

Abstract
The study of protein conformations using molecular dynamics (MD) simulations has been in place for decades. A major contribution to the structural stability and native conformation of a protein is made by the primary sequence and disulfide bonds formed during the folding process. Here, we investigated μ-conotoxins GIIIA, KIIIA, PIIIA, SIIIA, and SmIIIA as model peptides possessing three disulfide bonds. Their NMR structures were used for MD simulations in a novel approach studying the conformations between the folded and the unfolded states by systematically breaking the distinct disulfide bonds and monitoring the conformational stability of the peptides. As an outcome, the use of a combination of the existing knowledge and results from the simulations to classify the studied peptides within the extreme models of disulfide folding pathways, namely the bovine pancreatic trypsin inhibitor pathway and the hirudin pathway, is demonstrated. Recommendations for the design and synthesis of cysteine-rich peptides with a reduced number of disulfide bonds conclude the study.
Introduction
Conotoxins are neuropeptides from the venom of marine cone snails, which interact with a wide range of biological targets (e.g., ion channels, transmembrane receptors, and transporters) and hence are of pharmaceutical interest and of great potential as molecular probes to study the specific subtypes of ion channels and receptors.1,2 Conotoxins consist of approximately 10–50 amino acid residues and are classified according to their cysteine patterns.3,4 The typical CC–C–C–CC pattern defines the framework for conotoxins of the M-superfamily comprised by ψ-, μ-, and κM-conotoxins.3,5,6 The family of the 27 currently known μ-conotoxins4,7 selectively binds to the ion channel pore of the voltage-gated sodium channels, thus blocking the influx of sodium ions into the cell. Therefore, these peptides also gained interest as useful tools for research studies in electrophysiology.100−9 μ-Conotoxins are cysteine-rich peptides consisting of 6 cysteines which can give rise to 15 conformational isomers of different disulfide connectivities in various combinations of disulfide bonds.9 However, the dominant isomer among them bears the disulfide linkage of C1–C4, C2–C5, and C3–C8, often referred to as the “native fold”.9 The molecular principles underlying the folding bias contributing to one particular dominant isomer is as yet unclear. This subject is of great importance because an accumulation of cysteines may also occur in distinct regions of larger peptides and proteins such as that observed in defensins,10,11 resistins,12 Kunitz serine protease inhibitors,13 and various growth factors.12,14,15 In such cases, any information on their preferred disulfide connectivity would be relevant especially in the absence of actual chemical or structural data.
Conotoxins represent a promising tool for studying the impact of disulfide bonds on the folding process owing to their small to medium size which makes them intermediates between peptides and proteins and also their high disulfide bond content.3,5,16 We studied the μ-conotoxins GIIIA, KIIIA, PIIIA, SIIIA, and SmIIIA (Figure 1) using unbiased all-atom molecular dynamics (MD) simulations performed on their NMR structures. The simulation output was correlated with the in vitro data from the oxidation reaction for all the μ-conotoxins that had been described earlier.100−9 The individual μ-conotoxins could be grouped based on the reaction product yield and the side product formation,17 which allowed for a functional comparison with the computational study. For the MD studies, a rather unconventional approach was pursued by analyzing the process of refolding; that is, the behavior and stability of an individual peptide was observed upon successive opening of disulfide bonds in the folded peptide NMR structures. The analyses conducted on the resulting MD trajectories gave rise to inferences on characteristic factors contributing to the conformational stability of the folded conopeptides. On the other hand, we used the distances sampled by the sulfur atoms between the reduced cysteine residues to observe and determine if the peptide with a disulfide bond removed in silico tries to refold or not. This enabled drawing a distinction between the influence of the disulfide bond and the properties exerted by the rest of the sequence to the maintenance of close-to-native backbone conformations in peptides with computationally reduced disulfide bonds. This consequently simulates the state at which the peptide is at its final stage of folding and in its near-native conformation.
Figure 1.

Structure and surface representations of the investigated μ-conotoxins. (A) NMR structures of the five μ-conotoxins (GIIIA, KIIIA, PIIIA, SIIIA, and SmIIIA) used in this study represented as a cartoon. The secondary structure elements, α-helix (purple), 3–10 helix (blue), turn (cyan), and coil (white) were generated by STRIDE19 in visual molecular dynamics (VMD). The cysteine residues forming the disulfide bonds (yellow) were labeled. (B) Molecular surface was generated by SURF20 in VMD, indicating the hydrophobic (white), basic (blue), acidic (red), and hydrophilic (green) regions. All structures were taken from the ConoServer database.7,21
The pathways for disulfide folding have been classified into two extreme models despite exhibiting a high degree of diversity.18 One model is represented by the bovine pancreatic trypsin inhibitor (BPTI)-like folding, where there is a predominance of native intermediates at various steps down the folding funnel, and the other extreme, the hirudin pathway, is defined by highly heterogeneous non-native intermediates. Interestingly, conotoxins are so far placed in between these two extreme models in a hybrid BPTI–hirudin model.18 In the present study, we thus try to improve the clarity regarding the classification of the peptides used herein considering the existing models. Finally, an attempt to relate the simulation inferences from these peptides with open disulfide bonds to their propensity (or) favorability to retain close-to-native conformations is presented via a qualitative grouping.
Results and Discussion
Studies of conotoxin folding include, in general, experimental approaches such as regioselective oxidation strategies, spontaneous oxidative self-folding, and optimization of the folding methods tested, recombinant expression, and so far available information from the biosynthesis of conotoxins in the venom duct of cone snails. However, it is still unresolved how exactly the cone snails produce properly folded peptide and protein toxins. In order to approach the mechanisms behind conotoxin folding, several studies focused on using computational strategies to provide valuable insights into the structural features important for the folding process, but often a combination of both experimental and theoretical work is missing or only one conotoxin was in the focus of most of these studies.
Oxidative Self-Folding of μ-Conotoxins
In order to provide the in-house experimental data for comparison with the computational results, equal amounts of each of the selected five μ-conotoxins (Figure 1, Table 1) were used for the oxidation reaction according to a protocol earlier described yielding an undirected and spontaneous formation of disulfide bonds.100−9 Under the conditions applied, oxidation of μ-SmIIIA and μ-PIIIA resulted in several peaks of fully oxidized product(s) as confirmed by mass spectrometry; that is, the disulfide connectivity of the individual fractions is different from the native (major) fold.9 In contrast, oxidation of all other peptides, that is, μ-GIIIA, μ-KIIIA, and μ-SIIIA, resulted in one major product confirming earlier reports (Figure 2).17,22,23
Table 1. Comparison of Sequence Characteristics of the μ-Conotoxins Investigated in This Studya.

Residues are highlighted according to their character: basic (blue), acidic (red), polar uncharged (green), and cysteine (yellow) (Z: pyroglutamate, O: 4-hydroxyproline). *All peptides were used as amides. Native μ-SmIIIA occurs as the C-terminal acid, however, is usually used as an amide.100,8 We used μ-SmIIIA as an amide for reasons of comparison because both structures were found to be identical.30#In general, the IC50 values determined for the toxins ion channel blocking activity at the skeletal muscle NaV1.4 expressed in Xenopus oocytes are given, with the exception of SmIIIA where only KD is available.
Figure 2.

High-performance liquid chromatography (HPLC) elution profiles of μ-conotoxins studied. Linear, reduced precursors of μ-conotoxins (A) and folded crude mixtures after 1 h (B), with the main product marked with an asterisk. Respective electrospray ionization (ESI) mass spectra of the oxidized peptides are shown in (C).
The formation of one major product for μ-GIIIA and μ-SIIIA can be explained by a rapid collapse into the favored native fold with the disulfide connectivity C1–C4/C2–C5/C3–C6 as introduced earlier.17 For μ-KIIIA, such a rapid collapse also results in one main product; however, the connectivity C1–C5/C2–C4/C3–C6 is preferred here, and the pattern C1–C4/C2–C5/C3–C6 is only present as a minor fraction. Small differences in the elution profiles of buffer-oxidized μ-KIIIA (the crude product), that is, product formation, might result from the differences in batch size and composition of the oxidation buffer compared to the results of Khoo et al.25 In the case of μ-PIIIA and μ-SmIIIA, a different folding mechanism indicative of a slower rearrangement results in the formation of several isomers. This can be attributed to more diverse noncovalent interactions and electrostatic forces compared to μ-GIIIA and μ-SIIIA. Here, it was suggested that the native isomer accumulates via reshuffling of disulfide bonds during the folding process and is dependent on the thermodynamic stability of the isomer formed.17,31 Although μ-SIIIA and μ-SmIIIA have a high sequence similarity (Table 1), the higher number of basic residues in μ-SmIIIA (six Arg) might cause the formation of multiple isomers, whereas in case of μ-SIIIA (two Arg and one Lys), only one isomer is preferred.17,32 μ-PIIIA forms multiple isomers possibly for the same reason.9 In contrast, the structure of μ-GIIIA tolerates a high number of basic residues and forms only one major product compared to that of μ-PIIIA and μ-SmIIIA.17 Several aspects influencing the folding, such as the number of hydroxyproline residues, the loop size between the two linked cysteines, or amidation of the C-terminus, are discussed controversially in the literature without a clear preference indicating the uniqueness of each sequence and the respective biological activity.17,28,33,34
Conformational Analysis Using Molecular Dynamics
The solution NMR structures used herein as initial conformations for starting MD simulations were derived from the ConoServer database (Figure 1, Table 1).4,7 Comparison, alignment, and structural differences of the μ-conotoxin structures were discussed previously by Yao et al. and Tietze et al.9,28 All μ-conotoxins, except μ-KIIIA, possess conserved structures that align significantly better in the C-terminal part compared to the N-terminal region.28 A high similarity was seen for the backbone conformations between the loop 2 and loop 3 regions of μ-KIIIA, μ-SIIIA, and μ-SmIIIA, which have a higher selectivity for blocking NaV1.2 over NaV1.4 channels (Table 1). On the other hand, μ-GIIIA and μ-PIIIA, which prefer NaV1.4 over NaV1.2, superimpose well in the second loop between C2 and C5.9 Besides contributing to the structural rigidity, the disulfide bridges cause cysteine residues to form a hydrophobic core, enveloped by other charged and hydrophilic residues (Figure 1). This hydrophobic effect plays a key role in the stability of the native fold.35
With three disulfide bridges present, there are six ways to open them one by one. The order of disulfide bond formation during the synthesis (and also in vivo) is not known, but as mentioned before, the process appears to be guided by thermodynamic aspects. Regarding the simulation strategy, it was decided to open the longest disulfide bridge first (bridging the longest sequence in between, see Table S1) as it is expected to instantly introduce the highest level of flexibility into the peptide backbone. The intention for this opening strategy was to increase the conformational entropy of the reduced version. In the case of μ-KIIIA (non-native connectivity), the shortest disulfide bridge was opened first. This would serve as a means to gauge the effect of loop size in retaining a stable structure. The loops in between the three disulfide bonds differ in size (Table S1) and thus needed to be considered in the analysis process.
It was observed during the simulations at room temperature that all peptides retained their initial conformation as demonstrated by the root-mean-square deviation (RMSD) of Cα atoms compared to the chosen starting NMR structure of each peptide (Table 1) with μ-KIIIA possessing the lowest RMSD of 1.1 Å and μ-SmIIIA the highest of 2.7 Å (Figure S1, Table S2). The RMSD of the Cα atoms, the root mean square of fluctuation (RMSF) of all atoms of each residue, and the radius of gyration (Rg) of the whole protein were computed from each simulation for all of the five peptides (Figure S1, Table S2).
The native structure of μ-GIIIA displayed a hydrophobic core formed by the cysteines and a salt bridge between R1 and D12. As the first disulfide bond C2–C5 was removed, the distance between the now unbound cysteines increased moderately. However, this did not affect the overall three-dimensional conformation of the backbone proved by only a 0.3 Å increase in the backbone RMSD, but it could be observed that its RMSD progression is inconsistent in the simulation especially between 30 and 50 ns. The 300 ns simulation showed that the peptide had a stable backbone indicated by a relatively unwavering RMSD curve between 100 and 300 ns of simulation time as shown in Figure 3.
Figure 3.

RMSD plots of disulfide bond opened versions of the five μ-conotoxins. The comparison of backbone stability between the peptides with the C2–C5 disulfide bond removed (black) and both the C2–C5 and C3–C6 bridges removed (red) between 100 and 300 ns of simulation time: (A) μ-GIIIA, (B) μ-KIIIA, (C) μ-PIIIA, (D) μ-SIIIA, and (E) μ-SmIIIA. Above the plots is a representation of two cases of disulfide connectivity discussed. Here, red represents the version with the single C1–C4 disulfide bond and black represents the C1–C4/C3–C6 disulfide connectivity.
Importantly, the residues significant for bioactivity experienced none to a very minimal increase in fluctuations (Figure 3, Table S4). Upon subsequent removal of the second disulfide bridge between C3–C6, an obvious stretch in the overall shape of the peptide was observed (Figure S3). The peptide had an excessive flexibility and adopted close to completely unfolded conformations during different intervals in the simulation (Figure S3). Interestingly, the cysteines forming the C3–C6 bond moved much closer to each other than the C2–C5 cysteine residues in the two disulfide bond opened structure. This is shown by the decrease in the RMSD of the C2–C5 and C3–C6 removed peptide between 60 and 70 ns (Figure S3). In longer time scales (300 ns), it was observed that the RMSD values dropped close to the ones with just one disulfide bond (C2–C5) removed, indicating that μ-GIIIA tends to fold back to retain its preferred native conformation (Figure 3). The Rg of μ-GIIIA followed an almost identical pattern of progression to the RMSD, peaking between 40 and 65 ns before falling back toward its initial values, representing an unfolding-(re)folding event. The fluctuations of the residues K8, K11, R13, K16, and R19, which were reported to be responsible for ion channel binding,3 did not show a significant change compared to the structure with all disulfide bonds intact (Table S3). With two disulfide bridges removed, the C-terminus including R19 and K16 showed a significant increase in fluctuations. It has been reported that K16 has a low priority for biological activity. Moreover, the exchange of this residue increased the binding affinity compared to the native toxin.36 Our findings suggest that if the C2–C5 disulfide bond alone was removed, the structure may adopt a conformation still representing a structure close to the native fold.
μ-KIIIA has the lowest RMSD of 1.1 Å on average with respect to the chosen starting structure among the five peptides in their natively folded form (Table S2). In 100 ns of simulation time, the removal of the C2–C4 disulfide bond, thereby altering the C4 residue, results in the loss of its helix and increases the RMSD by 0.7 Å. However, at the 300 ns time scale, conformations sampled by μ-KIIIA showed the reappearance of its native helix (Figure 5). The remaining C1–C5 and C3–C6 disulfide bonds were sufficient to retain the backbone stability and conformation, respectively, of μ-KIIIA. The progression of RMSD and Rg for μ-KIIIA followed the same scheme as observed for μ-GIIIA, that is, with the structure possessing two reduced disulfides revealing the largest variation in the conformational flexibility. It was observed from the 300 ns simulations that unlike μ-GIIIA, the two disulfide-deficient versions of μ-KIIIA did not regain the backbone stability of its one disulfide bond removed counterpart as shown in Figure 3.
Figure 5.
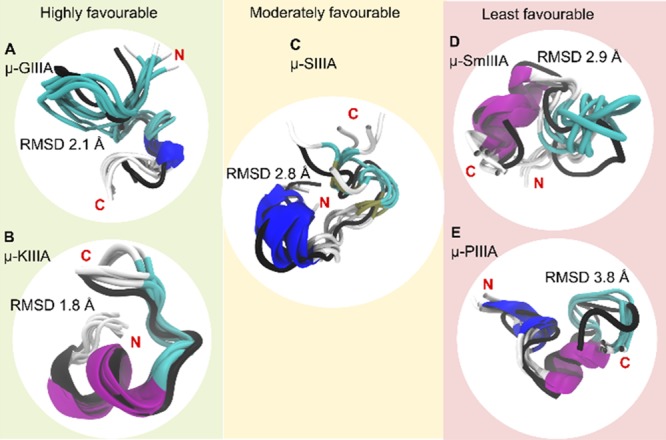
Grouping of the peptides based on the favorability of two disulfide bond stability. The C2–C5 disulfide-deficient conformations (cartoon representations colored to distinguish secondary structural elements) of the studied μ-conotoxins (A) GIIIA, (B) KIIIA, (C) SIIIA, (D) SmIIIA, and (E) PIIIA superimposed on their energy-minimized native structure (black cartoon representation). From the 300 ns trajectory, five conformations (one every 60 ns) have been used. The average RMSD of these conformations in comparison to the reference native structure is shown in Å. Besides displaying the regions of similarity and dissimilarity between the native and the C2–C5-deficient versions, the figure also provides a grouping for the five peptides in terms of favorability of the disulfide-deficient version retaining structural characteristics of the native peptide (based on RMSD, RMSF, and Rg).
Although the average RMSD of native μ-PIIIA was a decent 2.3 Å compared to the selected starting structure over the course of the simulation, the backbone was constantly subjected to changes as can be seen from the RMSD plot (Figure S1). The removal of the C2–C5 disulfide bond resulted in a lesser fluctuating RMSD progression, although it came at the expense of a 1.5 Å increase in RMSD in the first 100 ns of simulation time. Conformations sampled by this peptide between 100 and 300 ns of simulation time showed the reappearance of its native helices (α-helix between O8 and S14 and 3_10 helix between L3 and C5). The structure of μ-PIIIA with both the C2–C5 and C3–C6 disulfide bonds removed showed the largest extent of structural variation among the five conopeptides with an average RMSD of 4.9 Å (Figure S5). The peptide did not tend to refold within 300 ns simulation time. With one disulfide bond removed, none of its functionally significant residues showed a pronounced increase in fluctuations, the highest of which was a 1.3 Å increase for R14 compared to the natively folded peptides. Unlike μ-GIIIA, the two disulfide-deficient versions of μ-PIIIA did not regain the backbone stability of its one disulfide bond removed counterpart. Meanwhile, the single disulfide-deficient version of μ-PIIIA adopted a very stable conformation, with the RMSD curve almost flat lining between 200 and 300 ns of the 300 ns simulation (Figure S3).
In contrast, almost all residues of μ-SIIIA displayed marginally higher RMSF values for the one and two bond-removed structures in comparison to the native structure containing all the three disulfide bonds. In the one disulfide-deficient version of μ-SIIIA, although the marginal increase in residue fluctuations was observed, the functionally significant restudies W12, R14, and H16 revealed only a minimal increase in the overall residue fluctuations. Another key residue R18, however, showed a larger mobility between the native and one disulfide-deficient versions. More importantly, the one disulfide-deficient version of μ-SIIIA retained its α-helical motif between K11 and H16, which is a significant aspect in terms of targeting sodium channels.37
We focused further on μ-GIIIA that forms a single oxidation product and μ-SmIIIA that forms multiple oxidation products during the synthesis to illustrate the phenomenon described above (Figure S3). The overall RMSD between the native connectivity and the structure with one disulfide bond removed was observed to be low in μ-GIIIA (2.1 Å) and high in μ-SmIIIA (2.9 Å) among the five peptides. Figure S3 shows average conformations for all the three 100 ns simulations of μ-GIIIA and μ-SmIIIA compared with their corresponding RMSD plots. An interesting observation from both peptides with an opened disulfide bond was the formation of new secondary structure elements that were not present in the native state. The structure of C2–C5-deficient μ-GIIIA achieved a reasonable equilibration between 200 and 300 ns of the simulation, and the inspection of the trajectory revealed that the conformations sampled by this peptide had a 3_10 helix between K16 and Q18 (Figure 3). In μ-SmIIIA with one opened disulfide bond, an α-helix was formed between residues R13 and H18 (Figure 2).
The stability of this helix through the entire course of the simulation can be accounted for by a combination of hydrogen bond formation and the presence of the bonded C15 in the center of the helix. In comparison with the native fold, the structure with the C2–C5 bond opened appeared well ordered. The distribution of the hydrogen bonds around the helix can be seen in Figure 4.
Figure 4.
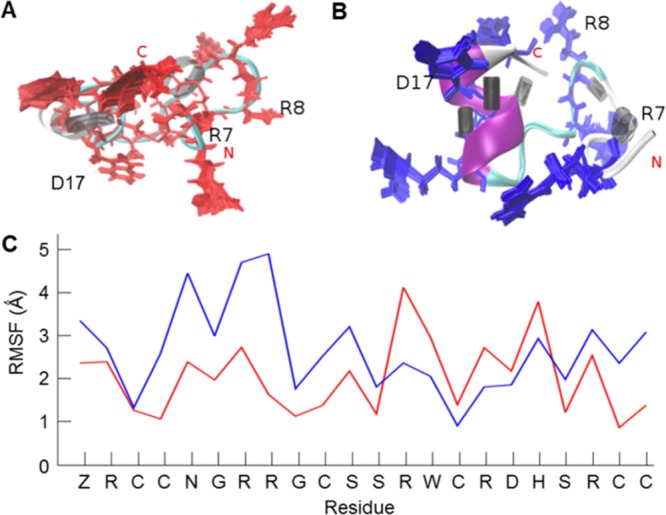
Comparison of μ-SmIIIA native fold and the structure with one disulfide bond opened. (A) Structure of the native peptide (completely oxidized, three disulfide bonds) with 100 conformations of its basic residues (red) superimposed with three residues marked as important for binding activity. The distribution of hydrogen bonds shows a sparse black area which indicates that the region surrounding it is relatively flexible. (B) Structure of the peptide containing one opened disulfide bond (C2–C5) showing (blue) its well-formed α-helix and the dense well-ordered hydrogen bonding illustrated as black cylinders. Higher rigidity inducing an improved structural stability of the peptide in (B) in comparison to the fully oxidized peptide in (A) is apparent from the reference RMSF plot (C). Despite the rigidity of the backbone, the orientations of the basic residues differ from the native structure.
The μ-SmIIIA structure with two disulfide bonds reduced formed a less stable 3_10 helix between residues R16 and H18, increasing the flexibility of the conformation. However, the functionality of a conotoxin is dependent on a stable backbone structure coupled with the favorable orientation of basic side chain residues for binding to their target and not solely on the flexibility of a distinct part of the peptide.3 In μ-SmIIIA, though the 3_10 helix formation reduced the Rg of the peptide, the overall peptide structure drifted significantly from that of the native state, and the orientations adopted by the side chains of its basic residues varied largely when compared to either the native or the structure with one disulfide bond opened. Even with a single disulfide bond removed, the functionally significant R7 showed a moderate increase in fluctuations (Figure 4, Table S4).
The 300 ns simulations for the disulfide-deficient species of both μ-SIIIA and μ-SmIIIA revealed similar trends based on the observed RMSD progression (Figure 3). In the C2–C5 disulfide removed version, between the two, μ-SmIIIA had a marginally more stable backbone than μ-SIIIA. However, with the C3–C6 disulfide bond also removed, both peptides had equally unstable backbones that did not show signs of refolding. Herewith, it is demonstrated how the individual disulfide bridges and the residues that occur within the loops affect the conformational stability on the basis of observed fluctuations in the backbone and the side chain residues.
A closer means of observing the folding behavior of the peptides in this study is to track the movement of the opened cysteine residues in the simulation. By tracking the distances spanned by the Sγ atoms of the cysteines, the tendency of the peptide to fold back to its original conformation or to explore completely new conformations can be identified. This behavior reflects on the underlying folding model that the peptide prefers to adopt. On the basis of this idea, we were able to find a clear correlation between the observations from the synthesis (Figure 2) and simulation (Figure 6). The peptides that preferred forming distinct main products (μ-GIIIA and μ-KIIIA) in the synthesis (Figure 2) also exhibited their preference to fold back to their original conformations as seen from the C2–C5 Sγ distance profiles in Figure 6. This is an indicator to the preference of the BPTI-like folding pathway. On the other hand, μ-PIIIA and μ-SmIIIA that formed a mixture of products in the synthesis (Figure 2) preferred moving away from their original conformations and exploring new conformations as indicated by the C2–C5 Sγ distance profiles in Figure 6. This reflects on the Hirudin-like folding pathway preferred by these peptides. The behavior of μ-SIIIA could be placed between these two extreme cases. The videos from the MD simulations where the distances between the opened CYS residues were tracked are provided for the cases representing the opposite extremes, namely, μ-GIIIA and μ-PIIIA are provided as a part of the Supporting Information.
Figure 6.

Relation between the unbound cysteine distances and the underlying folding pathway. Top: the distances between the Sγ atoms of the cysteine residues from the peptides with a single disulfide opened plotted over the 300 ns simulation for (A) μ-GIIIA, (B) μ-KIIIA, (C) μ-PIIIA, (D) μ-SIIIA, and (E) μ-SmIIIA. Bottom: a schematic representation of the placement of the five μ-conotoxins within the established BPTI-like and hirudin-like models.18 The NMR structures of hirudin (the gray cartoon) and BPTI (the blue cartoon) with their three disulfide bridges shown as yellow sticks. This classification is based on the observations from this study.
Conclusions
The folding of smaller disulfide-rich peptides and oligopeptides is a less well-understood folding event because of a much higher degree of flexibility and often a lower extent of structure-forming elements. MD simulations are increasingly used to assist the experimental work for understanding and predicting the folding process. Additionally, they have been routinely used in structure–activity relationship studies, drug discovery, and design pipelines.38−42 Previous studies using the five μ-conotoxins investigated herein gave insights into the folding and binding modes adopted by these peptides.9,25,28,32,36,43 Simulating the complete oxidative folding pathway following the formation of non-native disulfide intermediates until the native disulfide bonds are formed as reported by previous studies44,45 using coarse-grained models is not within the scope of this study. Our work, however, aimed at determining how a particular disulfide bond contributes to the stability of the peptide. Consequently, this approach reviews the validity of the logic that the removal of a disulfide bridge, that is, herein C2–C5, represents a reduction of the backbone stability when considering RMSD and RMSF values.
With the C2–C5 disulfide bond removed, only μ-SmIIIA revealed a noticeable increase in the average fluctuations of its functionally significant residues. In addition, the fact that secondary structural elements such as α-helices were formed in some peptides containing only two disulfide bonds suggests that in distinct cases (e.g., μ-GIIIA), a greater structural rigidity of the backbone may be achieved if one disulfide bridge is removed. This helix-induced stability while strengthening the backbone might reduce the extent of overall fluctuations of the basic residues responsible for binding activity. The biological activity and selectivity of disulfide-deficient mutants might differ from the native conformation as shown for μ-GIIIA recently. Fifteen different disulfide isomers are possible for a peptide containing six cysteines, and still three different isomers (ribbon, bead, and globular) might occur in case of four cysteines. It has, however, not been mentioned in the report by Han et al.46 which isomer of the μ-GIIIA analogs has been tested because the structural characterization of the respective products was not performed. Apart from the reports on μ-GIIIA regarding the disulfide-deficient variants, another study by Khoo et al.47 provides an insight into the removal of disulfide bridge C1–C9 in μ-KIIIA, resulting in only a minimal change in the biological activity against NaV1.2 and NaV1.4. In contrast, there are no experimental data for the disulfide-deficient species of μ-conotoxins PIIIA, SIIIA, and SmIIIA available so far.
With respect to drug design and synthesis, the simplification to two disulfide bonds would be a clear benefit for disulfide-rich peptides and proteins. A similar study by Yu et al. on α-conotoxin cVc1.1 complements our idea of the reduction of the number of disulfide bonds.49 In this respect, we can conclude from our MD simulations that two disulfide bridges could be sufficient to maintain a stable backbone for the majority of the μ-conotoxins studied. However, it is important that the deficient structure is sufficiently supported by at least one pair of cross-linked disulfide bridges that span to almost either ends of the sequence. From the results obtained, a rank order of the five peptides can be provided: μ-GIIIA and μ-KIIIA fall in the highly favorable category, μ-SIIIA falls in the moderately favorable group, and μ-PIIIA and μ-SmIIIA fall into the least favorable group (Figure 5). We also conclude that the C3–C6 disulfide bridge plays the greatest role in retaining the backbone stability for the current strategy of disulfide bond removal employed.
As expected, the removal of two disulfide bridges led to an increased backbone flexibility, the formation of a series of intermediate conformations, and a less stable peptide. The increased fluctuations of basic side chain residues responsible for the interactions with the sodium ion channels are in this case unlikely to stay in favorable orientations for binding. Furthermore, the formation of different disulfide isomers for μ-KIIIA, μ-PIIIA, and μ-SmIIIA in the experimental self-folding approach indicates a difference in their folding behavior,9,17,25,29 which cannot be explained unambiguously by simulations and disulfide removal. Finally, the observations of the structural stability of the backbone observed and the extent to which the peptide tries to fold back gives us clues on which disulfide folding model a particular peptide tends to likely prefer. It must be said that this distinction is still not completely in black and white but can be ranked or ordered relative to each other between the extreme models (BPTI and hirudin). Our attempt to classify the peptides between the BPTI and hirudin folding models is illustrated in Figure 6. The correlation of observations from the experiment and simulation as discussed earlier in the results adds further validity to this classification proposed in Figures 5 and 6. Thus, we have been able to demonstrate the usefulness of molecular simulations in applications beyond the observation of the structural behavior of a peptide in solution to being used as a tool for the generalized assignment of peptides to established folding models.
Materials and Methods
MD Simulations
Disulfide bonds were systematically removed to yield a partially folded conformation as the starting structure for the simulation. From an atomic perspective, this translates to changing the bonded cysteines to nonbonded cysteines by protonating the sulfur atoms. This step was done by pdb2gmx program within the GROMACS 5.1.4 package.50,51 This approach induces the least changes in the coordinate file to create a disulfide-deficient species as opposed to the usually performed mutation studies where the Cys residues are replaced by the Ala or Ser residues. It further eliminates the errors arising from the manual manipulation of the coordinate file. Four out of five μ-conotoxins in this study possess the native disulfide connectivity (C1–C4/C2–C5/C3–C6), whereas μ-KIIIA adopts a C1–C5/C2–C4/C3–C6 connection as the stable conformation.25 The following structures were used herein: PDB ID 1TCG (μ-GIIIA),22 PDB ID 2LXG (μ-KIIIA),25 S00159 (μ-PIIIA),9 BMRB 20025 (μ-SIIIA),28 and PDB ID 1Q2J (μ-SmIIIA).29 The NMR ensembles of μ-GIIIA, μ-KIIIA, μ-SIIIA, and μ-SmIIIA had 20 structures and μ-PIIIA had 15 structures in the respective PDB file. The first model was selected as the best representative structure except for SmIIIA, whereas model 13 was chosen as mentioned in the PDB file as the best model for this peptide.
GROMACS 5.1.450,51 was used for all the MD simulations in this study. An individual peptide was placed in the center of a cubic box that evolved to a final volume of 2.5 × 2.5 × 2.5 nm3. TIP3P52 water model was used as the solvent to fill the box. Appropriate amounts of Cl– ions were added to balance the positive charge of the μ-conotoxins. Simulations were run using the AMBER99SB-ILDN53 force field, which was chosen based on its better agreement with the NMR data and an accurate modeling of helical proteins in comparative studies.54−56 In the process of preparing the peptide for the production MD simulation, energy minimization simulations were with 10 000 steps of the steepest descents minimization protocol and convergence reached when the maximum force on any atom is no greater than 100 kJ/mol/nm. A thermal equilibration in the NVT ensemble at 300 K using the velocity-rescaling modified Berendsen thermostat57 and a constant pressure equilibration in the NPT ensemble using the Parrinello–Rahman barostat58,59 at 1 atm were carried out for 20 ns each, prior to production MD. During both the temperature and the pressure ensemble simulations, positional restraints on the peptides were applied using the LINCS60 algorithm. Each peptide was subjected to three production runs, and on each run, preprocessing and equilibration were performed independently. First, the conformation with all the three disulfide bonds was considered as the control simulation. In the second simulation, the link between C2–C5 (C2–C4 for KIIIA) was removed leaving the two other disulfide bonds intact. In the third simulation, both the C2–C5 (C2–C4 for KIIIA) and the C3–C6 disulfide bonds were removed, leaving the peptide constrained originally by three disulfide bonds now supported by a single disulfide bonds. The production MD was done for 100 ns for all the peptides, and for instances with the opening of a single or two disulfide bonds, a total of 300 ns of simulations were carried out by running an extended 200 ns simulation from the final checkpoint of the 100 ns trajectory. The extended 200 ns simulations were conducted to allow sufficient sampling for the observation of events from the refolding process such as the possibility of the unbound cysteines coming close to each other. All simulations were conducted with a 2 fs time step and data written to the logs and trajectory at every 5 ps. Periodic boundary conditions were applied to the system. Long-range electrostatics were accounted by the particle mesh Ewald method.61,62 For every 100 ns of simulation, 20 000 frames were written to the trajectory. The effect of periodic boundary conditions was adjusted by the suppression of center of mass movement from the trajectory prior to analysis. Visualizations of conformations for the analysis and creation of images were performed using VMD.63 The RMSD, RMSF, and Rg plots were created using the program Grace (version 5.1.25), whereas the distances between the unbound cysteines were plotted using the tools within VMD. The RMSD curves were plotted for every 10 ps (10 000 frames), whereas the distance curves were plotted for every 5 ps (20 000 frames).
Chemical Synthesis and Purification of μ-Conotoxins
Peptides were produced by an automated solid-phase peptide synthesis using a standard Fmoc (N-(9-fluorenyl) methoxycarbonyl)-protocol and an EPS 221 peptide synthesizer (Intavis Bioanalytical Instruments AG, Cologne, Germany) as described earlier and purified by preparative reversed-phase (RP) HPLC (Shimadzu LC-8A system, Duisburg, Germany). The gradient used was 0–50% eluent B in 120 min with 0.1% trifluoroacetic acid (TFA) in water (eluent A) and 0.1% TFA in acetonitrile/water (9:1) (eluent B) on a C18 column (Knauer Eurosphere 100, Berlin, Germany) with the dimensions 50 mm × 300 mm (5 mm particle size, 100 Å pore size). Reduced and oxidized peptides were analyzed on a LC–ESI micrOTOF-Q III mass spectrometer (Bruker Daltonics GmbH, Bremen, Germany) coupled with Dionex Ultimate 3000 (Thermo Scientific, Dreieich, Germany) equipped with a EC100/2 Nucleoshell RP18 Gravity 2.7 μm column (Macherey-Nagel, Düren, Germany). Analysis of the MS data was performed using Bruker Compass Data Analysis 4.1. The LC conditions used were as follows: eluent A was water with 0.1% acetic acid, whereas eluent B was acetonitrile containing 0.1% acetic acid. A gradient of 0–60% of eluent B in 12 min was used, and detection was at 220 nm.
Oxidation of Reduced μ-Conotoxin Precursors
Oxidative folding of the linear μ-conotoxins GIIIA, KIIIA, PIIIA, SIIIA, and SmIIIA in a buffer system containing redox agents was performed as described earlier. Each μ-conotoxin (1 mg) was subjected to oxidation, and fractions of the reaction mixture were monitored over time by RP HPLC using a Shimadzu LC-10AT system (Duisburg, Germany) equipped with a C18 column (Vydac 218TP54, Worms, Germany, 4.6 mm × 25 mm, 5 mm particle size, 300 Å pore size) and the gradient 0–60% eluent B in 60 min with 0.1% TFA in water (eluent A) and 0.1% TFA in acetonitrile (eluent B). Reaction control was performed over a time period of 24 h, and oxidation was stopped by adding 1% TFA in water. Monitoring revealed that the oxidation reactions were completed within the first 60 min of the reaction time. The fractions were collected for each peptide and subjected to LC–ESI mass spectrometry for the confirmation of the molar mass corresponding to the oxidized products.
Acknowledgments
The authors would like to thank Veda Thota for technical assistance. Financial support by the University of Bonn (to D.I.) is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01465.
RMSD, RMSF, and Rg of the five μ-conotoxins; backbone stability of the five μ-conotoxins with three, two, and one disulfide bridge; backbone conformation of μ-GIIIA and μ-SmIIIA and helix formation; backbone conformation of μ-PIIIA and loss of helix by disulfide bond removal; comparison of disulfide loop length of the five μ-conotoxins; assessment of the stability using RMSD and Rg for 100 ns MD simulation ; and impact of the C2–C5 and C3–C6 disulfide bridge on the backbone RMSF and the functionally stable residues (PDF)
Dynamics of the peptide μ-GIIIA with the C2–C5 disulfide bond removed (AVI)
Dynamics of the peptide μ-PIIIA with the C2–C5 disulfide bond removed (AVI)
Author Present Address
# Bachem AG, Hauptstraße 144, 4416 Bubendorf (Switzerland).
Author Contributions
A.A.P.G., P.H., and D.I. conceived the presented idea and designed and planned the experimental studies. P.H. performed the synthesis, purification, and analytical characterization of the peptides. A.A.P.G., A.M., J.H., M.H.-A., and A.B. developed the theory. A.A.P.G. performed the computations. All authors discussed the results and contributed to the final manuscript.
The authors declare no competing financial interest.
This paper was published ASAP on October 1, 2018, with errors in the references. The corrected paper was reposted on October 4, 2018.
Supplementary Material
References
- Hille B.The Superfamily of Voltage-gated Channels. Ion Channel Excitable Membranes, 3rd ed.; Sinauer Associates, Inc.: Sunderland, Massachusetts, 2001; pp 1–92. [Google Scholar]
- Cruz L. J.; Gray W. R.; Yoshikami D.; Olivera B. M. Conus Venoms: A Rich Source of Neuroactive Peptides. J. Toxicol., Toxin Rev. 1985, 4, 107–132. 10.3109/15569548509014416. [DOI] [Google Scholar]
- Akondi K. B.; Muttenthaler M.; Dutertre S.; Kaas Q.; Craik D. J.; Lewis R. J.; Alewood P. F. Discovery , Synthesis, and Structure – Activity Relationships of Conotoxins. Chem. Rev. 2014, 114, 5815–5847. 10.1021/cr400401e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas Q.; Yu R.; Jin A.-H.; Dutertre S.; Craik D. J. ConoServer: Updated Content, Knowledge, and Discovery Tools in the Conopeptide Database. Nucleic Acids Res. 2012, 40, D325–D330. 10.1093/nar/gkr886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipold E.; DeBie H.; Zorn S.; Adolfo B.; Olivera B. M.; Terlau H.; Heinemann S. H. μO-Conotoxins Inhibit NaVChannels by Interfering with their Voltage Sensors in Domain-2. Channels 2007, 1, 253–262. 10.4161/chan.4847. [DOI] [PubMed] [Google Scholar]
- Jacob R. B.; McDougal O. M. The M-Superfamily of Conotoxins: A Review. Cell. Mol. Life Sci. 2010, 67, 17–27. 10.1007/s00018-009-0125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas Q.; Westermann J.-C.; Halai R.; Wang C. K. L.; Craik D. J. ConoServer, a Database for Conopeptide Sequences and Structures. Bioinformatics 2008, 24, 445–446. 10.1093/bioinformatics/btm596. [DOI] [PubMed] [Google Scholar]
- Heimer P.; Tietze A. A.; Böhm M.; Giernoth R.; Kuchenbuch A.; Stark A.; Leipold E.; Heinemann S. H.; Kandt C.; Imhof D. Application of Room Temperature Aprotic and Protic Ionic Liquids for Oxidative Folding of Cysteine-Rich Peptides. ChemBioChem 2014, 15 (18), 2754–2765. 10.1002/cbic.201402356. [DOI] [PubMed] [Google Scholar]
- Tietze D.; Leipold E.; Heimer P.; Böhm M.; Winschel W.; Imhof D.; Heinemann S. H.; Tietze A. A. Molecular interaction of δ-conopeptide EVIA with voltage-gated Na+ channels. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860, 2053–2063. 10.1016/j.bbagen.2016.06.013. [DOI] [PubMed] [Google Scholar]
- Tietze A. A.; Tietze D.; Ohlenschläger O.; Leipold E.; Ullrich F.; Kühl T.; Mischo A.; Buntkowsky G.; Görlach M.; Heinemann S. H.; et al. Structurally Diverse μ-Conotoxin PIIIA Isomers Block Sodium Channel NaV1.4. Angew. Chem., Int. Ed. 2012, 51, 4058–4061. 10.1002/anie.201107011. [DOI] [PubMed] [Google Scholar]
- Winter J.; Wenghoefer M. Human Defensins: Potential Tools for Clinical Applications. Polymers 2012, 4, 691–709. 10.3390/polym4010691. [DOI] [Google Scholar]
- Szyk A.; Wu Z.; Tucker K.; Yang D.; Lu W.; Lubkowski J. Crystal structures of human α-defensins HNP4, HD5, and HD6. Protein Sci. 2006, 15, 2749–2760. 10.1110/ps.062336606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons M. S.; Bell B.; Stainier D.; Peters K. G. Isolation of the zebrafish homologues for the tie1 and tie2 endothelium-specific receptor tyrosine kinases. Dev. Dyn. 1998, 212, 133–140. . [DOI] [PubMed] [Google Scholar]
- Ranasinghe S.; McManus D. P. Structure and Function of Invertebrate Kunitz Serine Protease Inhibitors. Dev. Comp. Immunol. 2013, 39, 219–227. 10.1016/j.dci.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Barnham K. J.; Torres A. M.; Norton R. S.; Alewood D.; Alewood P. F.; Domagala T.; Nice E. C. Role of the 6-20 Disulfide Bridge in the Structure and Activity of Epidermal Growth Factor. Protein Sci. 1998, 7, 1738–1749. 10.1002/pro.5560070808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christinger H. W.; Fuh G.; de Vos A. M.; Wiesmann C. The Crystal Structure of Placental Growth Factor in Complex with Domain 2 of Vascular Endothelial Growth Factor Receptor-1. J. Biol. Chem. 2004, 279, 10382–10388. 10.1074/jbc.m313237200. [DOI] [PubMed] [Google Scholar]
- Góngora-Benítez M.; Tulla-Puche J.; Albericio F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- Fuller E.; Green B. R.; Catlin P.; Buczek O.; Nielsen J. S.; Olivera B. M.; Bulaj G. Oxidative Folding of Conotoxins Sharing an Identical Disulfide Bridging Framework. FEBS J. 2005, 272, 1727–1738. 10.1111/j.1742-4658.2005.04602.x. [DOI] [PubMed] [Google Scholar]
- Chang J.-Y. Diverse Pathways of Oxidative Folding of Disulfide Proteins: Underlying Causes and Folding Models. Biochemistry 2011, 50, 3414–3431. 10.1021/bi200131j. [DOI] [PubMed] [Google Scholar]
- Frishman D.; Argos P. Knowledge-Based Protein Secondary Structure Assignment. Proteins 1995, 23, 566–579. 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- Varshney A.; Brooks F. P.; Wright W. V. Computing Smooth Molecular Surfaces. IEEE Comput. Graph. Appl. Mag. 1994, 14, 19–25. 10.1109/38.310720. [DOI] [Google Scholar]
- Kaas Q.; Yu R.; Jin A.-H.; Dutertre S.; Craik D. J. ConoServer: Updated Content, Knowledge, and Discovery Tools in the Conopeptide Database. Nucleic Acids Res. 2012, 40, D325–D330. 10.1093/nar/gkr886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu K.; Kohda D.; Hatanaka H.; Lancelin J. M.; Ishida Y.; Oya M.; Nakamura H.; Inagaki F.; Sato K. Structure-activity relationships of .mu.-conotoxin GIIIA: structure determination of active and inactive sodium channel blocker peptides by NMR and simulated annealing calculations. Biochemistry 1992, 31, 12577–12584. 10.1021/bi00165a006. [DOI] [PubMed] [Google Scholar]
- Patel D.; Mahdavi S.; Kuyucak S. Computational Study of Binding of μ-Conotoxin GIIIA to Bacterial Sodium Channels NaVAb and NaVRh. Biochemistry 2016, 55, 1929–1938. 10.1021/acs.biochem.5b01324. [DOI] [PubMed] [Google Scholar]
- Wilson M. J.; Yoshikami D.; Azam L.; Gajewiak J.; Olivera B. M.; Bulaj G.; Zhang M.-M. -Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10302–10307. 10.1073/pnas.1107027108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo K. K.; Gupta K.; Green B. R.; Zhang M.-M.; Watkins M.; Olivera B. M.; Balaram P.; Yoshikami D.; Bulaj G.; Norton R. S. Distinct Disulfide Isomers of μ-Conotoxins KIIIA and KIIIB Block Voltage-Gated Sodium Channels. Biochemistry 2012, 51, 9826–9835. 10.1021/bi301256s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.-M.; Green B. R.; Catlin P.; Fiedler B.; Azam L.; Chadwick A.; Terlau H.; McArthur J. R.; French R. J.; Gulyas J.; et al. Structure/Function Characterization of μ-Conotoxin KIIIA, an Analgesic, Nearly Irreversible Blocker of Mammalian Neuronal Sodium Channels. J. Biol. Chem. 2007, 282, 30699–30706. 10.1074/jbc.m704616200. [DOI] [PubMed] [Google Scholar]
- Tietze A. A.; Tietze D.; Ohlenschläger O.; Leipold E.; Ullrich F.; Kühl T.; Mischo A.; Buntkowsky G.; Görlach M.; Heinemann S. H.; et al. Structurally Diverse μ-Conotoxin PIIIA Isomers Block Sodium Channel NaV1.4. Angew. Chem., Int. Ed. 2012, 51, 4058–4061. 10.1002/anie.201107011. [DOI] [PubMed] [Google Scholar]
- Yao S.; Zhang M.-M.; Yoshikami D.; Azam L.; Olivera B. M.; Bulaj G.; Norton R. S. Structure, Dynamics, and Selectivity of the Sodium Channel Blocker μ-Conotoxin SIIIA. Biochemistry 2008, 47, 10940–10949. 10.1021/bi801010u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keizer D. W.; West P. J.; Lee E. F.; Yoshikami D.; Olivera B. M.; Bulaj G.; Norton R. S. Structural Basis for Tetrodotoxin-resistant Sodium Channel Binding by μ-Conotoxin SmIIIA. J. Biol. Chem. 2003, 278, 46805–46813. 10.1074/jbc.m309222200. [DOI] [PubMed] [Google Scholar]
- Fuller E.; Green B. R.; Catlin P.; Buczek O.; Nielsen J. S.; Olivera B. M.; Bulaj G. Oxidative Folding of Conotoxins Sharing an Identical Disulfide Bridging Framework. FEBS J. 2005, 272, 1727–1738. 10.1111/j.1742-4658.2005.04602.x. [DOI] [PubMed] [Google Scholar]
- Walewska A.; Skalicky J. J.; Davis D. R.; Zhang M.-M.; Lopez-Vera E.; Watkins M.; Han T. S.; Yoshikami D.; Olivera B. M.; Bulaj G. NMR-Based Mapping of Disulfide Bridges in Cysteine-Rich Peptides: Application to the μ-Conotoxin SxIIIA. J. Am. Chem. Soc. 2008, 130, 14280–14286. 10.1021/ja804303p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Vera E.; Walewska A.; Skalicky J. J.; Olivera B. M.; Bulaj G. Role of Hydroxyprolines in the in Vitro Oxidative Folding and Biological Activity of Conotoxins. Biochemistry 2008, 47, 1741–1751. 10.1021/bi701934m. [DOI] [PubMed] [Google Scholar]
- Bulaj G.; Olivera B. M. Folding of Conotoxins: Formation of the Native Disulfide Bridges during Chemical Synthesis and Biosynthesis of Conus Peptides. Antioxid. Redox Signaling 2008, 10, 141–156. 10.1089/ars.2007.1856. [DOI] [PubMed] [Google Scholar]
- Kang T. S.; Kini R. M. Structural Determinants of Protein Folding. Cell. Mol. Life Sci. 2009, 66, 2341–2361. 10.1007/s00018-009-0023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace C. N.; Shirley B. A.; McNutt M.; Gajiwala K. Forces Contributing to the Conformational Stability of Proteins. FASEB J. 1996, 10, 75–83. 10.1096/fasebj.10.1.8566551. [DOI] [PubMed] [Google Scholar]
- Choudhary G.; Aliste M. P.; Tieleman D. P.; French R. J.; Dudley S. C. Jr. Docking of μ-Conotoxin GIIIA in the Sodium Channel Outer Vestibule. Channels 2007, 1, 344–352. 10.4161/chan.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder C. I.; Ekberg J.; Nielsen K. J.; Adams D.; Loughnan M. L.; Thomas L.; Adams D. J.; Alewood P. F.; Lewis R. J. Neuronally Selective μ-Conotoxins from Conus striatus Utilize an α-Helical Motif to Target Mammalian Sodium Channels. J. Biol. Chem. 2008, 283, 21621–21628. 10.1074/jbc.m802852200. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 71. 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Caflisch A. Molecular Dynamics in Drug Design. Eur. J. Med. Chem. 2015, 91, 4–14. 10.1016/j.ejmech.2014.08.004. [DOI] [PubMed] [Google Scholar]
- De Vivo M.; Masetti M.; Bottegoni G.; Cavalli A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- Nair P. C.; Miners J. O. Molecular Dynamics Simulations: From Structure Function Relationships to Drug Discovery. In Silico Pharmacol. 2014, 2, 1–4. 10.1186/s40203-014-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salsbury F. R. Molecular Dynamics Simulations of Protein Dynamics and Their Relevance to Drug Discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipold E.; Markgraf R.; Miloslavina A.; Kijas M.; Schirmeyer J.; Imhof D.; Heinemann S. H. Molecular determinants for the subtype specificity of μ-conotoxin SIIIA targeting neuronal voltage-gated sodium channels. Neuropharmacology 2011, 61, 105–111. 10.1016/j.neuropharm.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Qin M.; Wang W.; Thirumalai D. Protein Folding Guides Disulfide Bond Formation. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 11241–11246. 10.1073/pnas.1503909112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchio M.; Czaplewski C.; Liwo A.; Ołdziej S.; Scheraga H. A. Dynamic Formation and Breaking of Disulfide Bonds in Molecular Dynamics Simulations with the UNRES Force Field. J. Chem. Theory Comput. 2007, 3, 1236–1248. 10.1021/ct7000842. [DOI] [PubMed] [Google Scholar]
- Han P.; Wang K.; Dai X.; Cao Y.; Liu S.; Jiang H.; Fan C.; Wu W.; Chen J. The Role of Individual Disulfide Bonds of μ-Conotoxin GIIIA in the Inhibition of Nav1.4. Mar. Drugs 2016, 14, 213. 10.3390/md14110213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo K. K.; Feng Z.-P.; Smith B. J.; Zhang M.-M.; Yoshikami D.; Olivera B. M.; Bulaj G.; Norton R. S. Structure of the Analgesic μ-Conotoxin KIIIA and Effects on the Structure and Function of Disulfide Deletion. Biochemistry 2009, 48, 1210–1219. 10.1021/bi801998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R.; Seymour V. A. L.; Berecki G.; Jia X.; Akcan M.; Adams D. J.; Kaas Q.; Craik D. J. Less Is More: Design of a Highly Stable Disulfide-Deleted Mutant of Analgesic Cyclic α-Conotoxin Vc1.1. Sci. Rep. 2015, 5, 13264. 10.1038/srep13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins: Struct., Funct., Bioinf. 2010, 78, 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Maragakis P.; Piana S.; Eastwood M. P.; Dror R. O.; Shaw D. E. Systematic Validation of Protein Force Fields against Experimental Data. PLoS One 2012, 7, e32131 10.1371/journal.pone.0032131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafeim A.-P.; Salamanos G.; Patapati K. K.; Glykos N. M. Sensitivity of Folding Molecular Dynamics Simulations to Even Minor Force Field Changes. J. Chem. Inf. Model. 2016, 56, 2035–2041. 10.1021/acs.jcim.6b00493. [DOI] [PubMed] [Google Scholar]
- Piana S.; Lindorff-Larsen K.; Shaw D. E. How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization?. Biophys. J. 2011, 100, L47–L49. 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Nosé S.; Klein M. L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. 10.1080/00268978300102851. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. . [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.