

Abstract
Background
Lipopolysaccharide (LPS) is generally associated with sepsis, which causes multiple system injuries and systemic inflammatory response. Mitochondrial DNA (mtDNA) is of great importance in mediation of inflammation. The aim of this study was to investigate the protective profiles of Cyclosporine-A (CsA) in LPS-induced acute lung injury (ALI) and systemic inflammation by the inhibition of mtDNA and Toll-like receptor.
Material/Methods
Twenty-four C57BL/6 mice were randomly assigned to 4 groups: a sham group (n=6); an experiment group (ALI induced through intraperitoneal injection of 10 mg/ml LPS, n=6); a low-CsA group (injection of 2.5 mg/kg of CsA 15 min after injection of LPS, n=6); and a high-CsA group (injection of 25 mg/kg of CsA 15 min after injection of LPS, n=6). Lung tissue, bronchoalveolar lavage fluid (BALF), and blood samples were collected at 6 h for further analyses.
Results
CsA treatment significantly attenuated LPS-induced lung histopathological changes (P<.05), myeloperoxidase (MPO) activity (P<.05) and lung wet-to-dry weight ratio (P<.05). In addition, injection of CsA decreased total cells (P<.05), neutrophils (P<.05), and total protein (P<.05) in BALF and inflammatory mediators, including tumor necrosis factor-α (TNF-α, P<.05) and interleukin-6 (IL-6, P<.05) in a dose-dependent manner. A significant decrease in mtDNA was observed in the CsA group when compared with controls (P<.05). Furthermore, we demonstrated that there was a significant difference between the high-CsA group and low-CsA group in lung injury score (P<.05), mtDNA (P<.05), and MPO (P<.05).
Conclusions
The evidence from this study suggests that CsA attenuated lung inflammation after LPS injection, and the protective mechanism may at least in part involve decreasing the release of inflammatory cytokines and mtDNA.
MeSH Keywords: Acute Lung Injury; Cyclosporine; DNA, Mitochondrial; Lipopolysaccharides
Background
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection [1]. It is a global public health concern. International data demonstrate that sepsis contributes to more than 1 million deaths annually and represents a significant financial burden to patients and society [2]. Lipopolysaccharide (LPS), endotoxins of gram-negative bacteria, are particularly inflammatory because they generate auto-amplificatory loops after activation of monocytes/macrophages, which cause overzealous production of inflammatory cytokines, leading to a cytokine storm and contributing to sepsis [3]. Development of organ dysfunction is the most important clinical event during sepsis, as it directly causes mortality and morbidity [4]. Over 40% of individuals with sepsis develop acute lung injury (ALI), a syndrome characterized by neutrophilic inflammation and pulmonary vascular hyperpermeability [5]. ALI leads to progressive loss of breathing capacity and hypoxemia, as well as pulmonary surfactant dysfunction, and is responsible for significant morbidity and mortality [6].
Mitochondria are double-membrane-bound organelles that are engaged in cellular energy production [7]. They are involved in numerous important cellular processes such as energy metabolism, the intrinsic apoptotic pathway, oxidative stress, and systemic inflammatory responses (SIRS) [8]. During stress and injury, certain molecules released from impaired mitochondria function as danger-associated molecular patterns (DAMPs) and recent data suggest that mitochondrial DNA (mtDNA) can also function as a DAMP in SIRS associated with acute trauma [9]. In addition, studies of both sepsis patients and septic animal models have indicated that circulating free mtDNA fragments are increased in response to pathogens [10,11].
Fragments of mtDNA are released from mitochondria upon opening of the mitochondrial permeability transition pore (mPTP) [12]. Sandrine Lemoine et al. [13] proved that Cyclosporine-A, an inhibitor of the mPTP, reduces opening of the mPTP and prevent the release of mtDNA through binding to cyclophilin D (cypD) in the model of ischemia-reperfusion. As a well-known immunosuppressant, CsA was demonstrated to suppress T cell activation and cytokine production, as well as allergen-induced lung inflammation [14]. However, to the best of our knowledge, no previous studies have investigated the effect of CsA on LPS-induced lung injury.
The aim of this study was to investigate inflammatory responses and lung injury in mice after lipopolysaccharide-induced sepsis with specific monitoring of plasma mtDNA concentration and other parameters of lung function. We also examined outcomes induced by different doses of CsA, which may suggest possible mechanisms. Our study presents a new pharmacological choice for the treatment of sepsis and the modulation of inflammation.
Material and Methods
Animals
Twenty-four C57BL/6 mice (male, adult, 18–25 g) were acclimatized to ventilated cages and environment before the experiments. All mice were fed standard chow with free access to water. The environment temperature was kept at 37°C under pathogen-free conditions. Mice were randomly assigned to either a sham group (n =6), an experiment group (n=6), a low-CsA group (n=6), or a high-CsA group (n=6). All the mice were fasted for 12 h before the experiment. This study was conducted in compliance with the Institutional Animal Care and Use Committee at West China Hospital, Sichuan University.
Acute lung injury model administration and experimental design
ALI was induced through intraperitoneal [IP] administration of 10 mg/kg lipopolysaccharide (LPS) 6 h before anesthesia. All the mice were deeply anesthetized by IP injection of 1% Pelltobarbitalum Natricum at the dose of 50 mg/kg and secured in a constructed template device. After 15 min, the 2 CsA groups were IP injected with corresponding volumes of CsA of 2.5 mg/kg in the low-CsA group and 25 mg/kg in the high-CsA group. At 24 h after LPS challenge, the mice were euthanized and plasma samples were collected.
Detection of mitochondrial DNA
Mouse plasma DNA was isolated using the DNeasy Blood and Tissue Kit (#69504, Qiagen, Hilden, Germany). Briefly, 50 μl of PBS was added to an equivalent volume of plasma sample and the mixture was centrifuged at 16 000 g for 15 min at 4°C. Then, 90 μl of the supernatant was retained, and the DNA was purified according to the manufacturer’s protocol. DNA was eluted in 200 μl of elution buffer. To determine the concentrations of plasma mtDNA in the samples, we used a SYBR-green dye-based reverse transcription polymerase chain reaction (RT-PCR) assay and a PRISM 7300 sequence detection system. mtDNA was detected using primers for the mouse NADH dehydrogenase 1 gene (forward: CGCCTGAC-CAATAGCCATAA; reverse: ATTCGACGTTA-AAGCCTGAGA). After serially diluting the mtDNA standards 10-fold, each sample was assayed for accurate quantification, and plasma mtDNA concentrations were converted to copy numbers through the DNA copy number calculator (http://cels.uri.edu/gsc/cndna.html; University of Rhode Island Genomics and Sequencing Center). The concentration of each sample of plasma mtDNA, defined as the number of copies per microliter of plasma, was calculated using the following formula:
where c means the concentration of plasma mtDNA (copies/μl), Q refers to the quantity of DNA measured by RT-PCR, VDNA is the total volume of extracted plasma DNA (200 μl in this study), VPCR represents the volume of extracted DNA used in the RT-PCR assay (1 μl in this study), and Vext is the initial volume of plasma (50 μl in this study).
Measurement of wet-to-dry ratio of the lungs
At 24 h after LPS challenge, the mice were euthanized and lungs were excised. We recorded the “wet” weight and the “dry” weight after placing lungs in an incubator at 80°C for 48 h.
Bronchoalveolar lavage
The lungs were washed in 5 ml of sterile phosphate-buffered saline (PBS) 3 times. The recovery ratio of the fluid was about 88±5%. Then, we immediately centrifuged the bronchoalveolar lavage fluid (BALF) at 3000 rpm for 10 min at 4°C and stored the cell-free supernatants at −80°C for cytokine analysis. We counted the total cells, neutrophils, and macrophages with hematoxylin and eosin (H&E)-stained smears after suspending the sediment cells in PBS, and total proteins in the BALF were determined using the Pierce™ BCA Protein Assay kit according to the manufacturer’s instructions.
Cytokine assay
We used ELISA kits to detect the concentrations of inflammatory mediators such as TNF-α and IL-6 in BALF following the kit instructions. The optical density of each well was set at 450 nm.
Myeloperoxidase assay in lung tissues
The activity of myeloperoxidase (MPO), as an indicator of polymorphonuclear leukocyte activation, was determined in the lung tissue by use of an MPO kit (Biolegend, Inc., San Diego, CA, USA). One hundred milligrams of lung tissue was homogenized and fluidized in extraction buffer to obtain 5% of the homogenate. We heated the sample, which consists of 0.9 ml homogenate and 0.1 ml of reaction buffer, to 37°C in a water bath for 15 min. Then, the enzymatic activity was determined by measuring the changes in absorbance at 460 nm using a 96-well plate reader.
Assessment of the lung injury
We excised the left lungs of mice to make slices, which were randomly submerged in containers filled with lung tissue and paraformaldehyde. We stained 5-μm tissue sections with hematoxylin and eosin (H&E) before the examination of 5 fields per slice under a microscope. Five slices were randomly selected from the container and examined and scored by 2 independent pathology specialists who were blind to the experimental groups. To determine the extent and severity of the lung tissue injury, a score from 0 to 4 was assigned for each of 3 different characteristics: (1) extension of leukocyte infiltration: 0=0%, 1=0–25%, 2=25–50%, 3=50–75%, and 4=75–100% of the section areas occupied by leukocytes; (2) amount of intra alveolar leukocytes: 0=no, 1=occasional, 2=several leukocytes in the alveoli, 3=alveoli almost full of leukocytes, and 4=alveoli distended by tightly packed leukocytes; and (3) amount of exudative debris, including fibrin, hyaline membranes, edema fluid, and meconium: 0=alveoli open, 1=exudate scarcely seen, 2=exudate clearly visible, 3=alveoli almost full of exudate, and 4=alveoli distended by exudate. The calculated total injury score is the sum of these scores, and an average score for each slice was calculated and documented.
Statistical analysis
All descriptive data are presented as the mean ±SD. Using the t test or one- or two-way analysis of variance together with Bonferroni post hoc tests for multiple testing correction, statistical significance was assessed. The relationship between continuous variables was assessed using the Pearson correlation coefficient test. Differences were considered significant at P<0.05.
Results
CsA attenuates the lung wet-to-dry (W/D) ratio
The lung wet-to-dry (W/D) ratio increase reflects the extent of pulmonary edema. At 24 h after injection of LPS, the lung W/D ratios were significantly higher compared with the control group (Figure 1, P<.05). CsA (2.5 and 25 mg/kg) significantly attenuated W/D ratios compared with the LPS group. There was significant difference of the W/D ratio in lung tissue between the control and low-CsA groups (P<.05), but no significant difference was found between the control and high-CsA group (P>.05) and between the low-CsA and high-CsA group.
Figure 1.
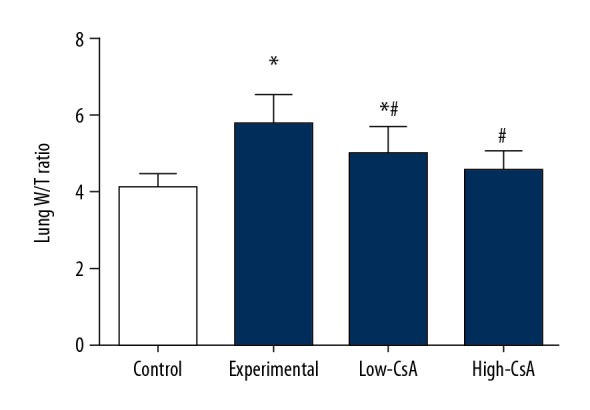
CsA Attenuates the Lung Wet-to-dry (W/D) Ratio. The lung wet-to-dry (W/D) ratio increase reflects pulmonary edema. CsA Significantly Attenuates the Lung Wet-to-dry Ratio. * P<0.05 vs. Control group, # P<0.05 vs. Experiment group. N=6.
CsA attenuates the LPS-induced myeloperoxidase
MPO activity was evaluated to indicate polymorphonuclear leukocyte accumulation in the lung. At 24 h after LPS injection, the MPO activity of lung tissues (Figure 2, P<.05) was significantly higher than in the control group. In the CsA (2.5 and 25 mg/kg) group, MPO activity was dramatically decreased compared with the LPS group (P<.05). There was a significant difference in MPO activity in lung tissues between the control and CsA groups (P<.05) and between the low CsA and high-CsA group (P>.05)
Figure 2.
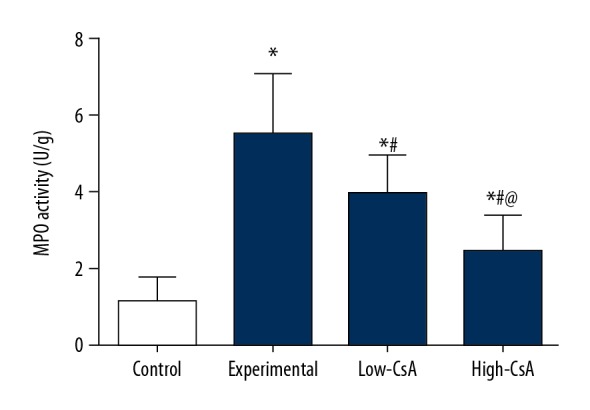
CsA Attenuates the LPS-Induced Myeloperoxidase. MPO activity was evaluated to indicate polymorphonuclear leukocyte accumulation in the lung. CsA significantly decreased the LPS-induced myeloperoxidase and there was a significant difference between the high- and low-CsA groups. * P<0.05 vs. Control group, # P<0.05 vs Experiment group, @ P<0.05 vs. Low-CsA group. N=6.
CsA Attenuates the LPS-induced lung injury
We used H&E staining and a lung injury score system to assess the pathological changes in lung tissues. In the control group, lung tissues (Figure 3A) showed a normal structure and no histopathological changes. The lung tissues from the experiment group (Figure 3B) demonstrated significant pathological alterations (P<.05). The high-CsA group (25 mg/kg, Figure 3D) had significantly attenuated destruction of lung structure and affected the lung injury score (Figure 3E, P<.05). However, no significant difference in lung injury scores was found between the low-CsA group (2.5 mg/kg, Figure 3C) and the experiment group (P>.05).
Figure 3.

CsA Attenuates the LPS-Induced Lung Injury. (A–D) Hematoxylin and eosin-stained images of lung tissues; (E) The lung injury score system was used to evaluate the severity of the lung injury. CsA significantly attenuated the LPS-induced lung injury score. * P<0.05 vs. Control group, # P<0.05 vs. Experiment group, @ P<0.05 vs. Low-CsA group. N=6.
CsA Decreases the LPS-induced induction of inflammatory cells and total proteins
There was an increase in the concentration of total protein and number of inflammatory cells in mice after LPS administration as compared to the control group. Both high- and low-CsA (2.5 and 25 mg/kg) significantly decreased the concentration of total cells (Figure 4A, P<.05) and the number of neutrophils (Figure 4B, P<.05) compared to those in the LPS group. The high-CsA group showed a significantly lower concentration of total cells (P<.05) and number of neutrophils (P<.05) compared to the low-CsA group. The high-CsA (25 mg/kg) group had a significantly decreased concentration of total protein (Figure 4C, P<.05). There was no significant difference in concentration of total protein in BALF between the low-CsA group and the experiment group (P>.05).
Figure 4.

CsA Decreased the LPS-Induced Induction of Inflammatory Cells and Total Proteins. (A–C) The total numbers of cell/neutrophils and the total protein were all significantly higher in the experiment group than in the control group. CsA significantly decreased the total number of cell/neutrophils in a dose-dependent manner. * P<0.05 vs. Control group, # P<0.05 vs. Experiment group, @ P<0.05 vs. Low-CsA group. N=6.
CsA Attenuates the LPS-Induced Mitochondrial DNA and Inflammatory Cytokines
To examine the possible anti-inflammatory mechanism of CsA, we assessed the concentration of mtDNA in plasma samples. As shown in Figure 5A, mice in the LPS group displayed significantly higher mtDNA concentrations compared with the control group. After the injection of CsA (2.5 mg/kg and 25 mg/kg), the concentration decreased significantly in a dose-dependent manner. To determine how CsA affects the LPS-induced cytokine production, we measured the concentration of TNF-α and IL-6 in BALF by ELISA. As illustrated in Figure 5, TNF-α and IL-6 levels were significantly higher in the experiment group than in the control group. CsA (2.5 and 25 mg/kg) efficiently reduced the production of TNF-α and IL-6 compared to those in the LPS group (Figure 5B, 5C, P<.05).
Figure 5.

CsA Attenuates the LPS-Induced Mitochondrial DNA and Inflammatory Cytokines. (A–C) Plasma interleukin-6, tumor necrosis factor-α, and mitochondrial DNA levels were significant higher in the experiment group than in the control group. CsA significantly attenuated the LPS-induced TNF-α, IL-6, and mitochondrial DNA in a dose-dependent manner. * P<0.05 vs. Control group, # P<0.05 vs. Experiment group, @ P<0.05 vs. Low-CsA group. N=6.
Discussion
In this study we demonstrated the protective role of CsA in LPS-induced ALI in mice. Administration of CsA attenuated lung inflammation after LPS injection, as demonstrated by decreased elevation of lung W/D weight ratio, total cells, neutrophils, macrophages, and MPO activity, together with reduced lung histological damage. To the best of our knowledge, this is the first study to confirm that CsA significantly decreases the pro-inflammatory cytokines in LPS-induced ALI in mice.
Mitochondria are essential for cell life and affect multiple cellular processes, including energy metabolism, calcium homeostasis, and signal transduction [15]. Mitochondria are both a major source of oxidants and a target for their damaging effects, which contribute greatly to cell apoptosis [16]. A previous study [17] has demonstrated the possible sites at which immunomodulatory interventions could be targeted in sepsis, which inhibits mitochondria respiration and induces apoptosis. According to current opinion, the mPTP is a protein channel that spans the 2 layers of mitochondria [18], composed of an outer-membrane voltage-dependent anion channel, the inner-membrane ANT, and CypD [19,20]. In unstressed cells, the mPTP is closed and has selective membrane permeability. When under apoptotic stress, massive ion influx occurs with sudden opening of the mPTP and the collapse of mitochondrial membrane potential [21]. Moreover, the mitochondria swell and depleat ATP, and there is a transient increase in reactive oxygen species (ROS) generation and the level of mtDNA [22]. Release of mtDNA into the circulation due to injury activates the neutrophil p38 MAPK signaling pathway via TLR9 and contributes to the development of post-traumatic SIRS [23]. Therefore, we conclude that mtDNA is a pro-inflammatory signal and new therapeutic approaches may be developed to prevent the release of mtDNA for inhibition of inflammation and post-traumatic injury.
Many studies [13,24,25] have found that Cyclosporine-A, an immunosuppresive agent, decreases the concentration of mtDNA since it can inhibit mitochondrial permeability transition pore (mPTP) opening. The mechanism is probably associated with inhibiting cyclophilin D (CypD) through dissociation of CypD from adenine nucleotide translocase (ANT), which is localized in the inner membrane, and acts as a structural component of the mPTP [19,25,26]. A protective effect of CsA through inhibiting the release of mtDNA has been observed in experimental ischemia-reperfusion in the heart [27], liver [28], brain [29], and kidneys [13], as well as in clinical acute myocardial infarction [29]. Gentaro Ikeda et al. [30] demonstrated that nanoparticle-mediated targeting of CsA enhances cardioprotection against ischemia-reperfusion injury through inhibition of mitochondrial permeability transition pore opening. Therefore, the present study aimed to test the hypothesis that CsA can alleviate LPS-induced ALI through preventing the release of mtDNA.
In the present study, sepsis was induced by LPS injection. The inflammatory consequences of sepsis are particularly apparent in the lungs [5]. Acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS), are important causes of morbidity and mortality in critically ill patients [31]. Damage to epithelial and endothelial cells is of great importance in the development of ALI/ARDS, such as overproduction of cytokines, increased vascular permeability, leukocyte recruitment, and dysfunction of surfactant, leading to interstitial and alveolar pulmonary edema, alveolar collapse, and hypoxemia [32]. In our study, we used lung W/D weight ratio, total cell count, neutrophil count, macrophage count, and MPO activity, together with lung histological damage score, to evaluate the level of ALI. In this study, we demonstrated that CsA ameliorates lung inflammation in LPS-induced ALI and that the inhibition of inflammation is correlated with inhibition of mtDNA release by CsA during sepsis.
Conclusions
The present study provides evidence that the CsA exerts protective effects on LPS-induced ALI in a dose-dependent manner by inhibiting mitochondrial damage and mtDNA release. While preliminary, our study suggests that CsA attenuates the LPS-induced inflammation, at least in part, through reducing the permeability of the mitochondrial membrane, which provides a new pharmacological target and novel agent for reducing LPS-induced ALI.
Footnotes
Source of support: The present study was funded by the National Natural Science Foundation of China (No. 81170288, 81700410) and the Chengdu Science and Technology Project (No. 2015-HM01-00587-SF)
Conflict of interests
None.
References
- 1.Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315(8):801–10. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tillmann B, Wunsch H. Epidemiology and outcomes. Crit Care Clin. 2018;34(1):15–27. doi: 10.1016/j.ccc.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Cavaillon JM. Exotoxins and endotoxins: Inducers of inflammatory cytokines. Toxicon. 2017;149:45–53. doi: 10.1016/j.toxicon.2017.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Pool R, Gomez H, Kellum JA. Mechanisms of organ dysfunction in sepsis. Crit Care Clin. 2018;34(1):63–80. doi: 10.1016/j.ccc.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt EP, Yang Y, Janssen WJ, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217–23. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su CF, Kao SJ, Chen HI. Acute respiratory distress syndrome and lung injury: Pathogenetic mechanism and therapeutic implication. World J Crit Care Med. 2012;1(2):50–60. doi: 10.5492/wjccm.v1.i2.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wojtczak L, Zablocki K. [Mitochondria in cell life, death and disease]. Postepy Biochem. 2008;54(2):129–41. [in Polish] [PubMed] [Google Scholar]
- 8.Zhang L, Deng S, Zhao S, et al. Intra-peritoneal administration of mitochondrial DNA provokes acute lung injury and systemic inflammation via toll-like receptor 9. Int J Mol Sci. 2016;17(9):1–16. doi: 10.3390/ijms17091425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krysko DV, Agostinis P, Krysko O, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32(4):157–64. doi: 10.1016/j.it.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pyle A, Burn DJ, Gordon C, et al. Fall in circulating mononuclear cell mitochondrial DNA content in human sepsis. Intensive Care Med. 2010;36(6):956–62. doi: 10.1007/s00134-010-1823-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patrushev M, Kasymov V, Patrusheva V, et al. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol LifeSci. 2004;61(24):3100–3. doi: 10.1007/s00018-004-4424-1. [DOI] [PubMed] [Google Scholar]
- 13.Lemoine S, Pillot B, Rognant N, et al. Postconditioning with cyclosporine a reduces early renal dysfunction by inhibiting mitochondrial permeability transition. Transplantation. 2015;99(4):717–23. doi: 10.1097/TP.0000000000000530. [DOI] [PubMed] [Google Scholar]
- 14.Kudo F, Ikutani M, Iseki M, Takaki S. Cyclosporin A indirectly attenuates activation of group 2 innate lymphoid cells in papain-induced lung inflammation. Cell Immunol. 2017;323:33–40. doi: 10.1016/j.cellimm.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Yu W, Zhang X, Liu J, et al. Cyclosporine A suppressed glucose oxidase induced P53 mitochondrial translocation and hepatic cell apoptosis through blocking mitochondrial permeability transition. Int J Biol Sci. 2016;12(2):198–209. doi: 10.7150/ijbs.13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fakharnia F, Khodagholi F, Dargahi L, Ahmadiani A. Prevention of cyclophilin D-mediated mPTP opening using cyclosporine-A alleviates the elevation of necroptosis, autophagy and apoptosis-related markers following global cerebral ischemia-reperfusion. J Mol Neurosci. 2017;61(1):52–60. doi: 10.1007/s12031-016-0843-3. [DOI] [PubMed] [Google Scholar]
- 17.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–49. [PMC free article] [PubMed] [Google Scholar]
- 18.Griffiths EJ, Halestrap AP. Further evidence that cyclosporin A protects mitochondria from calcium overload by inhibiting a matrix peptidyl-prolyl cis-trans isomerase. Implications for the immunosuppressive and toxic effects of cyclosporin. BiochemicalJ. 1991;274(Pt 2):611–14. doi: 10.1042/bj2740611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Argaud L, Gateau-Roesch O, Raisky O, et al. Postconditioning inhibits mitochondrial permeability transition. Circulation. 2005;111(2):194–97. doi: 10.1161/01.CIR.0000151290.04952.3B. [DOI] [PubMed] [Google Scholar]
- 20.Leducq N, Delmas-Beauvieux MC, Bourdel-Marchasson I, et al. Mitochondrial and energetic dysfunctions of the liver during normothermic reperfusion: Protective effect of cyclosporine and role of the mitochondrial permeability transition pore. Transplant Proc. 2000;32(2):479–80. doi: 10.1016/s0041-1345(00)00822-8. [DOI] [PubMed] [Google Scholar]
- 21.Ichas F, Jouaville LS, Sidash SS, et al. Mitochondrial calcium spiking: A transduction mechanism based on calcium-induced permeability transition involved in cell calcium signalling. FEBS Lett. 1994;348(2):211–15. doi: 10.1016/0014-5793(94)00615-6. [DOI] [PubMed] [Google Scholar]
- 22.Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359(5):473–81. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 23.Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Mitochondria in control of cell fate. Circ Res. 2012;110(4):526–29. doi: 10.1161/RES.0b013e31824ae5c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sousa CA, Soares EV. Mitochondria are the main source and one of the targets of Pb (lead)-induced oxidative stress in the yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2014;98(11):5153–60. doi: 10.1007/s00253-014-5631-9. [DOI] [PubMed] [Google Scholar]
- 25.Lemaout C, Gonzalez H, Aboab J, Annane D. [Pathophysiology of septic shock]. Press Med. 2006;35(3 Pt 2):521–27. doi: 10.1016/s0755-4982(06)74628-0. [in French] [DOI] [PubMed] [Google Scholar]
- 26.Bernardi P, Forte M. The mitochondrial permeability transition pore. Novartis Foundation symposium. 2007;287:157–64. discussion 164–69. [PubMed] [Google Scholar]
- 27.Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: Another view. Biochimie. 2002;84(2–3):153–66. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- 28.Halestrap AP, Richardson AP. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol. 2015;78:129–41. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 29.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion – a target for cardioprotection. Cardiovasc Res. 2004;61(3):372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 32.Tomashefski JF., Jr Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med. 2000;21(3):435–66. doi: 10.1016/s0272-5231(05)70158-1. [DOI] [PubMed] [Google Scholar]