this context, chronic inflammation is likely to be triggered by abnormal protein deposition, by signals elicited by injured neurons and synapses or by impaired pro‐ and anti‐inflammatory regulatory mechanisms that ultimately exacerbate the neurodegenerative process 21. Dysfunctional microglial responses are believed to worsen CNS diseases 22; nevertheless, their impact during the neuroinflammatory processes remains largely obscure.
In recent years, single‐cell RNA sequencing investigations have emerged as a remarkable method to depict heterogeneous cell populations and measure cell‐to‐cell expression variability of thousands of genes 23, 24, 25. In the murine and human brains, single‐cell RNA sequencing analyses have revealed neural and glial cell heterogeneity 26, 27, 28, 29, 30. Similarly, the complexity of immune cell types has been recently unravelled 31. However, although recent studies have elucidated microglia signatures associated with inflammatory conditions at the bulk level 4, 16, 32, it is still not clear whether all microglial cells uniformly react to the inflammatory stimuli.
To elucidate the heterogeneity of microglial responses towards systemic inflammation, we here analysed the effect of a peripheral injection of the Gram‐negative bacterial endotoxin lipopolysaccharide (LPS) in 3‐ to 4‐month‐old C57BL/6N mice using a combination of multicolour flow cytometry and single‐cell RNA sequencing analyses. LPS is a well‐known immunostimulant used to mimic inflammatory and infectious conditions inducing immune responses associated with sickness behaviour in mice and humans 33, 34. Notably, it has been shown that repeated peripheral injections of LPS in mice induce neurodegeneration, while a single‐dose injection of LPS induces acute inflammatory, but not neurodegenerative processes 35. By our approach, we have identified distinct microglial activated profiles under acute inflammatory conditions, which differ from the recently described disease‐associated phenotypes 14.
Understanding the specific molecular triggers and the subsequent genetic programmes defining microglia under homeostatic, inflammatory and neurodegenerative conditions at the single‐cell level is a fundamental step to further uncover the multifaceted nature of microglia, thus opening new windows to design novel therapeutic strategies to restore, for example, efficient inflammatory immune responses in CNS diseases.
Results and Discussion
Acutely isolated CD11b+CD45int cells express high levels of microglial homeostatic genes and represent a specific resident immune cell population
Cell‐specific transcriptomic analyses are critically dependent on isolation protocols to obtain pure populations resembling their physiological profiles. To characterize microglia close to their proper environment, mouse brains were mechanically dissociated into single‐cell suspension with all the steps performed at 4°C 36. Since microglia in the mouse brain represent only 10% of the cells, CD11b+CD45int microglia were purified from other CNS and immune cells, including CD11b+CD45high macrophages and CD11b−CD45high lymphocytes, by FACS, as described previously (Figs 1A and EV1) 37. To verify accurate microglial enrichment, we compared gene expression levels of specific CNS cell type markers between RNA extracted from unsorted total brain cells and CD11b+CD45int sorted microglia (Fig 1B). We analysed the expression levels of microglial homeostatic genes (Olfml3, Fcrls, Tmem119, Siglech, Gpr34, P2ry12) as well as astrocytic (Gfap, Gjb6, Ntsr2, Aldh1l1), oligodendrocytic (Mobp, Mog, Cldn11) and neuronal (Tubb3, Vglut1, NeuN) markers. As expected, microglial markers were highly expressed in CD11b+CD45int sorted cells, whereas astrocytic, oligodendrocytic and neuronal markers were undetectable or detectable at background levels (Figs 1B and EV1). We next investigated whether CD11b+CD45int population contained resident non‐parenchymal macrophages, such as perivascular macrophages. This was inferred using CD206 as an additional marker for resident macrophages 38. Under homeostatic conditions, CD11b+CD45int microglia contained only 0.04 ± 0.02% CD206+ cells, while CD11b+CD45high cells contained 24.7 ± 3.8% CD206+ resident macrophages (Fig 1C and D). Similar results were obtained for the dendritic cell marker CD11c and the monocytic markers Ly6C and CCR2 (Fig EV1). Taken together, these results show that our approach highly discriminates pure and not activated microglial populations from other resident CNS cells.
Figure 1. Characterization of acutely isolated CD11b+ CD45int cells.
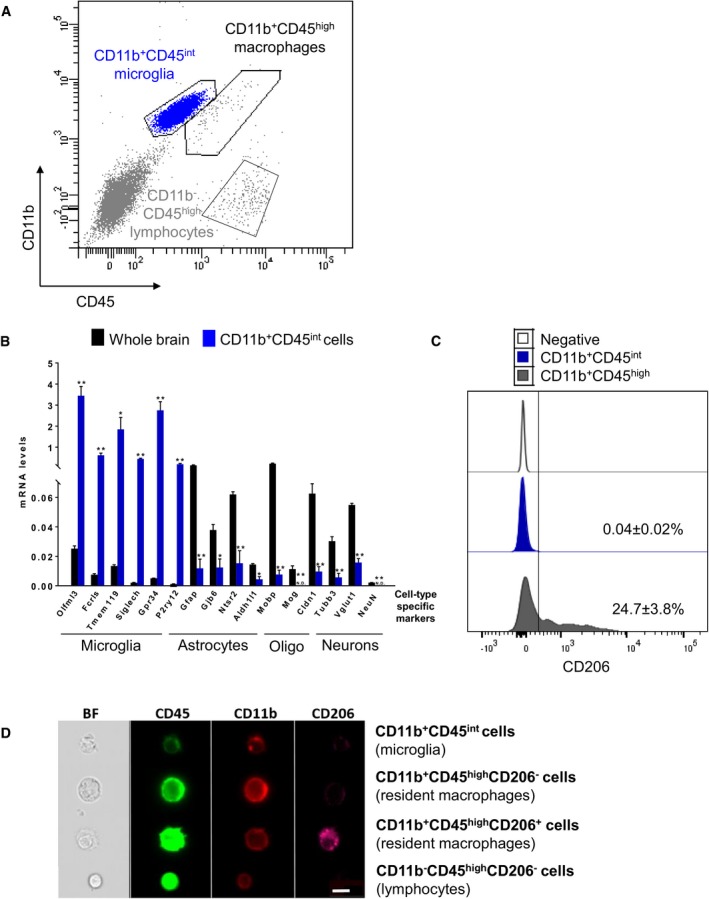
- FACS gating strategy representative of five independent experiments adopted to sort CD11b+CD45int microglia distinctly from CD11b+CD45high resident macrophages and CD11b−CD45high lymphocytes.
- Analysis of relative transcript levels of CD11b+CD45int FACS‐sorted microglia compared with whole brain tissue by qPCR. Gene expression levels of microglia (Olfml3, Fcrls, Tmem119, Siglech, Gpr34, P2ry12), astrocyte (Gfap, Gjb6, Ntsr2, Aldh1l1), oligodendrocyte (Mobp, Mog, Cldn1) and neuron (Tubb3, Vglut1, NeuN) markers. Bars represent mean (n = 4; pool of one female and one male per biological replicate) of relative expression (Gapdh as housekeeping gene) ± SEM (*P < 0.05; **P < 0.01 by two‐tailed Student's t‐test). N.D., not detected.
- Representative quantification of CD206 expression in CD11b+CD45int microglia and CD11b+CD45high resident macrophages. Values denote the percentage of the mean ± SEM of five independent experiments.
- Representative images of two independent experiments showing microglia, resident macrophages and lymphocytes acquired with ImageStream imaging cytometer (Amnis) based on CD45, CD11b and CD206 expression levels (scale bar represents 7 μm).
Figure EV1. Microglial isolation and characterization.
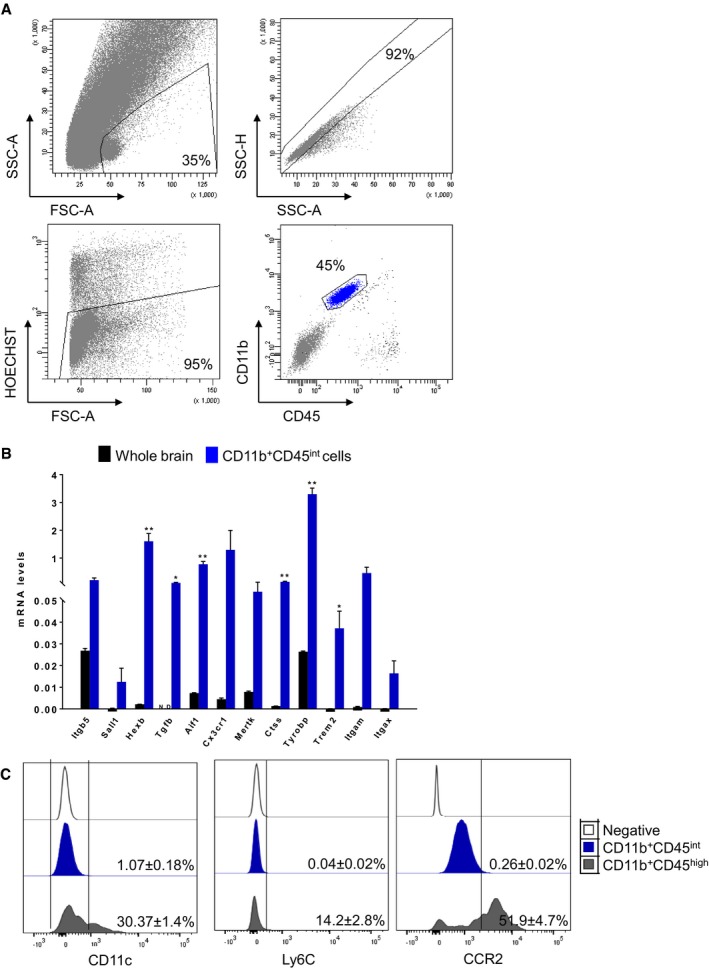
- FACS gating strategy representative of five independent experiments adopted to sort CD11b+CD45int microglial. Cells were distinguished from debris using forward (FSC‐A) and side (SSC‐A) scatters, followed by cell doublet and aggregate elimination (SSC‐H/SSC‐A). Dead cells were gated out by their strong positivity for the dead cell discrimination marker Hoechst. Single viable microglial cells were gated as CD11b+CD45int.
- Analysis of microglial purity by qPCR. Gene expression levels of microglial‐specific genes (Itgb5, Sall1, Hexb, Tgfb, Aif1, Cx3cr1, Mertk, Ctss, Tyrobp, Trem2, Itgam, Itgax) in purified microglia compared to whole brain. Bars represent mean (n = 4; one female and one male per sample) of relative expression (Gapdh as housekeeping gene) ± SEM (*P < 0.05; **P < 0.01 by two‐tailed Student's t‐test).
- Quantification of CD11c, Ly6C and CCR2 expression in CD11b+CD45int microglia and CD11b+CD45high resident macrophages representative of five independent experiments. Values denote the percentage of the mean ± SEM of five independent experiments.
Microglia isolated from LPS‐injected mice show a classical activated pro‐inflammatory profile accompanied by a decreased homeostatic signature
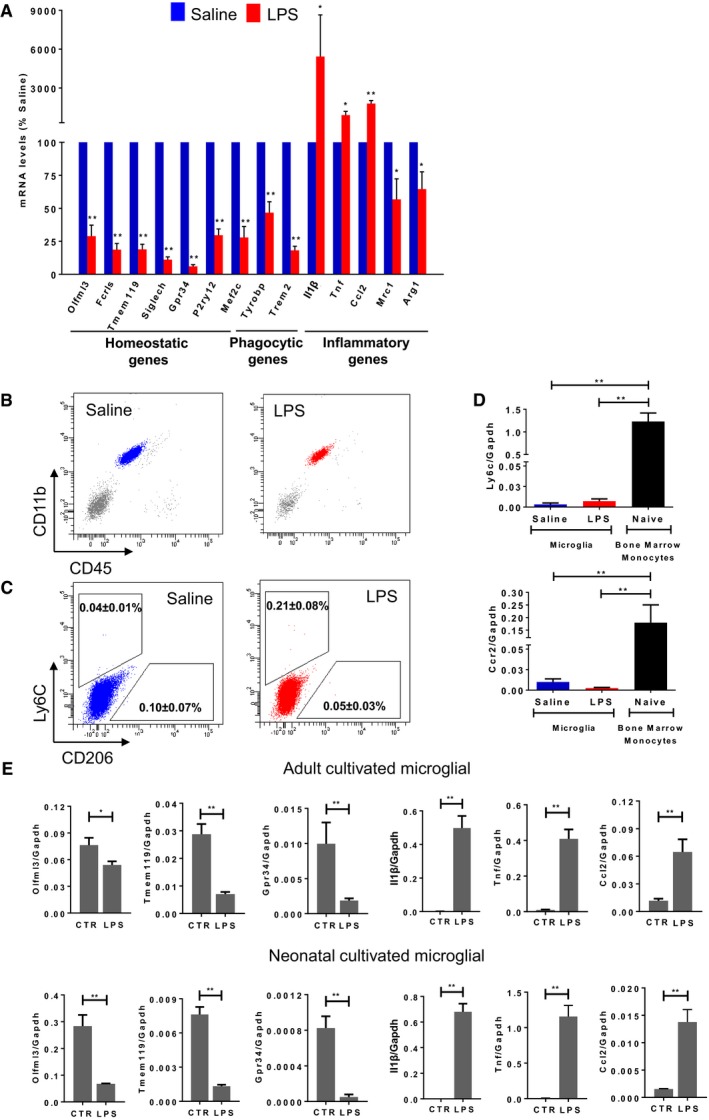
The response of microglia towards specific pro‐ or anti‐inflammatory cues in vitro has been extensively studied 39. Treatment of primary microglial cells with TGF‐β, LPS or IL‐4 generates, respectively, the so‐called M0 homeostatic, M1 pro‐inflammatory and M2 anti‐inflammatory states defined by specific gene signatures 5, 40. However, our understanding towards the reaction of microglia under inflammatory conditions in vivo is only starting to emerge. To comprehensively investigate the effect of a systemic inflammatory and/or infectious state on microglia, we peripherally injected mice with LPS (4 μg/g body) 24 h prior analysis. It has been shown that a single‐dose injection of LPS induces acute inflammatory, but not neurodegenerative processes 35. We isolated CD11b+CD45int cells from LPS‐injected mice and compared mRNA levels of specific genes to the corresponding cells isolated from saline‐injected control mice by qPCR. In agreement with previous studies 32, 41, the expression levels of homeostatic (e.g. Olfml3, Fcrls, Tmem119, Siglech, Gpr34, P2ry12, Mef2c), phagocytic (Tyrobp and Trem2) and anti‐inflammatory genes (e.g. Mrc1 and Arg1) were highly decreased in microglia isolated from LPS‐injected mice compared to untreated mice, while the classical pro‐inflammatory genes (e.g. Il1b, Tnf and Ccl2) were markedly increased (Figs 2A and EV2). Notably, it has been recently shown that signals from the CNS microenvironment have considerable influence in shaping, maintaining and reinforcing microglial identity by regulating expression and establishing distinct chromatin landscapes surrounding enhancer regions 42, 43, 44. Changes in chromatin remodelers associate with changes in the expression of nearby genes. Specifically, MEF2C binding sites were shown to be over‐enriched in enhancer regions of microglial‐specific genes 42 and the loss of MEF2C was associated with priming of microglia 45. In line with these observations, Mef2c expression levels were highly decreased in microglia isolated from LPS‐injected mice compared to naïve mice.
Figure 2. LPS stimulation induces an intrinsic loss of the microglia homeostatic signature.
-
A–DThree‐ to four‐month‐old C57BL/6N mice were treated with an acute dose of LPS (4 μg/g body) or vehicle (saline). Microglia (pool of two mice per group per replicate; one female and one male) were FACS‐sorted 24 h later. (A) Gene expression levels of microglial homeostatic (Olfml3, Fcrls, Tmem119, Siglech, Gpr34, P2ry12, Mef2c), phagocytic (Tyrobp, Trem2) and inflammatory (Il1b, Tnf, Ccl2, Mrc1, Arg1) markers were analysed by qPCR. Bars represent mean of relative expression (% of saline; Gapdh as housekeeping gene) ± SEM (*P < 0.05; **P < 0.01 by two‐tailed Student's t‐test; n = 4). (B) Representative multicolour flow cytometry analysis of five independent experiments showing CD11b‐ and CD45‐positive populations in single viable cells in saline or LPS‐injected mouse brains. (C) Representative multicolour flow cytometry analysis showing the percentage of the mean ± SEM of five independent experiments of Ly6C‐ and CD206‐expressing cells in CD11b+CD45int cells from saline or LPS‐injected mice. (D) Gene expression levels of the monocytic markers Ly6c1 and Ccr2 in purified microglia (n = 4) and isolated bone marrow monocytes (n = 2) by qPCR. Bars represent mean of relative expression (Gapdh as housekeeping gene) ± SEM (**P < 0.01 by two‐tailed Student's t‐test).
-
EPrimary adult microglia were cultivated in the presence of TGF‐β (50 μg/ml) and M‐CSF (10 ng/ml), while neonatal cells were stimulated for 24 h with TGF‐β (50 μg/ml) followed by 6 h of stimulation with LPS (1 ng/ml) or left untreated. Expression levels of microglial homeostatic (Olfml3, Tmem119, Gpr34) and inflammatory (Il1b, Tnf, Ccl2) genes were analysed by qPCR. Bars represent mean of relative expression (Gapdh as housekeeping gene) ± SEM (*P < 0.05; **P < 0.01 by two‐tailed Student's t‐test).
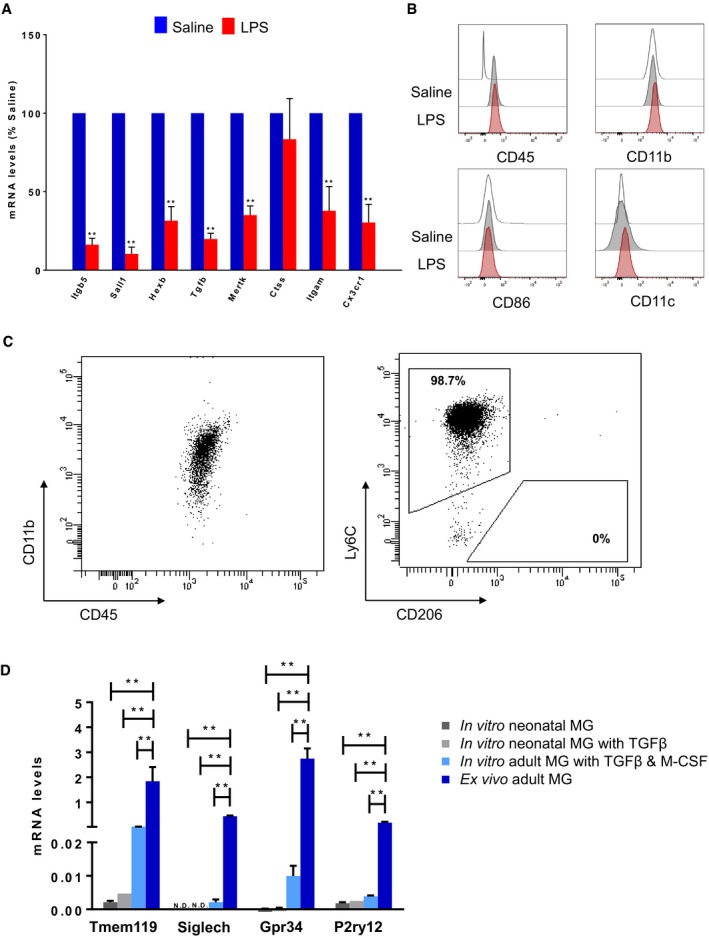
Figure EV2. Ex vivo and in vitro microglial characterization.
-
A, BThree‐ to four‐month‐old mice were treated with an acute dose of LPS (4 μg/g body) or vehicle (saline). Microglial (pool of two mice per group; one female and one male) were isolated 24 h later. (A) Gene expression levels of microglial homeostatic genes (Itgb5, Sall1, Hexb, Tgfb, Mertk, Ctss, Itgam, Cx3cr1) were analysed by qPCR. Bars represent mean of relative expression (% of saline; Gapdh as housekeeping gene) ± SEM (**P < 0.01 by two‐tailed Student's t‐test; n = 4). (B) Representative multicolour flow cytometry analysis of five independent experiments showing CD45, CD11b, CD86 and CD11c expression levels in CD11b+CD45int microglia of saline or LPS‐injected mouse brains.
-
CRepresentative results of two independent experiments showing the purity of MACS‐isolated bone marrow monocytes based on the expression levels of the monocytic marker Ly6C.
-
DComparison of the homeostatic signature (Tmem119, Siglech, Gpr34, P2ry12) between primary and acutely isolated microglia. Primary adult microglia were cultivated in the presence of TGF‐β (50 μg/ml) and M‐CSF (10 ng/ml), while neonatal cells were treated for 24 h with TGF‐β or left untreated. Gene expression levels were analysed by qPCR and normalized using Gapdh as housekeeping gene. Bars represent mean ± SEM (**P < 0.01 by two‐tailed Student's t‐test; n = 3).
We verified that this signature is microglia‐specific, and it is not affected by LPS‐activated immune peripheral cells, such as lymphocytes (CD11b−CD45high cells) and peripheral monocytes/macrophages (CD11b+CD45high cells), as no significant differences were detected between cellular populations present in brains of saline‐ and LPS‐injected mice (Figs 2B and EV2). Importantly, CD11b+CD45int FACS‐gated cells contained very rare (< 0.25%) Ly6C+ putative monocytes and (< 0.1%) CD206+ putative resident macrophages (Fig 2C). Also, the expression of monocytic markers Ly6c1 and Ccr2 was very low in CD11b+CD45int microglia compared to bone marrow‐isolated monocytes with no significant differences under LPS exposure (Figs 2D and EV2). In order to further assess that the decrease in the homeostatic signature under inflammatory conditions is not due to the presence of other immune cell types, but it is an intrinsic property of microglial cells, we also analysed the effect of LPS on cultivated microglial from adult and neonatal mice. As expected, the expression level of the homeostatic genes was markedly decreased in cultivated cells when compared to acutely isolated microglia (Fig EV2) 5. Thus, we cultivated adult microglia in the presence of TGF‐β (50 μg/ml) and M‐CSF (10 ng/ml) or neonatal cells with TGF‐β 24 h prior treatment with LPS to induce the expression of the homeostatic genes, although at a lower extent than in ex vivo isolated cells (Fig EV2). Cells treated with LPS (1 ng/ml) for 6 h showed a dramatic decrease of the expression levels of the homeostatic gene markers, such as Olfml3, Tmem119 and Gpr34, accompanied by enhanced expression levels of inflammatory marker genes, such as Il1b, Tnf and Ccl2 both in adult and in neonatal microglia when compared to cells treated with TGF‐β only (Fig 2E). In the healthy brain, TGF‐β is expressed at low levels by both neurons and glial cells 46, 47, while its expression is increased upon injury 48, 49, hypoxia–ischaemia 50 and neurodegeneration 51, 52. SMAD and signal transducer and activator of transcription (STAT) proteins are key signal transducers and transcription factors controlling TGF‐β downstream signalling 53. Specifically, STAT3 and suppressor of cytokine signalling 3 (SOCS3) regulate inflammatory responses 54. The binding of SOCS3 to both JAK kinase and the cytokine receptor results in the inhibition of STAT3 activation. In our analysis, microglial cells treated with LPS showed increased amounts of STAT3 phosphorylation along with upregulation of Socs3 expression levels compared to untreated cells (Appendix Fig S1). Taking advantage of the “harmonizome” collection of databases 55, we attested that more than 1/3 of the top 100 sensome genes 4 possess STAT3‐binding sites in their promoter region. Hence, we hypothesized that the SOCS3‐STAT3 antagonistic signalling may be responsible for the suppression of the homeostatic microglia signature and the concomitant shift towards the inflammatory profile 56.
These results show that microglia isolated from LPS‐injected mice display a classical activated pro‐inflammatory profile associated with a decrease in the expression of the homeostatic genes. The decrease in the homeostatic signature under inflammatory conditions is an inherent facet of microglial in vivo and in vitro.
Single‐cell mRNA sequencing of CD11b+CD45int microglia isolated from LPS‐injected mice reveals a global transcriptional shift and increased heterogeneity compared to steady state conditions
Based on the observed differences in the targeted qPCR approach under steady state and LPS conditions, we next aimed to investigate microglial states at the genome‐wide level and infer their transcriptomic heterogeneity at single‐cell resolution, since studying a population of cells masks the differences among individual cells. For this purpose, FACS‐sorted CD11b+CD45int cells from saline‐ or LPS‐injected mice were analysed using the recently developed high‐throughput droplet‐based Drop‐seq method 23. In Drop‐seq, single cells and functionalized barcoded beads as cell identifiers are co‐encapsulated into droplets followed by cDNA synthesis, amplification, library preparation and next‐generation sequencing. First, we sought for differentially expressed genes between all LPS and all naïve/saline cells using MAST 57. We identified 2,405 differentially expressed genes between these two conditions with a false discovery rate (FDR) cut‐off of 5% (Dataset EV1) and exemplified the top 100 differentially expressed genes in a heatmap (Fig 3A). Second, principal component analysis followed by two‐dimensional t‐distributed stochastic neighbour embedding (2D‐tSNE) of the overall gene expression data of 1,247 analysed cells identified two main cell clusters that were independent of the 2D‐tSNE parameters and library sizes (Appendix Fig S2). Microglia isolated from LPS‐injected mice distinctly clustered from the corresponding steady state microglia presenting discrete gene expression signatures (Fig 3B; Dataset EV1). Intriguingly, we noticed from both analyses that, although most of the activated cells clustered together, a small group of cells assembled closer to the control cells, thus highlighting the existence of potential subpopulations under inflammatory conditions, which we characterized later. Gene set enrichment analysis (GO) of upregulated genes in microglia isolated from LPS‐injected mice using DAVID 58, 59 uncovered significant involvement (P < 2.5 × 10−9) in “translation”, “protein folding”, “ribosome biogenesis” and “immune system process”, thus reflecting highly activated cells. On the other hand, GO of the corresponding downregulated genes identified, among others, significant enrichment (P < 4.9 × 10−5) in “regulation of transforming growth factor beta receptor signalling pathway”, thus reflecting that TGF‐β signalling is among the most affected pathways in microglia exposed to LPS (Appendix Fig S3). In line with gene expression results obtained at the bulk level, microglial homeostatic genes (e.g. Tmem119, Mef2c, P2ry13, P2ry12, Siglech) were among the top downregulated genes and classical pro‐inflammatory genes (e.g. Ccl2, Gpr84, Nfkbia) were mainly upregulated also at the single‐cell level (Appendix Fig S2). We further investigated individual gene expressions at single‐cell level using 2D‐tSNE to show specific homeostatic and inflammatory gene expression levels. For example, Tmem119, Siglech and P2ry12 genes were consistently expressed under steady state, but were downregulated in microglia isolated from LPS‐injected mice, while Ccl2 and Gpr84 were largely upregulated in most of the cells exposed to LPS compared to saline conditions (Fig 3C; Appendix Fig S4). Notably, a prominent decrease in TMEM119 and P2RY12 expression was further confirmed at the protein level by flow cytometry (Fig 3D).
Figure 3. Characterization of microglial activation at the single‐cell level.
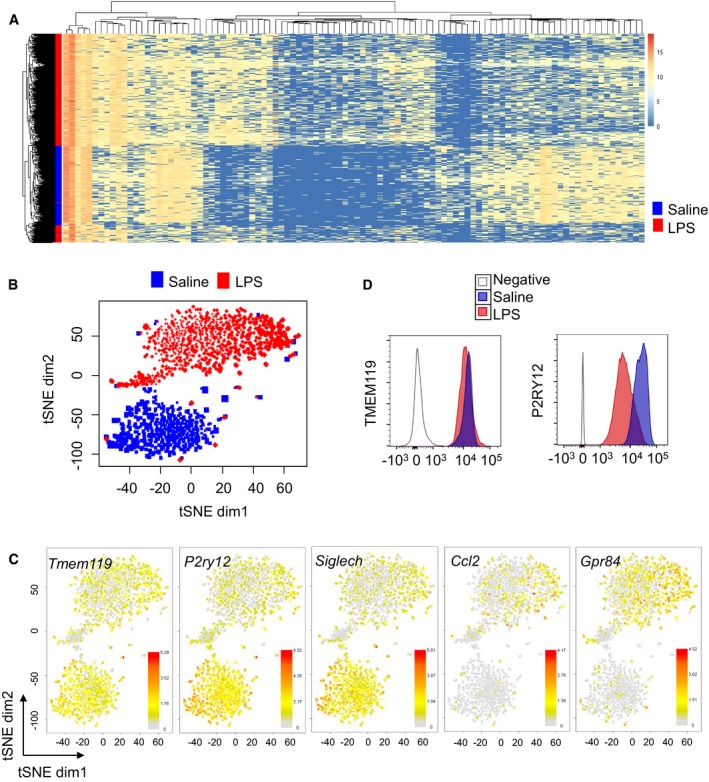
- Heatmap showing clustering analysis of 1,247 single cells, featuring 100 most variable genes (FDR < 0.05). Single‐cell RNA‐seq results are obtained from two mice per group (one female and one male each). Values denote a score based on gene expression rank.
- 2D‐tSNE representation of all single cells included in the study (n = 1,247) depicting the separation of microglia isolated from LPS‐injected mice (770 cells in red) and steady state (477 cells in blue) in two main clusters.
- Expression of specific homeostatic (Tmem119, P2ry12, Siglech) and inflammatory (Ccl2, Gpr84) genes overlaid on the 2D‐tSNE space. Bars represent log2 (Count + 1).
- Representative multicolour flow cytometry analysis of two independent experiments showing TMEM119 and P2RY12 expression levels in CD11b+CD45int microglia of saline or LPS‐injected mouse brains. For the unconjugated TMEM119 antibody, negative stands for primary antibody without secondary antibody. For P2RY12 antibody, negative represents isotype PE control.
Although microglial activation is a common hallmark under inflammatory and neurodegenerative conditions 22, microglia transcriptional signatures have been shown to be different. For example, Chiu et al 16 demonstrated that acutely isolated microglia from the SOD1G93A mouse model of amyotrophic lateral sclerosis (ALS) differed from LPS‐activated microglia, defining an ALS‐specific phenotype. Following the recent description of a novel disease‐associated microglial (DAM) phenotype identified under neurodegenerative conditions at single‐cell resolution 14, we here compared our inflammatory‐associated microglia (IAM) signature to DAM. The scatterplot showing the fold change of genes between microglia isolated from LPS‐injected mice (2,405 genes; Dataset EV1) versus DAM (1,660 genes; Dataset EV2) compared to homeostatic microglia (FDR < 0.05) disclosed 1,826 unique genes affected by the LPS treatment (e.g. Tnf, Irf1), 1,081 distinct genes in DAM (e.g. Itgax, Axl) and 579 shared genes between the two populations (e.g. Gpr84, Tmem119), thus highlighting that these cells mainly display a unique expression profile (Fig EV3). Specifically, only 215 upregulated genes (12.1%) and 364 downregulated genes (21.2%) were shared between the two groups (Fig EV3). Gene set enrichment analysis (GO) and identification of key genes being discriminative between inflammatory microglia and DAM revealed a high inflammatory reactivity upon LPS treatment and a phagocytic/lysosomal gene signature in DAM (Fig EV3). For instances, Trem2 and Tyrobp expression levels were highly decreased in IAM, whereas an elevation of both genes was reported in DAM. TREM2 associates with the immunoreceptor tyrosine‐based activation motif (ITAM)‐containing adaptor protein TYROBP (DAP12), in which signalling involves the recruitment of tyrosine kinase Syk that further phosphorylates downstream pathways inducing cell activation. TREM2 is required for phagocytosis of apoptotic neurons, microglial proliferation and survival 56, 60, 61, 62. These subtle differences in perceiving different signals induced by CNS perturbations support the microglial critical role in modulating specific functional activities. In fact, it is intuitive to consider that sensing inflammatory environments to maintain a homeostatic neuronal network (e.g. through the expression of Clec4a and Clec5a genes that are exclusively upregulated in our dataset) or recognizing and clearing pathogenic factors (e.g. by expressing Clec7a/Dectin‐1 in DAM), such as β‐amyloid aggregates in AD, require distinct activated phenotypes. In a different context, it has been recently shown that myelin pieces are gradually released from ageing myelin sheaths and are subsequently cleared by microglia 63. Age‐related myelin fragmentation is substantial, leading to lysosomal storage and contributing to microglial senescence and immune dysfunction in ageing 63. It could be then hypothesized that a similar accentuated mechanism may be encountered by microglia surrounding β‐amyloid plaques, which become dystrophic at a late stage of the disease 64. Interestingly, genes described to be associated with neurological diseases, such as Cd33, Cd9, Sod1, Ctsd, and Hif1a, were also downregulated in our signature in comparison with DAM.
Figure EV3. Inflammatory‐ versus disease‐associated signatures.
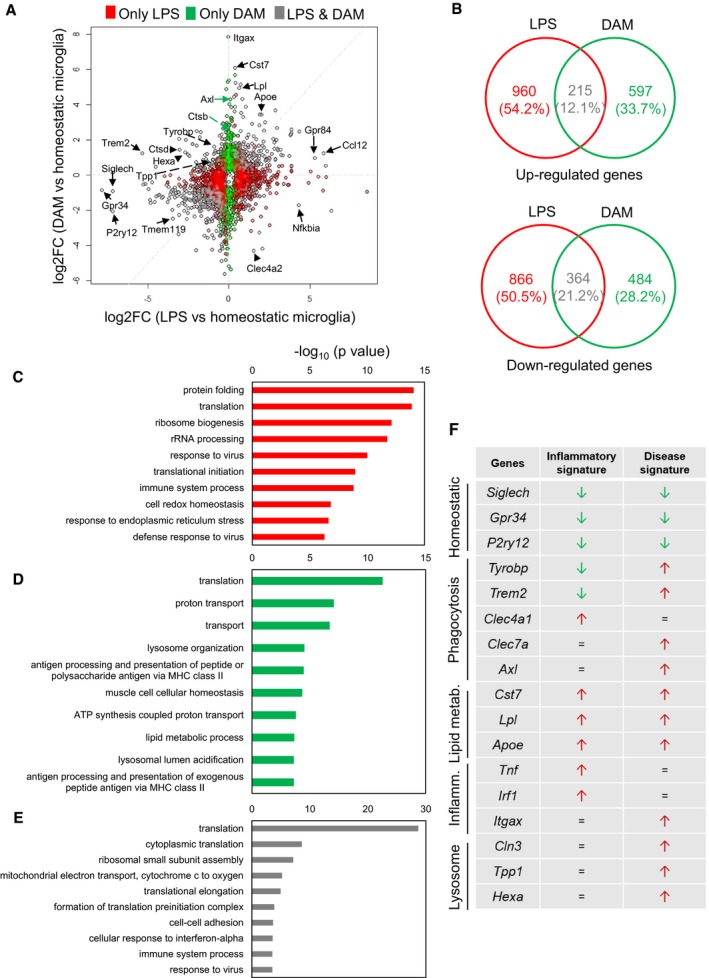
-
AScatterplot comparing the fold change of genes (log2 scale) between microglia isolated from LPS‐injected mice (x‐axis) versus DAM (y‐axis) compared to homeostatic microglia (FDR < 0.05).
-
BVenn diagrams showing the number of genes upregulated (left) or downregulated (right) uniquely under LPS treatment, exclusively by DAM and shared between LPS and DAM compared to their corresponding controls (FDR < 0.05).
-
C–ETop 10 biological processes identified by Database for Annotation, Visualization and Integrated Discovery (DAVID) resulting from (C) 960 uniquely upregulated genes under LPS treatment, (D) 597 increased genes specific for DAM and (E) 215 shared genes versus control conditions.
-
FInflammatory‐ and disease‐associated signatures identified by distinct genes corresponding to specific microglial functions (homeostasis, phagocytosis, lipid metabolism, inflammation and lysosomal activity).
Taken together, these results suggest that microglia under acute systemic inflammation present a highly activated state, which is heterogeneous and distinct from neurodegenerative disease‐associated profiles.
Microglia present distinct activated signatures under inflammatory conditions
Next, we aimed to elucidate whether the response to LPS was heterogeneous across microglial cells. Based on our previous observation (Figs 3A and B), we further analysed the identified subclusters by 2D‐tSNE representation (Fig 4A). Based on the obtained 2D representation, a specific LPS subgroup (“subset LPS”, in yellow) distinct from the core LPS cluster (“main LPS”, in red) was identified closer to naïve microglial cluster. Thus, we hypothesized that these cells may correspond to a microglial subset that is less sensitive to inflammatory stimuli or a cluster of cells which already partly recovered from their activated state following the prominent pro‐inflammatory immune response. We obtained differentially expressed genes between the “main LPS” (Dataset EV3) and the “subset LPS” (Dataset EV4) clusters compared to the corresponding control conditions (FDR < 0.05). We represented the top 100 differentially expressed genes among the identified clusters in a heatmap (FDR < 0.05; Appendix Fig S5). To elucidate the transcriptional signature of the LPS subgroups, we showed differentially expressed genes between “main LPS” and “subset LPS” clusters compared to the corresponding control conditions (FDR < 0.05). In line with their activated state, the main pro‐inflammatory genes (e.g. Ccl2, Tnf, Irg1, Gpr84) were upregulated (Fig 4B) and the microglial homeostatic genes (e.g. Siglech, P2ry12, Fcrls, Gpr34) were downregulated in both populations (Fig 4C), although at a lesser extent in “subset LPS”, compared to steady state conditions. Investigation of the top differentially expressed genes unique to “main LPS” or “subset LPS” compared to naïve cells (FDR < 0.05; Log2FC ≥ 3 or Log2FC ≤ −3; Table 1) identified, for example, Manf (a growth factor that promotes neuroprotection and tissue repair 65) and C5ar1 among the top upregulated genes in “main LPS” and Stab 1 as well as Ash1l (which suppresses the production of pro‐inflammatory mediators, such as IL‐6 and TNF 66), among the enhanced genes in “subset LPS”. Downregulated genes were, for example, the homeostatic gene marker Mef2c, which restrains the microglial inflammatory response 45 in “main LPS” and genes associated with endosomes/lysosomes in both “main LPS” (Maf) and “subset LPS” (Lamp1) (Figs 4D and EV4), thus potentially providing some mechanistic insights regarding the less activated state of “subset LPS” compared to the “main LPS” cluster. Further analysis of unique differentially expressed genes (FDR < 0.05) characterizing the two LPS subpopulations based on microglial functions and properties showed a dramatic increase in genes associated with the major histocompatibility complex (e.g. H2‐D1 and H2‐K1) exclusively in the “main LPS” group and a decrease in the complement system (e.g. C1qa, C1qb and C1qc) in the “subset LPS” group when compared to steady state (Table EV1).
Figure 4. Identification of microglial subpopulations under inflammatory conditions.
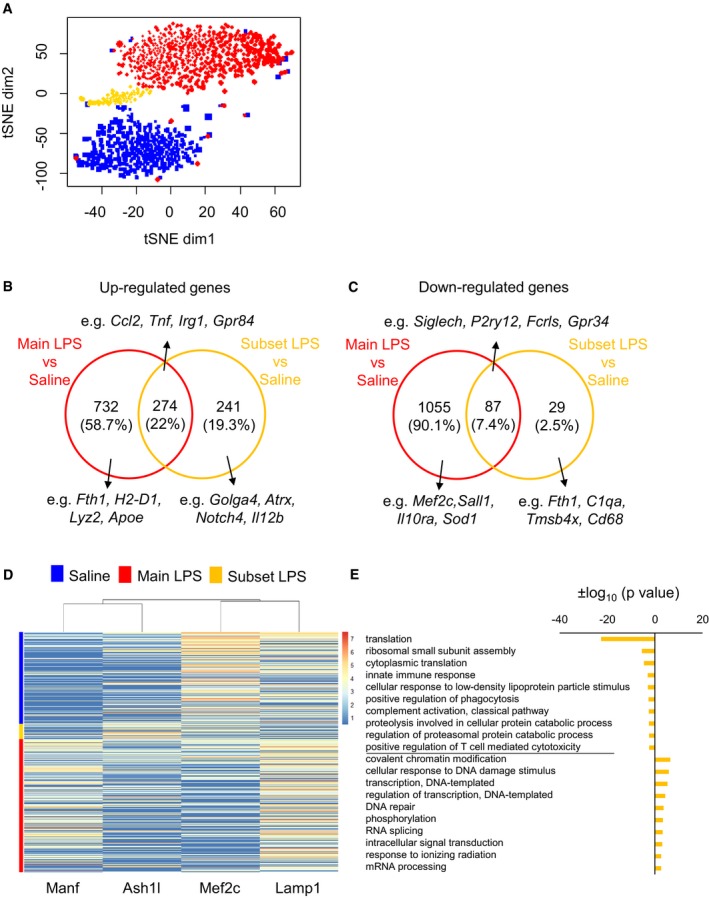
- 2D‐tSNE representation of 1,247 single cells isolated from naïve (blue)‐ and LPS‐treated mice showing two distinct subpopulations among the 770 cells isolated from LPS‐injected mice (n = 703, red; n = 67, yellow).
- Venn diagram showing 732 genes uniquely upregulated in the “main LPS” cluster (red) and 241 genes exclusively increased in the “subset LPS” (yellow) compared to their corresponding controls (blue) (FDR < 0.05). A total of 274 genes were shared between the two LPS populations.
- Venn diagram showing 1,055 genes uniquely downregulated in the “main LPS” cluster (red) and 29 genes exclusively decreased in the “subset LPS” (yellow) compared to their corresponding controls (blue) (FDR < 0.05). A total of 87 genes were shared between the two LPS populations.
- Heatmap showing examples of specific genes mainly upregulated in “main LPS” (Manf) or “subset LPS” (Ash1l) and downregulated in “main LPS” (Mef2c) or “subset LPS” (Lamp1) overlaid on the 2D‐tSNE space. Bars represent log2 (Count + 1).
- Gene set enrichment analysis (GO, top 10 biological processes) of 99 downregulated and 397 upregulated genes distinguishing cells in “subset LPS” versus “main LPS” (FDR < 0.05).
Table 1.
List of top differentially expressed genes unique to “main LPS” or “subset LPS” versus PBS (FDR < 0.05; upregulated genes, Log2FC ≥ 3; downregulated genes, Log2FC ≤ −3)
| Top upregulated genes | Top downregulated genes | ||
|---|---|---|---|
| “Main LPS” | “Subset LPS” | “Main LPS” | “Subset LPS” |
| Rplp0 | Gm26924 | Tanc2 | Lamp1 |
| Rps2 | Golga4 | Pde3b | Gm17087 |
| Cd52 | Zfc3h1 | Maf | Cd68 |
| Cd63 | RP24‐312B12.1 | Rasgrp3 | Rps14 |
| Ctsl | Stab 1 | Zfhx3 | C1qc |
| Manf | Cacna1d | 4632428N05Rik | Itm2c |
| Pdia4 | Ash1l | Mef2c | Eif1 |
| Calm1 | Ascc3 | Qk | H3f3b |
| Rps19 | Atrx | Ivns1abp | Cd81 |
| Fth1 | Ptprc | Pmepa1 | Ubb |
| Rps5 | Ttc14 | Lrrc58 | |
| Pdia6 | Chd7 | ||
| C5ar1 | Myo9a | ||
| Ptplb | |||
| Rpl32 | |||
| Gnl3 | |||
Figure EV4. Representation of mainly top deregulated genes in “main LPS” and “subset LPS”.
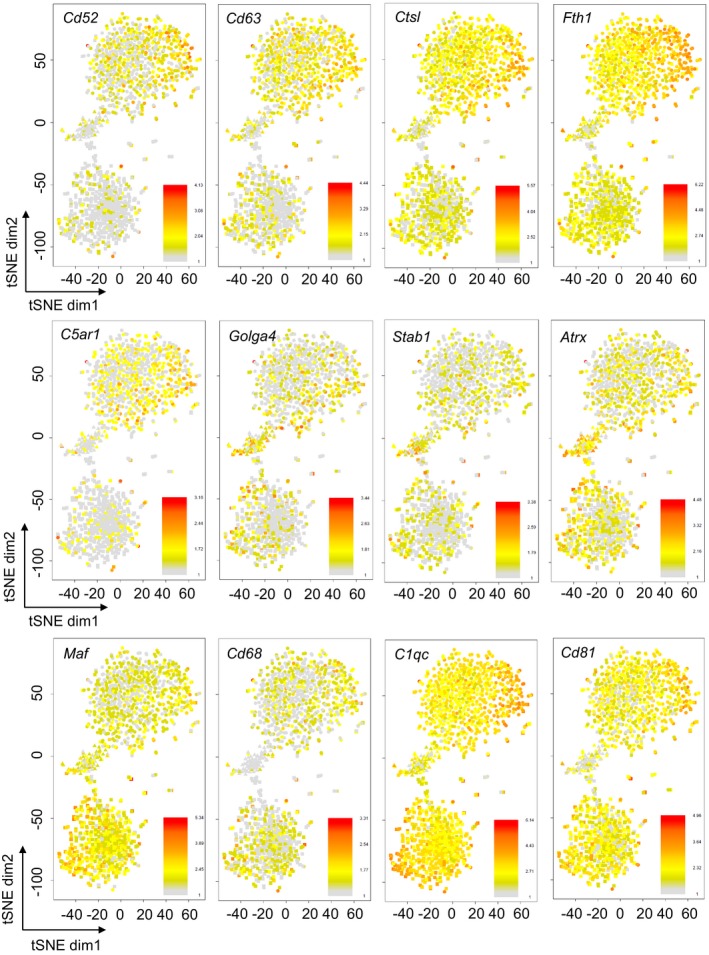
Expression of specific genes uniquely upregulated in “main LPS” (Cd52, Cd63, Ctsl, Fth1, C5ar1) or “subset LPS” (Golga4, Stab 1, Atrx) and downregulated in “main LPS” (Maf) or “subset LPS” (Cd68, C1qc, Cd81) overlaid on the 2D‐tSNE space. Bars represent log2 (Count + 1).
Notably, we characterized membrane markers corresponding to specific genes identified at single‐cell resolution by flow cytometry to analyse the expression levels of markers upregulated in both LPS groups (e.g. CD44), only in “main LPS” (e.g. CD274) or only in “subset LPS” (e.g. NOTCH4). Although three markers used simultaneously did not allow to clearly discriminate the “subset LPS” from the “main LPS” population, changes in the proportion of marker‐positive cells were in line with the scRNA‐seq data. Upon LPS treatment, a smaller proportion of NOTCH4‐positive cells (saline 5.4%; LPS 18.9%) compared to CD44 (saline 65.2%; LPS 97.5%) and CD274 (saline 48.7%; LPS 88.1%) were detected (Fig EV5). We confirmed this pattern by immunohistochemistry, showing that NOTCH4‐positive cells were evenly distributed throughout the brain, thus indicating that these cells were not associated with a specific brain region (Fig EV5).
Figure EV5. FACS and immunohistochemistry analyses of “main LPS” and “subset LPS” upregulated genes and further comparison with DAM .
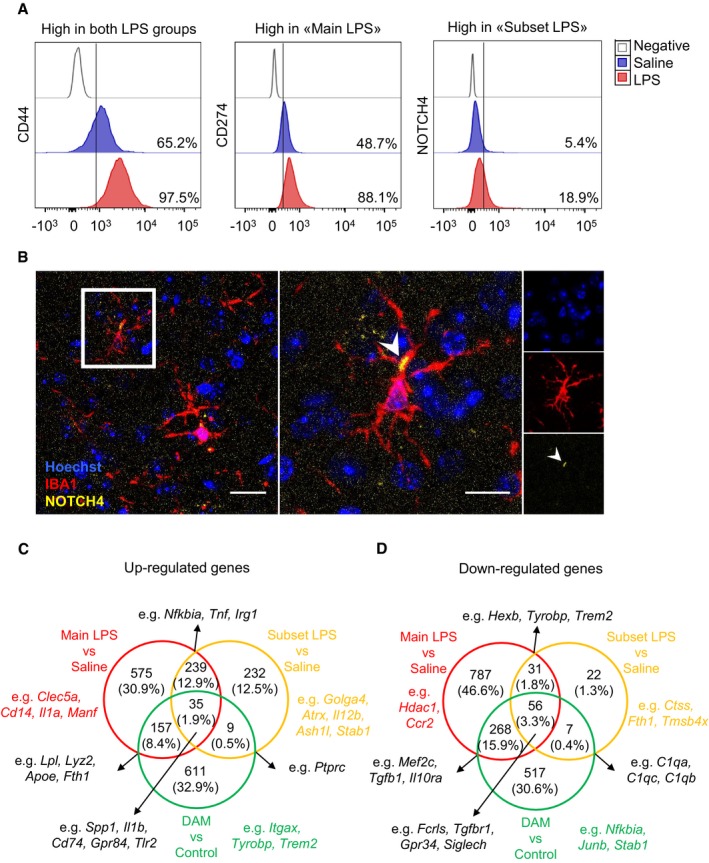
-
AQuantification of CD44, CD274 and NOTCH4 expression in CD11b+CD45int microglia under homeostatic (in blue) and inflammatory (in red) conditions. Values denote the percentage of cells obtained from one representative experiment.
-
BImmunohistochemistry for “subset LPS” marker (NOTCH4) in brain sections of tissue isolated from LPS‐injected mice after 24 h (IBA1 in red and NOTCH4 in yellow). Cell nuclei were counterstained with Hoechst (in blue). Scale bars: left panel 10 μm, right panel 20 μm. Arrows show NOTCH4 expression in IBA1+ cells.
-
C, DVenn diagrams showing unique and commonly (C) upregulated and (D) downregulated genes among “main LPS” cluster (red), “subset LPS” (yellow) and DAM (green) (FDR < 0.05).
Gene set enrichment analysis of downregulated genes characterizing “subset LPS” compared to “main LPS” confirmed “innate immune response” and “complement activation, classical pathway” as decreased terms, thus highlighting a less pronounced activated state of the “subset LPS”. Intriguingly, these cells revealed significant over‐representation (P < 0.05) of “covalent chromatin modification” and “DNA repair” that may indicate cells recovering from their acute activated state or a subset of cells with specific chromatin states and DNA repair properties conveying an attenuated activated phenotype than the main population (Fig 4E). In order to further corroborate the existence of the identified microglial subpopulations under inflammatory conditions, the corresponding 770 cells were subjected to the “SC3” method 67. With two clusters, we found a very high concordance between the subcluster obtained with “SC3” and the LPS subset identified by 2D‐tSNE, thus supporting the existence of the detected subpopulations. We represented the top 50 differentially expressed genes driving the segregation of cells into the two clusters in a heatmap (adjusted P‐value < 0.05; Appendix Fig S6).
Lastly, we used Venn diagrams to show unique and common upregulated and downregulated (Fig EV5) genes among “main LPS” cluster, “subset LPS” and DAM (FDR < 0.05). Among the deregulated genes, for example, Spp1, Il1b and Tlr2 were commonly upregulated, while Fcrls, Tgfbr1 and Siglech were downregulated in the three groups. Intriguingly, genes of the complement system (e.g. C1qa, C1qb and C1qc) were downregulated in both “subset LPS” and DAM, but not in “main LPS”. Further analysis of the top differentially expressed genes unique to the three groups compared to naïve cells (FDR < 0.05; Log2FC ≥ 3 or Log2FC ≤ −3; Appendix Table S1) showed that the previously identified genes, such as Manf and C5ar1 are uniquely upregulated in “main LPS”, while Stab 1 as well as Ash1l is among the increased genes only in “subset LPS”.
Overall, these results highlight the existence of specific microglial subpopulations under inflammatory conditions, which are distinct from neurodegenerative‐associated phenotypes. These findings emphasize heterogeneity of microglial activated states in vivo reflecting specific functional activities related to their corresponding environment.
Pseudotime analysis of LPS‐activated microglia uncovers “subset LPS” as an intermediate activated state
Although further analyses at different time points should be performed in the future to resolve the dynamic process of activation, to further investigate the activation process, the heterogeneity within LPS‐activated microglia and, specifically, the properties of “subset LPS” compared to “main LPS”, we applied branch expression analysis modelling (BEAM) and corresponding pseudotime analysis implemented in Monocle 2 68. Since the more subtle differences during the activation process would be dominated by the large differences between naïve and LPS conditions, we applied the branching analysis to the LPS‐activated microglia only. This more sensitive analysis revealed nine different states, with the largest difference of “subset LPS” to all others, in accordance with the previous tSNE and “SC3” analyses. Interestingly, cells assigned to “subset LPS” exhibit a dense core, but also a significant number of cells towards the other main clusters (Fig 5A). Given this more sensitive clustering and corresponding pseudotime analysis, we then investigated the characteristics of “subset LPS” with respect to their activation state and their relation with the other states. For this purpose, we plotted the estimated pseudotime of each cell in the state space indicating a delayed activation pattern of “subset LPS” (Fig 5B). Lastly, we investigated inflammatory (e.g. Ccl12, Ccl2, Gpr84) and homeostatic (e.g. Mef2c, P2ry12, Siglech) gene expression profiles in dependence on pseudotime, further indicating the delayed activation state of “subset LPS” by smaller pseudotimes (Fig 5C). By comparing the dynamics of the inflammatory and homeostatic genes along the activation process, we observed that inflammatory genes were upregulated first, while the homeostatic markers were only subsequently downregulated. Thus, “subset LPS” may correspond to an intermediate state where the gene expression levels of the inflammatory mediators are increased, but the homeostatic gene markers, such as Mef2c, are still partly preserved. In conclusion, from this analysis, we hypothesized that these cells may correspond to a microglial subpopulation that is less sensitive to inflammatory stimuli.
Figure 5. Pseudotime analysis.
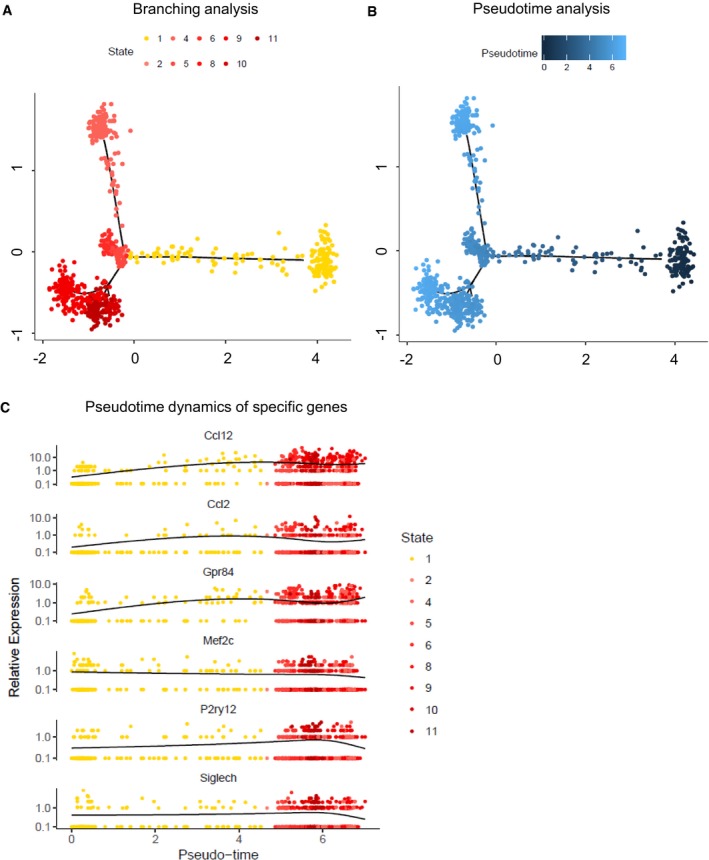
- Branching analysis of LPS‐activated microglia by Monocle 2 leads to nine distinct clusters in a two‐dimensional state space inferred by generalized regression modelling (see Materials and Methods) showing the major difference of “subset LPS” (in yellow) compared to the other clusters corresponding to “main LPS” (in red).
- Monocle estimated a pseudotime for each cell along the inferred cell trajectory within the state space showing a delayed activation pattern of “subset LPS” compared to the other fractions.
- Pseudotime dynamics of inflammatory (Ccl12, Ccl2, Gpr84) and homeostatic (Mef2c, P2ry12, Siglech) genes in dependence on inferred cell states.
In summary, our work elucidated an extensive picture of microglial profiles in steady state and upon inflammatory conditions, including unforeseen heterogeneity in their states of activation. We believe that our findings, together with the recent single‐cell RNA sequencing studies of microglia in Alzheimer's disease 14, present a comprehensive transcriptomic view of microglia under acute inflammatory conditions and a comparison with neurodegenerative processes. These results could then pave the way to design new therapeutic approaches to restore abnormal or detrimental microglial phenotypes found in several CNS disorders.
Materials and Methods
Animals
Three‐ to four‐month‐old C57BL/6N male and female mice were obtained from Charles River Laboratories (France). Mice were housed in 12‐h light/dark cycle, with sterile food and water ad libitum. All animal procedures were approved by the University of Luxembourg Animal Experimentation Ethics Committee and by appropriate government agencies. The animal work of the present study has been conducted and reported in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines to improve the design, analysis and reporting of research using animals, maximizing information published and minimizing unnecessary studies.
Acute microglial isolation and purification by multicolour flow cytometry
Mice were treated with a single intraperitoneal injection of LPS (4 μg LPS/g body weight) or with PBS (saline) as vehicle control. Twenty‐four hours later, mice were deeply anaesthetized with a combination of ketamine (100 mg/ml; Nimatek Vet)–dorbene (medetomidine hydrochloride; 1 mg/ml; Dorbene Vet) and perfused transcardially with ice‐cold PBS. Further processing was performed at 4°C and no‐break centrifugations. Brains were rapidly removed, stored in ice‐cold HBSS (Gibco/Life Technologies) with 1 M HEPES (Gibco/Life Technologies) and 0.5% D‐(+)‐glucose (Sigma‐Aldrich), mechanically homogenized in a potter homogenizer and centrifuged at 900 rpm for 10 min. Myelin was removed from cell suspension with the Myelin Removal Kit (Miltenyi Biotec) according to the manufacturer's protocol. Prior to the FACS, the cell suspension was resuspended in ice‐cold HBSS with 2% FBS and 10 mM HEPES, pH 7.4 and filtered through a 70‐μm nylon mesh (CellTrics). For multicolour staining, cells were incubated for 15 min with Fc receptor binding inhibitor (anti‐mouse CD16/CD32 monoclonal antibody; 1:100; eBioscience) to reduce binding of non‐specific Fc‐gamma receptors, and then stained with fluorochrome‐conjugated antibodies (Appendix Table S2) or their corresponding isotopic controls for 45 min at 4°C in dark. After washing, cells were pelleted at 300 g for 10 min at 4°C and resuspended in 200 μl of the appropriated buffer. Hoechst (0.1 μg/ml; Sigma) or Sytox Red (1:1,000; Thermo Fisher Scientific) were added shortly before flow cytometry measurements for dead cell discrimination. Cells were sorted with FACSAria™ SORP cytometer (BD Biosciences) fitted with a 640 nm (30 mW) red laser, a 355 nm (60 mW) UV laser, a 405 nm (50 mW) violet laser, a 488 nm (100 mW) blue laser and a 561 nm (50 mW) yellow/green laser. Data were analysed with FACSDiva software (Becton Dickinson) and FlowJo software (version 7.6.5; Tree Star). Imaging flow cytometry was performed with an ImageStream imaging cytometer (Amnis) fitted with a 375 UV laser, a 488 blue laser, a 561 yellow‐green laser, a 642 red laser and a 785 nm infrared laser. Acquisition was performed with the INSPIRE® software, and analysis was performed using IDEAS® image analysis software. Pictures were taken at 60× magnification at low speed, high sensitivity mode.
Isolation of bone marrow monocytes
Monocytes were isolated from mouse bone marrow cells by using the Monocyte Isolation Kit (Miltenyi Biotec) according to the manufacturer's protocol.
Primary adult mouse microglial culture
Adult microglia were isolated from brains of C57BL/6N mice at age 6–10 weeks by magnetic separation. Mice were transcardially perfused with ice‐cold PBS under anaesthesia, and brains were dissociated using the Neural Dissociation Kit P (MACS Miltenyi Biotec) according to the manufacturer's instruction. Microglia were enriched by magnetic separation using CD11b+ beads (MACS Miltenyi Biotec). Briefly, 1 × 107 cells were resuspended in 90 μl of MACS buffer [Hank's balanced salt solution (HBSS); Lonza] supplemented with 0.5% BSA (Sigma‐Aldrich) and 2 mM EDTA and 10 μl of CD11b MicroBeads (MACS Miltenyi Biotec). The cell suspension was incubated at 4°C for 20 min, washed and pelleted in 500 μl of MACS buffer at a density of 1 × 108 cells. The cell suspension was applied into LS columns (MACS Miltenyi Biotec), and the CD11b+ fraction was eluted. Primary adult microglia were plated in 24‐well plates coated in poly‐L‐lysine (0.1 mg/ml solution; Sigma‐Aldrich) at a density of 2 × 105 cells/ml and grown in microglial culture medium [Dulbecco's modified Eagle's medium (DMEM‐F12 w/L‐glutamine w/15 mM HEPES; Biowest)] supplemented with 10% foetal bovine serum (FBS; Gibco/Life Technologies), pen‐strep (100 U/ml/100 μg/ml; Gibco/Life Technologies), human recombinant TGF‐β (PeproTech) at a final concentration of 50 μg/ml and mouse recombinant M‐CSF (R&D Systems) at a final concentration of 10 ng/ml. Cells were cultured for 5 days without changing media. After 9 days of culture, cells were stimulated with lipopolysaccharide (LPS from Escherichia coli 055:B5; Sigma‐Aldrich) at a final concentration of 1 ng/ml for 6 h.
Primary newborn mouse microglial culture
Murine primary microglial cells were isolated from newborn (P1–P4) C57BL/6N mouse brains as previously described 69. Brains were dissected on ice. Subsequently, meninges and large blood vessels were carefully removed and brains were pooled and minced in cold Dulbecco's phosphate buffered saline (PBS; Lonza). Tissue dissociation was completed by 10 min of incubation in 2 mM EDTA (Sigma‐Aldrich). Cells were washed, centrifuged, seeded into six‐well plates coated with poly‐L‐lysine and allowed to attach and grow in complete medium DMEM (Gibco/Life Technologies) supplemented with 10% FBS and pen‐strep at 37°C in a water‐saturated atmosphere containing 5% CO2. The culture medium was renewed after 3 days of culture. After 10 days, when cells reached confluence, the mixed glial monolayer was trypsinated (0.05% Trypsin–EDTA; Gibco/Life Technologies) and microglial cells were purified by magnetic cell sorting (MACS Miltenyi Biotec) following the manufacturer's instructions. Primary microglia were plated in 12‐well plates coated with poly‐L‐lysine (Sigma‐Aldrich) at a density of 4 × 105 cells/ml. Twenty‐four hours after plating, cells were activated with different compounds: LPS at a final concentration of 1 ng/ml, TGF‐β at a final concentration of 50 μg/ml and M‐CSF at a final concentration of 10 ng/ml.
RNA isolation and RT–PCR
CD11b+CD45int cells were FACS‐sorted directly to TRIzol® LS, and total RNA was extracted according to the manufacturer's protocol (Life Technologies). RNA from primary cells was extracted using the RNeasy Mini Kit (QIAGEN), according to the manufacturer's instructions. RNA concentration was quantified by NanoDrop (NanoDrop Technologies) and the quality assessed by the quotient of the 28S to 18S ribosomal RNA electropherogram peak using a bioanalyser (Agilent 2100; Agilent Technologies) using a RNA Pico Chip (Agilent Technologies; only samples with RIN ≥ 7 were further analysed). For cDNA synthesis, RNA was reverse‐transcribed using SuperScript™ III reverse transcriptase (10,000 U; Invitrogen/Life Technologies) with 1 μl (50 μM)/reaction oligo(dT)20 (25 μM; Invitrogen/Life Technologies) as primer according to the manufacturer's instructions. Reverse transcription was performed at 50°C for 60 min. Gene expression reaction mixtures contained 2 μl of diluted cDNA, 10 μl of Fast SYBR Green Master Mix (Applied Biosystems/Thermo Fisher Scientific) and 0.5 μl of each 10 μM forward and reverse primers. PCRs were carried out in 96‐well plates on a ViiA™ 7 real‐time PCR system (Applied Biosystems/Thermo Fisher Scientific) using the following programme: 95°C for 20 s, 40 cycles at 95°C for 1 s and 60°C for 20 s. The sequences of the primers designed using Primer‐Blast tool are listed in Appendix Table S3. Samples were run in duplicates, and the mean C t (threshold cycle) values were used to calculate the relative amount of product by the ΔΔC t method using Gapdh as housekeeping gene.
Immunohistochemistry
Under deep ketamine–dorbene anaesthesia, mice were transcardially perfused with ice‐cold PBS, post‐fixed in 4% paraformaldehyde (PFA) for 48 h and stored at 4°C in 0.02% sodium azide/PBS as preservative. Serialized parasagittal free‐floating 50‐μm‐thick sections were generated with a vibratome (Leica; VT‐1000S) and collected in cryoprotective medium [PBS containing 1‐1 ethylene glycol (Sigma‐Aldrich) and 1% w/v polyvinylpyrrolidone (Sigma‐Aldrich)]. Sections were stored at −20°C in tubes, each containing a series of every 4th section.
For immunofluorescence, a standard protocol was used with minor modifications 70. Briefly, sections were washed (PBS with 0.1% Triton X‐100), permeabilized (PBS with 1.5% Triton X‐100), blocked (PBS with 5% BSA) and incubated with primary antibodies (PBS with 0.3% Triton X‐100 and 2% BSA): rabbit anti‐Iba1 (1:1,000; Wako) and pre‐conjugated PE anti‐mouse Notch4 (1:80; BioLegend). Iba1 antibody was visualized using goat anti‐rabbit IgG Molecular Probes Alexa Fluor 555 (Thermo Fisher Scientific) secondary antibody. Cell nuclei were counterstained with Hoechst (1 μg/ml; Sigma). Sections were mounted on glass slides coverslipped using Fluoromount™ Aqueous Mounting Medium (Sigma). Microscopic images were obtained using confocal microscopy (Zeiss LSM880).
SDS–PAGE and Western Blotting analysis
Heat‐denatured protein samples were separated on 4–12% BisTris–polyacrylamide gel electrophoresis followed by transfer to nitrocellulose membranes 0.2 μm (Bio‐Rad). After blocking with 5% (wt/vol) dry milk in TBST for STAT3 and 3% BSA in TBST for Phospho‐STAT3, respectively, the membrane was incubated overnight at 4°C in primary anti‐STAT3 antibody from mouse (Cell Signaling) diluted 1:1,000 in 5% (wt/vol) dry milk in TBST and in primary anti‐Phospho‐STAT3 antibody (Cell Signaling) diluted 1:500 in 3% BSA in TBST with constant shaking. After three washing steps with TBS containing 0.1% Tween‐20, the membrane was incubated with anti‐rabbit antibody or anti‐mouse respectively, coupled to horseradish peroxidase and revealed by chemoluminescence using the Pierce™ ECL detection reagents (Thermo Fisher Scientific).
Single‐cell RNA sequencing using Drop‐seq
Cell preparation
FACS‐sorted CD11b+CD45int cells were collected in pre‐cooled HBSS and 0.5% BSA and transferred directly for subsequent Drop‐seq analysis. The cells were stored on ice until the start of the Drop‐seq experiment (tissue harvest to running of Drop‐seq was < 1 h). Prior to cell loading on the Drop‐seq chips, the cell viability was verified and the concentration was adjusted to ~150 cells/μl. This was optimal based on Poissonian statistics to achieve single‐cell encapsulation within each droplet of ~800–900 pl droplet size. All samples analysed in this work had a cell viability > 95%.
Microfluidics fabrication
Microfluidics devices were generated using a previously published design 23. Soft lithography was performed using SU‐8 2050 photoresist (MicroChem) on 4” silicon substrate to obtain a feature aspect depth of 100 μm. After overnight silanization (using chlorotrimethylsilane; Sigma), the wafer masks were used for microfluidics fabrication. Drop‐seq chips were fabricated using silicon‐based polymerization chemistry, with the previously published protocol 71. Briefly, polydimethylsiloxane (PDMS) base and crosslinker (Dow Corning) were mixed at a 10:1 ratio, mixed and degassed before pouring onto the Drop‐seq master template. PDMS was cured on the master template, at 80°C for 2 h. After incubation and cooling, PDMS monoliths were cut and the inlet/outlet ports were punched with 1.25‐mm biopsy punchers (World Precision Instruments). The PDMS monolith was plasma‐bonded to a clean microscopic glass slide using a Harrick plasma cleaner. Immediately after pairing the plasma‐treated surfaces of the PDMS monolith and the glass slide, flow channels of the Drop‐seq chip were subjected to a hydrophobicity treatment using 1H,1H,2H,2H‐perfluorodecyltrichlorosilane (in 2% v/v in FC‐40 oil; Alfa Aesar/Sigma). After 5 min of treatment, excessive silane was blown through the inlet/outlet ports. Chips were further incubated at 80°C for 15 min.
Single‐cell droplet encapsulation
Experiments followed the original Drop‐seq protocol 23 with minor changes described below. Synthesized barcoded beads (ChemGenes Corp., USA) were co‐encapsulated with cells inside the droplets containing lysis reagents using an optimal bead concentration of 200 beads/μl in Drop‐seq Lysis buffer medium. Cellular mRNA was captured on the beads via barcoded oligo (dT) handles synthesized on the surface.
For cell encapsulation, bead suspensions and cell suspension were loaded into 3‐ml syringes (BD). To keep beads in homogenous suspension, a micro‐stirrer was used (VP Scientific). The QX200 carrier oil (Bio‐Rad) used as continuous phase in the droplet generation was loaded into a 20‐ml syringe (BD). For droplet generation, 3.6 ml/h and 13 ml/h flowrates were used in KD Scientific Legato Syringe Pumps for the dispersed and continuous phase flows, respectively. After stabilization of droplet formation, the droplet suspension was collected into a 50‐ml Falcon tube. Collection of the emulsion was carried out until 1 ml of the single‐cell suspension was dispensed. Droplet consistency and stability were evaluated by bright‐field microscopy using INCYTO C‐Chip Disposable Hemacytometer (Thermo Fisher Scientific). Bead occupancy within droplets was carefully monitored to avoid multiple beads per droplet.
The subsequent steps of droplet breakage, bead harvesting, reverse transcription and exonuclease treatment were carried out in accordance with the Drop‐seq method 23. RT buffer contained 1× Maxima RT buffer, 4% Ficoll PM‐400 (Sigma), 1 μM dNTPs (Thermo Fisher Scientific), 1 U/ml RNase Inhibitor (Lucigen), 2.5 μM Template Switch Oligo 23 and 10 U/ml Maxima H‐RT (Thermo Fisher Scientific). After Exo‐I treatment, the bead counts were estimated using INCYTO C‐Chip Disposable Hemacytometer, and 5,000–8,000 beads were aliquoted in 0.2 ml Eppendorf PCR tubes. PCR mix was dispensed in a volume of 50 μl using 1× HiFi HotStart ReadyMix (Kapa Biosystems) and 0.8 mM Template Switch PCR primer. The thermocycling programme for the PCR amplification was modified for the final PCR cycles by 95°C (3 min), four cycles of 98°C (20 s), 65°C (45 s), 72°C (3 min) and 16 cycles of 98°C (20 s), 67°C (20 s), 72°C (3 min), followed by a final extension step of 72°C for 5 min. After PCR amplification, libraries were purified with 0.6× Agencourt AMPure XP beads (Beckman Coulter), according to the manufacturer's protocol. Finally, the purified libraries were eluted in 20 μl RNase/DNase‐free Molecular Grade Water. Quality and concentration of the sequencing libraries were assessed using Bioanalyzer High Sensitivity Chip (Agilent Technologies).
NGS preparation for Drop‐seq libraries
The 3′ end enriched cDNA libraries were prepared by tagmentation reaction of 600 pg cDNA library using the standard Nextera XT tagmentation kit (Illumina). Reactions were performed according to the manufacturer's instructions. The PCR amplification cycling programme used was 95°C 30 s, and fourteen cycles of 95°C (10 s), 55°C (30 s) and 72°C (30 s), followed by a final extension step of 72°C (5 min). Libraries were purified twice to reduce primers and short DNA fragments with 0.6× and 1× Agencourt AMPure XP beads (Beckman Coulter), respectively, in accordance with the manufacturer's protocol. Finally, purified libraries were eluted in 15 μl Molecular Grade Water. Quality and quantity of the tagmented cDNA library were evaluated using Bioanalyzer High Sensitivity DNA Chip. The average size of the tagmented libraries prior to sequencing was between 400 and 700 bps.
Purified Drop‐seq cDNA libraries were sequenced using Illumina NextSeq 500 with the recommended sequencing protocol except for 6pM of custom primer (GCCTGTCCGCGGAAGCAGTGGTATCAACGCAGAGTAC) applied for priming of read 1. Paired‐end sequencing was performed for the read 1 of 20 bases (covering the random cell barcode 1–12 bases and the rest 13–20 bases of random unique molecular identifier (UMI) and for read 2 of 50 bases of the genes.
Bioinformatics processing and data analysis
The FASTQ files were assembled from the raw BCL files using Illumina's bcl2fastq converter and ran through the FASTQC codes (Babraham bioinformatics; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to check for consistency in library qualities. The monitored quality assessment parameters were (i) quality per base sequence (especially for the read 2 of the gene), (ii) per base N content, (iii) per base sequence content, and (iv) over‐represented sequences. Libraries that showed significant deviation were re‐sequenced. The FASTQ files were then merged and converted into binaries using PICARD's FastqToSam algorithm. The sequencing reads were converted into a digital gene expression matrix using the Drop‐seq bioinformatics pipeline 23.
Data analysis was done in R. Cells with less than 1,000 counts and genes with zero count in all cells were excluded from subsequent analyses, resulting in 1,247 cells (477 from the saline control and 770 from LPS‐injected mice) and 12,369 genes. PCA (prcomp function with scaling) was used for dimensionality reduction, and PCA results were projected onto a two‐dimensional (2D) space using t‐distributed stochastic neighbour embedding (tSNE, tsne package, v.0.1‐3). As the first principal component was strongly correlated to the total number of UMI (reads) per cell, it was not included in the 2D‐tSNE analysis. Differential expression analysis was performed with MAST 57. P‐values were adjusted for multiple testing using false discovery rate (FDR) 72. Prior to MAST analysis, counts were converted into counts per million and log2‐transformed. For subpopulation identification, two approaches were used: (i) based on visual inspection of 2D‐tSNE plot, cells were divided into three clusters: one cluster contained almost exclusively cells isolated from control mice, another cluster contained mainly cells harvested from LPS‐injected mice, and the last cluster was constituted of a small subset of LPS‐derived cells. Clusters were pruned to keep only cells coming from the predominant sample in the group. Comparisons of gene expression between different groups were done with the Kruskal–Wallis H‐test. P‐values were corrected with FDR 72; (ii) each condition was analysed separately with the “SC3” package 67. Branching analysis was performed by Monocle 2.4.0 in R (version 3.4.4) with standard parameters 68, 73. The branching method orders cells along an estimated cell trajectory within a gene expression state space based on gene expression similarities estimated by generalized linear regression models.
Statistical analysis
Statistical analyses for qPCRs and FACS experiments were performed using GraphPad Prism 7 software. Comparisons of two groups were performed with a two‐tailed Student's t‐test. Comparisons involving more than two groups were performed using one‐way ANOVA followed by the Bonferroni correction for multiple testing. All differences were considered significantly different at P < 0.05. Further statistical analysis details are reported in the figure legends.
Data availability
Single‐cell RNA sequencing data have been deposited in Gene Expression Omnibus (GEO) database under the accession number GSE115571 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115571).
Author contributions
CS and AM designed the project; KB, RB and SPN involved in the experimental design; CS, SKP, YP‐A and AM performed experiments; CS, AG, SKP, TK, SM, AS and AM analysed experiments; DC provided animals; FA supervised the bioinformatics analyses of the single‐cell RNA‐seq; CS and AM wrote the manuscript; and AG, SKP, TK, DC, YP‐A, SM, FA, AS, RB, KB and SPN edited and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Appendix
Review Process File
Acknowledgements
We thank Dr. Coralie Guérin and Dr. Léa Guyonnet for helping with flow cytometry experiments as well as Eliane Klein for Western blot analyses. We are grateful to Dr. Tony Heurtaux for technical assistance with cultivated microglial as well as Oihane Uriarte and Dr. Manuel Buttini for immunohistochemistry analyses. C.S. was supported by the Luxembourg National Research Fund (AFR project reference 6916713) and the Fondation du Pélican de Mie et Pierre Hippert‐Faber Under the Aegis of Fondation de Luxembourg. Y.P.A. and S.M. were supported by the Luxembourg National Research Fund (PRIDE15/10675146 and PRIDE16/10907093, respectively). A.S. was supported by the C14/BM/7975668/CaSCAD project as well as by the National Biomedical Computation Resource (NBCR) through the NIH P41 GM103426 grant from the National Institutes of Health. We acknowledge financial support by the Luxembourg Institute of Health (MIGLISYS) and the Luxembourg Centre for Systems Biomedicine.
EMBO Reports (2018) 19: e46171
References
- 1. Prinz M, Priller J (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15: 300–312 [DOI] [PubMed] [Google Scholar]
- 2. Sousa C, Biber K, Michelucci A (2017) Cellular and molecular characterization of microglia: a unique immune cell population. Front Immunol 8: 198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S et al (2012) Gene‐expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13: 1118–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16: 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE et al (2014) Identification of a unique TGF‐beta‐dependent molecular and functional signature in microglia. Nat Neurosci 17: 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N et al (2014) An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z, Greco DJ, Wu PM, Doykan CE, Kiner O et al (2015) Targeting miR‐155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol 77: 75–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elliott R, Li F, Dragomir I, Chua MM, Gregory BD, Weiss SR (2013) Analysis of the host transcriptome from demyelinating spinal cord of murine coronavirus‐infected mice. PLoS One 8: e75346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Moller T, Orre M, Kamphuis W et al (2015) Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co‐expression meta‐analysis. Acta Neuropathol Commun 3: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lewis ND, Hill JD, Juchem KW, Stefanopoulos DE, Modis LK (2014) RNA sequencing of microglia and monocyte‐derived macrophages from mice with experimental autoimmune encephalomyelitis illustrates a changing phenotype with disease course. J Neuroimmunol 277: 26–38 [DOI] [PubMed] [Google Scholar]
- 11. Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, Boddeke HW (2012) Identification of a microglia phenotype supportive of remyelination. Glia 60: 306–321 [DOI] [PubMed] [Google Scholar]
- 12. Verheijden S, Beckers L, Casazza A, Butovsky O, Mazzone M, Baes M (2015) Identification of a chronic non‐neurodegenerative microglia activation state in a mouse model of peroxisomal beta‐oxidation deficiency. Glia 63: 1606–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH et al (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160: 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK, David E, Baruch K, Lara‐Astaiso D, Toth B et al (2017) A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169: 1276–1290 e17 [DOI] [PubMed] [Google Scholar]
- 15. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z et al (2017) The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47: 566–581 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S et al (2013) A neurodegeneration‐specific gene‐expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 4: 385–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, Hol EM (2014) Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 35: 2746–2760 [DOI] [PubMed] [Google Scholar]
- 18. Norden DM, Godbout JP (2013) Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 39: 19–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crotti A, Ransohoff RM (2016) Microglial physiology and pathophysiology: insights from genome‐wide transcriptional profiling. Immunity 44: 505–515 [DOI] [PubMed] [Google Scholar]
- 20. Fullerton JN, Gilroy DW (2016) Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov 15: 551–567 [DOI] [PubMed] [Google Scholar]
- 21. Wyss‐Coray T, Mucke L (2002) Inflammation in neurodegenerative disease–a double‐edged sword. Neuron 35: 419–432 [DOI] [PubMed] [Google Scholar]
- 22. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140: 918–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM et al (2015) Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D et al (2013) Single‐cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498: 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A et al (2009) mRNA‐Seq whole‐transcriptome analysis of a single cell. Nat Methods 6: 377–382 [DOI] [PubMed] [Google Scholar]
- 26. Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T et al (2016) Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci 19: 335–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, Tsai LH (2017) Temporal tracking of microglia activation in neurodegeneration at single‐cell resolution. Cell Rep 21: 366–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dulken BW, Leeman DS, Boutet SC, Hebestreit K, Brunet A (2017) Single‐cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Rep 18: 777–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen R, Wu X, Jiang L, Zhang Y (2017) Single‐cell RNA‐seq reveals hypothalamic cell diversity. Cell Rep 18: 3227–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Artegiani B, Lyubimova A, Muraro M, van Es JH, van Oudenaarden A, Clevers H (2017) A single‐cell RNA sequencing study reveals cellular and molecular dynamics of the hippocampal neurogenic niche. Cell Rep 21: 3271–3284 [DOI] [PubMed] [Google Scholar]
- 31. Jaitin DA, Weiner A, Yofe I, Lara‐Astiaso D, Keren‐Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, Amit I (2016) Dissecting immune circuits by linking CRISPR‐pooled screens with single‐cell RNA‐seq. Cell 167: 1883–1896 e15 [DOI] [PubMed] [Google Scholar]
- 32. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A et al (2016) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA 113: E1738–E1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55: 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dantzer R (2001) Cytokine‐induced sickness behavior: mechanisms and implications. Ann N Y Acad Sci 933: 222–234 [DOI] [PubMed] [Google Scholar]
- 35. Bodea LG, Wang Y, Linnartz‐Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, Kaoma T, Muller A, Vallar L, Di Monte DA et al (2014) Neurodegeneration by activation of the microglial complement‐phagosome pathway. J Neurosci 34: 8546–8556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19: 987–991 [DOI] [PubMed] [Google Scholar]
- 37. Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V (1991) Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA 88: 7438–7442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M et al (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17: 797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P (2009) Characterization of the microglial phenotype under specific pro‐inflammatory and anti‐inflammatory conditions: effects of oligomeric and fibrillar amyloid‐beta. J Neuroimmunol 210: 3–12 [DOI] [PubMed] [Google Scholar]
- 40. Beutner C, Linnartz‐Gerlach B, Schmidt SV, Beyer M, Mallmann MR, Staratschek‐Jox A, Schultze JL, Neumann H (2013) Unique transcriptome signature of mouse microglia. Glia 61: 1429–1442 [DOI] [PubMed] [Google Scholar]
- 41. Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 9: 1512–1519 [DOI] [PubMed] [Google Scholar]
- 42. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F et al (2014) Environment drives selection and function of enhancers controlling tissue‐specific macrophage identities. Cell 159: 1327–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lavin Y, Winter D, Blecher‐Gonen R, David E, Keren‐Shaul H, Merad M, Jung S, Amit I (2014) Tissue‐resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159: 1312–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matcovitch‐Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben‐Yehuda H, David E, Zelada Gonzalez F, Perrin P et al (2016) Microglia development follows a stepwise program to regulate brain homeostasis. Science 353: aad8670 [DOI] [PubMed] [Google Scholar]
- 45. Deczkowska A, Matcovitch‐Natan O, Tsitsou‐Kampeli A, Ben‐Hamo S, Dvir‐Szternfeld R, Spinrad A, Singer O, David E, Winter DR, Smith LK et al (2017) Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN‐I‐dependent manner. Nat Commun 8: 717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Flanders KC, Ludecke G, Engels S, Cissel DS, Roberts AB, Kondaiah P, Lafyatis R, Sporn MB, Unsicker K (1991) Localization and actions of transforming growth factor‐beta s in the embryonic nervous system. Development 113: 183–191 [DOI] [PubMed] [Google Scholar]
- 47. Hamby ME, Hewett JA, Hewett SJ (2010) Smad3‐dependent signaling underlies the TGF‐beta1‐mediated enhancement in astrocytic iNOS expression. Glia 58: 1282–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H (1992) Transforming growth factor‐beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol 117: 395–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X, Chen W, Liu W, Wu J, Shao Y, Zhang X (2009) The role of thrombospondin‐1 and transforming growth factor‐beta after spinal cord injury in the rat. J Clin Neurosci 16: 818–821 [DOI] [PubMed] [Google Scholar]
- 50. Klempt ND, Sirimanne E, Gunn AJ, Klempt M, Singh K, Williams C, Gluckman PD (1992) Hypoxia‐ischemia induces transforming growth factor beta 1 mRNA in the infant rat brain. Brain Res Mol Brain Res 13: 93–101 [DOI] [PubMed] [Google Scholar]
- 51. Flanders KC, Ren RF, Lippa CF (1998) Transforming growth factor‐betas in neurodegenerative disease. Prog Neurobiol 54: 71–85 [DOI] [PubMed] [Google Scholar]
- 52. Dobolyi A, Vincze C, Pal G, Lovas G (2012) The neuroprotective functions of transforming growth factor beta proteins. Int J Mol Sci 13: 8219–8258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang G, Yu Y, Sun C, Liu T, Liang T, Zhan L, Lin X, Feng XH (2016) STAT3 selectively interacts with Smad3 to antagonize TGF‐beta. Oncogene 35: 4388–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carow B, Rottenberg ME (2014) SOCS3, a major regulator of infection and inflammation. Front Immunol 5: 58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma'ayan A (2016) The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016: baw100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Colonna M, Butovsky O (2017) Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 35: 441–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M et al (2015) MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single‐cell RNA sequencing data. Genome Biol 16: 278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang DW, Sherman BT, Lempicki RA (2008) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44 [DOI] [PubMed] [Google Scholar]
- 59. Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 109: 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takahashi K, Rochford CD, Neumann H (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells‐2. J Exp Med 201: 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zheng H, Jia L, Liu CC, Rong Z, Zhong L, Yang L, Chen XF, Fryer JD, Wang X, Zhang YW et al (2017) TREM2 promotes microglial survival by activating Wnt/beta‐Catenin pathway. J Neurosci 37: 1772–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, Edinger AL, Jung S, Rossner MJ, Simons M (2016) Age‐related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci 19: 995–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Streit WJ, Braak H, Xue QS, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol 118: 475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Neves J, Zhu J, Sousa‐Victor P, Konjikusic M, Riley R, Chew S, Qi Y, Jasper H, Lamba DA (2016) Immune modulation by MANF promotes tissue repair and regenerative success in the retina. Science 353: aaf3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xia M, Liu J, Wu X, Liu S, Li G, Han C, Song L, Li Z, Wang Q, Wang J et al (2013) Histone methyltransferase Ash1 l suppresses interleukin‐6 production and inflammatory autoimmune diseases by inducing the ubiquitin‐editing enzyme A20. Immunity 39: 470–481 [DOI] [PubMed] [Google Scholar]
- 67. Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, Natarajan KN, Reik W, Barahona M, Green AR et al (2017) SC3: consensus clustering of single‐cell RNA‐seq data. Nat Methods 14: 483–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qiu X, Hill A, Packer J, Lin D, Ma YA, Trapnell C (2017) Single‐cell mRNA quantification and differential analysis with Census. Nat Methods 14: 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Losciuto S, Dorban G, Gabel S, Gustin A, Hoenen C, Grandbarbe L, Heuschling P, Heurtaux T (2012) An efficient method to limit microglia‐dependent effects in astroglial cultures. J Neurosci Methods 207: 59–71 [DOI] [PubMed] [Google Scholar]
- 70. Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss‐Coray T, Mucke L, Mahley RW (1999) Expression of human apolipoprotein E3 or E4 in the brains of Apoe‐/‐ mice: isoform‐specific effects on neurodegeneration. J Neurosci 19: 4867–4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mazutis L, Gilbert J, Ung WL, Weitz DA, Griffiths AD, Heyman JA (2013) Single‐cell analysis and sorting using droplet‐based microfluidics. Nat Protoc 8: 870–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 57: 289–300 [Google Scholar]
- 73. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32: 381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Appendix
Review Process File
Data Availability Statement
Single‐cell RNA sequencing data have been deposited in Gene Expression Omnibus (GEO) database under the accession number GSE115571 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115571).